

Abstract
To investigate the evolutionary history of the current pandemic outbreak of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), a total of 137 genomes of coronavirus strains with release dates between January 2019 and 25 March 2020, were analyzed. To investigate the potential intermediate host of the SARS‐CoV‐2, we analyzed spike glycoprotein sequences from different animals, with particular emphasis on bats. We performed phylogenetic analysis and structural reconstruction of the spike glycoproteins with subsequent alignment and comparison. Our phylogenetic results revealed that SARS‐CoV‐2 was more similar to the bats' betacoronavirus isolates: HKU5‐related from Pipistrellus abramus and HKU4‐related from Tylonycteris pachypus. We also identified a yak betacoronavirus strain, YAK/HY24/CH/2017, as the closest match in the comparison of the structural models of spike glycoproteins. Interestingly, a set of unique features has been described for this particular strain of the yak betacoronavirus. Therefore, our results suggest that the human SARS‐CoV‐2, responsible for the current outbreak of COVID‐19, could also come from yak as an intermediate host.
Keywords: betacoronavirus, COVID‐19, SARS‐CoV‐2, spike glycoprotein
Highlights
Spike glycoprotein sequences analysis of the set of recent corona viruses compared to SARS‐CoV‐2.
Phylogenetic and structural analysis of the spike glycoproteins sequences.
Phylogenetic study shows close clustering of SARS‐CoV‐2 and bats' coronavirus sequences.
Structures comparison suggests yak (Bos grunniens) as an intermediate host for the SARS‐CoV‐2.
The N‐terminal domain binds to carcinoembryonic antigen‐related cell adhesion molecule 1 (CEACAM1) in mouse hepatitis coronavirus, and binds sugar in porcine transmissible gastroenteritis virus.
1. INTRODUCTION
The novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), is associated with the current pandemic outbreak of COVID‐19. The virus emerged in Wuhan (China) and it can be transmitted from person to person. 1 The patients visited a local seafood market selling various live animals, from where this zoonotic disease was suspected to have spread. 2 Till date, there have been 1 812 734 confirmed cases and 113 675 deaths across 202 countries according to WHO (https://www.who.int/).
Successful drug and vaccine development require a deep understanding of the virus phylogeny, evolutionary origin, and the source of zoonotic transmission. Also, it should be a top‐priority task for researchers to prevent outbreaks of this type in the future. Substantial efforts have been made to identify the animal source of SARS‐CoV‐2. Several recent studies have revealed the origin of SARS‐CoV‐2 from bats 3 ; however, some other intermediate hosts were also suggested such as snake, 4 pangolin, 5 or some mammals and birds. 6
The genome of SARS‐CoV‐2 contains two open reading frames (ORFs), ORF1 and ORF2 encoding two polyproteins, which are responsible for viral genome maintenance after cleavage. There is also a set of so‐called structural proteins such as spike glycoproteins, an envelope protein, membrane proteins, and the nucleocapsid. One of the structural proteins, the spike surface glycoprotein (“spike glycoprotein”), is one of the primary therapeutic targets. This protein plays an important role in binding to receptors on the host cell, fusion of the host and viral membranes, and as a target for antibodies (reviewed in Fung and Liu 7 ).
In this study, we examined 137 genomes to establish the relationship between spike glycoproteins from different coronaviruses. This was done with a combination of the phylogenetic tree analysis (reconstructed from the translated sequences) and a comparison of the structural models. The phylogenetic study has confirmed close relatedness of SARS‐CoV‐2 to bats' coronaviruses. In contrast, a comparison of the structural models has found yak (Bos grunniens) betacoronavirus as the closest match.
2. METHODS
2.1. Sequence retrieval and phylogenetic analysis
Recently released complete genome sequences were downloaded for the analysis from NCBI (listed in Table S1). Furthermore, spike glycoprotein ORFs (nucleotide and protein sequences) were retrieved from NCBI ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/) and conserved domains were checked with CD‐Search (NCBI), respectively. Complete translated ORFs were used for multiple sequence alignments performed using MUSCLE. The tests of substitution models and phylogenetic analysis were carried out using the MEGA X software. 8 The neighbor‐joining method and JTT substitution models were selected assuming an estimated proportion of invariant sites and 4‐gamma‐distributed rate categories to account for rate heterogeneity across sites. The gamma shape parameter was estimated directly from the data. Reliability for the internal branch was assessed using the bootstrapping method (1000 bootstrap replicates). Two spike glycoproteins from the gammacoronaviruses were used as an outgroup.
2.2. Structure modeling and comparison
Structural models of the spike glycoproteins (Table S1, marked with *) were built using SWISS‐MODEL. 9 Predicted structures were refined with an online tool 3Drefine (http://sysbio.rnet.missouri.edu/3Drefine/) and verified using QMEAN (https://swissmodel.expasy.org/qmean/). iPBA web server was used for pdb structure alignment (https://www.dsimb.inserm.fr/dsimb_tools/ipba/index.php). The quality of the structure alignments was evaluated using root mean square deviation and normalized score. Chimera software 10 was used for structure visualization.
3. RESULTS
3.1. Domain architecture
Altogether, 137 genomes were analyzed. Our primary focus was on the severe acute respiratory syndrome‐related coronavirus (92 genomes), released from 2019 till 25 March 2020 (NCBI). The Middle East respiratory syndrome‐related coronavirus genomes (n = 18) (further shortened to MERS) were used to check the reliability of our in silico approaches. A set of different coronaviruses (taxonomical origin, hosts, and release date) was used in our analyses (Table S1).
Four conserved domains were identified: spike glycoprotein N‐terminal domain (pfam16451), spike receptor‐binding domain (pfam09408), coronavirus S1 glycoprotein (pfam01600), and coronavirus S2 glycoprotein (pfam01601) (listed from N‐terminal to C‐terminal direction) (Figure S1). Surprisingly, none of the analyzed genomes exhibited such domain architecture, as is known, for example, for the viruses studied earlier (top line in Figures S1 and S2). As a common feature, we noticed that alphacoronaviruses and gammacoronaviruses share domain architecture represented by two domains: coronavirus S1 glycoprotein along with coronavirus S2 glycoprotein. In contrast, all analyzed betacoronaviruses have a full or partial spike receptor‐binding domain. Interestingly, only betacoronaviruses with humans and yaks as hosts have spike glycoprotein N‐terminal domain, which is absent in all other analyzed hosts.
3.2. Phylogenetic analysis
To gain insight into the phylogenetic relationships between spike glycoproteins from alphacoronaviruses, betacoronaviruses, and gammacoronaviruses of different hosts, a robust phylogenetic tree after multiple alignments of the 137 extracted sequences (Figure S3) was generated. The tree was rooted in the outgroup (gammacoronaviruses). As expected, all SARS‐CoV‐2 spike glycoproteins (with humans as a host) were almost identical and formed separate clusters. Similarly, all the MERS (with humans as a host) were nearly identical and clustered together with camels' MERS (isolates from Kenya), with closely related MERS isolates from the Egyptian camel. Two betacoronavirus isolates from bats, HKU5‐ and HKU4‐related (MN611520.1 Pipistrellus abramus and MN611519.1 Tylonycteris pachypus, respectively), were located between SARS‐CoV‐2 and MERS clusters.
Two other branches formed two well‐distinguished clusters for alphacoronaviruses and betacoronaviruses isolates. Alphacoronaviruses have host‐dependent subclusters (humans and bats). In contrast, other betacoronaviruses (HKU1 and OC43) are closely related to the yak isolate.
3.3. Comparison of structural models
Furthermore, to better understand the relationships between spike glycoproteins on the structural level, we built protein models with homology‐based server SWISS‐MODEL 9 (Table S1, marked with *). Obtained models were evaluated and verified (Table S2). In the next step, iPBA web server, with a local backbone conformation comparison similarity algorithm, was used to match human host‐delivered SARS‐CoV‐2 and MERS spike glycoproteins to models delivered from other coronaviruses and hosts (Table 1). MERS betacoronavirus was used as a control data set as it is well‐studied in both hosts (human and camel), and as their close genetic relationship is well‐characterized. 11 As expected, human host‐delivered‐MERS showed the highest similarity to that from camels. SARS‐CoV‐2's spike glycoprotein, on the contrary, has shown the highest similarity to the yak‐delivered betacoronavirus (Table 1 and Figure 1B).
Table 1.
Comparison of the spike glycoprotein models
| SARS‐CoV‐2 a | MERS b | ||||
|---|---|---|---|---|---|
| Virus species | Host | Normalized score | RMSD | Normalized score | RMSD |
| Betacoronavirus 1 | Bos grunniens | 196.04 | 2.65 | −62.33 | 2.71 |
| Pipistrellus abramus bat coronavirus HKU5‐related | P abramus | 147.33 | 2.55 | 57.17 | 2.41 |
| Tylonycteris pachypus bat coronavirus HKU4‐related | T pachypus | 141.90 | 1.60 | −60.25 | 2.85 |
| Middle East respiratory syndrome‐related coronavirus | Camelus dromedarius | −81.54 | 2.70 | 606.53 | 0.03 |
| Miniopterus pusillus bat coronavirus HKU8‐related | M pusillus | 50.15 | 2.99 | −125.14 | 3.17 |
| Hipposideros pomona bat coronavirus HKU10‐related | H pomona | −45.27 | 3.01 | −110.03 | 2.92 |
| Avian coronavirus | Tadorna tadornoides | 87.22 | 2.79 | −133.14 | 3.11 |
| Miniopterus schreibersii bat coronavirus 1‐related | M schreibersii | 23.50 | 3.01 | −154.62 | 2.73 |
| Hipposideros pomona bat coronavirus CHB25 | H larvatus | 40.54 | 2.93 | −125.29 | 3.13 |
| Scotophilus kuhlii bat coronavirus 512‐related | S kuhlii | 51.53 | 2.74 | −136.46 | 3.23 |
Abbreviation: RMSD, root mean square deviation.
Severe acute respiratory syndrome‐related coronavirus, host: Homo sapiens.
Middle East respiratory syndrome‐related coronavirus, host: H sapiens.
Figure 1.
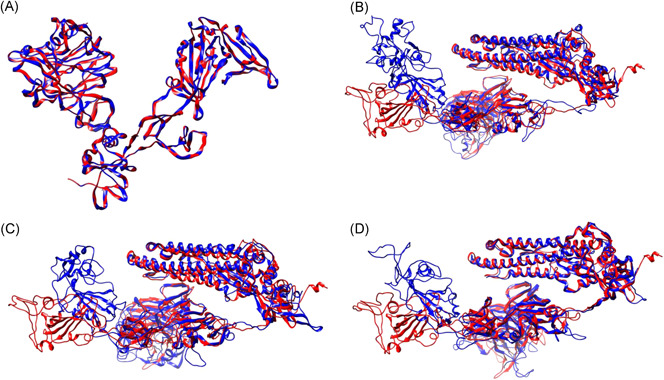
Graphical representation of compared structural models. A, MERS from the human host (red), MERS from camel host (blue); (B) SARS‐CoV‐2 from the human host (red), betacoronavirus from Bos grunniens (blue); (C) SARS‐CoV‐2 from the human host (red), Pipistrellus abramus bat coronavirus HKU5‐related (blue); (D) SARS‐CoV‐2 from the human host (red), Tylonycteris pachypus bat coronavirus HKU4‐related (blue). MERS, Middle East respiratory syndrome; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
4. DISCUSSION
Proper domain classification and identification remain to be a matter of discussion. Earlier papers refer to the spike glycoprotein as cleaved in the middle and forming S1 and S2 domains, further subdividing them into N‐terminal and C‐terminal domains in each. 12 Based on the current Conserved Domain Database (CDD) output, we concluded that N‐terminal S1 corresponds to the spike glycoprotein N‐terminal domain (pfam16451), C‐terminal S1 corresponds to the spike receptor‐binding domain (pfam09408), and S2 domain contains coronavirus S1 glycoprotein (pfam01600), and coronavirus S2 glycoprotein (pfam01601) as subdomains (Figure S1).
Canonical spike glycoprotein contains four domains, each of which plays a specific function. It is known that both spike glycoprotein N‐terminal domain (pfam16451) and spike receptor‐binding domain (pfam09408) participate in specific receptor binding. The N‐terminal domain binds to carcinoembryonic antigen‐related cell adhesion molecule 1 (CEACAM1) in mouse hepatitis coronavirus, and binds sugar in porcine transmissible gastroenteritis virus. 13 Spike receptor‐binding domain binds to the aminopeptidase N or angiotensin‐converting enzyme 2 (ACE2) in coronaviruses. 14 An interplay between coronavirus S1 glycoprotein (pfam01600) and coronavirus S2 glycoprotein (pfam01601) is required for the attachment of spike to susceptible tissues and subsequent fusion. 15
The phylogenetic data reported above show that the new human‐delivered SARS‐CoV‐2 spike glycoproteins cluster with two betacoronaviruses, HKU4‐ and HKU5‐related, delivered from the hosts T pachypus and P abramus, respectively. Also, as seen from the phylogenetic tree (Figure S1), these two sequences deviate from the other bat coronavirus sequences, suggesting that these bat coronaviruses are homologous and genetically more similar to human‐delivered SARS‐CoV‐2 than to the other bats' coronaviruses. In general, these data support phylogenetic results obtained by previous researches based on (a) whole‐genome; (b) nonstructural proteins NS7b and NS8; (c) spike glycoprotein, and (d) nucleocapsid protein. 16
For the next experiment, based on the alignment and comparison of the structures, MERS has been set as a control data set, due to the well‐characterized close relationship between human‐ and camel‐delivered strains. 11 , 17 In contrast, alignment and comparison of the SARS‐CoV‐2 structural models have revealed close relationship to the yak betacoronavirus (B grunniens) (Table 1), while bat‐delivered HKU4‐ and HKU5‐related betacoronaviruses had a lower matching score. Bovine coronavirus is a worldwide spread zoonotically transmissible infection in domestic and wild ruminants, that is known to cause severe diarrhoea in neonatal, dysentery in adult, and respiratory diseases in animals of all ages and could also infect humans. 18 Our conclusion is also well‐supported by a recent report 19 on the ACE2‐spike glycoprotein complexes which suggests Bovidae as one of the potential intermediate hosts. Interestingly, the identified bovine coronavirus strain (strain YAK/HY24/CH/2017) has unique amino acid variation in the S gene, that represents an uncommon adaptive evolution pathway with unknown biological meaning. 20
It should be noted that many bat species have been identified in Europe that are natural hosts for many viruses, including coronaviruses, in particular, SARS‐like. 21 Unexplored natural reservoirs of viruses could pose potential threats for public health. 22 This possibility also raises the question of unique transmission channels specific for each region. 23
5. CONCLUSION
In conclusion, the results of our phylogenetic study support the fact that the infection originated from bats. Additionally, results from the comparison structural models propose an additional intermediate host, yak (B grunniens), that could transmit bat coronavirus to human hosts. We also wish to emphasize the importance of further investigations into the evolution of the spike glycoprotein. Such work could render a positive impact on the current SARS‐CoV‐2 transmission and prevent zoonotic disease outbreaks of this type in the future.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
Supporting information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Dabravolski SA, Kavalionak YK. SARS‐CoV‐2: Structural diversity, phylogeny, and potential animal host identification of spike glycoprotein. J Med Virol. 2020;92:1690–1694. 10.1002/jmv.25976
REFERENCES
- 1. Li Q, Guan X, Wu P, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus‐infected pneumonia. N Engl J Med. 2020;382:1199‐1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hui DS, I Azhar E, Madani TA, et al. The continuing 2019‐nCoV epidemic threat of novel coronaviruses to global health—the latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis. 2020;91:264‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ji W, Wang W, Zhao X, Zai J, Li X. Cross‐species transmission of the newly identified coronavirus 2019‐nCoV. J Med Virol. 2020;92:433‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu P, Chen W, Chen J‐P. Viral metagenomics revealed Sendai virus and coronavirus infection of Malayan pangolins (Manis javanica). Viruses. 2019;11:979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang C, Zheng W, Huang X, Bell EW, Zhou X, Zhang Y. Protein structure and sequence reanalysis of 2019‐nCoV genome refutes snakes as its intermediate host and the unique similarity between its spike protein insertions and HIV‐1. J Proteome Res. 2020;19:1351‐1360. 10.1021/acs.jproteome.0c00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fung TS, Liu DX. Human coronavirus: host‐pathogen interaction. Annu Rev Microbiol. 2019;73:529‐557. [DOI] [PubMed] [Google Scholar]
- 8. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol. 2018;35:1547‐1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Waterhouse A, Bertoni M, Bienert S, et al. SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296‐W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605‐1612. [DOI] [PubMed] [Google Scholar]
- 11. Müller MA, Corman VM, Jores J, et al. MERS coronavirus neutralizing antibodies in camels, Eastern Africa, 1983–1997. Emerg Infect Dis. 2014;20:2093‐2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Promkuntod N, Wickramasinghe INA, de Vrieze G, Gröne A, Verheije MH. Contributions of the S2 spike ectodomain to attachment and host range of infectious bronchitis virus. Virus Res. 2013;177:127‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peng G, Sun D, Rajashankar KR, Qian Z, Holmes KV, Li F. Crystal structure of mouse coronavirus receptor‐binding domain complexed with its murine receptor. Proc Natl Acad Sci U S A. 2011;108:10696‐10701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu K, Li W, Peng G, Li F. Crystal structure of NL63 respiratory coronavirus receptor‐binding domain complexed with its human receptor. Proc Natl Acad Sci U S A. 2009;106:19970‐19974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. 2016;3:237‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Benvenuto D, Giovanetti M, Ciccozzi A, Spoto S, Angeletti S, Ciccozzi M. The 2019‐new coronavirus epidemic: evidence for virus evolution. J Med Virol. 2020;92:455‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ommeh S, Zhang W, Zohaib A, et al. Genetic evidence of Middle East respiratory syndrome coronavirus (MERS‐Cov) and widespread seroprevalence among camels in Kenya. Virol Sin. 2018;33:484‐492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suzuki T, Otake Y, Uchimoto S, Hasebe A, Goto Y. Genomic characterization and phylogenetic classification of bovine coronaviruses through whole genome sequence analysis. Viruses. 2020;12:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luan J, Jin X, Lu Y, Zhang L. SARS‐CoV‐2 spike protein favors ACE2 from Bovidae and Cricetidae. J Med Virol. 2020. 10.1002/jmv.25817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. He Q, Guo Z, Zhang B, Yue H, Tang C. First detection of bovine coronavirus in Yak (Bos grunniens) and a bovine coronavirus genome with a recombinant HE gene. J Gen Virol. 2019;100:793‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Balboni A, Gallina L, Palladini A, Prosperi S, Battilani M. A real‐time PCR assay for bat SARS‐like coronavirus detection and its application to Italian greater horseshoe bat faecal sample surveys. ScientificWorldJournal. 2012;2012:989514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rizzo F, Edenborough KM, Toffoli R, et al. Coronavirus and paramyxovirus in bats from Northwest Italy. BMC Vet Res. 2017;13:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chirumbolo S. Might the many positive COVID19 subjects in Italy have been caused by resident bat‐derived zoonotic β‐coronaviruses instead of the Wuhan (China) outbreak? J Med Virol. 2020. 10.1002/jmv.25777 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information