

Abstract
Myotubular myopathy, also called X-linked centronuclear myopathy (XL-CNM), is a severe congenital disease targeted for therapeutic trials. To date, biomarkers to monitor disease progression and therapy efficacy are lacking. The Mtm1−/y mouse is a faithful model for XL-CNM, due to myotubularin 1 (MTM1) loss-of-function mutations. Using both an unbiased approach (RNA sequencing [RNA-seq]) and a directed approach (qRT-PCR and protein level), we identified decreased Mstn levels in Mtm1−/y muscle, leading to low levels of myostatin in muscle and plasma. Myostatin (Mstn or growth differentiation factor 8 [Gdf8]) is a protein released by myocytes and inhibiting muscle growth and differentiation. Decreasing Dnm2 by genetic cross with Dnm2+/− mice or by antisense oligonucleotides blocked or postponed disease progression and resulted in an increase in circulating myostatin. In addition, plasma myostatin levels inversely correlated with disease severity and with Dnm2 mRNA levels in muscles. Altered Mstn levels were associated with a generalized disruption of the myostatin pathway. Importantly, in two different forms of CNMs we identified reduced circulating myostatin levels in plasma from patients. This provides evidence of a blood-based biomarker that may be used to monitor disease state in XL-CNM mice and patients and supports monitoring circulating myostatin during clinical trials for myotubular myopathy.
Keywords: centronuclear myopathies, myotubular myopathy, dynamin, antisense oligonucleotides, therapy, GDF8, MSTN, biomarker
Graphical Abstract
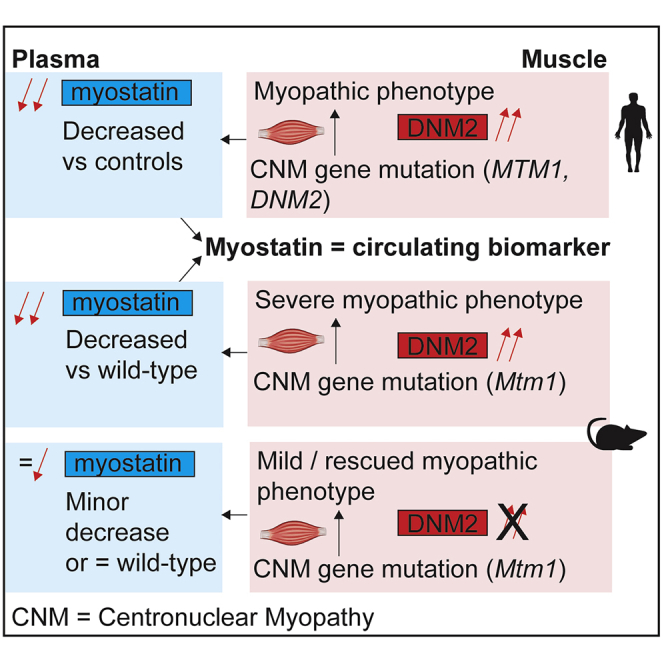
X-linked centronuclear myopathy (XL-CNM) is a severe congenital disease targeted for therapeutic trials. The authors identified decreased plasma myostatin levels in XL-CNM patients and mice. Decreasing Dnm2 in mice blocked or postponed disease progression and resulted in an increase in circulating myostatin, supporting monitoring circulating myostatin in XL-CNM clinical trials.
Introduction
Many potential therapies for neuromuscular diseases have moved from proof-of-concept toward clinical trials in the past decade.1 However, in many cases, reaching predefined endpoints to show efficacy in clinical trials can be challenging. Simple and practical biomarkers that are highly sensitive and respond rapidly to treatment are urgently required. Blood-based biomarkers can be time and cost efficient, and importantly, provide less-invasive and more global means of sampling from the patient than, for example, a muscle biopsy. In a research setting, mouse models of disease provide an excellent tool for biomarker discovery. Mouse lines that recapitulate the disease phenotype can be used to identify potential blood-based biomarkers and map in a temporal and dose-dependent manner response to treatment. This is especially true in the context of rare diseases, where large collection of samples is challenging and the absence of approved medication make sensitivity to positive change impossible to validate.
Myostatin (Mstn or growth differentiation factor 8[Gdf8]) is a protein produced and released by myocytes. Myostatin acts in an autocrine function to inhibit muscle growth and differentiation.2 Low levels of myostatin were identified in muscle biopsies and in serum from patients with different myopathies.3 Myostatin was also recently shown to be reduced in muscle biopsies from Mtm1−/y mice, a faithful mouse model for X-linked centronuclear myopathy due to MTM1 mutations.3 Centronuclear myopathies (CNMs) are non-dystrophic, debilitating rare congenital diseases, associated with muscle weakness and abnormally located nuclei in skeletal muscle.4 Several forms of CNMs have been characterized. The main forms are: X-linked CNM (XL-CNM, OMIM: 310400), also called myotubular myopathy, due to mutations in the phosphoinositides phosphatase myotubularin (MTM1),5 autosomal recessive and dominant CNM (OMIM: 255200) caused by mutations in the membrane remodeling protein amphiphysin 2 (BIN1),6,7 autosomal dominant CNM (AD-CNM, OMIM: 160150) due to mutations in dynamin 2 (DNM2),8 and autosomal recessive CNM-like disease due to mutations in ryanodine receptor (RYR1).9 There are approximately 4,000 living patients with CNM, in the USA, EU, Japan, and Australia.10
Mtm1−/y mice faithfully recapitulate XL-CNM, where mice display a severe myopathic phenotype and a reduced lifespan.11 Recent studies suggested that increased DNM2 was largely responsible for the centronuclear myopathy phenotype observed in mice and patients.11, 12, 13 Reduction of DNM2 was recently shown to rescue X-linked CNM in mice (Mtm1−/y mice) by genetic cross,11 systemic delivery of antisense oligonucleotides,14 and by reducing DNM2 using an AAV-mediated shRNA approach targeting Dnm2.15 Reduction of DNM2 in Bin1−/− mice, a model for the autosomal recessive CNM (AR-CNM, OMIM: 255200), also rescued lifespan and phenotype.16 Reduction of DNM2 by antisense oligonucleotides (ASOs) or AAV-shRNA approaches was also able to rescue the CNM phenotypes observed in a mouse model of AD-CNM due to the most common R465W mutation in the DNM2 gene.17 These results highlighted targeting DNM2 as a therapeutic potential for several centronuclear myopathy forms.
Blood-based biomarkers for monitoring disease progression or treatment efficacy have not been published for any forms of CNMs. Here we focused on myostatin, to investigate whether circulating levels of myostatin are altered in Mtm1−/y mice and whether these levels respond to treatment by reducing Dnm2.
Results
Circulating Myostatin Is Reduced in Mtm1−/y Mice and Improved in Response to Antisense Oligonucleotide Mediated Reduction of Dnm2
Here we investigated myostatin levels in plasma from Mtm1−/y mice, a mouse model for XL-CNM. Circulating plasma levels were significantly reduced in Mtm1−/y mice compared to wild-type (WT) controls (Figure 1A). To investigate whether circulating myostatin levels were altered in response to treatment, we tested ASOs previously shown to reduce Dnm2 expression and improve the disease phenotype in Mtm1−/y mice.14 Weekly injections were performed from 2–7 weeks of age in mice with ASOs targeting Dnm2 (DYN101-m) or control ASOs not targeting any known mouse genes (ASO-Ctrl; study 1, Table 1). Indeed, a significant reduction in DNM2 expression was observed at the completion of this study (Figures 1B and 1C; Figure S1A). Furthermore the reduction of DNM2 resulted in a significant improvement in the disease phenotype (Figure S1B), with Mtm1−/y mice injected with DYN101-m displaying a maximum disease severity score of 1, where the only obvious clinical difference with WT mice was a difference in body weight. A rescue was observed in muscle mass (partially) and whole-body strength (fully)(Figure 1D; Figure S1C). Importantly, in these mice a significant improvement in plasma myostatin levels was observed in Mtm1−/y DYN101-m mice, suggesting that myostatin levels respond to reduction of Dnm2 (Figure 1E). Of note, myostatin levels in Mtm1−/y mice significantly correlated with the hanging test ability (Figure 1F, Pearson correlation analysis R = 0.6296, p = 0.012) and disease severity Figure S1D, Pearson correlation analysis R = −0.5931, p = 0.0198) in these mice. Overall circulating myostatin levels were reduced in Mtm1−/y mice and were significantly improved in response to treatment.
Figure 1.

Circulating Myostatin Is Reduced in Mtm1−/y Mice and Improved in Response to Antisense Oligonucleotides Mediated Reduction of Dnm2
(A) Plasma myostatin protein levels (ng/mL) from WT and Mtm1−/y mice. (B) Representative immunoblot of DNM2 protein expression, and GAPDH loading control in TA skeletal muscles, from WT and Mtm1−/y mice, treated with DYN101-m targeting Dnm2 mRNA or ASO control (ASO-Ctrl), at 7 weeks of age. (C) Densitometry analysis from immunoblot for protein expression of DNM2 and GAPDH.. (D) Hanging test performed at 7 weeks of age, maximum time 60 s. Each point represents one mouse. (E) Circulating myostatin protein levels in plasma (ng/mL). (F) Linear regression analysis was performed between plasma myostatin levels from (E) and hanging time (D) in Mtm1−/y mice following ASO-Ctrl (black dots) or DYN101-m administration (red dots). Line of best fit shown, slope = 0.40 ± 0.14, 95% confidence interval (CI) 0.10–0.70, p value displayed. Pearson correlation analysis was also performed (r = 0.6296, ∗p = 0.0119). Each point represents one mouse, n ≥ 5 per group. Results represented as violin plots (A and C–E). Mann-Whitney test performed for DSS analysis. ∗p < 0.0125, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. All results from study 1.
Table 1.
Summary of Individual Studies Analyzed in This Manuscript
| Study | Laboratory | Study Design | Results Presented |
|---|---|---|---|
| 1 | A | weekly ASO injections, from 2/3–7 weeks of age (WT ASO-Ctrl, Mtm1−/y ASO-Ctrl, Mtm1−/y DYN101-m), 25 mg/kg | Figures 1, 2, 3, 4; Figures S1, S4, S5 |
| 2 | B | as for (1), 2nd independent cohort, 25 mg/kg | Figure 4; Figures S4 and S5 |
| 3 | A | single ASO injection at 3 weeks, analysis 1, 2, 4, 8 weeks post injection (WT ASO-Ctrl, Mtm1−/y ASO-Ctrl, Mtm1−/y DYN101-m), 25 mg/kg | Figure 2; Figures S2, S4, S5 |
| 4 | B | genetic cross cohort, analyzed at 2 weeks (WT, Mtm1−/y, Mtm1−/yDnm2+/−) | Figure 4; Figures S4 and S5 |
| 5 | A | weekly ASO injections, from 5 to 12 weeks of age (WT ASO-Ctrl, Mtm1−/y ASO-Ctrl, Mtm1−/y DYN101-m), 6.25, 12.5, and 25 mg/kg | Figure 2; Figure S3 |
| 6 | A | weekly ASO injections, from 8 to 12 weeks of age (WT ASO-Ctrl, Dnm2R465W/+ ASO-Ctrl, Dnm2R465W/+DYN101-m), 25 mg/kg | Figure S6 |
Circulating Myostatin Levels Respond to DNM2 Therapy in a Time-Dependent Manner, Correlating with Reduced Disease Severity
We next wanted to determine whether myostatin levels correlated with disease severity and reduction of Dnm2, in a temporal manner. To do this, we first established a protocol to correlate Dnm2 levels and disease phenotype in mice over time. We performed a single ASO injection in WT or Mtm1−/y mice at 3 weeks of age, near the onset of the disease phenotype (study 3). Indeed, a single injection of DYN101-m significantly delayed the progression of the disease phenotype in mice by approximately 1 month (Figure 2A). A significant reduction in Dnm2 mRNA was observed in tibialis anterior (TA) muscles of Mtm1−/y DYN101-m mice both 1 and 2 weeks post injection relative to age-matched ASO-Ctrl injected Mtm1−/y mice, which returned to WT levels 4 weeks post injection (Figure 2B; Figure S2A), coinciding with the timing of decline in the clinical phenotype in these mice. Plasma myostatin levels in untreated Mtm1−/y mice were decreased at all time points measured compared to WT mice (1–4 weeks post injection; Figure 2C), corresponding with increased severity of disease. However, Mtm1−/y mice injected with DYN101-m displayed a significant relative myostatin increase compared to Mtm1−/y control mice 2 and 4 weeks post injection (Figure 2C). The increase was apparent 1 week after ASO administration resulting in Dnm2 mRNA reduction, and at a time where mice displayed a significant improvement in disease phenotype (Figures 2A and 2B). Furthermore, myostatin levels did not purely reflect body weight or muscle mass (Figures S2B and S2C) suggesting myostatin levels are not altered solely due to a change in muscle mass.
Figure 2.

Circulating Myostatin Levels Respond to Dnm2 Reduction in a Time-Dependent Manner, Correlating with Reduced Disease Severity in Mtm1−/y Mice
(A) Combined hanging time (seconds) from all cohorts of Mtm1−/y mice following single injection of antisense oligonucleotide targeting Dnm2 (DYN101-m, red line) or ASO-Ctrl (black line). Individual cohorts were then sacrificed 1, 2, 4, 8, 12, or 16 weeks post injection, n = 18–30 mice. (B) Dnm2 mRNA expression quantified by qRT-PCR analysis, relative to Rpl27 expression, from tibialis anterior (TA) muscles, represented as mean + SD. (C) Circulating myostatin protein levels in plasma (ng/mL), represented as mean + SD. The asterisk (∗) indicates significant difference between Mtm1−/y mice with ASO-Ctrl versus DYN101-m administration for (B) and (C). (D) Hematoxylin and eosin (H&E, upper panel) and succinate dehydrogenase (SDH, lower panel) staining of TA muscles, 4 weeks post single injection. (E) Analysis of fiber size, represented as mean + SEM, n = 2–4 mice per group. (F) Analysis of fibers with altered nuclei positioning, represented as a violin plot, individual mouse data shown. ∗p < 0.0125, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. All results from study 3, (D–F) 4 week post single injection cohort. (G) ROC curve from studies 1, 3, and 5. This plot shows, for different thresholds of myostatin levels (indicated on graph), and hanging time performance in 10 s intervals, the rate of true positives (indicating myostatin level correctly classifies hanging test ability at the identified cut-off value, y axis) and the corresponding rate of false positives (where myostatin incorrectly classifies hanging test performance, x axis). 37 ng/mL was identified for all hanging times to have a true positive rate over 0.8 (see Table S4 for details).
We analyzed one cohort in detail, at 4 weeks post single injection, a time point where Mtm1−/y mice were still alive, and Mtm1−/y mice treated with DYN101-m displayed a significant improvement in phenotypes. Mtm1−/y muscles from this cohort were clearly affected by disease, with smaller fibers containing centralized nuclei, and an abnormal intracellular SDH staining pattern with accumulation observed in the center and/or the periphery of affected fibers (Figures 2D–2F). Muscles from Mtm1−/y mice treated with DYN101-m were clearly improved, with the majority of nuclei at the periphery of the fiber, and SDH staining greatly improved (Figures 2D and 2F). However, no improvement in fiber size was observed in response to a single injection of DYN101-m (Figures 2D and 2E), in contrast to the rescue observed upon repeated injections,14 and further suggesting the increase in myostatin may be reflective of improved muscle strength rather than muscle mass.
To further understand the relationship between circulating myostatin levels and disease rescue, we performed a dose-response study. Three doses were tested in Mtm1−/y mice, and phenotype and myostatin analysis was performed (study 5). All doses resulted in improved survival, with the middle and high doses resulting in similar levels of disease improvement at 12 weeks of age (Figures S3A and S3B). Correspondingly, myostatin levels were increased in all 3 doses compared to untreated 7-week-old mice (Figure 1E; Figure S3C). However, a clear dose response was not observed, suggesting a more complex mechanism of action. Notably, when a combined analysis of myostatin levels relative to hanging test ability was completed across studies (studies 1, 3, 5), myostatin was identified as a predictor for hanging test ability, with a myostatin value of 37 ng/mL corresponding to a true positive rate of over 0.8 for hanging test ability of 10 s or more (Figure 2G; Table S4). Combined, these results show circulating myostatin levels respond to treatment, in alignment with Dnm2 reduction and improvement in disease phenotype from baseline, suggesting that myostatin may be a useful biomarker for disease severity or for monitoring treatment efficacy.
Underlying Mechanisms Leading to Myostatin Alteration in Mtm1−/y Mice
We next wanted to investigate the molecular mechanism supporting reduced circulating myostatin levels in Mtm1−/y mice and increase in response to Dnm2 reduction. To do this, we analyzed the effects of Dnm2 reduction mediated by ASO administration or genetic cross, in Mtm1−/y mice at different ages from 4 independent studies including multiple cohorts performed in parallel from different mouse colonies (studies 1–4, Table 1, see Methods section for details). The level of Mstn mRNA was reduced in TA skeletal muscles from Mtm1−/y mice from all 4 studies including independent cohorts (Table 2; Figure S4), supported also by transcriptomics data from Mtm1−/y mice (Table S3). This is as expected given that myostatin is a myokine produced from muscles. This defect was limited to the TA muscles as Mstn mRNA in cardiac muscle was unaltered in Mtm1−/y mice. Surprisingly, despite the clear and rapid increase in circulating myostatin levels correlating with improved disease phenotype upon reduction of Dnm2, Mstn mRNA did not increase in TA muscles in response to Dnm2 reduction either through ASO treatment or genetic cross with Dnm2+/− mice in any cohort (Table 2; Figure S4). The TA muscle is one of the most severely affected muscles in Mtm1−/y mice,11 and variable myostatin levels have been observed in different muscles from 2 XL-CNM mouse models.18,19 Therefore we next hypothesized that the increase in circulating myostatin may be produced from other skeletal muscle tissues. Mstn production from gastrocnemius, diaphragm, and extensor digitorum longus (EDL) muscles confirmed a clear reduction in Mstn mRNA in Mtm1−/y mice, akin to TA muscles (Table 2; Figure S4). No significant increase (p < 0.059) in Mstn mRNA levels was detected in gastrocnemius muscles from Mtm1−/y mice upon reduction of Dnm2, however a significant increase was observed in the diaphragm and EDL skeletal muscles. Of note, a significant improvement in the diaphragm muscle histology in response to DNM2 reduction through a genetic cross was previously observed.11 These results suggest the increased circulating myostatin may come from increased transcription of Mstn in certain skeletal muscles (Figure S4). We next investigated myostatin protein levels. Myostatin is produced in skeletal muscles with an N-terminal prodomain.20 This prodomain was found in reduced levels in TA muscles from Mtm1−/y mice and was not altered in response to Dnm2 reduction following repeated ASO injections (Figures 3A and 3B), corresponding with mRNA analysis (Table 2; Figure S4). This immature protein is then secreted into the bloodstream, where a strong decrease was observed, and a significant amelioration upon treatment was observed (Figures 3C and 3D). Therefore, the increase in circulating myostatin levels in response to Dnm2 therapy may be the result of increased myostatin production from certain skeletal muscles only and/or from a compensatory mechanism at the protein level.
Table 2.
mRNA Analysis of Mstn Levels across Cohorts
| Study Number | Tissue | Group |
||
|---|---|---|---|---|
| WT | Mtm1−/y | Mtm1−/y+ Reduced Dnm2 | ||
| 1 | TA | 1.04 ± 0.09 (10) | 0.19 ± 0.03 (5)a | 0.25 ± 0.03 (9)a |
| GAS | 1.03 ± 0.13 (5) | 0.27 ± 0.01 (2)a | 0.83 ± 0.22 (5) | |
| heart | 1.43 ± 0.50 (5) | 0.92 ± 0.25 (2) | 1.56 ± 0.29 (5) | |
| 2 | TA | 1.00 ± 0.17 (6) | 0.32 ± 0.09 (4)a | 0.42 ± 0.07 (8)a |
| 3 | TA | 1.01 ± 0.14 (2) | 0.20 ± 0.03 (5) | 0.22 ± 0.03 (5) |
| GAS | 1.02 ± 0.19 (2) | 0.45 ± 0.04 (5) | 0.64 ± 0.19 (5) | |
| heart | 1.14 ± 0.55 (2) | 1.24 ± 0.35 (5) | 0.80 ± 0.12 (5) | |
| soleus | 1.03 ± 0.25 (2) | 0.79 ± 0.37 (5) | 0.98 ± 0.22 (5) | |
| diaphragm | 1.03 ± 0.23 (2) | 0.22 ± 0.06 (5)a | 0.53 ± 0.03 (5)a,b | |
| EDL | 1.00 ± 0.07 (2) | 0.15 ± 0.03 (5)a | 0.36 ± 0.04 (5)a,b | |
| 4, analyzed at 2 weeks | TA | 1.00 ± 0.03 (4) | 0.68 ± 0.08 (4)a | 0.63 ± 0.08 (4)a |
| 4, analyzed at 7 weeks | TA | 1.00 ± 0.10 (5) | 0.27 ± 0.06 (5)a | 0.50 ± 0.06 (4)a |
Mstn mRNA analysis relative to Rpl27 expression from various cohorts. Mean ± SEM represented, number of mice (n) indicated. See Figure S4 for violin plots.
Significant difference from WT.
Significant difference from Mtm1−/y.
Figure 3.

Underlying Mechanisms Leading to Myostatin Alteration in Mtm1−/y Mice
(A) Immunoblot for protein expression of Myostatin prodomain and GAPDH (protein loading control) in TA muscles. (B) Myostatin prodomain expression quantified relative to GAPDH loading control from (A). (C) Immunoblot for protein expression of Myostatin prodomain and ponceau (protein loading control) in plasma. (D) Densitometry analysis of myostatin prodomain expression from (C). (E) mRNA analysis of Pre-miR-27a relative to Rpl27 expression from TA skeletal muscles of WT and Mtm1−/y mice, following weekly injections of DYN101-m targeting Dnm2 or ASO-Ctrl. (F) Mature miR-27 analysis relative to U6+Snord61. Results presented as violin plots, individual mouse data shown. ∗p < 0.0125, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. All results from study 1.
To explore further potential alterations in the myostatin pathway leading to decreased circulating myostatin in disease, we investigated regulators of myostatin transcription. MicroRNAs (miRNAs) are small non-coding RNAs involved in regulating RNA silencing and post-transcriptional gene expression. miR-27a binds to the 3′ UTR of Mstn and regulates post-transcriptional expression of myostatin.21 We observed an increase in pre-miR-27a in Mtm1−/y TA skeletal muscles that was partially normalized in response to genetic and ASO-mediated reduction of Dnm2, both by qRT-PCR and RNA sequencing (RNA-seq) analyses in several cohorts (Figure 3E; Figure S5; Table S3). However mature miR-27 levels were not altered (Figure 3F), suggesting other cellular mechanisms may be involved in the reduction in myostatin in Mtm1−/y mice. The CCAAT enhancer binding proteins C/EBP regulate MSTN expression.22 Transcriptomics data suggests Cebpb and Cebpd are unchanged, whereas Cebpg mRNA was reduced in pre- and post-symptomatic stages in the Mtm1−/y muscle and partially normalized through Dnm2 reduction by genetic cross (Table S3). Similarly, c-Myc, another regulator of myostatin expression potentially acting through miR-27, was altered in Mtm1−/y muscle (Table S3).23, 24, 25 Overall, decreased expression of the myostatin gene, potentially due to alteration in its transcriptional regulators, may result in decreased circulating myostatin.
General Alterations of the Myostatin Pathway in Mtm1-/y Mice
To gain a better insight into the alterations of the myostatin pathway, we investigated growth differentiation factor 11 (GDF11), due to the high degree of similarity to myostatin (GDF8).2 Gdf11 levels were significantly increased in Mtm1−/y mice and variable following Dnm2 reduction (Figure 4A), supporting downregulation of the myostatin pathway in Mtm1−/y mice. Myostatin binds to the activin IIb receptor (AcvRIIb) on the sarcolemma of skeletal muscles to activate signaling pathways.26 AcvRIIb levels were not altered in Mtm1−/y mice (Figure 4B; Figure S5), consistent with previous results.3 Follistatin is a protein that acts in an antagonistic manner to myostatin through direct binding.26 Fstn mRNA levels were increased in Mtm1−/y mice (Table S3; Figure 4C; Figure S5) in the inverse direction to Mstn (Figure S4); however, levels did not return to WT upon reduction of Dnm2 by genetic cross or ASO injections.
Figure 4.

General Alterations of the Myostatin Pathway in Mtm1−/y Mice
(A–C) mRNA analysis relative to Rpl27 expression from TA skeletal muscles of wild-type (WT) and Mtm1−/y mice, treated with repeated injections of DYN101-m targeting Dnm2 or ASO-Ctrl, analyzed at 7 weeks of age, for Gdf11 (A), AcvRIIb (B), or Fstn (C). (D) Pre-miR-206 expression relative to Rpl27 expression, in WT, Mtm1−/y mice, and Mtm1−/yDnm2+/− mice at 7 weeks of age. (E) mRNA analysis of pre-miR-206 relative to Rpl27 expression from TA skeletal muscles of WT and Mtm1−/y mice, following weekly injections of DYN101-m targeting Dnm2 or ASO-Ctrl. (F) Mature miR-206 analysis relative to U6+Snord61, following repeated weekly injections of DYN101-m targeting Dnm2 or ASO-Ctrl. Each point represents one mouse, n ≥ 5 per group. Results presented as violin plots; individual mouse data shown. ∗p < 0.0125, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. (A–C and F) Study 1 (weekly ASO injections), (E) study 2 (second independent cohort), and (D) study 4 (genetic cross).
We next looked downstream of the myostatin pathway. Myostatin activates SMAD2/3 phosphorylation and atrophic signaling pathways (reviewed in Walker et al.27). While transcriptomics data identified changes in transcription of Smad family members, at the functional level we did not observe a change in total or phosphorylated levels of SMAD2/3 in any cohorts and groups (Figure S5; Table S3), suggesting myostatin-mediated effects may be modulated independent of SMAD2/3 signaling pathway in Mtm1−/y mice. Skeletal muscle miRNAs or “MyomiRs” play a role in physiological and pathological muscle functions. The MyomiR miR-206 is found predominantly in skeletal muscle,28 and its expression may be negatively regulated by the myostatin pathway.29 Accordingly in Mtm1−/y mice where myostatin levels are reduced, pre-miR-206 was increased both during early (Figure S5) and late (Figures 4D, E) stages of disease. This level was reduced in response to reduction of Dnm2 via genetic cross or ASO-mediated reduction of Dnm2 (Figures 4D and 4E; Figure S5). These changes were confirmed by analyzing expression of the mature miR-206 (Figure 4F). Furthermore, downstream dysregulation of the myostatin pathway was observed, in particular myogenin and Murf1 levels were altered, which were partially or fully rescued by reducing Dnm2 (Table S3). These results suggest that the myostatin pathway signaling is globally modified both in disease state (Figure 5) and in response to treatment.
Figure 5.

Dysregulation of the Myostatin Pathway in Mtm1−/y Mice
The members of the myostatin pathway analyzed in this manuscript at the mRNA or protein levels are indicated in this figure. Genes regulation in Mtm1−/y mice is represented by color-coding as follows; upregulated genes (green), downregulated genes (red), or unchanged compared to WT (blue).
Taken together, these results suggest that myostatin is altered early during development of disease pathogenesis, and these changes results in global downstream dysregulation of the myostatin pathway in mice.
Myostatin Levels Are Reduced in Patients with X-linked and Autosomal Dominant Forms of Centronuclear Myopathy
As circulating plasma myostatin levels are clearly altered in Mtm1−/y mice and respond rapidly to treatment, we next tested whether myostatin levels were also altered in patients with CNM. Circulating myostatin levels from plasma of X-linked CNM patients were shown to be dramatically reduced (3-fold) compared to age-matched control samples (Figure 6, control 3,588+328 pg/mL versus 1,122 ± 445 pg/mL in XL-CNM), suggesting the results generated in mice from this study may be translatable to patients.
Figure 6.

Plasma Myostatin Levels Are Reduced in Patients with X-linked and Autosomal Dominant Forms of Centronuclear Myopathy
Plasma myostatin protein levels (pg/mL) from adult X-linked CNM (XL-CNM, MTM1 mutations) patients and autosomal dominant CNM (AD-CNM, DNM2 mutations; ages 19–61), and adult human control samples (aged 17–72). Each point represents one patient or control, n ≥ 4 per group. Results presented as violin plots. ∗p < 0.025, ∗∗∗∗p < 0.0001.
We next wanted to see whether myostatin levels were also altered in an autosomal form of CNM. A faithful murine model for AD-CNM due to DNM2 mutations has been created (Dnm2RW/+ mice), which recapitulates the adult-onset form of the disease linked to the most common R465W mutation.30,31 No significant change in circulating myostatin levels were detected at the symptomatic age of 12 weeks (study 6, Table 1; Figure S6), which may be due to the mild phenotype presentation in this model,17,31 with minimal reduction in fiber size (14%, Figure S6). Importantly, circulating myostatin levels were significantly reduced (>4-fold) in patients with AD-CNM compared to age-matched controls (Figure 6, control 3,588+328 pg/mL versus 823 ± 367 pg/mL in AD-CNM). Overall these results suggest myostatin may be a relevant biomarker for disease in patients with several forms of CNMs.
Discussion
The myokine myostatin is known to inhibit muscle growth, and based on this concept, several anti-myostatin therapeutic clinical trials have been launched for neuromuscular disease patients, with the goal of inhibiting the myostatin pathway and therefore improving muscle mass and function. The publicly released data from these trials are so far disappointing (https://clinicaltrials.gov/; NCT02310763, NCT02515669). Recent data, including data presented here, suggest a common mechanism whereby myostatin levels in neuromuscular disease patients are naturally reduced,3,32 which may help explain in part the lack of positive data generated in clinical trials to date, and the limited therapeutic efficacy observed following AcvRIIb inhibition in two XL-CNM mouse models.18,19 Results generated here from several cohorts in independent laboratories now support that circulating myostatin is a biomarker for disease progression and severity in a mouse model of myotubular myopathy and is responsive to upstream treatment. The reduction in circulating myostatin is consistent with observations in XL-CNM and AD-CNM patients.
Defects in the Myostatin Pathway in XL-CNM
Myostatin was previously shown to be reduced in serum from patients with different neuromuscular disorders;3 however, this had not been investigated in plasma or serum from patients or animal models of centronuclear myopathies. Mstn mRNA levels were previously shown to be reduced in skeletal muscles from XL-CNM mice and dogs.3,33 In this study, we confirmed a reduction of myostatin at the mRNA level in muscle biopsies from XL-CNM mice and identified low levels of circulating myostatin in plasma. The shutdown of the myostatin pathway may be linked to a parallel increase in follistatin, an inhibitor of myostatin, and/or altered transcriptional regulation. While Fstn levels were inversely increased in our model, AcvRIIb levels were unaltered, in contrast to Duchenne Muscular Dystrophy patient muscles where AcvRIIb but not Fstn levels were altered,3 suggesting differential regulation of the myostatin pathway between neuromuscular disorders. Our data support a hypothesis where altered myostatin regulation occurs in the myopathic tissue, where myostatin levels are reduced due at least in part to direct signaling mechanisms altering transcription of Mstn and activation of the receptor and associated pathway. Furthermore, our data support a specific downregulation of myostatin (Gdf8), as transcription of the highly homologous family member Gdf11 is inversely increased in Mtm1−/y mice. Increased Gdf11 has previously been associated with aging muscles;34 however, the significance of this in Mtm1−/y mice is unclear. Interestingly this was not observed in other neuromuscular disorders,3 suggesting a specific regulation in XL-CNM.
A resulting increase in the MyomiR-206 in response to reduced myostatin in Mtm1−/y mice was observed, supporting previous data that showed an increase in the MyomiR-206 in response to loss of myostatin29 and in newly formed muscle fibers of mdx mice.35 MyomiRs have been previously shown to be altered from XL-CNM patient muscle biopsies.36 While a decrease in myostatin would in principle promote muscle hypertrophy, the muscles of the Mtm1−/y mice are very hypotrophic. Our results suggest the net loss in muscle mass alone is not responsible for the reduction in circulating myostatin levels observed in animal models of neuromuscular disease and that an overall dysregulation of the myostatin pathway occurs in Mtm1−/y mice (Figure 5).
Circulating Myostatin as a Biomarker for Disease Severity and Therapy Efficacy
Because centronuclear myopathies are non-dystrophic structural muscle diseases, blood-based markers such as creatine kinase levels are not informative. Here we identify that circulating myostatin levels are decreased in both the XL-CNM mouse model (Mtm1−/y mice) and in patients with XL or AD-CNM. In addition, in Mtm1−/y mice myostatin levels correlated with the progression of the disease severity across several studies. Therefore, myostatin is a potential biomarker to follow disease progression. Sampling patients at different disease stages and with different disease severity will be needed to confirm the relevance in humans. Patient age/gender will need to be taken into account when interpreting myostatin levels in clinic. Moreover, the differential regulation of this pathway in XL-CNM may suggest novel therapeutic targets.18 Given the rarity of the disease, a formal validation of the biomarker on large cohort of patients is not possible, but it is noteworthy that the different clinical programs in CNMs (https://clinicaltrials.gov; NCT04033159, NCT03199469) will help to verify whether the increase of myostatin level observed in treated animals translates into humans.
Therapeutic Dnm2 reduction resulted in a rapid increase in circulating myostatin, again supporting myostatin as a potential blood-based biomarker for XL-CNM. An increase in Mstn mRNA was observed in several skeletal muscles, akin to previously published data testing another therapeutic approach in mice and dogs,3,33 suggesting a possible molecular mechanism for increased plasma myostatin levels. These results suggest that circulating myostatin may be a relevant biomarker that may be used to monitor therapeutic efficacy in animal models of disease with different treatments. Of note, a correlation was observed between myostatin and increased whole-body strength even when muscle size was unaltered (Figure 2). Given the natural low level, myostatin is probably not a good treatment target but could be considered as a possible target in treated patients. These results confirm improvement in disease phenotype in animal models of CNMs by reduction of DNM2.14, 17, 37 Here we show that myostatin levels respond rapidly to treatment coinciding with improved muscle function and reduced Dnm2 mRNA from muscle biopsies in a temporal manner and that myostatin may act as a surrogate marker for treatment efficacy.
In conclusion, we show that reducing dynamin 2 increased survival and improved the myopathy phenotype in a time-dependent manner in Mtm1−/y mice, a mouse model for XL-CNM. In addition, we show here that repeated treatments may be required for patients with CNMs. Reduction of Dnm2 to treat CNMs is now being developed toward the clinic; however, supportive blood-based biomarkers are not yet available for monitoring of non-dystrophic muscle diseases. While skeletal muscles are the primary tissue affected by the disease, repeated muscle biopsies to monitor disease progression and efficacy of treatment are highly invasive for patients and not only result in permanent damage to muscles but also place a heavy burden on patients and the health care system. Here we show that centronuclear myopathies display a significant reduction in circulating myostatin. Our data therefore suggest myostatin as a novel blood-based biomarker for CNM that responds to treatment and may therefore be used to monitor disease state and rescue in X-linked centronuclear myopathy.
Materials and Methods
Generation of Mtm1−/y Mice
Mtm1−/y or WT 129SvPAS mice were previously generated and characterized by crossing Mtm1 heterozygous females obtained by homologous recombination with WT males.14,38 Animals were housed in a temperature-controlled room (19°C–22°C) with a 12:12 h light/dark cycle, with free access to food. Animal experimentation was approved by the institutional ethical committee Com’Eth IGBMC-ICS; APAFIS#5453-2016052510176016 and APAFIS#3026-201512041851445. Mice were humanely sacrificed when required according to national and European legislations on animal experimentation. Male mice were analyzed in this study.
ASOs
ASOs were chemically modified with phosphorothioate in the backbone and contrained ethyl (cEt) modifications on the wings with a deoxy gap (3-10-3 design). ASOs were synthesized by IONIS Pharmaceuticals using an Applied Biosystems 380B automated DNA synthesizer (PerkinElmer Life and Analytical Sciences-Applied Biosystems, Waltham, MA, USA) and purified.39 The 16 nucleotide ASO candidate (DYN101-m) was previously tested and shown to efficiently reduce Dnm2 expression levels in vivo, while the random control sequence has no homology to the mouse genome and does not reduce Dnm2 RNA nor protein.14,17 The sequence of each ASO is listed in Table S1. ASOs were dissolved in filtered and autoclaved sterile D-PBS (Life Technologies, #14190-144). Intraperitoneal injections of up to 25 mg/kg of ASOs were performed in Mtm1−/y, Dnm2R465W/+ or WT male mice.
Generation of Mouse Cohorts
Six independent mouse experiments were performed. Study 1: weekly ASO intraperitoneal injections of DYN101-m targeting Dnm2 mRNA or ASO control (ASO-Ctrl) were administered to WT and Mtm1−/y mice, from 2/3–7 weeks of age (25 mg/kg, laboratory A, Dynacure). Study 2: experiment performed as for study 1 (ASO injections weeks 3–7), this second experiment was performed from an independent colony of mice (laboratory B, IGBMC), thus further validating the protocol for study 1. Study 3: single injection of ASO targeting Dnm2 (DYN101-m) or ASO control (ASO-Ctrl) was performed at 3 weeks of age in WT and Mtm1−/y mice. Individual cohorts (6) were sacrificed 1, 2, 4, 8, 12, or 16 weeks post injection (laboratory A). Study 4: WT, Dnm2+/−, Mtm1−/y, and Mtm1−/yDnm2+/− mice were generated and sacrificed at 2 or 7 weeks of age (laboratory B). Study 5: weekly ASO intraperitoneal injections of DYN101-m (6.25, 12.5, 25 mg/kg) targeting Dnm2 mRNA or ASO control (ASO-Ctrl, 25 mg/kg) were administered to WT and Mtm1−/y mice, from 5 to 12 weeks of age (25 mg/kg, laboratory A). Study 6: weekly ASO injections (25 mg/kg) were performed from 8 to 12 weeks of age in WT or Dnm2R465W/+ mice (WT ASO-Ctrl, Dnm2R465W/+ ASO-Ctrl, Dnm2R465W/+DYN101-m, laboratory A). A summary of all studies can be found in Table 1.
Disease Severity Score (DSS)
DSS was performed to monitor the clinical appearance of mice. The DSS was designed to evaluate the clinical evolution of six centronuclear myopathy features; body weight, hanging test ability, kyphosis, hindlimb position while walking, breathing ability, and ptosis, as described previously.14
Hanging Test
Mice were placed on a grid (cage lid) and then turned upside down; the suspending animal should hold on to the grid to avoid falling when the grid was then inverted. The latency to fall was measured three times for each mouse. The three trials were performed with a minimum of 5 min interval to allow a recovery period. The latency time measurements began from the point when the mouse was hanging free on the wire and ended with the animal falling to the cage underneath the wire or grid. The maximum time measured was 60 s. The data were expressed as an average of three trials.
Blood Collection
Blood samples were collected on EDTA-coated tubes (Microvette 500 K3E, Sarstedt) or Heparin-coated tubes (Microvette 500 LH, Sarstedt), by mandibular puncture or by cardiac puncture on anaesthetized mice. Samples were centrifuged at +4°C during 10 min at 2,000 × g to collect plasma, and then plasma was stored at −20°C until analysis.
Myostatin ELISA
Myostatin plasma levels were quantified using a Quantikine ELISA GDF-8/Myostatin Immunoassay kit and according to the manufacturer’s instructions (catalog #DGDF80, RnDSystems). The latent form of myostatin was dissociated with an acid activation with 1 N HCl followed by a neutralization step (1.2 N NaOH/0.5 M HEPES). Total pool of plasma myostatin was then quantified with the quantitative sandwich enzyme immunoassay technique. The optical density was measured at 450 and 540 nm with a Multiskan Go (Thermo Fisher Scientific).
Quantitative RT-PCR
Studies 1 and 3: RNA was isolated from organs using NucleoSpin RNA kit (Macherey Nagel). For muscle tissue, the manufacturer “isolation of RNA from fibrous tissue” supplemental protocol was used. Reverse transcription was carried out on 500 ng aliquot using SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific). qPCR was done with PowerUp SYBR Green Master Mix (Thermo Fisher Scientific) in a Quant Studio 3 Real-Time PCR System (Thermo Fisher Scientific). Studies 2 and 4: Trizol reagent (Invitrogen, UK) was used to extract total RNA from mouse TA muscles, then 1 μg of total RNA was used for reverse transcription using Superscript IV reverse transcriptase (Thermo Fisher Scientific). qPCR was performed in Lightcycler 480 (Roche) using primers mixed in SybrGreen (QIAGEN). The list of primers used is available in Table S2. The relative expression of mRNAs was normalized to Rpl27. Rpl27 was selected as the normalizing gene after testing of four different candidate genes (Hprt, Rpl27, Rpl41, and P0) in 2 different tissues (liver and TA skeletal muscle) of untreated and treated WT and Mtm1−/y mice. The Thermo Fisher cloud application was used to select the gene with the best score. This process follows most MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) standards.
Total RNA Extraction and miRNA Quantification
Total RNA including miRNA was isolated from muscle tissues with the NucleoSpin miRNA kit (Macherey Nagel) using the manufacturer’s “isolation of RNA from fibrous tissue” supplemental protocol. Reverse transcription of mature miRNA was carried out on 500 ng using the miScript II RT kit (QIAGEN). qPCR was performed using miScript SYBR Green PCR Kit (QIAGEN), miRNA specific forward primers (miR-27 5′-TTCACAGTGGCTAAGTTCCGC-3′; miR-206 5′- TGGAATGTAAGGAAGTGTGTGG-3′) and miScript universal reverse primer (QIAGEN). The expression of mature miRNAs was normalized by the average of Rnu6-2 and Snord61 small non-coding RNAs. Four different candidate genes (Rnu6-2, Snord61, Snord68, and Snord70) were tested, and Rnu6-2 and Snord61 were selected as the normalizing genes based on PCR efficiency and the Thermo Fisher cloud application score (miScript primer assays catalog #MS00033740 and #MS00033705, QIAGEN).
Western Blotting
Tissues were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris Base, 100 mM NaCl, 1 mM EGTA, 0.5% NP40, 0.5% Triton X-100, 0.1% SDS) supplemented with 1 mM PMSF, 1 mM DTT, and complete mini EDTA-free protease inhibitor cocktail (Roche Diagnostic). Protein concentration was determined with the BioRad DC protein Assay kit. Samples were diluted in NuPAGE buffer (Invitrogen) and denatured at 95°C for 5 min. 10 or 20 μg of protein were loaded, separated in 4%–15% precast polyacrylamide gels by electrophoresis (Bio-Rad) and transferred on nitrocellulose membranes for 7 min (Trans-Blot Turbo Transfer System, BioRad). Membranes were stained with Ponceau red (catalog #P7170, Sigma Aldrich) and imaged with a Fusion FX imager (Vilber) before an incubation for 1 h at room temperature in blocking buffer (TBS containing 5% non-fat dry milk and 0.1% Tween 20). The following primary antibodies were diluted in blocking buffer and incubated for 1 h at room temperature: mouse monoclonal antibody against DNM2 (1:250, clone 6A6, home-made), mouse GDF-8/Myostatin Propeptide (1:500, catalog #AF1539, R&D Systems), rabbit monoclonal anti-SMAD2/3 (1:2,000, catalog #04-914, Sigma Aldrich), rabbit polyclonal anti-phospho-SMAD2/3 (1:2,000, catalog #SAB4504208, Sigma Aldrich), GAPDH (1:5,000 for organs, catalog #MAB374, Millipore), or GAPDH-HRP (1:10,000 for muscle, catalog #MA5-15738-HRP, Thermo Fisher Scientific). Goat anti-mouse secondary antibody coupled to horseradish peroxidase (1:5,000, catalog #115-036-068, Jackson ImmunoResearch) or goat anti-rabbit coupled to horseradish peroxidase (1:2,000, catalog #111-036-045, Jackson ImmunoResearch) was incubated for 1 h at room temperature. Immobilon ECL (catalog #WBKLS0100, Millipore) or Pierce ECL (catalog #11517271, Thermo Fisher Scientific) were used and membranes were visualized with a Fusion FX imager (Vilber). Images were quantified using the FIJI software.
Histological Analysis of Skeletal Muscle
Air-dried transverse cryosections (8 μm) were fixed and stained with hematoxylin and eosin (H&E) or succinate dehydrogenase (SDH), and image acquisition performed with a slide scanner NanoZoomer 2 HT equipped with the fluorescence module L11600-21 (Hamamatsu Photonics, Japan). Cross-sectional area (CSA) was analyzed in WGA sections from TA mouse skeletal muscle, using a plugin developed in ImageJ. CSA (μm2) was calculated in >500 fibers per mouse. The percentage of TA muscle fibers with centralized or internalized nuclei was counted in >500 fibers using the cell counter plugin in ImageJ (Rasband, W.S., ImageJ, National Institutes of Health, Bethesda, MD, USA, http://rsb.info.nih.gov/ij/, 1997–2009) or FIJI analysis software. Qualitative SDH staining analysis was performed. Fiber to fiber variation in intensity in SDH staining is normal and is indicative of the oxidative state of the fiber. SDH staining is normally relatively homogeneous within each individual fiber. Accumulation within the center or the periphery of a fiber of SDH staining indicates an abnormal distribution.
Human Sample Collection and Analysis
30 healthy control K2 EDTA plasma samples were purchased from Zenbio (catalog #SER-PLE2ML), including 15 men and 15 women, aged from 19 to 61 years old. Plasma were obtained from consenting adult donors undergoing an elective procedure in the United States who had signed an IRB or FDA validated donor consent form that specifically lists both the intended uses for non-clinical research and confirms the procedures for processing the samples are Standard Operating Procedure (SOP) managed GLP protocols in compliance with ethical regulations. Plasma samples from 4 MTM1 and 13 DNM2 mutated patients between 17–72 years old were obtained from the NatHis-CNM Natural History study40. The clinical study has been approved by the ERB Paris VI, local ERB in Belgium and Germany, and by the French Regulatory Agency ANSM. It was recorded on clinicaltrials.gov NCT02057705 / NCT03351270, IDRCB 2013-A00974-41 (France). Blood was collected on K2 EDTA tubes and centrifuged at 2,000 × g for 10 min. Plasma was collected and stored at −20°C or lower.
Statistics
Statistical analyses were performed using ANOVA unless otherwise stated in the figure legend (one-way, or two-way if matched genotype/treatment groups), with p values of <0.05 considered significant. Multiple comparisons were done using t test with the significance threshold corrected by the number of comparisons (0.05/nb comparisons). t tests were also performed when comparing only two groups, unless otherwise stated in the figure legend. Correlations were done using Pearson’s method.
ROC Method
For different thresholds of hanging time (10 s intervals from 10 s to 60 s), a ROC curve was constructed based on the level of myostatin. This descriptive analysis allows us to represent the capability of the predictor (myostatin) to correctly classify the phenotypic reaction (hanging time). The ROC curves translate the true positive rate (well-classified mice with good phenotypic reaction) and the false positive rate (rate of mice classified as having good phenotypic reaction when not the case).
Author Contributions
B.S.C. and J.L. designed the study. C. Koch, S.B., A.M., S.D., and A.R. performed experiments, A.R. performed statistical analysis, C. Kretz, R.G.O., and M.D. provided technical support, L.T. provided scientific advice, L.S. and NatHis-CNM study group provided samples from XL-CNM and AD-CNM patients, J.L. and B.S.C. directed the research, and B.S.C. wrote the manuscript with input from coauthors.
Conflicts of Interest
B.S.C. and J.L. are coinventors of a patent on targeting DNM2 for the treatment of centronuclear myopathies, and cofounders of Dynacure. B.S.C., C. Koch, S.B., A.M., A.R., R.G.O., M.D., and L.T. are currently employed by Dynacure, and L.S. is on the medical advisory board.
Consortia
NatHis-CNM study group contributors: Mélanie Annoussamy, Andreea Seferian, Jonathan Baets, Nicole Voermans, Antony Behin, U Schara, Adele D'Amico, Arturo Hernandez, Capucine de Lattre, Jean-Michel Arnal, Michèle Mayer, Jean-Marie Cuisset, Carole Vuillerot, Stéphanie Fontaine, Rémy Bellance. Please see supplemental information for NatHis-CNM study group author affiliations.
Acknowledgments
We thank the animal house and histology platform of the IGBMC for support. Thanks to Thomas Voit for advice and suggestions, Pascal Kessler and Doulaye Dembele for technical support and/or support for statistical analysis, Shuling Guo and Brett Monia for providing reagents and advice, and Kristin Geenen for support for patient samples. This study was supported by a Be Est Projets d’Avenir’ du programme d’Investissement Avenir 3 awarded grant (coDyn101) to B.C., the grant ANR-10-LABX-0030-INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame program Investissements d’Avenir ANR-10-IDEX-0002-02, and the Muscular Dystrophy Association USA and AFM-Téléthon to J.L.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.04.022.
Contributor Information
Belinda S. Cowling, Email: belinda.cowling@dynacure.com.
the NatHis-CNM Study Group:
Mélanie Annoussamy, Andreea Seferian, Jonathan Baets, Nicole Voermans, Antony Behin, U. Schara, Adele D’Amico, Arturo Hernandez, Capucine de Lattre, Jean-Michel Arnal, Michèle Mayer, Jean-Marie Cuisset, Carole Vuillerot, Stéphanie Fontaine, and Rémy Bellance
Supplemental Information
References
- 1.Cowling B.S., Thielemans L. Translational medicine in neuromuscular disorders: from academia to industry. Dis. Model. Mech. 2019;13 doi: 10.1242/dmm.041434. dmm041434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McPherron A.C., Lawler A.M., Lee S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 3.Mariot V., Joubert R., Hourdé C., Féasson L., Hanna M., Muntoni F., Maisonobe T., Servais L., Bogni C., Le Panse R. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat. Commun. 2017;8:1859. doi: 10.1038/s41467-017-01486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ravenscroft G., Laing N.G., Bönnemann C.G. Pathophysiological concepts in the congenital myopathies: blurring the boundaries, sharpening the focus. Brain. 2015;138:246–268. doi: 10.1093/brain/awu368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laporte J., Hu L.J., Kretz C., Mandel J.L., Kioschis P., Coy J.F., Klauck S.M., Poustka A., Dahl N. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat. Genet. 1996;13:175–182. doi: 10.1038/ng0696-175. [DOI] [PubMed] [Google Scholar]
- 6.Nicot A.S., Toussaint A., Tosch V., Kretz C., Wallgren-Pettersson C., Iwarsson E., Kingston H., Garnier J.M., Biancalana V., Oldfors A. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat. Genet. 2007;39:1134–1139. doi: 10.1038/ng2086. [DOI] [PubMed] [Google Scholar]
- 7.Böhm J., Biancalana V., Malfatti E., Dondaine N., Koch C., Vasli N., Kress W., Strittmatter M., Taratuto A.L., Gonorazky H. Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain. 2014;137:3160–3170. doi: 10.1093/brain/awu272. [DOI] [PubMed] [Google Scholar]
- 8.Bitoun M., Maugenre S., Jeannet P.Y., Lacène E., Ferrer X., Laforêt P., Martin J.J., Laporte J., Lochmüller H., Beggs A.H. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat. Genet. 2005;37:1207–1209. doi: 10.1038/ng1657. [DOI] [PubMed] [Google Scholar]
- 9.Wilmshurst J.M., Lillis S., Zhou H., Pillay K., Henderson H., Kress W., Müller C.R., Ndondo A., Cloke V., Cullup T. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann. Neurol. 2010;68:717–726. doi: 10.1002/ana.22119. [DOI] [PubMed] [Google Scholar]
- 10.Vandersmissen I., Biancalana V., Servais L., Dowling J.J., Vander Stichele G., Van Rooijen S., Thielemans L. An Integrated Modelling Methodology for Estimating the Prevalence of Centronuclear Myopathy. Neuromuscul. Disord. 2018;28:766–777. doi: 10.1016/j.nmd.2018.06.012. [DOI] [PubMed] [Google Scholar]
- 11.Cowling B.S., Chevremont T., Prokic I., Kretz C., Ferry A., Coirault C., Koutsopoulos O., Laugel V., Romero N.B., Laporte J. Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J. Clin. Invest. 2014;124:1350–1363. doi: 10.1172/JCI71206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cowling B.S., Toussaint A., Amoasii L., Koebel P., Ferry A., Davignon L., Nishino I., Mandel J.L., Laporte J. Increased expression of wild-type or a centronuclear myopathy mutant of dynamin 2 in skeletal muscle of adult mice leads to structural defects and muscle weakness. Am. J. Pathol. 2011;178:2224–2235. doi: 10.1016/j.ajpath.2011.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu N., Bezprozvannaya S., Shelton J.M., Frisard M.I., Hulver M.W., McMillan R.P., Wu Y., Voelker K.A., Grange R.W., Richardson J.A. Mice lacking microRNA 133a develop dynamin 2–dependent centronuclear myopathy. J. Clin. Invest. 2011;121:3258–3268. doi: 10.1172/JCI46267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tasfaout H., Buono S., Guo S., Kretz C., Messaddeq N., Booten S., Greenlee S., Monia B.P., Cowling B.S., Laporte J. Antisense oligonucleotide-mediated Dnm2 knockdown prevents and reverts myotubular myopathy in mice. Nat. Commun. 2017;8:15661. doi: 10.1038/ncomms15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tasfaout H., Lionello V.M., Kretz C., Koebel P., Messaddeq N., Bitz D., Laporte J., Cowling B.S. Single Intramuscular Injection of AAV-shRNA Reduces DNM2 and Prevents Myotubular Myopathy in Mice. Mol. Ther. 2018;26:1082–1092. doi: 10.1016/j.ymthe.2018.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cowling B.S., Prokic I., Tasfaout H., Rabai A., Humbert F., Rinaldi B., Nicot A.S., Kretz C., Friant S., Roux A., Laporte J. Amphiphysin (BIN1) negatively regulates dynamin 2 for normal muscle maturation. J. Clin. Invest. 2017;127:4477–4487. doi: 10.1172/JCI90542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buono S., Ross J.A., Tasfaout H., Levy Y., Kretz C., Tayefeh L., Matson J., Guo S., Kessler P., Monia B.P. Reducing dynamin 2 (DNM2) rescues DNM2-related dominant centronuclear myopathy. Proc. Natl. Acad. Sci. USA. 2018;115:11066–11071. doi: 10.1073/pnas.1808170115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawlor M.W., Read B.P., Edelstein R., Yang N., Pierson C.R., Stein M.J., Wermer-Colan A., Buj-Bello A., Lachey J.L., Seehra J.S., Beggs A.H. Inhibition of activin receptor type IIB increases strength and lifespan in myotubularin-deficient mice. Am. J. Pathol. 2011;178:784–793. doi: 10.1016/j.ajpath.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lawlor M.W., Viola M.G., Meng H., Edelstein R.V., Liu F., Yan K., Luna E.J., Lerch-Gaggl A., Hoffmann R.G., Pierson C.R. Differential muscle hypertrophy is associated with satellite cell numbers and Akt pathway activation following activin type IIB receptor inhibition in Mtm1 p.R69C mice. Am. J. Pathol. 2014;184:1831–1842. doi: 10.1016/j.ajpath.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson S.B., Goldberg A.L., Whitman M. Identification of a novel pool of extracellular pro-myostatin in skeletal muscle. J. Biol. Chem. 2008;283:7027–7035. doi: 10.1074/jbc.M706678200. [DOI] [PubMed] [Google Scholar]
- 21.Allen D.L., Loh A.S. Posttranscriptional mechanisms involving microRNA-27a and b contribute to fast-specific and glucocorticoid-mediated myostatin expression in skeletal muscle. Am. J. Physiol. Cell Physiol. 2011;300:C124–C137. doi: 10.1152/ajpcell.00142.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen D.L., Cleary A.S., Hanson A.M., Lindsay S.F., Reed J.M. CCAAT/enhancer binding protein-delta expression is increased in fast skeletal muscle by food deprivation and regulates myostatin transcription in vitro. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010;299:R1592–R1601. doi: 10.1152/ajpregu.00247.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.West D.W., Baehr L.M., Marcotte G.R., Chason C.M., Tolento L., Gomes A.V., Bodine S.C., Baar K. Acute resistance exercise activates rapamycin-sensitive and -insensitive mechanisms that control translational activity and capacity in skeletal muscle. J. Physiol. 2016;594:453–468. doi: 10.1113/JP271365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang H., Li T.W., Zhou Y., Peng H., Liu T., Zandi E., Martínez-Chantar M.L., Mato J.M., Lu S.C. Activation of a novel c-Myc-miR27-prohibitin 1 circuitry in cholestatic liver injury inhibits glutathione synthesis in mice. Antioxid. Redox Signal. 2015;22:259–274. doi: 10.1089/ars.2014.6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X., Xu M., Ding L., Tang J. MiR-27a: A Novel Biomarker and Potential Therapeutic Target in Tumors. J. Cancer. 2019;10:2836–2848. doi: 10.7150/jca.31361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee S.J., McPherron A.C. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA. 2001;98:9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker R.G., Poggioli T., Katsimpardi L., Buchanan S.M., Oh J., Wattrus S., Heidecker B., Fong Y.W., Rubin L.L., Ganz P. Biochemistry and Biology of GDF11 and Myostatin: Similarities, Differences, and Questions for Future Investigation. Circ. Res. 2016;118:1125–1141, discussion 1142. doi: 10.1161/CIRCRESAHA.116.308391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sempere L.F., Freemantle S., Pitha-Rowe I., Moss E., Dmitrovsky E., Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004;5:R13. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rachagani S., Cheng Y., Reecy J.M. Myostatin genotype regulates muscle-specific miRNA expression in mouse pectoralis muscle. BMC Res. Notes. 2010;3:297. doi: 10.1186/1756-0500-3-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Böhm J., Biancalana V., Dechene E.T., Bitoun M., Pierson C.R., Schaefer E., Karasoy H., Dempsey M.A., Klein F., Dondaine N. Mutation spectrum in the large GTPase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum. Mutat. 2012;33:949–959. doi: 10.1002/humu.22067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Durieux A.C., Vignaud A., Prudhon B., Viou M.T., Beuvin M., Vassilopoulos S., Fraysse B., Ferry A., Lainé J., Romero N.B. A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum. Mol. Genet. 2010;19:4820–4836. doi: 10.1093/hmg/ddq413. [DOI] [PubMed] [Google Scholar]
- 32.Burch P.M., Pogoryelova O., Palandra J., Goldstein R., Bennett D., Fitz L., Guglieri M., Bettolo C.M., Straub V., Evangelista T. Reduced serum myostatin concentrations associated with genetic muscle disease progression. J. Neurol. 2017;264:541–553. doi: 10.1007/s00415-016-8379-6. [DOI] [PubMed] [Google Scholar]
- 33.Dupont J.B., Guo J., Renaud-Gabardos E., Poulard K., Latournerie V., Lawlor M.W., Grange R.W., Gray J.T., Buj-Bello A., Childers M.K., Mack D.L. AAV-Mediated Gene Transfer Restores a Normal Muscle Transcriptome in a Canine Model of X-Linked Myotubular Myopathy. Mol. Ther. 2020;28:382–393. doi: 10.1016/j.ymthe.2019.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Egerman M.A., Cadena S.M., Gilbert J.A., Meyer A., Nelson H.N., Swalley S.E., Mallozzi C., Jacobi C., Jennings L.L., Clay I. GDF11 Increases with Age and Inhibits Skeletal Muscle Regeneration. Cell Metab. 2015;22:164–174. doi: 10.1016/j.cmet.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuasa K., Hagiwara Y., Ando M., Nakamura A., Takeda S., Hijikata T. MicroRNA-206 is highly expressed in newly formed muscle fibers: implications regarding potential for muscle regeneration and maturation in muscular dystrophy. Cell Struct. Funct. 2008;33:163–169. doi: 10.1247/csf.08022. [DOI] [PubMed] [Google Scholar]
- 36.Bachmann C., Jungbluth H., Muntoni F., Manzur A.Y., Zorzato F., Treves S. Cellular, biochemical and molecular changes in muscles from patients with X-linked myotubular myopathy due to MTM1 mutations. Hum. Mol. Genet. 2017;26:320–332. doi: 10.1093/hmg/ddw388. [DOI] [PubMed] [Google Scholar]
- 37.Maani N., Sabha N., Rezai K., Ramani A., Groom L., Eltayeb N., Mavandadnejad F., Pang A., Russo G., Brudno M. Tamoxifen therapy in a murine model of myotubular myopathy. Nat. Commun. 2018;9:4849. doi: 10.1038/s41467-018-07057-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buj-Bello A., Laugel V., Messaddeq N., Zahreddine H., Laporte J., Pellissier J.F., Mandel J.L. The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc. Natl. Acad. Sci. USA. 2002;99:15060–15065. doi: 10.1073/pnas.212498399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seth P.P., Siwkowski A., Allerson C.R., Vasquez G., Lee S., Prakash T.P., Kinberger G., Migawa M.T., Gaus H., Bhat B., Swayze E.E. Design, synthesis and evaluation of constrained methoxyethyl (cMOE) and constrained ethyl (cEt) nucleoside analogs. Nucleic Acids Symp. Ser. (Oxf) 2008;52:553–554. doi: 10.1093/nass/nrn280. [DOI] [PubMed] [Google Scholar]
- 40.Annoussamy M., Lilien C., Gidaro T., Gargaun E., Chê V., Schara U., Gangfuß A., D’Amico A., Dowling J.J., Darras B.T. X-linked myotubular myopathy: A prospective international natural history study. Neurology. 2019;92:e1852–e1867. doi: 10.1212/WNL.0000000000007319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.