

Summary
Endothelial cell (EC)-derived signals contribute to organ regeneration, but angiocrine metabolic communication is not described. We found that EC-specific loss of the glycolytic regulator pfkfb3 reduced ischemic hindlimb revascularization and impaired muscle regeneration. This was caused by the reduced ability of macrophages to adopt a proangiogenic and proregenerative M2-like phenotype. Mechanistically, loss of pfkfb3 reduced lactate secretion by ECs and lowered lactate levels in the ischemic muscle. Addition of lactate to pfkfb3-deficient ECs restored M2-like polarization in an MCT1-dependent fashion. Lactate shuttling by ECs enabled macrophages to promote proliferation and fusion of muscle progenitors. Moreover, VEGF production by lactate-polarized macrophages was increased, resulting in a positive feedback loop that further stimulated angiogenesis. Finally, increasing lactate levels during ischemia rescued macrophage polarization and improved muscle reperfusion and regeneration, whereas macrophage-specific mct1 deletion prevented M2-like polarization. In summary, ECs exploit glycolysis for angiocrine lactate shuttling to steer muscle regeneration from ischemia.
Keywords: angiogenesis, metabolism, angriocrine signals, lactate, MCT1, ischemia, macrophage polarization, muscle regeneration, endothelial cells
Graphical Abstract
Highlights
-
•
Endothelial loss of pfkfb3 impairs ischemic muscle revascularization and regeneration
-
•
EC-derived lactate instructs MCT1-dependent macrophage functional polarization
-
•
Lactate-polarized macrophages promote muscle revascularization and regeneration
-
•
Restoring lactate levels improves macrophage polarization and recovery from ischemia
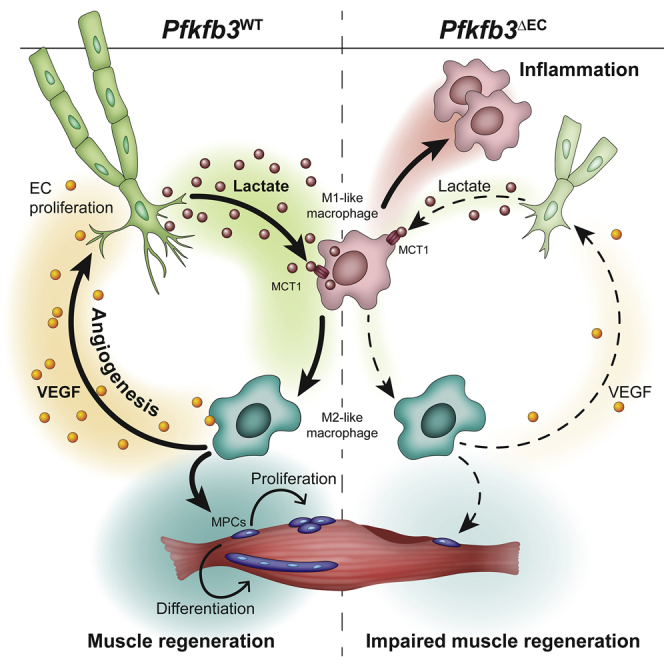
Endothelial cells (ECs) critically control muscle recovery from ischemia by secreting lactate. Angiocrine lactate is taken up and oxidized by macrophages in an MCT1-dependent fashion. Lactate-mediated macrophage polarization promotes revascularization and muscle regeneration. Consequently, EC-specific loss of pfkfb3 lowers muscle lactate levels and impairs muscle recovery from ischemia.
Context and Significance
Skeletal muscle regeneration from ischemia is coordinated by strictly timed interactions between several cell types. These interactions are poorly understood but can lead to the development of therapies for regenerative medicine or peripheral artery disease. Zhang et al. show that endothelial cells (the main cell type of blood vessels) play a crucial role during muscle regeneration. Besides restoring oxygen and nutrient supply, endothelial cells directly control the function of macrophages (specialized immune cells) by releasing lactate. Endothelial cell-derived lactate is taken up by macrophages via the MCT1-lactate transporter. Upon lactate stimulation, macrophages actively support the formation of new muscle fibers and further stimulate blood vessel formation. Our findings imply that endothelial cells use lactate to actively control muscle regeneration.
Introduction
Endothelial cells (ECs) cover the inner wall of blood vessels and act as gatekeepers of metabolism by adapting oxygen and nutrient delivery to the metabolic needs of tissues through angiogenesis (Adams and Alitalo, 2007, Potente et al., 2011). Recent studies have indicated that ECs possess specific metabolic characteristics (Fitzgerald et al., 2018, Potente and Carmeliet, 2017). Even under quiescent conditions, ECs generate the majority of their energy via the glycolytic conversion of glucose to lactate (Culic et al., 1997, De Bock et al., 2013, Krützfeldt et al., 1990). In addition, ECs further upregulate glycolysis to fuel migration and proliferation during angiogenesis. The increase in glycolysis upon EC activation is mediated by the glycolytic regulator phosphofructokinase-2/fructose-2,6-bisphosphatase isoform 3 (PFKFB3) (De Bock et al., 2013, Schoors et al., 2014). Deletion of pfkfb3 in ECs (pfkfb3ΔEC) prevents blood vessel growth during development as well as in various models of pathological angiogenesis (Cantelmo et al., 2016, De Bock et al., 2013, Schoors et al., 2014, Xu et al., 2014). Although the exact reasons for the dependence of ECs on glycolysis remain to be fully elucidated, it has been hypothesized that high glycolysis would support efficient oxygen transfer to surrounding tissues or allow ECs to rapidly invade avascular and hypoxic areas during vessel formation. However, whether ECs exploit glycolysis to engage in metabolic crosstalk with other cells within their microenvironment is unknown.
It has become increasingly recognized that ECs also regulate tissue homeostasis in an angiogenesis-independent manner through the production and release of angiocrine factors. Those factors are actively involved in the maintenance as well in the activation, specification, and guidance of organ regeneration, often in an organotypic and context-dependent manner (Augustin and Koh, 2017, Rafii et al., 2016). For instance, angiocrine growth factors promote liver and lung regeneration by orchestrating self-renewal and differentiation of tissue-specific resident stem and progenitor cells into functional organs (Ding et al., 2010, Ding et al., 2011, Hu et al., 2014, LeCouter et al., 2003). Angiocrine signals also ensure neuronal stem cell quiescence (Delgado et al., 2014, Ottone et al., 2014), while during regenerative neurogenesis, they promote neuronal stem cell proliferation, activation, and differentiation (Rafii et al., 2016).
Skeletal muscle is a highly vascularized tissue and is characterized by a remarkable capacity for regeneration (Almada and Wagers, 2016, Itagaki et al., 1995). Muscle regeneration is dependent on the activation and proliferation of resident muscle stem cells that give rise to a population of proliferating myogenic progenitor cells (MPCs) (Almada and Wagers, 2016). A subset of these MPCs self-renews to replenish the muscle stem cell pool, whereas the other MPCs enter myogenic differentiation and fuse with each other or with remaining myofibers to repair the damaged muscle (Yin et al., 2013). This sequence of events is coordinated by strictly timed cellular interactions between several cell types within the muscle microenvironment, including macrophages (Bentzinger et al., 2013). Macrophages initially exhibit a more pro-inflammatory M1-like phenotype but soon thereafter functionally repolarize toward an M2-like phenotype to actively support muscle regeneration (Arnold et al., 2007). Blood vessels also undergo profound alterations during muscle regeneration (Latroche et al., 2015a, Latroche et al., 2015b), and this is particularly true after ischemia-induced muscle damage, where muscle regeneration coincides with the formation of new blood vessels. Angiogenesis restores oxygen and nutrient delivery to the regenerating muscle tissue (Adams and Alitalo, 2007, Potente et al., 2011), and angiocrine growth factors promote the proliferation of muscle progenitor cells (Arsic et al., 2004, Borselli et al., 2010, Latroche et al., 2017). ECs also control macrophage differentiation and maturation via angiocrine Notch signaling to promote arteriogenesis during hindlimb ischemia (Krishnasamy et al., 2017). However, whether ECs engage into metabolic angiocrine crosstalk to control ischemia-induced muscle regeneration is unknown.
Here, we show that ECs exploit their glycolytic capacity to steer muscle regeneration during ischemia. Upon EC activation, the angiocrine release of lactate, the main metabolic product of glycolytic glucose catabolism, initiates a lactate shuttle that promotes an MCT1-dependent oxidative switch in macrophages and instructs their polarization toward a pro-regenerative M2-like phenotype. These macrophages promote muscle regeneration by stimulating the proliferation and differentiation of MPCs. Moreover, lactate-polarized macrophages also upregulate the expression of vascular endothelial growth factor (VEGF), thereby creating a positive feedback loop that further stimulates angiogenesis.
Results
PFKFB3 Controls Glycolysis in Muscle ECs
To study the metabolic angiocrine properties of ECs, we generated pfkfb3LoxP/LoxP mice and intercrossed them with EC-specific inducible pdgfb-CreERT2 mice (Claxton et al., 2008), hereafter referred to as pfkfb3ΔEC mice (Figure 1A). Ten days after the last tamoxifen injection, we isolated ECs from calf muscle (m. gastrocnemius and m. soleus) (mECs) and confirmed a decrease in pfkfb3 mRNA levels in mECs from pfkfb3ΔEC mice (hereafter termed mECsΔpfkfb3) when compared to mECs isolated from pfkfb3WT mice (hereafter termed mECsWT). This resulted in an almost complete loss of PFKFB3 protein content (Figures S1A and S1B). Loss of endothelial pfkfb3 did not affect baseline muscle vascular density (Figures S1C and S1D) nor did it affect the number of CD31+CD45− mECs (Figure S1E). Isolated mECsΔpfkfb3 showed lower extracellular acidification rate (ECAR) (Figure S1F) and a reduction in glycolytic flux (Figure S1G), consistent with previous data using short-hairpin-mediated knockdown of pfkfb3 in human umbilical vein endothelial cells (De Bock et al., 2013). Collectively, these data demonstrate that PFKFB3 controls glycolysis in mECs.
Figure 1.

Endothelial PFKFB3 Controls EC Glycolysis and Ischemia-Induced Revascularization
(A) Scheme showing the generation of pdgfb-CreERT2 × pfkfb3LoxP/LoxP (pfkfb3ΔEC) mice and experimental setup.
(B and C) Representative images (B) and quantification (C) of hindlimb blood perfusion in pfkfb3WT and pfkfb3ΔEC mice using laser Doppler imaging before and after HLI surgery (n = 7).
(D) Distribution of necrotic toes per paw 14 days after HLI (n = 7).
(E) Representative images of Ki67 (green), CD31 (red), and hoechst (blue) immunostainings and quantification of Ki67+CD31+ ECs in muscles at 3 days (n = 3; scale bar, 50 μm).
(F and G) Representative CD31 immunofluorescent images on muscle cross sections (F) at the indicated times (scale bar, 50 μm) and whole mounted muscle bundles (G) at 28 days (scale bar, 10 μm) in pfkfb3WT and pfkfb3ΔEC mice.
(H and I) Representative hematoxylin-eosin (H&E) staining images (H) at the indicated times and quantification of regenerating area 28 days after HLI (I). Scale bar, 10 μm.
(J) Muscle fiber size distribution at 28 days (n = 4).
(K) Representative images of pimonidazole staining (yellow) and hoechst (blue) at 3 days (scale bar, 50 μm).
(L and M) Muscle VEGF protein content in pfkfb3WT and pfkfb3ΔEC mice (L) or in vegfWT and vegfΔMac mice (M) 72 h after HLI.
Student’s t test (two-tailed, unpaired) in (I) (∗p ≤ 0.05). Two-way ANOVA with Tukey’s multiple comparisons test in (J), (L), and (M) (∗p < 0.05). Two-way repeated-measures ANOVA with Sidak’s multiple comparisons test in (C) (∗p < 0.05). Each dot represents a single mouse ([I], [L], and [M]). Bar graphs represents mean ± SEM. See also Figure S1.
Deletion of Endothelial pfkfb3 Impairs Hindlimb Ischemia (HLI)-Induced Revascularization
To induce hindlimb ischemia (HLI), we ligated the femoral artery, which leads to reduction of blood flow by >80%, causing profound ischemia-induced muscle damage. This damage is followed by robust revascularization and muscle regeneration, leading to full recovery of muscle function after four weeks (Limbourg et al., 2009, Zhang et al., 2017) (Figure 1A). Laser Doppler imaging showed that blood flow upon HLI was similarly reduced in both pfkfb3WT and pfkfb3ΔEC mice. However, although it gradually recovered in pfkfb3WT mice during the 4-week follow up period, blood flow remained severely impaired in pfkfb3ΔEC mice (Figures 1B and 1C). Pfkfb3ΔEC mice also displayed a higher frequency of necrotic toes (Figure 1D). Reduced revascularization was not caused by differences in EC apoptosis after HLI (Figures S1H–S1J). Instead, mECsΔpfkfb3 proliferated less, as indicated by fewer Ki67+CD31+ cells (Figure 1E), which was associated with reduced vascular density in pfkfb3ΔEC muscle 7 days after HLI (Figures 1F and S1K). Morphological analysis on whole-mounted muscle bundles additionally showed that lower vascular density coincided with reduced intercapillary anastomosis (Figures 1G and S1L). Concomitantly, pfkfb3ΔEC mice developed increased muscle necrosis that resolved slower over time (Figures 1H and S1M–S1O). Importantly, muscle damage 1 day after HLI was similar between pfkfb3ΔEC and pfkfb3WT mice (Figures 1H, S1M and S1N), showing that impaired regeneration was not secondary to differences in initial muscle damage. The regenerative area in wild-type (WT) animals reached a maximum 7 days after HLI, and normal muscle morphology was almost completely restored after 4 weeks, whereas pfkfb3ΔEC mice still showed large areas of intense regeneration (Figures 1H, 1I, S1M, and S1O). Consequently, the muscle fiber cross-sectional area was lower in pfkfb3ΔEC mice (Figure 1J). Thus, endothelial PFKFB3 is required for ischemia-induced muscle revascularization and regeneration.
We next performed pimonidazole injections 3 days after HLI to detect hypoxic areas within the injured muscles. As expected, reduced revascularization in pfkfb3ΔEC mice coincided with extensive and widespread hypoxia (Figures 1K and S1P). Despite this, we surprisingly measured less VEGF in the ischemic hindlimb of pfkfb3ΔEC mice (Figure 1L). Vegf is highly expressed in MPCs (Verma et al., 2018), but we did not find differences between genotypes (Figure S1Q). Macrophages also produce and respond to VEGF (Casazza et al., 2013, Ganta et al., 2019, He et al., 2012). Deletion of vegf in macrophages by using the myeloid-specific LysM-Cre line (vegfΔMac) (Stockmann et al., 2008) reduced muscle VEGF levels upon HLI (Figure 1M). We thus decided to investigate whether endothelial pfkfb3 affects macrophage function in the muscle upon ischemia.
Endothelial PFKFB3 Is Crucial for M2-like Polarization of Macrophages in the Muscle
Macrophages play a crucial role during skeletal muscle regeneration after injury (Chazaud, 2014, Tidball, 2017). To study whether loss of endothelial pfkfb3 affects immune cell recruitment/infiltration and macrophage differentiation, we first confirmed with a Rosa26mTmG fate-tracing mouse line and mRNA measurements that pdgfb-CreERT2 activity was restricted to ECs and was largely absent in macrophages and the total CD45+ immune-cell compartment (Figures S2A and S2B). Next, we performed a time course experiment where we evaluated the dynamics of various myeloid cell populations within the hindlimb upon HLI. Initially, muscle damage leads to the infiltration of neutrophils and CX3CR1+Ly-6Chigh monocytes, most of which also expressed CCR2 (Figures 2A, 2B, S2C–S2E). These monocytes gradually differentiate into macrophages indicated by downregulation of Ly-6C and upregulation of F4/80 and MERTK expression (Figures S2C–S2E) (Krishnasamy et al., 2017). Although the number of monocytes, neutrophils, and macrophages increased comparably in the ischemic muscle of pfkfb3WT and pfkfb3ΔEC mice until day 2, it was more pronounced in pfkfb3ΔEC mice at day 3 (Figures 2A–2C), reflecting a general and maintained pro-inflammatory state. Notably, we found no difference in monocyte proliferation (Figures 2D and S2F) between pfkfb3WT and pfkfb3ΔEC mice, suggesting that the enhanced number of monocytes in pfkfb3ΔEC mice is the result of increased recruitment. The increased macrophage number 3 days after HLI was confirmed by immunofluorescent detection of F4/80+ cells in the ischemic area (Figures 2E and 2F). We also detected a small and transient delay in monocyte-to-macrophage differentiation since the gradual decline of Ly-6C versus the increase in F4/80, and MERTK expression was slightly lower in pfkfb3ΔEC muscle at day 2 but recovered to pfkfb3WT levels by day 3 (Figure 2G).
Figure 2.

Endothelial PFKFB3 Is Crucial for M2-like Polarization of Macrophages in the Muscle
(A–C) Total number of neutrophils (A), monocytes (B), and macrophages (C) in muscle at the indicated times after HLI determined by flow cytometry.
(D) Quantification of EdU+ monocytes (% of total monocytes) at the indicated times after HLI.
(E and F) Representative images of F4/80 immunostainings (red) and hoechst (blue) (E) and quantification of F4/80+ area (F) on muscle at 3 days.
(G) Mean fluorescent intensity (MFI) of Ly-6C, F4/80, and MERTK in muscle macrophages at the indicated times after HLI determined by flow cytometry.
(H) Representative flow cytometric analysis of CD206+ and Relmα+ cells in muscle 3 days after HLI.
(I and J) Quantification of Relmα+CD206+ (I) and Relmα−CD206+ (J) F4/80+CD11b+ macrophages.
(K and L) Representative histograms (K) and quantification of CD206 MFI (L) in the total muscle macrophage population 3 days after HLI.
(M and N) Representative immunostainings for F4/80 (green) and CD206 (red) on CD45+ cells (M) sorted from muscle 3 d after HLI and quantification of CD206 MFI (N).
(O and P) Representative images of CD206 immunostainings (red) and hoechst (blue) (O) and quantification of CD206+ macrophages number (P) 3 days after HLI.
(Q) Quantification of EdU+CD206+ macrophages (% of CD206+ macrophages) at 3 days after HLI determined by flow cytometry
(R and S). Principal-component analysis (R) and Volcano plot from RNA-seq analysis showing differential gene expression (S) of macrophages isolated from pfkfb3WT and pfkfb3ΔEC muscles 3 days after HLI (n = 3).
(T) Expression pattern of pro-inflammatory and anti-inflammatory markers genes in muscle macrophages 3 days after HLI (n = 3).
Arrowheads point at CD206+ cells (O). Scale bar, 50 μm. Student’s t test (two-tailed, unpaired) in (F), (I), (J), (L), (N), (P), and (Q) (∗p < 0.05). Two-way ANOVA with Tukey’s multiple comparisons test in (A), (B), (C), (D), and (G) (∗p < 0.05). Each dot represents a single mouse ([A], [B], [C], [D], [F], [G], [I], [J], [L], [N], [P], and [Q]). Bar graphs represent mean ± SEM. See also Figures S2 and S3.
Further characterization of the macrophage population at 3 days after HLI showed a strong reduction in the relative number of Relmα+CD206+ as well as Relmα−CD206+ M2-like macrophage in pfkfb3ΔEC muscle (Figures 2H–2J) and lower CD206 protein expression in the total macrophage population (Figures 2K and 2L). CD206 staining on FACS-sorted CD45+ cells (Figures 2M and 2N) or on tissue sections (Figures 2O and 2P) confirmed that, despite the increase in total macrophage number, there were fewer CD206+ M2-like macrophages in pfkfb3ΔEC muscle 3 days after HLI. Proliferation of macrophages in the muscle was very low, and even though CD206+ macrophages proliferated more than the CD206− macrophages, their proliferation rate was not affected in pfkfb3ΔEC mice (Figures 2Q, S2G, and S2H), indicating that loss of endothelial pfkfb3 impairs development rather than proliferation of M2-like macrophages. To evaluate whether the difference in expression of typical M2 markers coincided with a transcriptional M2-like signature, we isolated macrophages from pfkfb3WT and pfkfb3ΔEC muscle 3 days after HLI and performed RNA-seq. Principal-component analysis showed that pfkfb3WT- and pfkfb3ΔEC-derived macrophages clustered separately (Figure 2R). Moreover, 1,989 genes were differentially regulated between genotypes (Figure 2S). There was a coordinated activation of M2 macrophage-related genes (Liu et al., 2017, Varga et al., 2016) in pfkfb3WT-derived macrophages whereas pfkfb3ΔEC-derived macrophages showed higher expression of M1-related genes (Figure 2T). Gene Ontology pathway analysis confirmed that pathways associated to an M2-like phenotype were more activated in in pfkfb3WT-derived macrophages (Figures S3A and S3B). Taken together, these data indicate that loss of endothelial pfkfb3 increases monocyte recruitment during ischemia but impairs macrophage polarization toward a M2-like phenotype.
Restoring M2 Macrophage Content in Muscle of pfkfb3ΔEC Mice Improves Perfusion and Regeneration
To dissect the functional relevance of impaired M2-like polarization to impaired recovery from ischemia in pfkfb3ΔEC mice, we performed adoptive transplantation experiments. We injected either unpolarized (ctrl) bone-marrow-derived macrophages (BMDMs) or IL-4-cultured BMDMs (classical M2, BMDMs(IL-4)) into the hindlimb of mice 3 days after HLI (Figure 3A). Hindlimb perfusion measurements showed lower perfusion in pfkfb3ΔEC mice upon transfer of unpolarized BMDMs in comparison to pfkfb3WT mice. However, transfer of BMDMs(IL-4) improved hindlimb reperfusion in pfkfb3ΔEC mice (Figures 3B and 3C) and increased vascularization (Figures 3D and 3E). BMDM(IL-4) transfer also sufficed to increase VEGF protein levels (Figure 3F), consistent with increased VEGF secretion by BMDM(IL-4) (Figure S4A). In addition, BMDM(IL-4) transfer led to a striking acceleration of muscle regeneration (Figures 3G–3I and S4B). These data show that loss of endothelial pfkfb3 controls muscle revascularization and regeneration at least partially via controlling macrophage polarization.
Figure 3.

Restoring M2 Macrophage Content in Muscle of pfkfb3ΔEC Mice Improves Muscle Perfusion and Regeneration
(A) Scheme showing macrophage transfer experiments.
(B and C) Representative laser Doppler images (B) and quantification of hindlimb perfusion (C) in pfkfb3WT and pfkfb3ΔEC mice upon BMDMs (ctrl) or BMDMs (IL-4) transfer (n = 5).
(D and E) Representative images of CD31 immunofluorescent staining (D) and quantification of CD31+ area (E) in muscle 12 days after HLI (scale bar, 50 μm).
(F) VEGF protein content in muscle 5 days after HLI.
(G and H) H&E staining of regenerating muscle (scale bar, 10 μm) (G) and quantification of regenerating area (H) at 12 days.
(I) Muscle fiber size distribution at 12 days (n = 4).
Two-way ANOVA with Tukey’s multiple comparisons test in (E), (F), and (H) (∗p < 0.05) and in (I) (∗p < 0.05 versus pfkfb3WT; #p < 0.05 versus ctrl). Two-way repeated-measures ANOVA with Sidak’s multiple comparisons test in (C) (∗p < 0.05). Each dot represents a single mouse ([E], [F], and [H]). Bar graphs represents mean ± SEM. See also Figure S4.
Endothelial Lactate Controls Macrophage Polarization upon Muscle Ischemia
To study whether mECs exploit angiocrine mechanisms to affect macrophage polarization, we isolated mECsWT and mECsΔpfkfb3 and co-cultured them with BMDMs (Figure S5A). Subsequently, CD11b+F4/80+ BMDMs were sorted and analyzed. Co-culturing BMDMs with mECsWT led to the upregulation of several M2-marker genes including mgl1 (CD301a), mgl2 (CD301b), arg1, and vegf, whereas co-culture of mECsΔpfkfb3 with BMDMs only modestly activated an M2-gene expression fingerprint (Figure S5B). The expression of genes associated with an M1-like phenotype (tnf-α and il-1β) was not different between BMDMs co-cultured with mECWT versus mECsΔpfkfb3 (Figure S5B). This was not dependent on physical contact between mECs and BMDMs, because the utilization of conditioned medium (CM) from mEC cultures led to similar results (Figures 4A and 4B), demonstrating that endothelial PFKFB3 controls macrophage polarization via a secreted factor. Interestingly, BMDM stimulation with mECs-CM did not fully recapitulate classical IL-4-mediated M2 polarization, because the activation of several M2-related genes was significantly lower upon mECs-CM administration when compared to IL-4 stimulation despite equal CD206 membrane expression (Figures S5C–S5E).
Figure 4.

Endothelial Lactate Controls Macrophage Polarization and Function upon Muscle Ischemia
(A) Scheme illustrating experimental set-up.
(B) Gene profiling of unstimulated BMDMs (vehicle) or BMDMs stimulated with mECswt-CM, mECsΔpfkfb3-CM.
(C and D) Gene expression analysis of arg1 (C) and mrc1 (D) in BMDMs stimulated with fractionated mECswt-CM and mECsΔpfkfb3-CM, supplemented with lactate (5 mM) where indicated.
(E) Lactate concentration in mECswt-CM and mECsΔpfkfb3-CM.
(F) Representative images of immunostainings of F4/80 (green), CD206 (red), and hoechst (blue) in BMDMs stimulated with mECswt-CM, mECsΔpfkfb3-CM with or without lactate supplementation and quantification of CD206 MFI (n = 3).
(G) OCR upon injection of oligomycin (oligo), FCCP, and rotenone plus antimycin A after mECswt-CM, mECsΔpfkfb3-CM, mECswt+lac-CM, and mECsΔpfkfb3+lac-CM stimulation (n = 4–5).
(H) RNaseq data showing OXPHOS gene expression in muscle macrophages 3 days after HLI (n = 3).
(I and J) Representative images (I) and quantification (J) of proliferating MPCs upon incubation with macrophage-derived CM, measured as percentage of EdU+ nuclei (red, EdU+; blue, hoechst).
(K and L) MPC fusion analysis: representative images of immunofluorescent DESMIN staining (K) (red, DESMIN; blue, hoechst) and quantification of the number of nuclei per DESMIN+ myotube (L).
(M) Vegf gene expression in CD45+ cells sorted from pfkfb3WT and pfkfb3ΔEC muscle at 3 d. (N) VEGF secretion by BMDMs after stimulation with mECswt-CM and mECsΔpfkfb3-CM with or without lactate supplementation.
Scale bar, 50 μm. Student’s t test (two-tailed, unpaired) in (E) and (M). One-way ANOVA with Tukey’s multiple comparisons test in (B). Two-way ANOVA with Tukey’s multiple comparisons test in (C), (D), (G), (J), (L), and (N) (∗p < 0.05) as well as in (F) (∗p < 0.05 versus mECswt-CM; #p < 0.05 versus ctrl). Each dot represents a single mouse (M) or the average of an independent experiment ([B], [C], [D], [E], [J], [L], and [N]). Bar graphs represent mean ± SEM. See also Figure S5 and Table S1.
Among other stimuli, macrophage polarization occurs in response to cytokine stimulation. We therefore analyzed the cytokine profile of CM from mECsWT and mECsΔpfkfb3 isolated from ischemic muscles but did not detect any differences (Figures S5F and S5G). Subsequently, we passed the CM from mECsWT and mECsΔpfkfb3 through a 3-kDa filter to concentrate the protein fraction and remove metabolites. Strikingly, CM of mECsWT containing only >3-kDa proteins (CM>3kDa) failed to induce arg1 and mrc1 expression in BMDMs (Figures 4C and 4D). In fact, arg1 and mrc1 expression by BMDMs was equally low after stimulation with CM>3kDa from either mECsWT or mECsΔpfkfb3. These data show that removal of metabolites from CM of mECs blunts their capacity to induce M2-like polarization. Moreover, the mEC-derived metabolite(s) that control macrophage polarization is/are not (or to a lesser extent) secreted by mECsΔpfkfb3.
Because PFKFB3 is a main glycolytic regulator in ECs and controls EC-derived lactate production, we hypothesized that EC-derived lactate might drive macrophage polarization. Indeed, in tumor-associated macrophages, lactate derived from highly glycolytic tumor cells promotes M2-like polarization through stabilizing HIF-1α in normoxia or via activating GPR132 (Chen et al., 2017, Colegio et al., 2014). However, a role for endothelial-derived lactate in determining macrophage functional polarization in vivo has not been described. In agreement with reduced glycolysis, we found lactate levels to be lower in CM derived from mECsΔpfkfb3 than in that derived from mECsWT (Figure 4E). We also performed metabolomic profiling of CM from mECsΔpfkfb3 and mECsWT focusing on metabolites that are known to play a role in macrophage function (Van den Bossche et al., 2017, Viola et al., 2019), but we did not find other metabolites that were reduced in mECsΔpfkfb3-CM (Table S1). Importantly, addition of lactate (5 mM) to mECsWT CM>3kDa, as well as to both CM and CM>3kDa from mECsΔpfkfb3, restored their capacity to induce arg1 and mrc1 (Figures 4C and 4D) as well as CD206 cell surface expression in BMDMs (Figure 4F). Similar observations were made for mgl1 and mgl2, although to a lesser extent in CM and CM>3kDa from mECsΔpfkfb3 (Figures S5H and S5I). Of note, adding lactate to mECswt-CM did not further increase arg1 and mrc1 expression (Figures 4C and 4D). Reducing endothelial lactate production via knocking down ldha or mct4 also reduced the activation of M2 marker genes (Figures S5J and S5K). Importantly, the ability of lactate to promote M2-like polarization required the presence of mECs-CM, because supplementing lactate (5–10 mM) to BMDMs in the absence of mECs-CM did not affect arg1 or CD206 expression (Figures S5L and S5M).
Alternatively, activated M2 macrophages display enhanced mitochondrial oxidative phosphorylation (OXPHOS) in comparison to M1 macrophages (Diskin and Pålsson-McDermott, 2018, O’Neill and Pearce, 2016). Because lactate, after conversion to pyruvate, can enter the tricarboxylic acid (TCA) cycle via pyruvate dehydrogenase, we also evaluated whether angiocrine lactate would promote oxygen consumption rate (OCR) in BMDMs. As expected, mECswt-CM-treated BMDMs showed higher OCR than mECsΔpfkfb3-CM-treated BMDMs. Addition of lactate to mECsΔpfkfb3-CM during the culture of the BMDMs restored OCR to similar levels when compared to mECswt-CM-treated BMDMs (Figures 4G and S5N). These data were confirmed by our transcriptomic data in primary isolated macrophage from ischemic muscle which displayed lower expression of OXPHOS-related genes in pfkfb3ΔEC (Figures 4H, S3A, and S3B).
Angiocrine-Lactate-Induced Macrophage Polarization Promotes Muscle Regeneration and VEGF Secretion
After muscle injury, macrophages that initially present with a pro-inflammatory M1-like phenotype need to switch to a repair-promoting M2-like state, and interference with this M1 to M2 fate switch impairs muscle regeneration (Arnold et al., 2007, Deng et al., 2012, Mounier et al., 2013). In fact, classically LPS/IFNγ-activated M1 macrophages promote MPC growth and proliferation but inhibit MPCdifferentiation and fusion (Arnold et al., 2007), whereas IL-4-stimulated M2 macrophages promote MPC differentiation and fusion (Saclier et al., 2013). To assess whether angiocrine-lactate-induced macrophage polarization also affects the ability of MPCs to promote muscle regeneration, we again incubated BMDMs with CM from mECsWT and mECsΔpfkfb3 to promote polarization (mECswt-CM → BMDMs and mECsΔpfkfb3-CM → BMDMs). Subsequently, mEC-derived CM was removed and BMDM-derived CM was generated (mECswt-CM → BMDMs-CM and mECsΔpfkfb3-CM → BMDMs-CM) and added to MPCs (Figure 4A). When compared to CM derived from unstimulated macrophages, mECswt-CM → BMDMs-CM stimulated MPC proliferation (Figures 4I and 4J) and improved MPCfusion into myotubes (Figures 4K and 4L). Interestingly, mECsΔpfkfb3-CM → BMDMs-CM failed to stimulate MPC proliferation and fusion. The addition of lactate to mECsΔpfkfb3-CM during polarization of BMDMs, however, restored the capacity of mECsΔpfkfb3+lac-CM → BMDMs-CM to drive MPC proliferation and fusion (Figures 4I–4L). Importantly, adding lactate to MPC culture medium did not affect proliferation or differentiation of MPCs (Figures S5O and S5P). Thus, angiocrine lactate controls the ability of macrophage to promote MPC proliferation and fusion.
Because we observed lower VEGF levels in pfkfb3ΔEC muscle, we also wondered whether angiocrine lactate would promote VEGF secretion from macrophages. Macrophages isolated from the ischemic hindlimb of pfkfb3ΔEC mice expressed lower vegf (Figure 4M), consistent with literature showing lower vegf expression by M1-like macrophages (He et al., 2012). Incubation of BMDMs with mECsΔpfkfb3-CM also resulted in lower VEGF secretion as compared to BMDMs stimulated with mECswt-CM, and this was rescued by adding lactate to the CM (Figure 4N). These data show that angiocrine lactate promotes M2-like macrophage functional polarization, which leads to increased VEGF secretion and creates a positive feedback loop to further stimulate angiogenesis.
Increasing Muscle Lactate Levels in pfkfb3ΔEC Mice Restores M2 Macrophage Content and Improves Muscle Reperfusion and Regeneration
We next asked whether angiocrine lactate could control macrophage polarization in the muscle during ischemia. To address this, we harvested ischemic muscle and assessed muscle lactate levels. Whereas induction of ischemia led to a pronounced increase in local muscle lactate levels in WT animals, this increase was almost completely abrogated in pfkfb3ΔEC mice (Figure 5A). Accordingly, circulating blood lactate levels after HLI were lower in pfkfb3ΔEC mice (Figure 5B). In agreement with these observations, lactate levels also rose more in muscle with higher vascular density (and low glycolytic potential) such as m. soleus when compared to muscles with low vascular density (and high glycolytic potential) (Figure 5C). Finally, activation of angiogenesis in the absence of muscle damage by injecting vegf-expressing myoblasts increased muscle vascular density (Figures S6A and S6B), and lactate levels were higher in pfkfb3WT muscle (Figure 5D).
Figure 5.

Increasing Muscle Lactate Levels in pfkfb3ΔEC Mice Restores M2 Macrophage Content and Improves Muscle Reperfusion and Regeneration
(A–C) Lactate levels in calf muscle (A), blood serum (B), and in m. extensor digitorum longus (EDL) and m. soleus (C) before and 12 h after HLI.
(D) Lactate concentration in tibialis anterior muscle 5 days after injection of vegf overexpressing myoblasts.
(E) Total number of macrophages determined by flow cytometry in muscle samples at 3 days.
(F and G) Representative histograms (F) and CD206 mean fluorescence intensity (MFI) (G) in total macrophages isolated from calf muscle 3 days after HLI.
(H and I) Representative laser Doppler perfusion images (H) and quantification of blood perfusion ratio (I) (n = 7) in pfkfb3WT and pfkfb3ΔEC mice with or without lactate (lac) at the indicated times.
(J) Representative images of CD31 immunofluorescent staining and quantification of CD31-positive area in muscle at 12 days (n = 5–7; scale bar, 50 μm).
(K) VEGF protein content in muscle at 12 days.
(L) H&E staining of regenerating muscle (scale bar, 10 μm).
(M) Muscle fiber size distribution 12 days after HLI (n = 4).
Student’s t test (two-tailed, unpaired) in (D). Two-way ANOVA with Tukey’s multiple comparisons test in (A), (B), (C), (E), (G), and (K) (∗p < 0.05) as well as in (J) and (M) (∗p < 0.05 versus pfkfb3WT; #p < 0.05 versus PBS). Two-way repeated-measures ANOVA with Sidak’s multiple comparisons test in (I) (∗p < 0.05). Each dot represents a single mouse ([A], [B], [C], [D], [E], [G], [K], and [M]). Bar graphs represents mean ± SEM. See also Figure S6.
Next, we tested whether lactate supplementation in pfkfb3ΔEC mice would restore M2-like polarization and muscle regeneration. We implanted growth-factor-reduced Matrigel plugs containing lactate in the subcutis of mice immediately after HLI (Figure S6C)). As previously described (Porporato et al., 2012), such plugs allow the slow release of lactate, leading to a small but consistent increase in blood lactate levels (Figure S6D). Three days after HLI, we found that lactate administration in pfkfb3ΔEC mice restored total numbers of macrophages and macrophage CD206 expression to similar levels in control mice (Figures 5E–5G). This confirms that lactate is sufficient to drive M2-like polarization in vivo. Moreover, lactate significantly improved but did not completely restore perfusion in pfkfb3ΔEC mice (Figures 5H and 5I). In agreement with the perfusion data, vascular density remained slightly lower (Figure 5J). Lactate sufficed to increase VEGF protein levels (Figure 5K). Harvesting muscle tissue at 12 days after ischemia showed that lactate led to a striking acceleration of muscle regeneration and higher muscle fiber cross-sectional area (Figures 5L–5M, S6E, and S6F). Thus, restoring lactate levels in muscle from pfkfb3ΔEC rescues macrophage polarization and muscle regeneration.
Lactate-Induced Macrophage Polarization Is MCT1 Dependent
To explore how angiocrine lactate affects macrophage polarization in muscle, we investigated whether EC-derived lactate is taken up by macrophages. We found that 14C-lactate uptake in freshly isolated primary muscle macrophages was higher in pfkfb3WT versus pfkfb3ΔEC mice 3 days after HLI (Figure 6A). This was confirmed in BMDMs, where mECswt-CM but not mECsΔpfkfb3-CM promoted lactate uptake (Figure 6B). MCT1 regulates lactate uptake in T cells, and inhibition of MCT1 during T lymphocyte activation results in selective and profound inhibition of the extremely rapid phase of T cell division essential for an effective immune response (Murray et al., 2005). We thus hypothesized that MCT1 could control lactate uptake in macrophages. We first confirmed that the MCT1 inhibitor AZD3965 reduced lactate uptake in BMDMs stimulated with mECswt-CM (Figure 6B). Based on higher OCR in lactate-stimulated macrophages, we next asked whether the incorporated lactate is actually used for oxidation. By using radioactive substrate tracing experiments, we found that M2-like macrophages oxidize lactate in an MCT1-dependent fashion, and lactate oxidation was lower in mECsΔpfkfb3-CM-treated BMDMs (Figure 6C). And third, MCT1-mediated lactate uptake is required for macrophage polarization because AZD3965 reduced the fraction of CD206+F4/80+ macrophages induced by mECswt-CM to similar levels compared to incubation with mECsΔpfkfb3-CM (Figure 6D). To confirm that MCT1-dependent lactate uptake also affected functional properties of macrophages, we used CM from BMDMs that were stimulated with mECswt-CM, mECsΔpfkfb3-CM either not supplemented with AZD3965 (mECswt+AZD-CM, mECsΔpfkfb3+AZD-CM), or ctrl (DMSO). mECswt+AZD-CM failed to stimulate MPC proliferation (Figures 6E and 6F) and differentiation (Figures 6G and 6H), as compared to stimulation with mECsWT CM → BMDMs-CM. Moreover, BMDMs stimulated with mECswt+AZD-CM secreted less VEGF (Figure 6I). Taken together, lactate uptake through MCT1 does not only promote M2-like polarization but also instructs M2-like macrophage-dependent functions.
Figure 6.

Lactate-Induced Macrophage Polarization Is MCT1 Dependent
(A) Lactate uptake in macrophages isolated from muscle 3 days after HLI.
(B) Lactate uptake in BMDMs after stimulation with mECs-CM. mECs-CM was supplemented with vehicle (ctrl), lactate, or MCT1 inhibitor AZD3965 (AZD) (mECswt+AZD-CM, mECsΔpfkfb3+AZD-CM).
(C) Lactate oxidation in BMDMs upon stimulation with mECswt-CM, mECsΔpfkfb3-CM, mECswt+lac-CM, mECsΔpfkfb3+lac-CM, mECswt+AZD-CM, and mECsΔpfkfb3+AZD-CM.
(D) Representative images of immunostainings for F4/80 (green), CD206 (red), and hoechst (blue) in BMDMs stimulated with mECswt+AZD-CM or mECsΔpfkfb3+AZD-CM and flow cytometry quantification of CD206 MFI.
(E and F) Representative images (red, EdU+; blue, hoechst) (E) and quantification (F) of EdU+ MPCs.
(G and H) Representative DESMIN staining (red, DESMIN; blue, hoechst) (G) and fusion analysis (H) upon stimulation with mECs-CM → BMDMs-CM.
(I) VEGF levels in mECs-CM → BMDMs-CM.
Scale bar, 50 μm. Student’s t test (two-tailed, unpaired) in (A) (∗p < 0.05). Two-way ANOVA with Tukey’s multiple comparisons test in (B), (C), (F), (H), and (I) (∗p < 0.05) test and in (D) (∗p < 0.05 versus mECswt-CM; #p < 0.05 versus ctrl). Each dot represents a single mouse (A) or the average of an independent experiment ([B], [C], [F], [H], and [I]). Bar graphs represent mean ± SEM.
Finally, we investigated whether lactate also controls macrophage polarization and muscle recovery from ischemia in an MCT1-dependent fashion in vivo. To this end, we generated mice lacking Mct1 in macrophages (mct1ΔMac mice; Figures 7A and 7B) (and neutrophils) by intercrossing mct1-floxed mice with myeloid-cell-specific LysM-Cre mice. Consistent with our observations in pfkfb3ΔEC mice, we found that 3 days after HLI, mct1ΔMac muscles contained more neutrophils, monocytes, and macrophages (Figure 7C). Moreover, mct1-deficient macrophages expressed less CD206 and Relmα (Figures 7D–7F). This shows that MCT1-dependent lactate uptake is required for M2-like functional polarization of macrophages upon muscle ischemia. In addition, hindlimb perfusion of mct1ΔMac mice recovered slower (Figures 7G and 7H), and CD31 stainings confirmed that mct1ΔMac reduced revascularization 28 days after HLI (Figure 7I). In agreement with our in vitro and in vivo data confirming the lack of M2-like functional repolarization upon MCT1 inhibition, mct1ΔMac muscle displayed reduced muscle regeneration. Indeed, mct1ΔMac mice had higher muscle necrosis 3 days after HLI (Figures 7J–7K and S67A), and even though the number of regenerating fibers was similar to their WT littermates, muscle fiber cross-sectional area was lower in mct1ΔMac mice (Figures 7J, 7L, S7A, and S7B). Also, mct1ΔMac muscle had lower VEGF levels (Figure 7M). Together, these results show that lactate uptake through MCT1 does not only promote M2-like polarization but also instructs M2-like macrophage-dependent revascularization upon HLI.
Figure 7.

Loss of MCT1 in Macrophages Impairs M2-like Macrophage Polarization and Muscle Recovery from Ischemia
(A) Scheme showing the generation of LysM-Cre × mct1Loxp/Loxp (mct1ΔMac) mice and HLI experiment.
(B) Mct1 mRNA expression in macrophages 3 days after HLI.
(C) Total number of neutrophils, monocytes, and macrophages determined by flow cytometry in muscle 3 days after HLI.
(D and E) Representative histograms showing CD206 (D) and Relmα (E) expression.
(F) Quantification of CD206 and Relmα MFI.
(G and H) Representative laser Doppler images (G) and quantification of hindlimb perfusion (H) in mct1WT and mct1ΔMac (n = 7).
(I) Representative images of CD31 immunostaining and quantification of CD31+ area 12 days after HLI (n = 6; scale bar, 50 μm).
(J and K) H&E staining of regenerating muscle (scale bar, 10 μm) at the indicated time (J) and quantification of necrotic area (K) 3 days after HLI.
(L) Muscle fiber size distribution at 28 days (n = 4).
(M) VEGF protein content at 3 days.
Student’s t test (two-tailed, unpaired) in (B), (C), (F), (K), and (M) (∗p < 0.05). Two-way ANOVA with Tukey’s multiple comparisons test in (L) (∗p < 0.05 versus mct1WT). Two-way repeated-measures ANOVA with Sidak’s multiple comparisons test in (H) (∗p < 0.05). Each dot represents a single mouse ([B], [C], [F], [K], and [M]). Bar graphs represent mean ± SEM. See also Figure S7.
Discussion
ECs contribute to tissue homeostasis and regeneration via angiocrine signaling (Augustin and Koh, 2017, Rafii et al., 2016). Known organotypic angiocrine signals include secreted growth factors as well as ligand/receptor interactions at the cell membrane. For instance, the release of HGF and TGF-β1 from liver sinusoidal ECs ensures spatiotemporal control of liver regeneration after partial hepatectomy (Ding et al., 2010, Hu et al., 2014, LeCouter et al., 2003). In the brain, neurotrophin-3 derived from brain ECs sustains neural stem cell quiescence in addition to membrane-bound Jagged-1 and Ephrin-B2 (Delgado et al., 2014, Ottone et al., 2014). Here, we show that ECs also exploit their unique metabolic features to engage in angiocrine communication. Within the muscle microenvironment, they shuttle lactate as a metabolic substrate to promote macrophage polarization. By doing so, ECs actively steer muscle regeneration after hindlimb ischemia.
ECs are highly glycolytic and generate the majority of their energy via the glycolytic breakdown of glucose to lactate (Culic et al., 1997, De Bock et al., 2013, Krützfeldt et al., 1990, Schoors et al., 2014, Xu et al., 2014, Yu et al., 2017). We found that mECs use lactate as a “ready-to-sprout” signal to increase VEGF secretion and promote muscle regeneration by instructing the polarization of macrophages toward an M2-like phenotype in a MCT1-dependent manner. Thus, through the release of lactate, ECs themselves shape a pro-angiogenic environment to allow optimal revascularization. Macrophage-derived VEGF subsequently consolidates a positive feedback loop, which leads to a further angiogenic activation of mECs. Interestingly, restoring VEGF levels in the muscle of pfkfb3ΔEC mice by promoting M2-like macrophage polarization (through M2 macrophage transfer or by increasing lactate levels) increased vascular density, although the latter failed to reach pfkfb3WT levels upon lactate administration. This indicates that the observed angiogenic deficit (at least in the ischemic muscle) is a combination of a direct inhibitory effect of PFKFB3 on endothelial migration/proliferation as well as reduced angiogenic stimulation by the muscle microenvironment.
It is possible that the delivery of lactate also had a direct pro-angiogenic effect in WT as well as pfkfb3ΔEC mice. In vitro, lactate renders ECs more responsive to VEGF by increasing VEGFR2 content due to enhanced HIF-1α stabilization (Sonveaux et al., 2008, Végran et al., 2011). Moreover, delivery of lactate promoted, and inhibition of lactate uptake by blocking MCTs reduced ischemia revascularization (Porporato et al., 2012). Similar observations have been made in tumors (Sonveaux et al., 2008). A potential contribution of macrophages to this pro-angiogenic effect in vivo has, to our knowledge, not been addressed. Although previous observations showed that lactate delivery resulted in higher muscle fiber area after ischemia (Porporato et al., 2012), our data revealed that lactate by itself does not promote MPCs proliferation or fusion, and it underscores the ability of lactate to reshape macrophages (and possibly other cells) in the muscle microenvironment to control optimal regeneration.
We made the striking observation that loss of endothelial pfkfb3 lowers muscle as well as plasma lactate levels upon hindlimb ischemia. If anything, we expected that the presence of highly glycolytic M1-like macrophages in combination with higher hypoxia would lead to higher lactate levels in pfkfb3ΔEC muscle. This suggests that mECs significantly contribute to muscle lactate levels, which is surprising given the modest volume of the endothelial population versus myofibers in muscle. In support of these observations, activation of angiogenesis in the absence of muscle damage (via increasing VEGF) also resulted into higher muscle lactate levels in pfkfb3WT when compared to pfkfb3ΔEC mice. Lactate is a main carbon source for energy production and can be shuttled between different cells or even organs (Brooks, 2018, Hui et al., 2017). In the muscle, the presence of a lactate shuttle from glycolytic to oxidative muscle fibers has been described for decades, predominantly in the context of exercise (Bergman et al., 1999, Brooks, 1986, Stanley et al., 1986). However, contribution of other cell types to lactate metabolism in muscle has not been studied. Here, we demonstrate the presence of a second lactate shuttling mechanism within the muscle, i.e., from the endothelium to macrophages.
Endothelial-derived lactate promoted the functional polarization of macrophages, and this was dependent on lactate uptake because its inhibition by using MCT1 inhibitors in vitro or deletion of mct1 from macrophages in vivo prevented M2-like polarization. Loss of MCT1 in macrophages decreased muscle VEGF content and reduced revascularization as well as muscle regeneration. Interestingly, angiocrine lactate sufficed to increase macrophage oxygen consumption, a typical feature of M2 macrophages (Pearce and Pearce, 2013). Also, M2-like macrophages oxidized lactate, and lactate oxidation rate was higher when compared to macrophages which were stimulated with mECsΔpfkfb3-CM. Gene profiling of macrophages isolated from ischemic hindlimb of pfkfb3WT mice confirmed that genes involved in OXPHOS were enriched, whereas this was not the case in pfkfb3ΔEC mice. This shows that endothelial glycolysis can contribute to metabolic reprogramming in macrophages by providing additional substrates for oxidative phosphorylation. We did not detect differences in baseline lactate levels in muscle. However, because lactate is continuously produced and consumed by different cells, even under fully aerobic conditions (Brooks, 1986, Hui et al., 2017), future research will be needed to unravel the exact contribution of endothelial lactate to muscle physiology.
The ability of lactate to control macrophage polarization required conditioned medium, indicating that the presence of other cytokines or metabolites was required for functional repolarization. Raising lactate concentration to 5–10 mM sufficed to induce M2-like skewing in the presence of conditioned medium but failed to do so in the absence thereof. This is in apparent contrast with previous observations in tumor settings, where lactate itself sufficed to induce macrophage skewing via MCT-dependent uptake (Colegio et al., 2014) or via acting as a signaling molecule through initiating GPR132 signaling (Chen et al., 2017). It is worthwhile noting that the lactate concentrations used in the present study (5 mM) are within physiological ranges: exercising at high intensity, even in mice (Ayachi et al., 2016), raises blood lactate levels above 5–10 mM. We found an approximately 3-fold increase in muscle lactate concentration 12 h after ischemia. In addition, although adding lactate to the conditioned medium restored the expression of CD206, not all M2 linked genes were completely rescued. Also, expression levels of many M2 genes were induced to a lower extent by lactate compared to IL-4 despite similar CD206 membrane expression. This is not surprising, as in vivo transcriptional profiling of macrophages upon cardiotoxin injury only showed partial overlap with canonical M1/M2 profiles (Varga et al., 2016). Nonetheless, lactate polarized macrophages recapitulated many functional properties of IL-4-stimulated macrophages, such as their pro-angiogenic properties (Jetten et al., 2014) as well as their ability to promote muscle regeneration (Saclier et al., 2013). Our current data therefore indicate that the metabolic and transcriptional alterations induced by lactate are sufficient to acquire pro-regenerative and pro-angiogenic properties.
Macrophages are key regulators of muscle regeneration (Dort et al., 2019). Initially, they have a pro-inflammatory M1-like phenotype, which is associated with the expression of several cytokines that further promote inflammation (Mosser and Edwards, 2008). Rapidly thereafter, those M1-like macrophages repolarize and change their phenotype toward a more anti-inflammatory, pro-angiogenic, and pro-regenerative M2-like phenotype (Arnold et al., 2007, Mosser and Edwards, 2008, Raes et al., 2002). This M1 to M2 repolarization is required for optimal muscle regeneration (Arnold et al., 2007, Deng et al., 2012, Tidball and Wehling-Henricks, 2007). The contribution of ECs to macrophage differentiation and maturation is poorly understood. ECs can promote macrophage polarization in a contact-dependent manner (He et al., 2012, Krishnasamy et al., 2017), but only limited evidence exists which supports metabolic crosstalk between macrophages and ECs. In the tumor microenvironment, low glucose availability forces macrophages and ECs to compete for glucose (Wenes et al., 2016). During hypoxia, tumor-associated macrophages shift toward a more oxidative metabolism and reduce glucose uptake. This increases glucose availability for ECs, thereby promoting excessive angiogenesis (Wenes et al., 2016). Although we cannot exclude a similar scenario being active in the ischemic hindlimb, our data suggest that ECs and macrophages metabolically collaborate to promote muscle regeneration. They efficiently share the available energetic substrate glucose via the glycolytic breakdown to lactate by ECs followed by lactate oxidation in macrophages.
In conclusion, we provide evidence that ECs exploit their unique metabolic characteristics to steer muscle regeneration during ischemia in an angiocrine fashion that is dependent on lactate shuttling to macrophages. Metabolic angiocrine signaling provides a novel mechanism through which ECs can contribute to tissue homeostasis and regeneration.
Limitations of Study
Although our study shows that angiocrine lactate controls M2-like macrophage polarization in an MCT1-dependent manner, the exact molecular mechanism through which lactate drives M2-like polarization remains to be unveiled. To further dissect the kinetics of angiocrine metabolic crosstalk, it would be insightful to have genetic mouse models to selectively uncouple endothelial lactate secretion from angiogenesis. Furthermore, arteriogenic collateral formation also contributes to muscle reperfusion upon hindlimb ischemia, but arteriogenesis was not evaluated in this study.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD45, BV785, clone 30-F11 | Biolegend | Cat#103149; RRID: AB_2564590 |
| Anti-mouse CD11b, PerCP-Cy5.5, clone M1/70 | Biolegend | Cat#101228; RRID: AB_893232 |
| Anti-mouse F4/80, Alexa Fluor 488, clone BM8 | Biolegend | Cat#123120; RRID: AB_893479 |
| Anti-mouse F4/80, Biotin, clone BM8 | Biolegend | Cat#123105; RRID: AB_893499 |
| Anti-mouse CD16/32, clone 2.4G2 | This paper | N/A |
| Anti-mouse CD11b, PerCP-Cy5.5, clone M1/70 | Biolegend | Cat#101228; RRID: AB_893232 |
| Anti-mouse CD11c, BV605, clone N418 | Biolegend | Cat#117334; RRID: AB_2562415 |
| Anti-mouse Ly-6G, BV421, clone 1A8 | Biolegend | Cat#127628; RRID: AB_2562567 |
| Anti-mouse CD45, APC/Fire™ 750, clone 30_F11 | Biolegend | Cat#103154; RRID: AB_2572115 |
| Anti-mouse CD31, Alexa Fluor 488, clone 390 | Biolegend | Cat#102414; RRID: AB_493408 |
| Anti-mouse CD31, PE, Clone 390 | Thermo Fisher Scientific | Cat#12-0311-82; RRID: AB_465632 |
| Anti-mouse Ly-6C, PE-Cy7, clone HK1.4 | Biolegend | Cat#128018; RRID: AB_1732082 |
| Anti-mouse CD206, PerCP-Cy5.5, clone C068C2 | Biolegend | Cat#141716; RRID: AB_2561992 |
| Anti-mouse CD206, Alexa Fluor 647, clone MR5D3 | BD Biosciences | Cat#565250; RRID: AB_2739133 |
| Goat Anti-Rabbit IgG(H+L), Mouse/Human ads-FITC | SouthernBiotech | Cat#4050-02; RRID: AB_2795952 |
| Anti-mouse CCR2, PE, Clone # 475301 | R&D Systems | Cat#FAB5538P-100; RRID: AB_10718414 |
| Anti-mouse MHC class II, BV510, clone M5/114.15.2 | Biolegend | Cat#107635; RRID: AB_2561397 |
| Anti-mouse CD64, APC, clone X54-5/7.1 | Biolegend | Cat#139306; RRID: AB_11219391 |
| Anti-mouse MERTK, FITC, clone 2B10C42 | Biolegend | Cat#151504; RRID: AB_2617035 |
| Anti-mouse CX3CR1, APC, clone SA011F11 | Biolegend | Cat#149008; RRID: AB_2564492 |
| Anti-Murine RELMα | Peprotech | Cat#500-P214; RRID: AB_1268332 |
| Ki-67 | Cell Signaling Technology | Cat#9129S; RRID: AB_2687446 |
| Cleaved Caspase-3 (Asp175) | Cell Signaling Technology | Cat#9661; RRID: AB_2341118 |
| CD31 | R&D Systems | Cat#3628; RRID: AB_2161028 |
| Anti-F4/80, clone A3-1 | Abcam | Cat#ab6640; RRID: AB_10770512 |
| Laminin | Thermo Fisher Scientific | Cat#PA1-16730; RRID: AB_2133633 |
| Anti-Mannose Receptor | Abcam | Cat#ab64693; RRID: AB_1523910 |
| Anti-PFKFB3 | Abcam | Cat#ab181861; RRID: N/A |
| β-actin | Cell Signaling Technology | Cat#ab181861; RRID: AB_2566811 |
| Desmin | Cell Signaling Technology | Cat#5332; RRID: AB_1903947 |
| Donkey anti-Goat IgG (H+L) AF 488 | Thermo Fisher Scientific | Cat#A-11055; RRID: AB_2534102 |
| Donkey anti-rabbit IgG (H+L) AF 568 | Thermo Fisher Scientific | Cat#A10042; RRID: AB_2534017 |
| Donkey anti-rabbit IgG (H+L) AF plus 647 | Thermo Fisher Scientific | Cat#A32795; RRID: AB_2762835 |
| Donkey anti-rat IgG (H+L) AF 488 | Thermo Fisher Scientific | Cat#A-21208; RRID: AB_2535794 |
| Donkey anti-rat IgG (H+L) DyLight 650 | Thermo Fisher Scientific | Cat#SA5-10029; RRID: AB_2556609 |
| rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat#7074S; RRID: AB_2099233 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Streptavidin-BV421 | Biolegend | Cat#405225 |
| Streptavidin-BV711 | BD Biosciences | Cat#563262 |
| eFluor® 780 | Thermo Fisher Scientific | Cat#65-0865-14 |
| EdU | Thermo Fisher Scientific | Cat#C10632 |
| AZD3965 | Cayman Chemical | Cat#1448671-31-5 |
| Tamoxifen | Sigma-Aldrich | Cat#T5648 |
| Collagenase IV | Thermo Fisher Scientific | Cat#17104019 |
| Dispase II | Sigma-Aldrich | Cat#D4693 |
| M-CSF | PeproTech | Cat#315-02 |
| Sodium L-lactate | Sigma-Aldrich | Cat#L7022 |
| Endothelial cell growth supplement | Sigma-Aldrich | Cat#E2759 |
| Hoechst | Thermo Fisher Scientific | Cat#62249 |
| SYTOX® Blue | Thermo Fisher Scientific | Cat#S34857 |
| SYTOX™ Red | Thermo Fisher Scientific | Cat#S34859 |
| Normal Donkey Serum | Jackson Immuoresearch | Cat#017-000-121 |
| FGF-Basic (AA 1-155) Recombinant Human Protein | Thermo Fisher Scientific | Cat#PHG0266 |
| Puromycin | Sigma-Aldrich | Cat#P8833 |
| Deoxyribonuclease I | Sigma-Aldrich | Cat#D4527 |
| Critical Commercial Assays | ||
| Mouse VEGF Quantikine ELISA Kit | R&D Systems | Cat#MMV00 |
| Proteome Profiler Mouse XL Cytokine Array | R&D Systems | Cat#ARY028 |
| Hypoxyprobe™ Plus Kit | Hypoxyprobe, Inc | Cat# HP2-100Kit |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat#103015-100 |
| DC™ Protein Assay Kit I | Bio-Rad | Cat#5000111 |
| Clarity Western ECL Substrate | Bio-Rad | Cat#1705061 |
| Lactate-Glo™ assay | Promega | Cat#J5021 |
| Click-iT™ Cell Reaction Buffer Kit | Thermo Fisher Scientific | Cat#C10269 |
| Experimental Models: Organisms/Strains | ||
| Mouse: mct1fl/fl | Cyagen Bioscience | N/A |
| Mouse: Rosa26mTmG | Muzumdar et al., 2007 | https://www.jax.org/strain/007676 |
| Mouse: LysMCre | Clausen BE et al.,1999 | N/A |
| Mouse: LysMCre-vegffl/fl | Stockmann et al.,2008 | N/A |
| Mouse: pfkfb3Loxp/Loxp | This paper | N/A |
| Mouse: pdgfb-CreERT2 | Claxton et al., 2008 | N/A |
| Metabolites | ||
| See Table S1 for LC-MS/MS mediated determination of metabolites. | This paper | N/A |
| Deposited Data | ||
| RNA-sequencing data | This paper | GSE148584 |
| Oligonucleotides | ||
| See Table S2 for oligonucleotide sequences used for real-time quantitative PCR. | This paper | N/A |
| Software and Algorithms | ||
| FlowJo Software (version 10.4.2) | Three Star | https://www.flowjo.com/ |
| ImageJ (for image analysis) | NIH | https://imagej.nih.gov/ij/ |
| Prism 8 (version 8.0.0) | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Adobe Illustrator CS6 (version 16.0.4) | Adobe | https://www.adobe.com/ |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Katrien De Bock (katrien-debock@ethz.ch).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The mouse RNA-seq data reported in this study are available at the Gene Expression Omnibus (GEO) repository under the accession number (GSE148584). All other data are available from the Lead Contact on request.
Experimental Models
Mice
Pfkfb3LoxP/LoxP mice were generated using homologous recombination in embryonic stem (ES) cells. The targeting vector was built in the pComTrue plasmid (Reich et al., 2011) (kindly provided by M. Baes, KU Leuven) with Frt sites flanking a neomycine resistance (neor) cassette, and containing from 5′ to 3′: a 1.78-kb NotI fragment comprising exon 2, a floxed 4.4-kb AscI fragment comprising exon 3, 4, 5 and 6 as 5′ homology arm, a 1.2-kb Frt flanked neor cassette, and a 2.38-kb BamHI fragment comprising exon 7 as 3′ homology arm. The construct was linearized with PmeI and electroporated into G4 ES cells of 129SvEv/C57BL6 origin (kind gift from A. Nagy, Toronto) as described (Aragonés et al., 2008). After positive-negative drug selection with 200 μg/mL G418 (Invitrogen), resistant clones were analyzed for correct homologous recombination by appropriate Southern blotting and PCR. Correctly targeted ES cells were then transiently electroporated with the Flp recombinase expressing plasmid pCAGGS-Flpe (Pieters et al., 2017) to excise the neor cassette, and used for morula aggregation to generate chimeric and germline pfkfb3+/LoxP offspring. Genotyping on genomic DNA from Flp-recombined ES cells and from mouse tails was done by PCR, using the forward primer 5′-cac ctg agc aac att gta act-3′ and reverse primer 5′-cag gcc cag acc aag gac agc −3′, revealing a 185-bp and 300-bp amplicon for the wild type and floxed pfkfb3 allele, respectively.
To obtain inducible EC-specific pfkfb3-knockout (pfkfb3ΔEC) mice, pfkfb3LoxP/LoxP mice were intercrossed with pdgfb-CreERT2 mice, an EC-selective inducible Cre-driver line (Claxton et al., 2008). To evaluate recombination specificity, pdgfb-CreERT2 mice were intercrossed with Rosa26mTmG mice (Muzumdar et al., 2007). Recombination was induced in 8-14 weeks old mice by daily intraperitoneal administration of 1mg tamoxifen (T5648, Sigma-Aldrich) dissolved in 1:10 ethanol: corn oil solution for 5 consecutive days. A wash out period of at least 7 days was allowed before starting the experiments. Tamoxifen-treated Cre-negative littermates were used as control for all experiments.
To obtain myeloid cell specific deletion of mct1 (Slc16a1), mice carrying floxed alleles for mct1 (exon 5, mct1 LoxP/LoxP) were generated by Cyagen, Santa Clara CA, USA in collaboration with L. Pellerin, and intercrossed with LysM-Cre mice (Clausen et al., 1999). LysM-Cre x Vegf LoxP/LoxP mice (Gerber et al., 1999, Stockmann et al., 2008) were obtained in collaboration with C. Stockmann.
Mice were randomly allocated to different treatment groups, and the investigator was blinded to the group allocation during the experiment as well as during the analysis. Mice were housed in individually ventilated cages at standard housing conditions (22°C, 12 h light/dark cycle, dark phase starting at 7pm), with ad libitum access to chow diet (18% proteins, 4.5% fibers, 4.5% fat, 6.3% ashes, Provimi Kliba SA) and water. Health status of all mouse lines was regularly monitored according to FELASA guidelines. All animal experiments were approved by the local animal ethics committee (Kantonales Veterinärsamt Zürich, licenses ZH123/17, and ZH014/16), and performed according to local guidelines (TschV, Zurich) and the Swiss animal protection law (TschG).
Cell Culture
Isolated primary mouse skeletal muscle endothelial cells (mECs) were cultured in Endothelial Cell Growth Medium-2 (EGM2) (CC-3162, Lonza, Basel, Switzerland) or in a 1:1 ratio of M199 ((11150059, ThermoFisher Scientific) supplemented with 20% fetal bovine serum (FBS) (10270-106, ThermoFisher Scientific), 2mM L-glutamine (25030081, ThermoFisher Scientific) and 30 mg/L endothelial cell growth factor supplements (EGCS) (E2759, Sigma-Aldrich)) and Endopan 3 (P04-0010K, PAN BIOTECH) (denoted as M/E). Freshly isolated MPCs were cultured in a 1:1 ratio of DMEM (ThermoFisher Scientific, 12320032) and Ham’s F-10 (1X) nutrient mix (22390058, ThermoFisher Scientific) supplemented with 10% horse serum (HS, 16050-122, ThermoFisher Scientific), 20% FBS and 10 ng/mL basic-FGF (PHG0266, ThermoFisher Scientific) on dishes coated with Matrigel Basement Membrane Matrix (#356237, Corning, 1:25 dilution). To induce myogenic differentiation MPCs were seeded at 70,000 cells/well in 12-well plates coated with Matrigel and cultured in differentiation medium (low-glucose DMEM (22320022, ThermoFisher Scientific) supplemented with 2% HS) for 2 days. Bone-marrow precursor cells were differentiated into BMDMs in RPMI-1640 medium (31870025, ThermoFisher Scientific) supplemented with 20 ng/mL recombinant M-CSF (315-02, PeproTech), 2mM L-glutamine (25030081, ThermoFisher Scientific), 10mM HEPES (15630056, ThermoFisher Scientific), 50 μm 2-Mercaptoethanol (31350010, ThermoFisher Scientific), 10% FBS. Medium was changed on day 3 and day 6. On day 7, adherent BMDMs were detached by washing the plates with cold DPBS (14190250, ThermoFisher Scientific) with 2mM EDTA (E8008, Sigma) and used in experiments. All media was supplemented with 100 units/mL penicillin and 100 μg/mL streptomycin (15140122, ThermoFisher Scientific). Cells were routinely cultured at 37°C in 21% O2 and 5% CO2. Cells were regularly tested for the presence of mycoplasma.
Method Details
Isolation of Endothelial Cells
Primary ECs from skeletal muscle (mECs) were isolated from adult pfkfb3ΔEC and pfkfb3WT littermates. Mice were euthanized, all hind-limb muscles were immediately dissected, and muscles were minced in a Petri dish on ice using a surgical blade. Next, the minced muscle tissue was enzymatically digested in digestion buffer containing 2 mg/mL Dispase II (D4693, Sigma-Aldrich, Steinheim, Germany), 2 mg/mL Collagenase IV (17104019, ThermoFisher Scientific, Massachusetts, USA) and 2 mM CaCl2 in PBS at 37°C for 40 min, with gentle shaking every 10 min. The reaction was stopped by adding an equal volume of 20% FBS in HBSS and the suspension was passed through a series of 100-μm cell strainers (#352360, Corning, New York, USA) and 70-μm cell strainers (#352350, Corning, New York, USA) to remove tissue debris. After a series of centrifugation and washing steps, the heterogeneous cell population was purified by FACS or by puromycin selection. For FACS, ECs were sorted based on positive CD31 staining and the absence of CD45 staining. For puromycin selection, the pellet was resuspended in EC culture medium and seeded in collagen type I (125-50, Sigma)-coated plates. Due to the higher expression of P-glycoprotein in ECs compared to other skeletal muscle cells, mECs were selected by adding 4 μg/mL puromycin (P8833, Sigma-Aldrich, St. Louis, USA) to the medium overnight. After 7 days in culture, the purity of mECs was determined by CD31 fluorescence staining and only cultures containing at least 85% of the cells positive for CD31 were used for further experiments.
Isolation Primary Myogenic Progenitor Cells (MPCs)
MPCs were extracted as described before (D’Hulst et al., 2020). In brief, muscle tissue was digested in HBSS supplemented with 1.5% bovine serum albumin (BSA) (9048-46-8, Pan Reac AppliChem) and 2 mg/mL collagenase type II (ThermoFisher Scientific, 17101015) for 1 h at 37°C. After centrifugation, the cell pellet was then filtered using 100 and 40 μm cell strainers and a heterogeneous cell population was purified by FACS. For FACS, MPCs were sorted based on positive alpha 7-integrin staining and the absence of Sca1, CD31, and CD45 staining.
Isolation of Bone Marrow-Derived Macrophages (BMDMs)
Bone-marrow-derived macrophages (BMDMs) were obtained from bone-marrow precursor cells by flushing the femur and tibiae of sex-matched 6 to 12-week old mice (Freigang et al., 2013).
Conditioned Medium (CM)
mEC-derived CM: mECs isolated from adult pfkfb3ΔEC and pfkfb3WT were cultured in 12-well plate and cultured until they reached confluency (7 days). Medium was then refreshed with M/E medium and mECs were cultured in this media for 48 h. After 48 h the culture medium was collected and filtered through a 0.2 μm filter (431219, brunschwig) to obtain conditioned medium (mECswt-CM and mECsΔpfkfb3-CM). Fractionation of the mECs-CM was achieved using Amicon Ultra centrifugal filters (3KUltracel, Millipore) following centrifugation at 4,000 rpm for 1 h. The > 3kDa fraction was resuspended in unsupplemented M/E to equal the pre-filtration volume of the CM. Macrophage-derived CM: BMDMs were seeded at 400,000 cells/well. When the cells were adherent (approximately 2 h later) medium was replaced with mEC-derived CM (see paragraph above) mixed in a 1:1 ratio with RPMI-1640 medium. In some experiments, mEC-derived CM was supplemented with 5mM Sodium L-lactate (L7022, Sigma) and/or 250nM AZD3965 (1448671-31-5, Cayman Chemical) or DMSO as a control. After 48 h, the polarized BMDMs were briefly washed, and MPC growth medium or differentiation medium was added for 24 h to generate the BMDM-derived CM. Thereafter, the culture medium (mECswt-CM→BMDMs-CM, mECsΔpfkfb3-CM→BMDMs-CM, mECswt+lac-CM→BMDMs-CM, mECsΔpfkfb3+lac-CM→BMDMs-CM, mECswt+AZD-CM→BMDMs-CM, mECsΔpfkfb3+AZD-CM →BMDMs-CM) was collected as described above.
MPCs Proliferation
MPCs were seeded at 100,000 cells/well per well in a 12-well plate in MPC growth media. As a measure of proliferation, incorporation of 5-ethynyl-2’-deoxyuridine (EdU) was assessed using the Click-iT Cell Reaction Buffer Kit (C10269, ThermoFisher Scientific, Massachusetts, USA) according to the manufacturer’s instructions. Briefly, MPCs were incubated under standard growth conditions with 10 μM EdU for 2 h. Thereafter, cells were fixed with 4% PFA for 10 min at room temperature and washed twice with 3% BSA in PBS. Cells were permeabilized for 20 min at room temperature in 0.5% Triton X-100 with 3% BSA in PBS, then washed twice with 3% BSA in PBS and incubated with the Click-iT reaction cocktail for 45 min in the dark at room temperature. Thereafter, cells were briefly washed and counterstained with Hoechst (#62249, ThermoFisher Scientific). MPCs were imaged using an AxioObserver.Z1 fluorescence microscope (Carl Zeiss, Oberkochen, Germany). The percentage of EdU-positive MPCs was calculated in at least 15 random fields.
Fusion Determination
MPCs were seeded at 70,000 cells/well per well in a 12-well plate and cultured in differentiation media for 48 h (see cell culture section). Myotubes were fixed with 2% PFA for 5 min at room temperature and washed twice with PBS. Cells were blocked for 30 min at room temperature in blocking buffer (PBS supplemented with 0.5% Triton X-100 and 2% BSA) and subsequently incubated with antibodies against DESMIN (5332, Cell Signaling, 1:100) diluted in blocking buffer for 1 h at room temperature. After washing, cells were incubated with secondary antibody (goat anti-rabbit conjugated to Alexa Fluor 568, A11011, ThermoFisher Scientific, 1:400) diluted in blocking buffer for 1 h. Thereafter, cells were counterstained with Hoechst. The number of nuclei per myotube was then determined by counting the number of Hoechst+ nuclei per DESMIN-positive myotube (only DEMSIN+ myotubes with 3 or more nuclei cells were considered).
Hindlimb Ischemia Model
Hind-limb ischemia experiments were performed as described before with minor modifications (Limbourg et al., 2009, Zhang et al., 2017). Briefly, mice were anesthetized with isoflurane, the hind limb was shaved, and, following a small incision in the skin, both the proximal end of the femoral artery and the distal portion of the saphenous artery were ligated. The artery and all side-branches were dissected free; after this, the femoral artery and attached side-branches were excised. Immediately after surgery, perfusion was measured by Laser Doppler Imaging of plantar regions of interest (Moor Instruments Ltd, Axminster, Devon, England) and calculated as ratio of left (ligated) versus right (unligated) values. For lactate rescue experiments in vivo, a 100 μL plug of growth factor-reduced Matrigel (BD Biosciences) supplemented with 150 μM rotenone (Sigma) containing 150 mM sodium L-lactate (Sigma) (Lactate) or an equal volume of PBS (control) was directly implanted adjacent to the lesion during the ligation surgery, as described (Porporato et al., 2012). For macrophage transfer experiments, BMDMs were freshly isolated (see isolation and culture of BMDM section) and stimulated with IL-4 (20ng/mL; PeproTech) or vehicle (0.1% BSA) for 2 days. Then, 5 × 105 cells diluted in 50μL DPBS were injected into recipient calf muscles 3 days after HLI. For EdU labeling, mice were i.p. injected with 1.25mg EdU (LifeTechnologies) 20 h before tissue collection.
For myoblast transfer experiments, VEGF or LacZ/control overexpressing myoblasts (Ozawa et al., 2004) were dissociated in trypsin and resuspended at a concentration of 108 cells/mL in sterile PBS with 0.5% BSA, and 106 myoblasts were injected into Tibialis anterior (TA) muscles of pfkfb3WT and pfkfb3ΔEC mice, respectively.
Immunohistochemistry and Histology
Calf muscle samples were harvested and embedded in Tissue-Tek and frozen in liquid N2-cooled isopentane. Skeletal muscle cryosections (10 μm) were fixed in ice-cold acetone for 5 min, washed twice with PBS and subsequently incubated for 1 h in blocking buffer (PBS with 1% BSA) at room temperature. Thereafter, samples were incubated overnight at 4°C with primary antibodies diluted in blocking buffer with or without addition of 0.1% Triton X-100. The following primary antibodies were used: anti-CD31 (AF3628, R&D Systems, 1:250), anti-F4/80 (ab6640, Abcam, 1:200), anti-laminin (PA1-16730, Thermo Fisher, 1:250), anti-MRC1/CD206 (ab64693, Abcam, 1:50), cleaved caspase-3 (#9661, Cell Signaling, 1:100), anti-Ki67 (#9129S, Cell Signaling, 1:200). Slides were subsequently washed in PBS and incubated for 1 h in blocking buffer with the appropriate secondary antibodies at 1:250 dilution. Nuclei were stained with Hoechst. Muscle hypoxia was detected with Hypoxyprobe™ Plus Kit (Hypoxyprobe, Inc, HP2-100Kit) according to the manufacturer’s protocol. Briefly, mice received an intraperitoneal injection of 100 mg/kg pimonidazole HCl (20 mg/mL in 0.9% NaCl). One h after injection, muscles were collected and frozen as described above. Hypoxyprobe was detected by incubating the samples with anti-pimonidazole FITC-conjugated mouse IgG1 monoclonal antibody (FITC-Mab1) and rabbit anti-FITC conjugated with horseradish peroxidase. H&E staining was used to quantify the necrotic and regenerating muscle fibers. Tissue necrosis was identified by morphological alterations of myofibers or loss of sarcolemmal integrity and by the presence of cellular debris and many mononuclear cell infiltrates in the surrounding interstitial space. Regenerating areas were identified by the dominant presence of fibers with centrally located nuclei and/or some remaining mononuclear cell infiltrates. For immunostaining of whole-mounted muscle bundles, gastrocnemius muscle was fixed in 2% PFA for 1 h at 4°C, and washed with PBST (0.2% Triton X-100 in PBS). A small bundle of muscle was carefully dissected by fine forceps from the center portion of the calf muscle. The muscle tissues were then blocked with 1% BSA in PBST for 1 h at room temperature. The tissues were incubated rocking at 4°C for 48 h with anti-CD31 (1:100) antibody. Subsequently, tissues were washed overnight in PBS at 4°C, followed by incubation with secondary antibodies (1:250) overnight at 4°C. Images were captured with a Zeiss Axio observer Z.1 or an Olympus confocal microscope (FV1200). Fiber cross-sectional area was automatically determined on laminin stained sections with the Muscle J plugin for ImageJ software (Mayeuf-Louchart et al., 2018). In the myoblast injection experiment, vascular density (% CD31+ area) was quantified within the areas of myoblast implantation with ImageJ software after threshold processing on 20x images acquired with a Nikon Eclipse Ti2 microscope (Nikon, Egg/Zürich, Switzerland).
RNA Extraction and Quantitative RT-PCR
RNA of mECs and BMDMs was extracted using a RNeasy Plus Micro Kit according to the manufacturer’s instructions (QIAGEN, 74034). RNA purity and concentration were assed via a spectrophotometer (Tecan, Spark). RNA was reverse-transcribed to cDNA by High Capacity cDNA Reverse Transcription Kit (Thermo Fisher, 43-688-13). A SYBR Green-based master mix (ThermoFisher Scientific, A25778) was used for real-time qPCR analysis with primers listed in Table S2. To compensate for variations in RNA input and efficiency of reverse-transcription, 18S was used as a housekeeping gene. The delta-delta CT method was used to normalize the data.
RNA Sequencing and Differential Gene Expression Analysis
RNA sequencing was performed by Funcional Genomics Center Zurich (FGCZ). The quality and quantity of isolated RNA and final libraries were determined using Qubit® (1.0) Fluorometer and the Tapestation (Agilent, Waldbronn, Germany). Sequencing libraries were prepared following SMARTer® Universal Low Input RNA Kit for Sequencing. Briefly, total RNA samples (0.25–10 ng) were reverse-transcribed using random priming into double-stranded cDNA in the presence of a template switch oligo (TSO). Ribosomal cDNA was cleaved by ZapR in the presence of the mammalian-specific R-Probes. Remaining fragments were enriched with a second round of PCR amplification using primers designed to match Illumina adapters. The product is a smear with an average fragment size of approximately 360 bp. The libraries were normalized to 10nM in Tris-Cl 10 mM, pH8.5 with 0.1% Tween 20. For mapping and trimming of FASTQ format sequences was performed using TrimGalore, and sequence quality control was assessed using FastQC. Alignment to the Ensembl reference genome GRCm38 was performed using the Hisat2 aligner. Gene expression values were computed with the function featureCounts from the R package Rsubread. A minimum expression of each gene (mean of counts > 1) was applied as a cut off before analysis. Differential gene expression was computed using Negative Binomial model implemented in the Bioconductor package DESeq. Significantly differentially expressed genes was defined as a pvalue < 0.01 with a false discovery ratio (FDR) < 0.1. FDR values were calculated using the Benjamini–Hochberg method. Pathway analysis was performed using the clusterProfiler R package.
Immunoblot Analysis
Cells were collected and lysed with [50 mM Tris–HCl pH 7.0, 270 mM sucrose, 5 mM EGTA, 1 mM EDTA, 1 mM sodium orthovanadate, 50 mM glycerophosphate, 5 mM sodium pyrophosphate, 50 mM sodium fluoride, 1 mM DTT, 0.1% Triton X-100 and a complete protease inhibitor tablet (Roche Applied Science)]. Lysates were centrifuged at 10000 g for 10 min at 4°C. Supernatant was collected, and protein concentration was measured using the DC protein assay kit (5000116, Bio-rad). 5-10 μg of total protein was loaded in a 10-well pre-casted gradient gel (456-8086, Bio-Rad). After electrophoresis, a picture of the gel was taken under UV-light to determine protein loading using stain-free technology. Proteins were transferred onto a PVDF membrane (Bio-rad, 170-4156) with a semi-dry system and subsequently blocked for 1 h at room temperature with 5% milk in 0.1% TBS-Tween. Membranes were incubated overnight at 4°C with primary antibodies listed in Key Resources Table. The appropriate HRP-linked secondary antibodies (see Key Resources Table) were used for chemiluminescent detection of proteins. Membranes were scanned with a Chemidoc imaging system (Bio-rad) and quantified using Image Lab 6 software (Bio-rad).
Enzyme-Linked Immunosorbent Assay (ELISA)
Skeletal muscle tissue samples (10-15 mg) were homogenized with a tissue homogenizer (Omni THq) in ice-cold lysis buffer (1:15 w/v) as described above. Homogenates were centrifuged at 10000 g for 10 min at 4°C, and VEGF was measured in the supernatants using the Mouse VEGF Quantikine ELISA Kit (R&D System, MMV00) according to the manufacturer’s protocol. The same kit was used to measure VEGF levels in BMDMs-derived CM.
In Vitro Chemokine Measurement
Chemokine expression levels in the culture supernatants were measured using the Proteome Profiler Mouse XL Cytokine Array (R&D, ARY028). 400 μL of culture supernatants were applied to each membrane. The results were further normalized to the protein concentrations of the endothelial cell lysates. Staining and exposure were performed according to the manufacturer’s instructions. Membranes were scanned with a Chemidoc imaging system (Bio-rad) and quantified using Image Lab 6 software (Bio-rad).
Metabolism Assays
Lactate levels: Lactate concentration in the medium, tissue or blood was determined using the Lactate-Glo Assay (Promega) or L- Lactate Assay kit (Abcam) according to the manufacturer’s protocol. Glycolytic flux: Glycolytic flux measurements were performed as previously described (Veys et al., 2019). Briefly, cells were incubated for 2 h in culture medium containing D-[5-3H(N)]-glucose (NET53100, PerkinElmer, Zaventem, Belgium) at a final concentration of 0.4 μCi/mL medium. The supernatant was then transferred into glass vials sealed with rubber stoppers, and 3H2O was captured in hanging wells using a H2O-soaked Whatman paper for 48 h at 37°C to reach saturation. Radioactivity in 3H-labeled paper was determined by liquid scintillation counting (LSC) and the glycolytic flux was measured by the rate of 3H2O production. 14C-Lactate uptake: Cells were incubated for 6 h in RPMI containing 1 μCi/mL L-[14C(U)] - Lactic Acid, Sodium Salt (NEC599050UC, PerkinElmer, Zaventem, Belgium) and 5 mM sodium lactate. Cells were washed and lysed with 1 N NaOH at room temperature for 30 min and the radioactivity in the homogenate was determined by LSC. 14C-Lactate oxidation: Cells were incubated for 6 h in RPMI containing 1 μCi/mL L-[14C(U)] - Lactic Acid, Sodium Salt (NEC599050UC, PerkinElmer, Zaventem, Belgium) and 5 mM sodium lactate. Thereafter, 2 M perchloric acid was added to each well to stop cellular metabolism, and wells were immediately covered with a 1 x hyamine hydroxide-saturated Whatman paper. Overnight absorption of 14CO2 released during the oxidation of lactate into the paper was performed at room temperature, and radioactivity in the paper was determined by LSC. Extracellular acidification rate (ECAR) and oxygen consumption (OCR) were determined using a Seahorse XF-96 Extracellular Flux Analyzer (Seahorse Bioscience). BMDMs were seeded at 100,000 cells/well in 96-well plates and treated with mEC-derived CM supplemented with lactate or AZD3965 overnight. The assay medium was unbuffered RPMI-1640 supplemented with 10 mM glucose, 1 mM pyruvate, and 2 mM L-glutamine, pH 7.4. The measurements were performed at 4.5-min intervals (30 s mixing, 1 min recovery, 3 min measuring) for 1.5 h. Baseline ECAR and OCR and their response to the indicated compounds were determined. Inhibitors were used at the following concentrations: 2.5 μM oligomycin, 1.5 μM FCCP, 500 nM rotenone, and 500 nM antimycin A (all from Seahorse Bioscience). Pathway analysis was performed using the clusterProfiler R package.
Mass Spectrometry Analysis
Metabolite abundances were analyzed by liquid chromatography–mass spectrometry as previously described (Elia et al., 2019). In brief, metabolites were resuspended in 60% acetonitrile. Metabolites were measured using a 1290 Infinity II HPLC (Agilent) coupled to a 6470 triple quadrupole mass spectrometer (Agilent). Samples were injected onto an iHILIC-Fusion(P) column with the above-mentioned solvents. The solvent, composed of acetonitrile and ammonium acetate (pH 9.3, 10 mM), was used at a flow rate of 0.100 mL min−1. Data analysis was performed with MSD Chemstation Data Analysis (v.E.02.0.2.1431) or Agilent MassHunter (v.B.0802 Build 8.2.8260.0) followed by an in-house-developed MATLAB script. Data is available in Table S1.
Flow Cytometry
Cells were incubated in PBS with the fixable viability dye eFluor® 780 (65-0865-14, eBioscience) before antibody staining. Prior to surface staining with antibodies, Fc gamma receptors were blocked by incubating cells with anti-CD16/CD32 antibodies (2.4G2, homemade). Thereafter, cells were incubated with the appropriate primary antibodies (CD45, CD11b, Ly-6G, Ly-6C, F4/80, MERTK, CCR2, CD64, MHCII, CD11c, and CX3CR1) diluted in FACS buffer (DPBS + 2% FCS) for 15 min. For CD206 and Relmα intracellular stainings, cells were fixed with 4% formalin, permeabilized in permeabilization buffer (DPBS + 2% FCS + 0.5% saponin), and subsequently incubated with antibodies for 30 min on ice. The anti-Relmα antibody was detected with an anti-rabbit secondary antibody labeled with FITC (Southern Biotech). For EdU proliferation experiments, cells from EdU-injected mice were first stained with antibodies for cell surface markers and subsequently labeled with the click-iT plus EdU Alexa Fluor® 488 Flow Cytometry Assay Kit (Life Technologies) according to the manufacturer’s instructions. Cells were analyzed with a LSRFortessa (BD Bioscience) flow cytometer or sorted using a FACS Aria III (BD Bioscience) sorter. Data were analyzed using FlowJo 10 software (Tree Star). A complete list of all antibodies and staining reagents used can be found in Key Resources Table. The gating strategies used for flow cytometry plots are shown in Figure S2.
Quantification and Statistical Analysis
The images presented in the manuscript are representative of the data (quantification of image is approximately the group average) and the image/staining quality. All data represent mean ± SEM. GraphPhad Prism software (version 8.0.0) was used for statistical analyses. Investigators were always blinded to group allocation. When comparing two group means, Student’s t test was used in an unpaired two-tailed fashion. For more than two groups, one-way ANOVA with Tukey’s multiple comparisons test was used and for experimental set-ups with a second variable, two-way ANOVA with Tukey’s multiple comparisons test was used. The statistical method used for each experiment is indicated in each figure legend. Asterisks in figure legends denote statistical significance. For Laser Doppler imaging experiments, mice were compared using a two-way repeated-measures ANOVA with Sidak’s multiple comparisons test. No experiment-wide multiple test correction was applied. p > 0.05 is considered non-significant (ns). p < 0.05 is considered significant (∗).
Acknowledgments
We thank Véronique Juvin from SciArtWork (http://www.sciartwork.com) for her help with the graphical abstract. We thank the Functional Genomics Center Zürich and ETH Flow Cytometry Core Facility for technical support. This project was funded by a European Research Council (ERC) Starting Grant (716140) and by the Swiss National Science Foundation (SNSF 31003A_176056). G.F. is supported by an ETH Research Grant (ETH-16 17-1). J.M. and M.K. are supported by an ETH Research Grant (ETH-23 16-2) and an SNF grant (SNF 310030B_182829). I.S.A. received a Fundacion Ramon Areces fellowship. The work of P.C. is supported by an ERC Advanced Research Grant (EU-ERC269073) and long-term structural Methusalem funding by the Flemish government. S.M.F. is supported by an ERC Consolidator Grant (771486), by FWO research projects, and by KU Leuven Methusalem co-funding. A.B. is supported by SNSF (SNSF 310030_182357). K.D.B. is endowed by the Schulthess Foundation.
Author Contributions
J.Z. conceptualized the study and designed and performed the experiments. J.M. performed macrophage analysis and contributed to the design of the experiments. G.F., I.S.A., T.G., G.D., Z.F., G.T., P.G., and E.M. contributed to data analysis and performing experiments. R.G.B. performed and analyzed myoblast experiments. M.P. performed metabolite analysis. T.W. performed the bioinformatics analysis under the supervision of C.W. P.C. generated pfkfb3LoxP/LoxP mice. L.P. generated mct1LoxP/LoxP mice. C.S. provided vegfΔMac mice and supervised the experiments. S.M.F. supervised the metabolite analysis. A.B. supervised the myoblast injection experiments and analyzed the results. M.K. contributed to conceptualization of the study and writing of the paper. K.D.B. conceptualized the study, supervised the experiments, acquired funding, and wrote the paper.
Declaration of interests
Authors declare no competing interests.
Published: June 2, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cmet.2020.05.004.
Supplemental Information
references
- Adams R.H., Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007;8:464–478. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- Almada A.E., Wagers A.J. Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease. Nat. Rev. Mol. Cell Biol. 2016;17:267–279. doi: 10.1038/nrm.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragonés J., Schneider M., Van Geyte K., Fraisl P., Dresselaers T., Mazzone M., Dirkx R., Zacchigna S., Lemieux H., Jeoung N.H. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat. Genet. 2008;40:170–180. doi: 10.1038/ng.2007.62. [DOI] [PubMed] [Google Scholar]
- Arnold L., Henry A., Poron F., Baba-Amer Y., van Rooijen N., Plonquet A., Gherardi R.K., Chazaud B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 2007;204:1057–1069. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsic N., Zacchigna S., Zentilin L., Ramirez-Correa G., Pattarini L., Salvi A., Sinagra G., Giacca M. Vascular endothelial growth factor stimulates skeletal muscle regeneration in vivo. Mol. Ther. 2004;10:844–854. doi: 10.1016/j.ymthe.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Augustin H.G., Koh G.Y. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science. 2017;357:357. doi: 10.1126/science.aal2379. [DOI] [PubMed] [Google Scholar]
- Ayachi M., Niel R., Momken I., Billat V.L., Mille-Hamard L. Validation of a Ramp Running Protocol for Determination of the True VO2max in Mice. Front. Physiol. 2016;7:372. doi: 10.3389/fphys.2016.00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzinger C.F., Wang Y.X., Dumont N.A., Rudnicki M.A. Cellular dynamics in the muscle satellite cell niche. EMBO Rep. 2013;14:1062–1072. doi: 10.1038/embor.2013.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman B.C., Wolfel E.E., Butterfield G.E., Lopaschuk G.D., Casazza G.A., Horning M.A., Brooks G.A. Active muscle and whole body lactate kinetics after endurance training in men. J. Appl. Physiol. 1999;87:1684–1696. doi: 10.1152/jappl.1999.87.5.1684. [DOI] [PubMed] [Google Scholar]
- Borselli C., Storrie H., Benesch-Lee F., Shvartsman D., Cezar C., Lichtman J.W., Vandenburgh H.H., Mooney D.J. Functional muscle regeneration with combined delivery of angiogenesis and myogenesis factors. Proc. Natl. Acad. Sci. USA. 2010;107:3287–3292. doi: 10.1073/pnas.0903875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks G.A. The lactate shuttle during exercise and recovery. Med. Sci. Sports Exerc. 1986;18:360–368. doi: 10.1249/00005768-198606000-00019. [DOI] [PubMed] [Google Scholar]
- Brooks G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018;27:757–785. doi: 10.1016/j.cmet.2018.03.008. [DOI] [PubMed] [Google Scholar]
- Cantelmo A.R., Conradi L.C., Brajic A., Goveia J., Kalucka J., Pircher A., Chaturvedi P., Hol J., Thienpont B., Teuwen L.A. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell. 2016;30:968–985. doi: 10.1016/j.ccell.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casazza A., Laoui D., Wenes M., Rizzolio S., Bassani N., Mambretti M., Deschoemaeker S., Van Ginderachter J.A., Tamagnone L., Mazzone M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013;24:695–709. doi: 10.1016/j.ccr.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Chazaud B. Macrophages: supportive cells for tissue repair and regeneration. Immunobiology. 2014;219:172–178. doi: 10.1016/j.imbio.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Chen P., Zuo H., Xiong H., Kolar M.J., Chu Q., Saghatelian A., Siegwart D.J., Wan Y. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad. Sci. USA. 2017;114:580–585. doi: 10.1073/pnas.1614035114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen B.E., Burkhardt C., Reith W., Renkawitz R., Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- Claxton S., Kostourou V., Jadeja S., Chambon P., Hodivala-Dilke K., Fruttiger M. Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis. 2008;46:74–80. doi: 10.1002/dvg.20367. [DOI] [PubMed] [Google Scholar]
- Colegio O.R., Chu N.Q., Szabo A.L., Chu T., Rhebergen A.M., Jairam V., Cyrus N., Brokowski C.E., Eisenbarth S.C., Phillips G.M. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culic O., Gruwel M.L., Schrader J. Energy turnover of vascular endothelial cells. Am. J. Physiol. 1997;273:C205–C213. doi: 10.1152/ajpcell.1997.273.1.C205. [DOI] [PubMed] [Google Scholar]
- D’Hulst G., Soro-Arnaiz I., Masschelein E., Veys K., Fitzgerald G., Smeuninx B., Kim S., Deldicque L., Blaauw B., Carmeliet P. PHD1 controls muscle mTORC1 in a hydroxylation-independent manner by stabilizing leucyl tRNA synthetase. Nat. Commun. 2020;11:174. doi: 10.1038/s41467-019-13889-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bock K., Georgiadou M., Schoors S., Kuchnio A., Wong B.W., Cantelmo A.R., Quaegebeur A., Ghesquière B., Cauwenberghs S., Eelen G. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- Delgado A.C., Ferrón S.R., Vicente D., Porlan E., Perez-Villalba A., Trujillo C.M., D’Ocón P., Fariñas I. Endothelial NT-3 delivered by vasculature and CSF promotes quiescence of subependymal neural stem cells through nitric oxide induction. Neuron. 2014;83:572–585. doi: 10.1016/j.neuron.2014.06.015. [DOI] [PubMed] [Google Scholar]
- Deng B., Wehling-Henricks M., Villalta S.A., Wang Y., Tidball J.G. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J. Immunol. 2012;189:3669–3680. doi: 10.4049/jimmunol.1103180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B.S., Nolan D.J., Butler J.M., James D., Babazadeh A.O., Rosenwaks Z., Mittal V., Kobayashi H., Shido K., Lyden D. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature. 2010;468:310–315. doi: 10.1038/nature09493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B.S., Nolan D.J., Guo P., Babazadeh A.O., Cao Z., Rosenwaks Z., Crystal R.G., Simons M., Sato T.N., Worgall S. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell. 2011;147:539–553. doi: 10.1016/j.cell.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diskin C., Pålsson-McDermott E.M. Metabolic Modulation in Macrophage Effector Function. Front. Immunol. 2018;9:270. doi: 10.3389/fimmu.2018.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dort J., Fabre P., Molina T., Dumont N.A. Macrophages Are Key Regulators of Stem Cells during Skeletal Muscle Regeneration and Diseases. Stem Cells Int. 2019;2019:4761427. doi: 10.1155/2019/4761427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia I., Rossi M., Stegen S., Broekaert D., Doglioni G., van Gorsel M., Boon R., Escalona-Noguero C., Torrekens S., Verfaillie C. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature. 2019;568:117–121. doi: 10.1038/s41586-019-0977-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald G., Soro-Arnaiz I., De Bock K. The Warburg Effect in Endothelial Cells and its Potential as an Anti-angiogenic Target in Cancer. Front. Cell Dev. Biol. 2018;6:100. doi: 10.3389/fcell.2018.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freigang S., Ampenberger F., Weiss A., Kanneganti T.D., Iwakura Y., Hersberger M., Kopf M. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat. Immunol. 2013;14:1045–1053. doi: 10.1038/ni.2704. [DOI] [PubMed] [Google Scholar]
- Ganta V.C., Choi M., Farber C.R., Annex B.H. Antiangiogenic VEGF165b Regulates Macrophage Polarization via S100A8/S100A9 in Peripheral Artery Disease. Circulation. 2019;139:226–242. doi: 10.1161/CIRCULATIONAHA.118.034165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber H.P., Vu T.H., Ryan A.M., Kowalski J., Werb Z., Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- He H., Xu J., Warren C.M., Duan D., Li X., Wu L., Iruela-Arispe M.L. Endothelial cells provide an instructive niche for the differentiation and functional polarization of M2-like macrophages. Blood. 2012;120:3152–3162. doi: 10.1182/blood-2012-04-422758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Srivastava K., Wieland M., Runge A., Mogler C., Besemfelder E., Terhardt D., Vogel M.J., Cao L., Korn C. Endothelial cell-derived angiopoietin-2 controls liver regeneration as a spatiotemporal rheostat. Science. 2014;343:416–419. doi: 10.1126/science.1244880. [DOI] [PubMed] [Google Scholar]
- Hui S., Ghergurovich J.M., Morscher R.J., Jang C., Teng X., Lu W., Esparza L.A., Reya T., Le Zhan, Yanxiang Guo J. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551:115–118. doi: 10.1038/nature24057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itagaki Y., Saida K., Iwamura K. Regenerative capacity of mdx mouse muscles after repeated applications of myo-necrotic bupivacaine. Acta Neuropathol. 1995;89:380–384. doi: 10.1007/BF00309633. [DOI] [PubMed] [Google Scholar]
- Jetten N., Verbruggen S., Gijbels M.J., Post M.J., De Winther M.P., Donners M.M. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis. 2014;17:109–118. doi: 10.1007/s10456-013-9381-6. [DOI] [PubMed] [Google Scholar]
- Krishnasamy K., Limbourg A., Kapanadze T., Gamrekelashvili J., Beger C., Häger C., Lozanovski V.J., Falk C.S., Napp L.C., Bauersachs J. Blood vessel control of macrophage maturation promotes arteriogenesis in ischemia. Nat. Commun. 2017;8:952. doi: 10.1038/s41467-017-00953-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krützfeldt A., Spahr R., Mertens S., Siegmund B., Piper H.M. Metabolism of exogenous substrates by coronary endothelial cells in culture. J. Mol. Cell. Cardiol. 1990;22:1393–1404. doi: 10.1016/0022-2828(90)90984-a. [DOI] [PubMed] [Google Scholar]
- Latroche C., Gitiaux C., Chrétien F., Desguerre I., Mounier R., Chazaud B. Skeletal Muscle Microvasculature: A Highly Dynamic Lifeline. Physiology (Bethesda) 2015;30:417–427. doi: 10.1152/physiol.00026.2015. [DOI] [PubMed] [Google Scholar]
- Latroche C., Matot B., Martins-Bach A., Briand D., Chazaud B., Wary C., Carlier P.G., Chrétien F., Jouvion G. Structural and Functional Alterations of Skeletal Muscle Microvasculature in Dystrophin-Deficient mdx Mice. Am. J. Pathol. 2015;185:2482–2494. doi: 10.1016/j.ajpath.2015.05.009. [DOI] [PubMed] [Google Scholar]
- Latroche C., Weiss-Gayet M., Muller L., Gitiaux C., Leblanc P., Liot S., Ben-Larbi S., Abou-Khalil R., Verger N., Bardot P. Coupling between Myogenesis and Angiogenesis during Skeletal Muscle Regeneration Is Stimulated by Restorative Macrophages. Stem Cell Reports. 2017;9:2018–2033. doi: 10.1016/j.stemcr.2017.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCouter J., Moritz D.R., Li B., Phillips G.L., Liang X.H., Gerber H.P., Hillan K.J., Ferrara N. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science. 2003;299:890–893. doi: 10.1126/science.1079562. [DOI] [PubMed] [Google Scholar]
- Limbourg A., Korff T., Napp L.C., Schaper W., Drexler H., Limbourg F.P. Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nat. Protoc. 2009;4:1737–1746. doi: 10.1038/nprot.2009.185. [DOI] [PubMed] [Google Scholar]
- Liu P.S., Wang H., Li X., Chao T., Teav T., Christen S., Di Conza G., Cheng W.C., Chou C.H., Vavakova M. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017;18:985–994. doi: 10.1038/ni.3796. [DOI] [PubMed] [Google Scholar]
- Mayeuf-Louchart A., Hardy D., Thorel Q., Roux P., Gueniot L., Briand D., Mazeraud A., Bouglé A., Shorte S.L., Staels B. MuscleJ: a high-content analysis method to study skeletal muscle with a new Fiji tool. Skelet. Muscle. 2018;8:25. doi: 10.1186/s13395-018-0171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser D.M., Edwards J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounier R., Théret M., Arnold L., Cuvellier S., Bultot L., Göransson O., Sanz N., Ferry A., Sakamoto K., Foretz M. AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 2013;18:251–264. doi: 10.1016/j.cmet.2013.06.017. [DOI] [PubMed] [Google Scholar]
- Murray C.M., Hutchinson R., Bantick J.R., Belfield G.P., Benjamin A.D., Brazma D., Bundick R.V., Cook I.D., Craggs R.I., Edwards S. Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat. Chem. Biol. 2005;1:371–376. doi: 10.1038/nchembio744. [DOI] [PubMed] [Google Scholar]
- Muzumdar M.D., Tasic B., Miyamichi K., Li L., Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- O’Neill L.A., Pearce E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016;213:15–23. doi: 10.1084/jem.20151570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottone C., Krusche B., Whitby A., Clements M., Quadrato G., Pitulescu M.E., Adams R.H., Parrinello S. Direct cell-cell contact with the vascular niche maintains quiescent neural stem cells. Nat. Cell Biol. 2014;16:1045–1056. doi: 10.1038/ncb3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa C.R., Banfi A., Glazer N.L., Thurston G., Springer M.L., Kraft P.E., McDonald D.M., Blau H.M. Microenvironmental VEGF concentration, not total dose, determines a threshold between normal and aberrant angiogenesis. J. Clin. Invest. 2004;113:516–527. doi: 10.1172/JCI18420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce E.L., Pearce E.J. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieters T., Haenebalcke L., Bruneel K., Vandamme N., Hochepied T., van Hengel J., Wirth D., Berx G., Haigh J.J., van Roy F., Goossens S. Structure-function Studies in Mouse Embryonic Stem Cells Using Recombinase-mediated Cassette Exchange. J. Vis. Exp. 2017;(122) doi: 10.3791/55575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porporato P.E., Payen V.L., De Saedeleer C.J., Préat V., Thissen J.P., Feron O., Sonveaux P. Lactate stimulates angiogenesis and accelerates the healing of superficial and ischemic wounds in mice. Angiogenesis. 2012;15:581–592. doi: 10.1007/s10456-012-9282-0. [DOI] [PubMed] [Google Scholar]
- Potente M., Carmeliet P. The Link Between Angiogenesis and Endothelial Metabolism. Annu. Rev. Physiol. 2017;79:43–66. doi: 10.1146/annurev-physiol-021115-105134. [DOI] [PubMed] [Google Scholar]
- Potente M., Gerhardt H., Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- Raes G., De Baetselier P., Noël W., Beschin A., Brombacher F., Hassanzadeh Gh G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J. Leukoc. Biol. 2002;71:597–602. [PubMed] [Google Scholar]
- Rafii S., Butler J.M., Ding B.S. Angiocrine functions of organ-specific endothelial cells. Nature. 2016;529:316–325. doi: 10.1038/nature17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich A., Spering C., Gertz K., Harms C., Gerhardt E., Kronenberg G., Nave K.A., Schwab M., Tauber S.C., Drinkut A. Fas/CD95 regulatory protein Faim2 is neuroprotective after transient brain ischemia. J. Neurosci. 2011;31:225–233. doi: 10.1523/JNEUROSCI.2188-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saclier M., Yacoub-Youssef H., Mackey A.L., Arnold L., Ardjoune H., Magnan M., Sailhan F., Chelly J., Pavlath G.K., Mounier R. Differentially activated macrophages orchestrate myogenic precursor cell fate during human skeletal muscle regeneration. Stem Cells. 2013;31:384–396. doi: 10.1002/stem.1288. [DOI] [PubMed] [Google Scholar]
- Schoors S., De Bock K., Cantelmo A.R., Georgiadou M., Ghesquière B., Cauwenberghs S., Kuchnio A., Wong B.W., Quaegebeur A., Goveia J. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014;19:37–48. doi: 10.1016/j.cmet.2013.11.008. [DOI] [PubMed] [Google Scholar]
- Sonveaux P., Végran F., Schroeder T., Wergin M.C., Verrax J., Rabbani Z.N., De Saedeleer C.J., Kennedy K.M., Diepart C., Jordan B.F. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest. 2008;118:3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley W.C., Gertz E.W., Wisneski J.A., Neese R.A., Morris D.L., Brooks G.A. Lactate extraction during net lactate release in legs of humans during exercise. J. Appl. Physiol. 1986;60:1116–1120. doi: 10.1152/jappl.1986.60.4.1116. [DOI] [PubMed] [Google Scholar]
- Stockmann C., Doedens A., Weidemann A., Zhang N., Takeda N., Greenberg J.I., Cheresh D.A., Johnson R.S. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008;456:814–818. doi: 10.1038/nature07445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball J.G. Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol. 2017;17:165–178. doi: 10.1038/nri.2016.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball J.G., Wehling-Henricks M. Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. J. Physiol. 2007;578:327–336. doi: 10.1113/jphysiol.2006.118265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bossche J., O’Neill L.A., Menon D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017;38:395–406. doi: 10.1016/j.it.2017.03.001. [DOI] [PubMed] [Google Scholar]
- Varga T., Mounier R., Horvath A., Cuvellier S., Dumont F., Poliska S., Ardjoune H., Juban G., Nagy L., Chazaud B. Highly Dynamic Transcriptional Signature of Distinct Macrophage Subsets during Sterile Inflammation, Resolution, and Tissue Repair. J. Immunol. 2016;196:4771–4782. doi: 10.4049/jimmunol.1502490. [DOI] [PubMed] [Google Scholar]
- Végran F., Boidot R., Michiels C., Sonveaux P., Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71:2550–2560. doi: 10.1158/0008-5472.CAN-10-2828. [DOI] [PubMed] [Google Scholar]
- Verma M., Asakura Y., Murakonda B.S.R., Pengo T., Latroche C., Chazaud B., McLoon L.K., Asakura A. Muscle Satellite Cell Cross-Talk with a Vascular Niche Maintains Quiescence via VEGF and Notch Signaling. Cell Stem Cell. 2018;23:530–543.e9, e539. doi: 10.1016/j.stem.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veys K., Alvarado-Diaz A., De Bock K. Measuring Glycolytic and Mitochondrial Fluxes in Endothelial Cells Using Radioactive Tracers. Methods Mol. Biol. 2019;1862:121–136. doi: 10.1007/978-1-4939-8769-6_9. [DOI] [PubMed] [Google Scholar]
- Viola A., Munari F., Sánchez-Rodríguez R., Scolaro T., Castegna A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019;10:1462. doi: 10.3389/fimmu.2019.01462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenes M., Shang M., Di Matteo M., Goveia J., Martín-Pérez R., Serneels J., Prenen H., Ghesquière B., Carmeliet P., Mazzone M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016;24:701–715. doi: 10.1016/j.cmet.2016.09.008. [DOI] [PubMed] [Google Scholar]
- Xu Y., An X., Guo X., Habtetsion T.G., Wang Y., Xu X., Kandala S., Li Q., Li H., Zhang C. Endothelial PFKFB3 plays a critical role in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2014;34:1231–1239. doi: 10.1161/ATVBAHA.113.303041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H., Price F., Rudnicki M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013;93:23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P., Wilhelm K., Dubrac A., Tung J.K., Alves T.C., Fang J.S., Xie Y., Zhu J., Chen Z., De Smet F. FGF-dependent metabolic control of vascular development. Nature. 2017;545:224–228. doi: 10.1038/nature22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Kasim V., Xie Y.D., Huang C., Sisjayawan J., Dwi Ariyanti A., Yan X.S., Wu X.Y., Liu C.P., Yang L. Inhibition of PHD3 by salidroside promotes neovascularization through cell-cell communications mediated by muscle-secreted angiogenic factors. Sci. Rep. 2017;7:43935. doi: 10.1038/srep43935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mouse RNA-seq data reported in this study are available at the Gene Expression Omnibus (GEO) repository under the accession number (GSE148584). All other data are available from the Lead Contact on request.