

Abstract
Allogeneic hematopoietic transplantation (allo-HCT) is a curative therapy for a variety of hematologic malignancies, primarily through immune-mediated clearance of malignant cells. This graft-versus-leukemia (GvL) effect is mediated by alloreactive donor T-cells against recipient malignant cells. Unfortunately, graft versus host disease is a potentially lethal complication of this procedure, also mediated by alloreactive donor T-cells against recipient normal tissues. Graft-versus-host disease (GVHD) remains a key contributor to nonrelapse mortality and long-term morbidity in patients undergoing allo-HCT. Reducing GVHD without interfering with – or ideally while enhancing – GvL, would improve outcomes and increase patient eligibility for allo-HCT. The JAK/STAT signaling pathway acts downstream of over 50 cytokines and is central to a wide variety of inflammatory pathways. These pathways play a role in the development and maintenance of GVHD throughout the disease process and within T-cells, B-cells, macrophages, neutrophils, and natural killer cells. Agents targeting JAK/STAT signaling pathways have shown clinical efficacy and gained US Food and Drug Administration approval for numerous diseases. Here, we review the preclinical and clinical evidence for the role of JAK/STAT signaling in the development and maintenance of GVHD and the utility of blocking agents at preventing and treating GVHD.
Keywords: Graft-versus-host disease, GVHD, JAK/STAT signaling
Introduction
Allo-HCT and GVHD
Allogeneic hematopoietic cell transplantation (allo-HCT) is a potentially curative therapy for an array of hematological malignancies. Efficacy is mediated, in large part, through the graft versus leukemia (GvL) effect, and alloreactivity of donor T-cells against host leukemic cells.1 Grafted donor immune cells recognize leukemic cells as foreign and eliminate them. Evidence for GvL comes from a number of observations: patients who relapse after allo-HCT can achieve remission after stopping immunosuppression or with donor lymphocyte infusions, allo-HCT with reduced intensity conditioning regimens can induce remission in acute myeloid leukemia (AML), T-cell depleted grafts have higher rates of relapse, and patients with AML who develop graft-versus-host disease (GVHD), especially chronic GVHD (cGVHD),2 after allo-HCT have a lower rate of relapse. Unfortunately, this donor cell reactivity against malignant cells is coupled with donor cell reactivity against normal host tissues, which leads to GVHD.
GVHD is one of the main sources of morbidity and mortality after allo-HCT, limiting the use of this potentially curative therapy. GVHD can be classified as acute GVHD (aGVHD), cGVHD, and overlap syndrome, which presents with features of both. aGVHD occurs in 25–55% of patients after allo-HCT, and cGVHD in 40% of patients. GVHD is life-threatening in 15–20% of patients. A Center for International Blood and Marrow Transplant Research study including 4224 patients with AML and 1517 patients with myelodysplastic syndrome, showed that patients who develop aGVHD after transplant have an increased risk of treatment-related mortality and lower overall survival.3 Risk factors for GVHD include intensive chemotherapy, previous viral infections, and advanced stage of leukemia.
The focus is on prevention of GVHD as an established disease remains notoriously difficult to treat. There are many regimens in use to prevent GVHD after allo-HCT, but in general they include combinations of agents that reduce T-cell activation (cyclosporine, tacrolimus, sirolimus), reduce T-cell proliferation (methotrexate, mycophenolate mofetil), and cause T-cell depletion (antithymocyte globulin, post-transplant cyclophosphamide).4 Treatment options for an established disease are limited, with steroids representing first line therapy for aGVHD and cGVHD. Outcomes in patients who do not respond to steroids are dismal, with steroid-refractory aGVHD patients having a long term survival rate of 5–30%.5 The first US Food and Drug Administration (FDA) approvals in this setting have come recently: ruxolitinib for steroid-refractory aGVHD (May 2019) and ibrutinib for steroid-refractory cGVHD (August 2017). An array of other agents are currently undergoing preclinical and clinical trials.
The JAK/STAT system
Janus kinases (JAK) and signal transducers and activation of transcription (STAT) were described in the early 1990s as a family of rapid membrane to nucleus signaling molecules that act downstream of over 50 cytokines.6 JAK kinases phosphorylate intracellular STAT family proteins in response to extracellular signaling (Table 1). Phosphorylated STATs then dimerize and translocate to the nucleus, where they bind to enhancers and promoters to regulate transcription of target genes, predominantly without the need of second messengers7 (Figure 1). There are four members of the JAK family – JAK1, JAK2, JAK3, and TYK2 – and seven members of the STAT family – STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6.8 Each JAK kinase is activated by multiple cytokines, and there is overlap in downstream effects. Furthermore, STATs have overlapping and at times competing effects on individual genes. An example is the Bcl-2 gene, which is modulated by STAT1 (which downregulates transcription) as well as STAT3, STAT5, and STAT6 (which upregulate transcription).9 It is possible that blockade of one JAK or STAT protein may allow dominant transcription modulation by another member of the JAK/STAT system, and the ultimate physiologic effects are difficult to predict.
Table 1.
Cytokine receptors and downstream JAKs and STATs.9
| Cytokine | JAKs | STATs |
|---|---|---|
| IFNγ | JAK1/JAK2 | STAT 1/STAT1 |
| IFNα/β | JAK1/TYK2 | STAT1/STAT2 |
| IL-6 | JAK1/JAK2 | STAT3/STAT3 |
| IL-12 | JAK2/TYK2 | STAT4/STAT4 |
| IL-3 | JAK2/JAK2 | STAT5a/STAT5b |
| GM-CSF | JAK2/JAK2 | STAT5a/STAT5b |
| IL-2 | JAK1/JAK3 | STAT5a/STAT5b |
| IL-4 | JAK1/JAK3 | STAT6/STAT6 |
| IL-13 | JAK1/JAK3 | STAT6/STAT6 |
| EPO | JAK2/JAK2 | STAT3/STAT5 |
| MPL | JAK2/JAK2 | STAT3/STAT5 |
EPO, erythropoietin; GM-CSF, Granulocyte-macrophage colony-stimulating factor; IFNα/β, interferon alpha/beta; IFNγ, interferon gamma; IL, interleukin; JAK, Janus kinases; MPL, myeloproliferative leukemia virus oncogene; STAT, signal transducers and activation of transcription.
Figure 1.

The JAK/STAT System. The JAK/STAT system is downstream of over 50 cytokine receptors. JAKs phosphorylate STATs, which then dimerize and translocate to the nucleus where they regulate gene transcription. Promiscuity and redundancy exists at multiple levels, including promiscuity of cytokine receptors and DNA STAT binding domains and overlap at the STAT phosphorylation level. Blockade of individual signaling elements may therefore have incomplete effects. Illustration courtesy of Alessandro Baliani © (2019).
EPO, erythropoietin; GM-CSF, Granulocyte-macrophage colony-stimulating factor; IFNα/β, interferon alpha/beta; IFNγ, interferon gamma; IL, interleukin; JAK, Janus kinases; MPL, myeloproliferative leukemia virus oncogene; STAT, signal transducers and activation of transcription.
It has long been known that both loss-of-function and gain-of-function mutations in the JAK/STAT system can have profound phenotypic effects.10 JAK1 and JAK2 loss results in perinatal and embryonic lethality, respectively. Immunodeficiency syndromes are caused by both JAK3 loss (JAK3-SCID) and TYK2, due to a lack of response to inflammatory cytokines including interferon gamma (IFNγ), interferon alpha/beta (IFNα/β), interleukin (IL)-6, IL-10, IL-12, and IL-23.11,12 Gain-of-function mutations are associated with myeloproliferative syndromes, including activating JAK2 in polycythemia vera, essential thrombocythemia, myelofibrosis, and hypereosinophilia.
Small molecule JAK inhibitors – or JAKinibs – have varying specificity for the JAK kinases.13 Given the ubiquity of the JAK/STAT system in inflammatory processes, modulation with JAKinibs has been studied in various settings, including rheumatologic disease,13,14 hematological malignancies,15,16 solid tumor malignancies,17 and septic shock.18 Currently, there are a large number of clinical trials involving JAK and STAT inhibitors in all of these disease areas. JAKinibs with FDA approvals or in clinical trials for hematologic diseases are partially listed in Table 2. Ruxolitinib is FDA-approved for treatment of steroid-refractory aGVHD. Baricitinib, itacitinib and ruxolitinib are undergoing clinical trials for treatment or prevention of aGVHD or cGVHD GVHD.
Table 2.
JAK/STAT inhibitors for hematologic diseases (GVHD indications highlighted).
| Drug | Target of inhibition | Disease setting | Clinical phase |
|---|---|---|---|
| FDA Approved: | |||
| Fedratinib | JAK2 | Myelofibrosis | FDA Approved |
| Ruxolitinib | JAK1/JAK2 | Myelofibrosis | FDA Approved |
| Polycythemia Vera | FDA Approved | ||
| Steroid Refractory Acute GVHD | FDA Approved | ||
| Baricitinib | JAK1/JAK2>JAK3/TYK2 | Rheumatoid Arthritis | FDA Approved |
| Under Clinical Investigation: | |||
| Baricitinib | JAK1/JAK2>JAK3, TYK2 | Chronic GVHD | Phase II (NCT02759731) |
| Prevention of acute GVHD | Phase I (NCT04131738) | ||
| Cerdulatinib | SYK/JAK | Non-Hodgkin lymphoma | Phase II (NCT01994382, NCT04021082) |
| Gandotinib | JAK2 | Myeloproliferative neoplasms | Phase II (NCT01594723) |
| Itacitinib | JAK1 | Prevention of acute GVHD | Phase I (NCT03320642, NCT03755414) Phase II (NCT04127721) |
| Acute GVHD | Phase I (NCT04070781, NCT03497273) Phase II (NCT03846479, NCT03721965) |
||
| Chronic GVHD | Phase II (NCT04200365) Phase III (NCT03584516) |
||
| Prevention of Cytokine Release Syndrome | Phase I (NCT04071366, NCT03755414) | ||
| Hodgkin Lymphoma | Phase II (NCT03697408) | ||
| Myelofibrosis | Phase II (NCT03144687) | ||
| Acute Lymphocytic Leukemia | Phase I (NCT03989466) | ||
| Lestaurtinib | JAK2 | Acute myeloid leukemia | Phase II (NCT02428543) |
| Momelotinib | JAK1/JAK2 | Myeloproliferative neoplasms | Phase III (NCT04173494) |
| Pacritinib | JAK2 | Prevention of GVHD | Phase II (NCT02891603) |
| Myeloproliferative neoplasms | Phase II (NCT03645824) | ||
| Lymphoproliferative Disorders | Phase I (NCT03601819) | ||
| Ruxolitinib | JAK1/JAK2 | Acute myeloid leukemia, Acute lymphoblastic leukemia, Acute GVHD, Chronic GVHD, Chronic myeloid leukemia, Hodgkin lymphoma, Non-Hodgkin Lymphoma, Hypereosinophilic disorders | Total 76 studies recruiting subjects. Eight studies recruiting in acute or chronic GVHD. |
FDA, US Food and Drug Administration; GVHD, graft-versus-host disease; JAK, Janus kinases; STAT, signal transducers and activation of transcription.
The remainder of this review focuses on the pathophysiology of aGVHD and cGVHD. Within each section, we will discuss the evidence for JAK/STAT system involvement as well as effects of inhibition (Figure 2).
Figure 2.
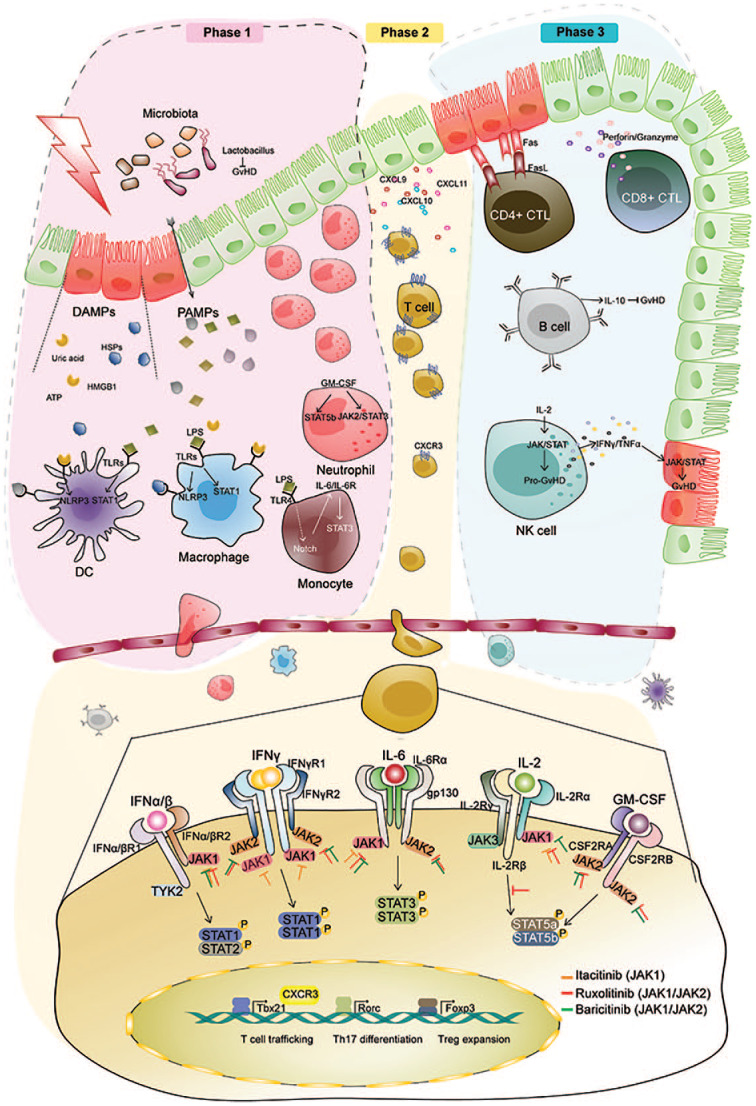
The role of the JAK/STAT system in graft-versus-host disease. Top: Three step model of acute GVHD. Bottom: Arrows with flat heads represent inhibition of JAKs by itacitinib (orange), ruxolitinib (red), and baricitinib (green).
Acute GVHD: three step model
aGVHD is traditionally characterized as a three step process: (a) tissue damage from disease and conditioning chemoradiotherapy, (b) donor T-cell activation, and (c) recruitment and activation of other immune cells.19 The JAK/STAT pathways are active in each step.20 The pathophysiology of each step is reviewed separately below, with a focus on evidence for JAK/STAT involvement and utility of pathway inhibition.
Phase I: tissue damage
Prior to allo-HCT, conditioning chemoradiotherapy causes tissue damage, inducing a systemic inflammatory state. Inflammatory cytokines such as TNFα and IL-1 are released, and higher levels are associated with GVHD.21 Higher intensity of conditioning has also been associated with more severe GVHD in both animal models and human studies.22,23 There are a number of innate and adaptive mechanisms through which tissue injury leads to immune activation, and many signal through the JAK/STAT system.24
Damage-associated molecular patterns
Damage-associated molecular patterns (DAMPs) are endogenous biomolecules, including ATP, HMGB1, uric acid and heat shock proteins that are released in the context of tissue injury. They lead to immune activation through an array of pathways, including toll-like receptors (TLRs),25 NLRP326 and STAT1.27 STAT1, specifically, may be a particularly important mediator of GVHD. In preclinical models in which STAT1 is knocked out in CD4+ T-cells, T-regulatory expansion is promoted and GVHD is reduced.28 STAT1 is downstream of two cytokine receptors, the IFNγ and IFNα/β receptors.29 These cytokines are known mediators of GVHD.28,30 The IFNγ and IFNα/β receptors signal through JAK1/JAK2 and JAK1/TYK2, respectively.29,31 Targeting GVHD at both the level of the IFNγ receptor and the JAK1/JAK2 level has been successful, and the combination may be synergistic. In murine allo-HCT models, inhibition of this axis with IFNγ-R blockers or JAKinibs alone or in combination prevents GVHD.28,30,32
Pathogen-associated molecular patterns and the gut microbiome
Pathogen-associated molecular patterns (PAMPs) are exogenous biomolecules, including bacterial lipopolysaccharides (LPSs). Gut injury from conditioning chemoradiotherapy increases translocation of bacteria, leading to release of PAMPs into tissue and blood. In step 3 of aGVHD (below), PAMPs activate primed antigen-presenting immune cells through TLRs. Therefore, the role of the gut microbiome in GVHD has long been an active area of research. Recently, rapid progress has been made possible by modern bacterial genome sequencing technologies. Pre-transplant microbiome diversity is associated with reduced mortality, and loss of diversity – worsened by prophylactic antibiotics and total parenteral nutrition (TPN) – is associated with increased GVHD and mortality.33,34 Furthermore, certain bacteria may be protective against GVHD, including Lactobacillus and Blautia genus. TLR activation by bacterial LPS induces Notch signaling in monocytes, which can be abrogated by STAT3 inhibition.35 STAT3 is downstream of the IL-6 receptor. This may represent an opportunity to break the positive feedback loop between steps one and three in the development of aGVHD. Consistent with this concept, several groups have shown promising clinical results targeting this signaling axis with tocilizumab, an IL-6 receptor blocking antibody, to prevent aGVHD.36,37
Macrophages and neutrophils
Neoangiogenesis and gastrointestinal infiltration by macrophages and neutrophils are among the earliest events in GVHD.5 Macrophages residing in target tissues secrete significant amounts of nitric oxide, causing direct tissue damage and inhibition of epithelial stem cell proliferation. Macrophage production of nitric oxide occurs through cooperative JAK2/STAT3 and PI3-K signaling.38 Neutrophils are recruited to the gastrointestinal tract by DAMPs and PAMPs, enhancing aGVHD through tissue damage.39 Granulocyte-macrophage colony-stimulating factor-activated neutrophils signal through the JAK2/STAT3 and STAT5b axis.40 Whether macrophage and neutrophil activity in GVHD can be prevented by targeting the JAK/STAT system is an ongoing area of research.
Phase II: donor T-cell activation
This state of systemic inflammation leads to an increase in major histocompatibility complex (MHC) expression by antigen-presenting cells (APCs) at the time of donor cell infusion. This leads to phase II in aGVHD: donor T-cell activation.
After infusion, donor T-cell receptors recognize allo-antigens presented on MHC molecules by host (direct) or donor (indirect) antigen presenting cells. This leads to rapid donor T-cell activation and secretion of an array of cytokines, including IL-2, IL-15 and IFNγ. Multiple models have demonstrated the correlation between elevated IFNγ signaling and GVHD. The dose effect of IL-2, however, has been less clear. Administration of low dose IL-2 increases the severity of GVHD in preclinical models,41 however high doses of IL-2 are protective in others,42 perhaps through inhibition of IFNγ signaling.43 A phase I–II clinical trial of an anti-IL-2 receptor antibody for steroid refractory aGVHD showed some responses,44 but this approach has not been taken forward in additional clinical trials.
Trafficking of activated donor T-cells to target organs is an essential step in the development of aGVHD. In response to activation by APCs, T-cells secrete IFNγ and initiate signaling through the IFNγ-receptor and JAK/STAT system.28 This results in increased T-cell expression of CXCR3, leading to T-cell trafficking to the gut, liver, and skin.28 This trafficking is reduced in IFNγ-receptor- or CXCR3-knockout models, and both trafficking and GVHD are reduced by JAKinibs in mouse models.28,30,45 Furthermore, dual inhibition of the IFNγ-receptor and IL-6-receptor, both genetically and pharmacologically, completely prevents aGVHD in murine models.30
Phase III: recruitment and activation of immune cells
Phase III of aGVHD involves recruitment of other cell types and propagation of tissue damage through cellular and inflammatory effectors.
Effector T-cells
Activated cytotoxic T-cells (CTLs) induce apoptosis of host tissues through multiple pathways. CD8+ CTLs predominantly use the perforin/granzyme pathway, while CD4+ CTLs use the Fas/Fas ligand pathway. Both pathways are important in aGVHD, and murine models deficient in either perforin-dependent killing or Fas ligand still develop GVHD, with differential severity in target tissues.30,46 IFNγ increases Fas ligand expression on CD4+ CTLs.47 Granzyme has different roles in GVHD and GVL, depending on the T-cell subset. For example, the lack of granzyme in the CD8 subset cells prevents GVHD and augments GVL, while the lack of granzyme in the CD4 subset cells aggravates GVHD and impairs GVL.48–50
B-cells
While T-cells have long been regarded as the main regulators of aGVHD and cGVHD, the role of B-cells has recently been examined more closely.51–57 B-cells are central to adaptive immune response, specializing in memory responses, antigen presentation and the formation of antibodies against foreign bacteria, viruses and peptide antigens. Cytokines classically attributed to B-cells include IL-6, TNFα and IL-10. In addition, B-cells can secrete IL-2, IFNγ, IL-12 and IL-4, causing differentiation of naïve T-cells into TH1 and TH2 effector subtypes.55 Other B-cell subsets have immunosuppressive functions and can induce anergy and T-cell deletion.52–54
Preclinical studies with B-cell targeting therapies for GVHD have shown opposing results, consistent with the ability of B-cells to either promote or inhibit cytotoxic T-cell function. aGVHD is reduced in B-cell deficient mice receiving mismatched B-cell depleted grafts.58 However, host B-cells can suppress GVHD through IL-10 secretion, and removing this B-cell-derived IL-10 through killing or gene editing B-cells leads to more severe aGVHD.51
Likewise, clinical experiences with anti-CD20 therapies have had mixed results. One report included patients treated with rituximab as part of conditioning, and rates of aGVHD were encouraging.59 A randomized phase II trial compared four doses of rituximab at day +21 then at day +175 with untreated patients showed no difference in aGVHD between the two groups. A third report described the use of rituximab in patients with steroid-refractory aGVHD with occasional complete responses observed.59 Together, these reports indicate that B-cell depletion may have a role in prophylaxis against aGVHD if used early in the peri-transplant period and also in the frontline treatment of aGVHD.
There is rationale for targeting the JAK/STAT system to modulate B-cell activity. Stimulation of the B-cell Receptor (BCR) leads to downstream phosphorylation of multiple STATs, including STAT1, STAT3 and STAT5.57 STAT phosphorylation can occur in both JAK-dependent and-independent manners. In a B-cell line, BCR activation by the Src kinase Lyn causes JAK-independent STAT phosphorylation, which is not inhibited by loss of JAK1 and JAK2 activity.60 In contrast, in chronic lymphocytic leukemia cells, BCR stimulation by anti-human IgM antibody caused STAT3 phosphorylation and shuttling to the nucleus.61 This effect was blocked by JAK1/2 inhibition with ruxolitinib, but not by Lyn kinase blockade with dasatinib.61 Therefore, the type and magnitude of BCR stimulation affects whether STAT3 signaling occurs in a JAK/STAT-dependent or -independent manner. The effect of JAKinibs on B-cells in the context of human aGVHD is a current area of investigation.
Natural killer cells
Natural killer (NK) cells are the first donor-derived lymphocyte subset to recover after allo-HCT. Similar to B-cells, NK cells can both inhibit and promote GVHD, depending on the physiologic milieu, as well as the specific NK cell subtype.62 Transplantation of activated human NK cells into SCID mice induces GVHD.63 Conversely, in a phase I trial, memory-like NK cells from haploidentical donors showed anti-leukemia activity in recipients without causing GVHD.64 The pro-GVHD effects of NK cells are mediated through inflammatory cytokines including IL-2, IFNγ and TNFα, which signal through the JAK/STAT system. Furthermore, disruption of these pathways can reduce cytotoxicity of NK cells in preclinical models. In a murine sarcoma model, TRAIL-mediated NK cell cytotoxicity was dependent on IFNγ.65 In multiple independent models, disruption of the IL-12/STAT4 axis through STAT4 deletion leads to impaired cytotoxicity of NK cells.66,67 The role of NK cells in GVHD, and the effects of JAKinibs on NK cells, are active areas of investigation. In patients with aGVHD treated with the JAK1 inhibitor itacitinib, those with signs of overactive NK cell activity had a higher response rate.68
Chronic GVHD
While cGVHD is a distinct syndrome from aGVHD, they share many immunologic underpinnings through which APC interactions with alloreactive T-cells lead to an immune response against recipient tissue.69,70 Features distinct to cGVHD include insufficient central tolerance due to thymic dysfunction, insufficient peripheral tolerance due to T-regulatory dysfunction and tissue fibrosis. The role of the JAK/STAT pathway is an area of current investigation. Ruxolitinib therapy reduces the number of circulating activated T (CD3+HLA-DR+) cells in patients with cGVHD.71 There are other novel T-cell signaling pathways that have been implicated in cGVHD. Recently, the Rho-associated kinase 2 (ROCK2) inhibitor KD025 was shown to reduce cGVHD end organ damage and induce a shift from T follicular helper cells to T follicular regulatory cells. There was a reduction in STAT3 and an increase in STAT5 phosphorylation.72 The role of ROCK2 was subsequently described as a feed-forward signal potentiating STAT3-related gene transcription through a ROCK2/STAT3/JAK2 complex.73 A clinical trial evaluating the safety and activity of KD025 in patients with cGVHD is underway (Clinical Trials.gov Identifier: NCT02841995). B-cell activity is important in cGVHD, and the evidence for JAK/STAT involvement in B-cell signaling is discussed above. Ibrutinib, a Bruton’s tyrosine kinase (BTK) inhibitor that inhibits B-cell activity, was FDA approved for steroid refractory cGVHD in 2017 based on the PCYC-1129-CA study showing a response rate of 67%.74 Ibrutinib is being evaluated in a phase III study as frontline therapy for cGVHD (Clinical Trials.gov Identifier: NCT02959944).
Clinical evidence of JAK/STAT inhibition for prevention and treatment of GVHD
JAK inhibition for the prevention and treatment of GVHD is an extremely active area of study. There are many open and recruiting clinical trial in this area (Table 2). A number of clinical trials have completed with results reported. One of these has led to an FDA approval.
Ruxolitinib, a JAK1/2 inhibitor, gained FDA approval in May 2019 for the treatment of steroid-refractory aGVHD. In the REACH1 trial, a single arm phase II study, the overall response rate for ruxolitinib in patients with steroid-refractory aGVHD was 55% by day 28 and 73% at any time. Responses were fast (median day 7) and durable (median duration of response 345 days) and occurred in lung, liver, intestines and skin.75 Ruxolitinib was shown to reduce levels of soluble IL-2 receptor, IL-6, and IFNγ.71,76 Of note, only 5.6% of patients had relapse of disease. Patients did experience hematologic toxicity, with cytopenias seen in all three lines. Infectious complications included cytomegalovirus, sepsis, and bacteremia. In another disease, a pilot study investigating the use of ruxolitinib for GVHD prophylaxis in patients with myelofibrosis undergoing allo-HCT has completed recruitment (Clinical Trials.gov Identifier: NCT02806375), with interval results indicating feasibility of the approach, although therapy may be associated with delayed engraftment.15
Itacitinib, a JAK1 inhibitor, is being studied for the treatment of aGVHD and cGVHD. A phase I study of itacitinib in patients with aGVHD revealed overall response rates of 83% and 64% respectively for treatment of naïve and steroid-refractory patients. Unfortunately, the Gravitas-301 phase III study evaluating itacitinib in combination with corticosteroids in patients with treatment-naïve aGVHD failed to meet its primary (overall response rate at say 28) and secondary (non-relapse mortality at 6 months) endpoints (Incyte Press Release 1/2/2020). The GRAVITAS-309 phase III study is currently evaluating itacitinib with corticosteroids versus corticosteroids alone as first-line treatment for moderate or severe cGVHD. Itacitinib is also being evaluated for prevention of aGVHD after haplo-HCT (Clinical Trials.gov Identifier: NCT03755414) and after cellular therapies (Clinical Trials.gov Identifier: NCT04071366).
Baricitinib is under investigation for the prevention of GVHD (Clinical Trials.gov Identifier: NCT04131738) and the treatment of cGVHD (Clinical Trials.gov Identifier: NCT02759731). Pacritinib is under investigation for the prevention of GVHD (Clinical Trials.gov Identifier: NCT02891603).
Interleukin 6, which is upstream of JAK1, JAK2, and STAT3, is upregulated after allo-HCT. Two groups have conducted early stage trials integrating the IL-6 receptor blocking agent tocilizumab into GVHD prophylaxis. The first trial used cyclosporine, methotrexate, and tocilizumab: the day 100 incidence of grade 2–4 aGVHD was 12% (95% CI 5–24) and the incidence of grade 3–4 aGVHD was 4% [95% confidence interval (CI) 1–13].37 The second trial used tacrolimus, methotrexate, and tocilizumab, and the cumulative incidences of grades II–IV and III–IV aGVHD were 14% (95% CI 5–30) and 3% (95% CI 0–11) at day 100.36 Both trials reported a reduction in aGVHD of the GI tract, consistent with known IL-6 signaling involvement in T-cell trafficking to the gut.28 There are two trials currently recruiting patients (Clinical Trials.gov Identifiers: NCT03699631 and NCT03434730) examining the role of tocilizumab to prevent GVHD after all-HCT.
Toxicity of long-term use of JAK inhibitors
As more patients are treated with JAK inhibitors for extended periods of time, a close eye must be kept on long-term toxicities. Much of the long-term safety data comes from the rheumatology literature, where JAK inhibitors are used as disease-modifying therapies.77 There is, however, growing experience using long-term JAK inhibition in myeloproliferative neoplasms. These data suggest there may be an increased incidence of cutaneous malignancies and infections.
In the COMFORT-II phase III trial of ruxolitinib versus best available therapy for myelofibrosis, 17% of patients treated with ruxolitinib developed non-melanoma skin cancers, compared with 2.7% in the control group.78 Furthermore, a recent case series described five cases of aggressive skin cancers – including one lentigo maligna melanoma and one metastatic undifferentiated pleomorphic sarcoma – in patients with myelofibrosis on ruxolitinib.79 In contrast, patients with rheumatoid arthritis (RA) treated with tofacitinib or baricitinib did not appear to be at increased risk of cutaneous malignancies or lymphoma. Patients with RA have a lower risk of malignancy than the general population, which may underpin some of this conflicting data.
A number of infectious complications appear to be increased in patient treated with JAK inhibitors. Herpes zoster infections are more common in patients treated tofacitinib, baricitinib, and ruxolitinib.77,80 There may also be a mild increase in pulmonary and urinary tract infections. In patients with GVHD, who are profoundly immunosuppressed and frequently suffer from infections, it may be especially difficult to determine the effect of JAK inhibition on infection rates.
These long-term complications may be specifically relevant to patients with cGVHD. JAK inhibition for prevention of GVHD or treatment of aGVHD may be of sufficiently limited duration, thus minimizing these toxicities.
Conclusion
The JAK/STAT signaling pathways have both inflammatory and immunoregulatory effects. JAKinibs with different JAK specificities and inhibitory capacities have the potential to produce very different clinical results. Both the dose and specificity of JAKinibs are important in determining clinical outcomes, with different diseases demanding different profiles of inhibition for optimal therapeutic activity. Both baricitinib and ruxolitinib are regarded as JAK1/2 inhibitors, yet there are significant differences in their effect on both GvL and GVHD.30 Indeed, there is evidence that GvL and GVHD could be uncoupled with the right approach. In a murine mismatched allo-HCT model, baricitinib caused greater GVHD reduction compared with ruxolitinib, with paradoxical improvement in GvL.30 Baricitinib causes less inhibition of STAT5 phosphorylation, and STAT5 plays a role in regulatory T-cell expansion, which in turn may suppress GVHD.14,30,81,82 This underscores the importance of dosing and specificity of JAKinibs in optimizing clinical outcomes for patients. This is exemplified in another systemic inflammatory disease: sepsis. In a mouse model of Candida sepsis, an intermediate dose of ruxolitinib led to superior survival compared with both high and low doses.18 Fungal burden was highest in mice treated with high doses of ruxolitinib, implying that death was dictated by immune over-suppression. This immune suppression, in the context of allo-HCT or cellular therapies, could reduce GvL or immune mediated tumor killing, respectively. Altogether, these data signal that the type and dose of JAK inhibition will dictate clinical outcomes, and great care must be taken in both JAKinib selection and timing of intervention.
These data support a “Goldilocks Effect” - both too little and too much JAK/STAT inhibition being suboptimal for the prevention and treatment of GVHD in the context of allo-HCT for malignant hematologic conditions. Immune over-suppression could lead to higher rates of relapse and lower rates of engraftment. Immune under-suppression could lead to a higher rate and severity of GVHD. JAKinibs may represent a new tool in balancing GVL and GVHD, allowing a customized approach for each patient based on disease status and comorbidities.
Footnotes
Author contributions: RA and JFD wrote the first version of the manuscript. JC, PR, MAS, and CNA wrote subsequent versions of the manuscript.
Conflict of interest statement: RA, JC, PR, MAS, CNA, and JFD have employment/salary from Washington University in St. Louis. JC has received research funding from Mallinckrodt Pharmaceuticals, served as a consultant for Daewoong Pharmaceutical, and received honorarium from Incyte Corporation. MAS has the following consulting/advisory committees: Amgen, Astellas, Dova pharmaceuticals, FlatIron Inc, Incyte, Partners Therapeutics, Pfizer, Sanofi Genzyme, Abbvie, Merck, Takeda. CNA has the following consulting/advisory committees: Bayer, Incyte, Jazz Pharma, NKarta, Novartis, Pfizer, Seattle Genetics, Tetraphase Pharmaceuticals. JFD has the following consulting/advisory committees: Rivervest, Bioline, Amphivena, Incyte, NeoImuneTech, Macrogenics, and ownership investment: Magenta, WUGEN. The authors have no other conflicts relevant to this publication.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: RA is supported by the American Society of Hematology Clinical Research Training Institute and the National Cancer Institute of the National Institutes of Health under Award Number R25 CA190190. JFD is supported by NIH/NCI: 1 P50 CA171963, (PI: Link, Project 4 Leader: DiPersio) and NIH/NCI: R35 1R35CA210084 NCI (PI: DiPersio).
ORCID iD: Ramzi Abboud  https://orcid.org/0000-0001-7786-4155
https://orcid.org/0000-0001-7786-4155
Contributor Information
Ramzi Abboud, Division of Oncology, Washington University School of Medicine, St. Louis, MO, USA.
Jaebok Choi, Division of Oncology, Washington University School of Medicine, St. Louis, MO, USA.
Peter Ruminski, Division of Oncology, Washington University School of Medicine, St. Louis, MO, USA.
Mark A. Schroeder, Division of Oncology, Washington University School of Medicine, St. Louis, MO, USA
Sena Kim, Division of Oncology, Washington University School of Medicine, St. Louis, MO, USA.
Camille N. Abboud, Division of Oncology, Washington University School of Medicine, St. Louis, MO, USA
John F. DiPersio, Virginia E. and Samuel J. Golman Professor, Chief, Division of Oncology, Deputy Director, Siteman Cancer Center, Washington University School of Medicine, 66o S. Euclid Avenue, CB 8007, Saint Louis, MO 63110, USA.
References
- 1. Warren EH. Biology of the human graft-versus-tumor response and how to exploit it. In: Stephen Forman J, Negrin RS, Antin JH, et al. (eds) Thomas’ hematopoietic cell transplantation. 5th ed. Chichester, UK: John Wiley & Sons, Ltd, 2016, pp. 166–181. [Google Scholar]
- 2. Horowitz MM, Gale RP, Sondel PM, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood 1990; 75: 555–562. [PubMed] [Google Scholar]
- 3. Weisdorf D, Zhang MJ, Arora M, et al. Graft-versus-host disease induced graft-versus-leukemia effect: greater impact on relapse and disease-free survival after reduced intensity conditioning. Biol Blood Marrow Transplant 2012; 18: 1727–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chao NJ. Pharmacologic prevention of acute graft-versus-host disease. In: Stephen Forman J, Negrin RS, Antin JH, et al. (eds) Thomas’ hematopoietic cell transplantation. Chichester, UK: John Wiley & Sons, Ltd, 2016, pp. 990–1006. [Google Scholar]
- 5. Zeiser R, Blazar BR. Acute graft-versus-host disease — biologic process, prevention, and therapy. N Engl J Med 2017; 377: 2167–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of JAK-STAT signaling in the immune system. Nat Immunol 2017; 18: 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science 2002; 296: 1653–1655. [DOI] [PubMed] [Google Scholar]
- 8. Darnell JE, Kerr IM, Stark GR. JAK-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994; 264: 1415–1421. [DOI] [PubMed] [Google Scholar]
- 9. Bousoik E, Montazeri Aliabadi H. “Do We Know Jack” about JAK? A closer look at JAK/STAT signaling pathway. Front Oncol 2018; 8: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of JAK–STAT signaling in the immune system. Nat Immunol 2017; 18: 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kreins AY, Ciancanelli MJ, Okada S, et al. Human TYK2 deficiency: mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med 2015; 212: 1641–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nemoto M, Hattori H, Maeda N, et al. Compound heterozygous TYK2 mutations underlie primary immunodeficiency with T-cell lymphopenia. Sci Rep 2018; 8: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fragoulis GE, Mcinnes IB, Siebert S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology (Oxford) 2019; 58: i43–i54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012; 36: 542–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morozova EV, Moiseev IS, Barabanshikova MV, et al. Graft-versus-host disease prophylaxis with posttransplantation cyclophosphamide and ruxolitinib in patients with myelofibrosis. Blood 2017; 130(Suppl. 1): 4492. [Google Scholar]
- 16. Jagasia M, Zeiser R, Arbushites M, et al. Ruxolitinib for the treatment of patients with steroid-refractory GVHD: an introduction to the REACH trials. Immunotherapy 2018; 10: 391–402. [DOI] [PubMed] [Google Scholar]
- 17. Buchert M, Burns CJ, Ernst M. Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene 2016; 35: 939–951. [DOI] [PubMed] [Google Scholar]
- 18. Tsirigotis P, Papanikolaou N, Elefanti A, et al. Treatment of experimental Candida sepsis with a Janus Kinase inhibitor controls inflammation and prolongs survival. Antimicrob Agents Chemother 2015; 59: 7367–7373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ferrara J, Antin J. The Pathophysiology of Graft-versus-Host Disease. In: Stephen FJ, Negrin RS, Antin JH, et al. (eds) Thomas’ hematopoietic cell transplantation. Chichester, UK: John Wiley & Sons Ltd, 2016, pp. 146–152. [Google Scholar]
- 20. Schroeder MA, Choi J, Staser K, et al. The role of janus kinase signaling in graft-versus-host disease and graft versus leukemia. Biol Blood Marrow Transplant 2018; 24: 1125–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Remberger M, Ringden O, Markling L. TNF alpha levels are increased during bone marrow transplantation conditioning in patients who develop acute GVHD. Bone Marrow Transplant 1995; 15: 99–104. [PubMed] [Google Scholar]
- 22. Hill GR, Crawford JM, Cooke KR, et al. Total body irradiation and acute graft-versus-host disease: the role of gastrointestinal damage and inflammatory cytokines. Blood 1997; 90: 3204–3213. [PubMed] [Google Scholar]
- 23. Nakasone H, Fukuda T, Kanda J, et al. Impact of conditioning intensity and TBI on acute GVHD after hematopoietic cell transplantation. Bone Marrow Transplant 2015; 50: 559–565. [DOI] [PubMed] [Google Scholar]
- 24. Magenau J, Runaas L, Reddy P. Advances in understanding the pathogenesis of graft-versus-host disease. Br J Haematol 2016; 173: 190–205. [DOI] [PubMed] [Google Scholar]
- 25. Brennan TV, Lin L, Huang X, et al. Heparan sulfate, an endogenous TLR4 agonist, promotes acute GVHD after allogeneic stem cell transplantation. Blood 2012; 120: 2899–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jankovic D, Ganesan J, Bscheider M, et al. The Nlrp3 inflammasome regulates acute graft-versus-host disease. J Exp Med 2013; 210: 1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wilhelm K, Ganesan J, Müller T, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med 2010; 16: 1434–1439. [DOI] [PubMed] [Google Scholar]
- 28. Choi J, Ziga ED, Ritchey J, et al. IFNγR signaling mediates alloreactive T-cell trafficking and GVHD. Blood 2012; 120: 4093–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol 2007; 178: 2623–2629. [DOI] [PubMed] [Google Scholar]
- 30. Choi J, Cooper ML, Staser K, et al. Baricitinib-induced blockade of interferon gamma receptor and interleukin-6 receptor for the prevention and treatment of graft-versus-host disease. Leukemia 2018; 32: 2483–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yoon P, Keylock KT, Hartman ME, et al. Macrophage hypo-responsiveness to interferon-γ in aged mice is associated with impaired signaling through JAK-STAT. Mech Ageing Dev 2004; 125: 137–143. [DOI] [PubMed] [Google Scholar]
- 32. Choi J, Cooper ML, Alahmari B, et al. Pharmacologic blockade of JAK1/JAK2 reduces GvHD and preserves the graft-versus-leukemia effect. PLoS One 2014; 9: 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jenq RR, Ubeda C, Taur Y, et al. Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J Exp Med 2012; 209: 903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Holler E, Butzhammer P, Schmid K, et al. Metagenomic analysis of the stool microbiome in patients receiving allogeneic stem cell transplantation: loss of diversity is associated with use of systemic antibiotics and more pronounced in gastrointestinal graft-versus-host disease. Biol Blood Marrow Transplant 2014; 20: 640–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hildebrand D, Uhle F, Sahin D, et al. The interplay of notch signaling and STAT3 in TLR-activated human primary monocytes. Front Cell Infect Microbiol 2018; 8: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Drobyski WR, Szabo A, Zhu F, et al. Tocilizumab, tacrolimus and methotrexate for the prevention of acute graft-versus-host disease: low incidence of lower gastrointestinal tract disease. Haematologica 2018; 103: 717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kennedy GA, Varelias A, Vuckovic S, et al. Addition of interleukin-6 inhibition with tocilizumab to standard graft-versus-host disease prophylaxis after allogeneic stem-cell transplantation: a phase 1/2 trial. Lancet Oncol 2014; 15: 1451–1459. [DOI] [PubMed] [Google Scholar]
- 38. Alblas J, Honing H, De CR, et al. Signal regulatory protein alpha ligation induces macrophage nitric oxide production through JAK/STAT- and phosphatidylinositol 3-kinase/Rac1/NAPDH oxidase/H2O2-dependent pathways. Mol Cell Biol 2005; 25: 7181–7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schwab L, Goroncy L, Palaniyandi S, et al. Neutrophil granulocytes recruited upon translocation of intestinal bacteria enhance graft-versus-host disease via tissue damage. Nat Med 2014; 20: 648–654. [DOI] [PubMed] [Google Scholar]
- 40. Al-shami A, Naccache PH. Granulocyte-macrophage colony-stimulating factor-activated signaling pathways in human neutrophils. J Biol Chem 1999; 274: 5333–5338. [DOI] [PubMed] [Google Scholar]
- 41. Malkovský M, Brenner MK, Hunt R, et al. T-cell depletion of allogeneic bone marrow prevents acceleration of graft-versus-host disease induced by exogenous interleukin 2. Cell Immunol 1986; 103: 476–480. [DOI] [PubMed] [Google Scholar]
- 42. Sykes M, Hoyles KA, Romick ML, et al. In vitro and in vivo analysis of bone marrow-derived CD3+, CD4–, CD8–, NK1.1+ cell lines. Cell Immunol 1990; 129: 478–493. [DOI] [PubMed] [Google Scholar]
- 43. Szebeni J, Wang MG, Pearson DA, et al. IL-2 inhibits early increases in serum gamma interferon levels associated with graft-versus-host-disease. Transplantation 1994; 58: 1385–1393. [PubMed] [Google Scholar]
- 44. Anasetti C, Martin PJ, Hansen JA, et al. A phase I–II study evaluating the murine anti-IL-2 receptor antibody 2A3 for treatment of acute graft-versus-host disease. Transplantation 1990; 50: 49–54. [DOI] [PubMed] [Google Scholar]
- 45. Carniti C, Gimondi S, Vendramin A, et al. Pharmacologic inhibition of JAK1/JAK2 signaling reduces experimental murine acute GVHD while preserving GVT effects. Clin Cancer Res 2015; 21: 3740–3749. [DOI] [PubMed] [Google Scholar]
- 46. Baker MB, Altman NH, Podack ER, et al. The role of cell-mediated cytotoxicity in acute GVHD after MHC-matched allogeneic bone marrow transplantation in mice. J Exp Med 1996; 183: 2645–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ueno Y, Ishii M, Yahagi K, et al. Fas-mediated cholangiopathy in the murine model of graft versus host disease. Hepatology 2000; 31: 966–974. [DOI] [PubMed] [Google Scholar]
- 48. Du W, Leigh ND, Bian G, et al. Granzyme B contributes to the optimal graft-versus-tumor effect mediated by conventional CD4+ T cells. J Immunol Res Ther 2016; 1: 22–28. [PMC free article] [PubMed] [Google Scholar]
- 49. Du W, Leigh ND, Bian G, et al. Granzyme B–mediated activation-induced death of CD4+ T cells inhibits murine acute graft-versus-host disease. J Immunol 2015; 195: 4514–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bian G, Ding X, Leigh ND, et al. Granzyme B–mediated damage of CD8+ T cells impairs graft-versus-tumor effect. J Immunol 2013; 190: 1341–1350. [DOI] [PubMed] [Google Scholar]
- 51. Rowe V, Banovic T, MacDonald KP, et al. Host B cells produce IL-10 following TBI and attenuate acute GVHD after allogeneic bone marrow transplantation. Blood 2006; 108: 2485–2492. [DOI] [PubMed] [Google Scholar]
- 52. Bennett SR, Carbone FR, Toy T, et al. B cells directly tolerize CD8+ T cells. J Exp Med 1998; 188: 1977–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Höllsberg P, Batra V, Dressel A, et al. Induction of anergy in CD8 T cells by B cell presentation of antigen. J Immunol 1996; 157: 5269–5276. [PubMed] [Google Scholar]
- 54. Tretter T, Venigalla RK, Eckstein V, et al. Induction of CD4+ T-cell anergy and apoptosis by activated human B cells. Blood 2008; 112: 4555–4564. [DOI] [PubMed] [Google Scholar]
- 55. Harris DP, Haynes L, Sayles PC, et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol 2000; 1: 475–482. [DOI] [PubMed] [Google Scholar]
- 56. Shimabukuro-Vornhagen A, Hallek MJ, Storb RF, et al. The role of B cells in the pathogenesis of graft-versus-host disease. Blood 2009; 114: 4919–4927. [DOI] [PubMed] [Google Scholar]
- 57. Papin JA, Palsson BO. The JAK-STAT signaling network in the human B-cell: an extreme signaling pathway analysis. Biophys J 2004; 87: 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schultz KR, Paquet J, Bader S, et al. Requirement for B cells in T cell priming to minor histocompatibility antigens and development of graft-versus-host disease. Bone Marrow Transplant 1995; 16: 289–295. [PubMed] [Google Scholar]
- 59. Christopeit M, Schütte V, Theurich S, et al. Rituximab reduces the incidence of acute graft-versus-host disease. Blood 2009; 113: 3130–3131. [DOI] [PubMed] [Google Scholar]
- 60. Wang L, Kurosaki T, Corey SJ. Engagement of the B-cell antigen receptor activates STAT through Lyn in a Jak-independent pathway. Oncogene 2007; 26: 2851–2859. [DOI] [PubMed] [Google Scholar]
- 61. Rozovski U, Wu JY, Harris DM, et al. Stimulation of the B-cell receptor activates the JAK2/STAT3 signaling pathway in chronic lymphocytic leukemia cells. Blood 2014; 123: 3797–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Simonetta F, Alvarez M, Negrin RS. Natural killer cells in graft-versus-host-disease after allogeneic hematopoietic cell transplantation. Front Immunol 2017; 8: 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Xun C, Brown SA, Jennings CD, et al. Acute graft-versus-host-like disease induced by transplantation of human activated natural killer cells into SCID mice. Transplantation 1993; 56: 409–417. [DOI] [PubMed] [Google Scholar]
- 64. Romee R, Rosario M, Berrien-Elliott MM, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med 2016; 8: 357ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Takeda K, Smyth MJ, Cretney E, et al. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J Exp Med 2002; 195: 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kaplan M, Sun YL, Hoey T, et al. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 1996; 382: 174–177. [DOI] [PubMed] [Google Scholar]
- 67. Thierfelder WE, van Deursent JM, Yamamoto K, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature 1996; 382: 171–174. [DOI] [PubMed] [Google Scholar]
- 68. Staser K, Choi J, Khoury J, et al. 21-color flow cytometry reveals immunophenotypes associated with response in acute graft-versus-host disease (aGVHD) patients treated with the Janus kinase (JAK) inhibitor INCB039110 (itacitinib). Presented at 22nd Congress European Hematology Association, 25 June 2017, Madrid, Spain, Abstract S794. [Google Scholar]
- 69. Carpenter PA, Flowers MED, Pavletic SZ. Chronic graft-versus-host disease - clinical manifestations and therapy. In: Stephen Forman J, Negrin RS, Antin JH, et al. (eds) Thomas’ hematopoietic cell transplantation. Chichester, UK: John Wiley & Sons, Ltd, 2016, pp. 1020–1039. [Google Scholar]
- 70. Lee SJ. Classification systems for chronic graft-versus-host disease. Blood 2017; 129: 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zeiser R, Burchert A, Lengerke C, et al. Ruxolitinib in corticosteroid-refractory graft-versus-host disease after allogeneic stem cell transplantation: a multicenter survey. Leukemia 2015; 29: 2062–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Flynn R, Paz K, Du J, et al. Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism. Blood 2016; 127: 2144–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen W, Nyuydzefe MS, Weiss JM, et al. ROCK2, but not ROCK1 interacts with phosphorylated STAT3 and co-occupies TH17/TFH gene promoters in TH17-activated human T cells. Sci Rep 2018; 8: 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jaglowski SM, Blazar BR. How ibrutinib, a B-cell malignancy drug, became an FDA-approved second-line therapy for steroid-resistant chronic GVHD. Blood Adv 2018; 2: 2012–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jagasia M, Ali H, Schroeder MA, et al. Ruxolitinib in combination with corticosteroids for the treatment of steroid-refractory acute graft-vs-host disease: results from the phase 2 REACH1 trial. Biol Blood Marrow Transplant 2019; 25: S52. [Google Scholar]
- 76. Spoerl S, Mathew NR, Bscheider M, et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood 2014; 123: 3832–3842. [DOI] [PubMed] [Google Scholar]
- 77. Harigai M. Growing evidence of the safety of JAK inhibitors in patients with rheumatoid arthritis. Rheumatol (Oxford) 2019; 58: i34–i42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016; 30: 1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Blechman AB, Cabell CE, Weinberger CH, et al. Aggressive skin cancers occurring in patients treated with the Janus Kinase inhibitor ruxolitinib. J Drugs Dermatology 2017; 16: 508–511. [PubMed] [Google Scholar]
- 80. Lussana F, Cattaneo M, Rambaldi A, et al. Ruxolitinib-associated infections: a systematic review and meta-analysis. Am J Hematol 2018; 93: 339–347. [DOI] [PubMed] [Google Scholar]
- 81. Zorn E, Nelson EA, Mohseni M, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006; 108: 1571–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yao Z, Kanno Y, Kerenyi M, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood 2007; 109: 4368–4375. [DOI] [PMC free article] [PubMed] [Google Scholar]