

Abstract
Myotonic dystrophy type 1 (DM1), the most common adult muscular dystrophy, is an autosomal dominant disorder caused by an expansion of a (CTG)n tract within the 3′ untranslated region (3′UTR) of the dystrophia myotonica protein kinase (DMPK) gene. Mutant DMPK mRNAs are toxic, present in nuclear RNA foci and correlated with a plethora of RNA splicing defects. Cardinal features of DM1 are myotonia and cardiac conduction abnormalities. Using transgenic mice, we have demonstrated that expression of the mutant DMPK 3′UTR is sufficient to elicit these features of DM1. Here, using these mice, we present a study of systemic treatment with an antisense oligonucleotide (ASO) (ISIS 486178) targeted to a non-CUG sequence within the 3′UTR of DMPK. RNA foci and DMPK 3′UTR mRNA levels were reduced in both the heart and skeletal muscles. This correlated with improvements in several splicing defects in skeletal and cardiac muscles. The treatment reduced myotonia and this correlated with increased Clcn1 expression. Furthermore, functional testing showed improvements in treadmill running. Of note, we demonstrate that the ASO treatment reversed the cardiac conduction abnormalities, and this correlated with restoration of Gja5 (connexin 40) expression in the heart. This is the first time that an ASO targeting a non-CUG sequence within the DMPK 3′UTR has demonstrated benefit on the key DM1 phenotypes of myotonia and cardiac conduction defects. Our data also shows for the first time that ASOs may be a viable option for treating cardiac pathology in DM1.
Introduction
Myotonic dystrophy type 1 (DM1) is one of the most common types of muscular dystrophy. DM1 is a multisystemic disorder characterized by a myriad of features including myotonia, progressive muscle wasting, cardiac conduction defects, and cognitive dysfunctions (1). DM1 is caused by an expanded (CTG)n repeat tract in the 3'UTR of the dystrophia myotonica protein kinase (DMPK) gene (2). The mutant RNA accumulates in the nuclei of DM1 cells (3), and affects RNA binding proteins such as muscleblind family members (MBNL1-3) and CUGBP Elav like family member 1 (CELF1) (4). These proteins are involved in multiple aspects of RNA processing and localization (5). Many other RNA-binding proteins including Staufen1, hnRNP (H and F), DEAD-box protein 6 (DDX6), RNA helicase p68/DDX5, TBPH (homolog of human TAR DNA-binding protein 43 or TDP-43) and BSF (Bicoid stability factor; homolog of human LRPPRC) have also been implicated in DM1 associated RNA toxicity (6–11). In addition, a number of signaling pathways and proteins including glycogen synthase kinase 3β (GSK3β), AKT, AMPK, and protein kinase C (PKC) have been suggested to play a role in DM1 pathogenesis (1). DM1 is caused by an expanded (CTG)n repeat tract in the 3'UTR of the dystrophia myotonica protein kinase (DMPK) gene (2). The mutant RNA accumulates in the nuclei of DM1 cells (3), and affects RNA binding proteins such as muscleblind family members (MBNL1-3) and CUGBP Elav like family member 1 (CELF1) (4). These proteins are involved in multiple aspects of RNA processing and localization (5). Many other RNA-binding proteins including Staufen1, hnRNP (H and F), DEAD-box protein 6 (DDX6), RNA helicase p68/DDX5, TBPH (homolog of human TAR DNA-binding protein 43 or TDP-43) and BSF (Bicoid stability factor; homolog of human LRPPRC) have also been implicated in DM1 associated RNA toxicity (12–14). Results from our lab also showed that the Fn14/TWEAK signaling pathway is altered in DM1 and blocking TWEAK-Fn14 is beneficial in improving skeletal muscle pathology in a mouse model of RNA toxicity (15).
Though the majority of research in DM1 has focused on skeletal muscle, cardiac involvement is prevalent in DM1 patients (16). This is principally a variety of cardiac conduction abnormalities including atrial arrhythmias, progressive sinus node dysfunction and progressive atrioventricular conduction blocks (17). There is also some evidence for myocardial contractile dysfunction (18, 19). Studies suggest that approximately 30% of the deaths in DM1 patients are due to cardiac complications (20, 21). Current clinical recommendations are for regular monitoring of cardiac conduction and function, and treatment is primarily symptom and complication driven, with various implantation devices used to monitor and control cardiac rhythm (22).
The first mouse model of cardiac conduction abnormalities associated with RNA toxicity came from our laboratory more than a decade ago (15). In this inducible model of RNA toxicity, expression of the toxic RNA results in cardiac conduction defects (15, 23, 24). Using this model, we also showed that these abnormalities were potentially reversible with silencing expression of, or by potentially targeting, the toxic RNA (15). Subsequently, others have made cardiac-specific models of RNA toxicity (25–27) or demonstrated abnormal electrical activity in RNA toxicity models (28) and also shown that mice with deficits in MBNL proteins or increased CELF1 in the heart can also manifest cardiac pathologies (29–33).
A number of approaches have been developed to reduce toxicity of CUG repeat RNAs in cell culture as well as in mouse models (34). Recent studies suggest that small molecules can also rescue some of the phenotypes in cells as well as in mouse models (35). Antisense strategy such as a viral vector producing antisense RNA (36), a hammerhead RNA (37), siRNAs (38), CAG antisense oligonucleotides (39–41) and Hu7-snRNA-CAG (42) has been used to correct some features of the disease in DM1 cells and mouse models. These strategies involved either targeted degradation of the mutant RNA or steric blocking of interactions of RNA-binding proteins with the mutant RNAs. More recently, a novel high affinity antisense oligonucleotide that contain 2′-4′-constrained ethyl (cEt) modifications has been shown to be effective in reducing DMPK or Dmpk expression in monkeys and mice when administered by subcutaneous injection (43, 44). However, its efficacy in alleviating the key clinical manifestations of DM1, namely, myotonia and cardiac conduction defects has not been established. This ASO, called ISIS 486178, is called a ‘gapmer’ and consists of a central core of 10 bases of deoxynucleotides flanked by 3 bases of cEt modified nucleotides on the 5′ and 3′ ends (44, 45). The intent is to enhance RNase H-mediated degradation of the target in the nucleus. The target sequence for ISIS 486178 is located 3′ of the (CUG)n tract in the DMPK 3'UTR about 50 bp from the polyadenylation signal sequence. Here, we use this ASO to validate a mouse model of RNA toxicity expressing the expanded DMPK 3'UTR and show, for the first time, the potential benefits of ASO therapy in treating cardiac abnormalities associated with RNA toxicity related to DM1.
Results
ISIS 486178 treatment improves myotonia and reduces expression of toxic RNAs in skeletal muscle
We have an inducible/reversible mouse model of RNA toxicity (called DM200) in which over-expression of an eGFP-DMPK3'UTR (CUG)200 mRNA results in many DM1 features including myotonia, RNA foci, RNA splicing defects and progressive cardiac conduction defects. To evaluate the effect of the ASO on skeletal muscle, we assessed myotonia by electromyography (EMG) using a scale previously described (46). Uninduced DM200 mice did not exhibit any myotonia. However, DM200 mice developed myotonia at the end of 2 weeks of transgene induction with 0.2% doxycycline in their drinking water (ad libidum), with a score of 1 or 2 (Fig. 1A). Only mice that had myotonia were used for analyses of the ASO. They were then divided into three groups [PBS, control ASO (MOE chemistry) and ISIS 486178 (cEt chemistry)]. The DM200-induced mice were treated with 25 mg/kg of ISIS 486178 or control ASO or PBS administered subcutaneously, twice a week for 6 weeks. Repeat EMGs were done at 3 and 6 weeks post-treatment. All experiments were done in a blinded fashion. After 6 weeks of treatment, the mice treated with ISIS 486178 had reduced myotonia scores (0 or 1), with more than 50% of them having no myotonia at all. In contrast, all the mice in the control ASO or PBS groups had persistent myotonia (scores 1–3) with many having increased in severity over the course of the experiment (Fig. 1A). These data show that the myotonia responded favorably to treatment with ISIS 486178 in the RNA toxicity mice.
Figure 1.

ISIS 486178 treatments improves myotonia in the RNA toxicity mice. (A) Graph shows the degree of myotonia in DM200 D+ mice before (at 2 weeks induced) and after 6 weeks of treatment with PBS, control ASO or ISIS 486178; PBS (n = 9), control ASO (n = 9) and ISIS 486178 (n = 7). (B) RT-PCR analysis of Clcn1 splicing in skeletal muscle (gluteus maximus) from DM200 D+ mice shows significant increased inclusion of exon7a (ex7a) as compared to DM200 D− mice. ISIS 486178 treatment has no significant effect on Clcn1 (ex7a) as compared to control ASO. N = 4–5 mice/group; *P ≤ 0.05; **P ≤ 0.01; student’s t-test; error bars are mean ± S.D. (C) Representative image of CLC-1 expression detected by immunofluorescence in quadriceps femoris shows a normal sarcolemmal CLC-1 staining pattern in wild-type (WT) and DM200 D− mice. HSA-LR and DM200 D+ mice treated with control ASO or PBS show loss of sarcolemmal CLC-1, whereas DM200 D+ mice treated with ISIS 486178 show restored sarcolemmal CLC-1. (D) qRT-PCR shows slightly decreased expression of total Clcn1 mRNA levels in skeletal muscle (gluteus maximus) of DM200 D− mice and significantly further decreases in DM200 D+ mice. qRT-PCR shows increased expression of total Clcn1 mRNA in DM200 D+ mice treated with ISIS 486178 as compared to mice treated with the control ASO. N = 4–5 mice/group; *P ≤ 0.05; **P ≤ 0.01; Student’s t-test; error bars are mean ± SEM.
Myotonia in DM1 is linked to reduced chloride channel expression (47, 48). To examine whether ISIS 486178 affects the expression of Clcn1 in skeletal muscle, we evaluated Clcn1 exon7a splicing in the DM200 mice, as this splicing event is associated with myotonia in DM1 (47, 48). The uninduced DM200 (DM200 D−) mice showed about 8.5% exon 7a inclusion, similar to the 7.4% inclusion seen in the wild-type control mice. Upon induction of RNA toxicity (DM200 D+), this increased to about 18% (P = 0.001) (Fig. 1B). This is significantly less that the 44% reported for HSA-LR mice (48) or the 43% we found by analyzing HSA-LR muscles in our laboratory (data not shown). Nevertheless, this was associated with myotonia in the DM200 mice, though the level of myotonia was less than that reported in the HSA-LR mice (46). Though the ISIS 486178 treatment did reduce the exon7a inclusion to about 12%, this barely reached statistical significance (P = 0.05) and was similar to the changes seen with PBS or control ASO treatment (Fig. 1B). This was surprising, since we did see functional rescue of myotonia. We then analyzed Clcn1 expression in skeletal muscle by immunofluorescence and qRT-PCR. Using immunofluorescence, we found normal sarcolemmal CLC-1 staining in wild-type and DM200 D− mice (Fig. 1C). The pattern observed in the DM200 D+ PBS- or control ASO-treated mice was similar to that seen in the HSA-LR mice, with patchy staining of the sarcolemma with many fibers lacking CLC-1 staining in the muscle. After treatment, we observed better and more uniform sarcolemmal CLC-1 staining in the ISIS 486178-treated group as compared to control ASO- or PBS-treated mice (Fig. 1C). Therefore, we assessed total Clcn1 mRNA levels as this has been reported to be reduced in the HSA-LR mice [down to 43% of wild-type mice; similar to the 50% reduction we found in HSA-LR mice (data not shown)] (48). We found that DM200 D− mice had a slight but significant reduction (P = 0.02) in total Clcn-1 mRNA levels to about 80% of wild-type mice. Induction of RNA toxicity (DM200 D+) reduced Clcn-1 mRNA levels further, to about 50–55% of wild-type mice (P = 0.0008) (Fig. 1D). This is similar to the changes in the HSA-LR mice. Of note, treatment of DM200 D+ mice with ISIS 486178 resulted in a significant increase in Clcn1 mRNA levels as compared to mice treated with the control ASO (P = 0.003), reaching to about 71% of wild-type levels, and was similar to the levels seen in the DM200 D− mice. This response was not in the DM200 D+ mice treated with PBS (52% of wild-type levels) or control ASO (56% of wild-type levels) (Fig. 1D).
We also evaluated the effect of ISIS 486178 on the expression of toxic RNAs by RNA fluorescence in situ hybridization (RNA-FISH) and qRT-PCR in DM200 skeletal muscles (quadriceps femoris). Six weeks of treatment with ISIS 486178 reduced the visible RNA foci in DM200 D+ mice as compared to mice treated with control ASO (Fig. 2A). qRT-PCR also showed a 53% reduction in the level of the toxic RNA in DM200 D+ mice treated with ISIS 486178 (P = 0.002) as compared to mice treated with the control ASO and a 37% reduction as compared to mice treated with PBS (P = 0.03) (Fig. 2B). This reduction in toxic RNA and the RNA foci was associated with a redistribution of MBNL1 protein from its sequestered state on the RNA foci to a more nucleoplasmic distribution (Fig. 2C).
Figure 2.

ISIS 486178 reduces toxic RNA levels in skeletal muscle. (A) RNA-FISH (red) in skeletal muscle (quadriceps femoris) shows reduced RNA foci in DM200 D+ mice treated with ISIS 486178. No foci were detected in wild-type mice. A few RNA foci were detected in the skeletal muscle of uninduced mice (DM200 D−) and more in the induced mice (DM200 D+). RNA foci detected with a Cy3-conugated CAG10 oligonucleotide probe and DAPI (blue) was used to stain nuclei. (B) qRT-PCR shows reduced toxic RNA levels in skeletal muscle (gluteus maximus) of mice treated with ISIS 486178 as compared to control ASO-treated mice. N = 5 mice/group; *P ≤ 0.05; **P ≤ 0.01; student’s t-test; error bars are mean ± SEM. (C) Representative merged image of RNA-FISH/MBNL1 immunofluorescence showing MBNL1 co-localization with RNA foci (yellow) in the skeletal muscle (quadriceps femoris) of DM200 D+ (control ASO) mice as compared to ISIS 486178-treated DM200 D+ mice where RNA foci are scarce and MBNL1 shows more nucleoplasmic staining. MBNL1 (green); RNA foci (red); DAPI (blue). Note, we have confirmed that gluteus maximus and quadriceps femoris give similar results in our assays (data not shown).
ISIS 486178 improves cardiac conduction defects in a mouse model of RNA toxicity
We also assessed cardiac conduction abnormalities, as the DM200 model is one of the few that shows both myotonia and cardiac conduction defects. All the DM200 mice used in this study were phenotypically normal by surface ECG, at 2 months of age (Table 1, D− mice). After 2 weeks of 0.2% doxycycline, the mice developed a variety of cardiac conduction defects similar to those seen in DM1 individuals, ranging from atrial arrhythmias to varying degrees of heart block (Table 1, Supplementary Material, Fig. S1). To test whether ISIS 486178 could rescue the cardiac conduction defects in the RNA toxicity mice, we treated the mice for 6 weeks as described above. We monitored the mice using surface ECGs at 3 weeks and 6 weeks post-treatment. Significant improvement in cardiac conduction was noted in all the ISIS 486178-treated mice after 6 weeks of treatment. In fact, all the mice went back to the normal cardiac rhythm noted prior to toxic RNA induction. In contrast, we observed no rescue in the control ASO- or PBS-treated groups (Table 1).
Table 1.
ECG results
| Treatment | Mouse ID | D− | D+(2 weeks) | After 6x ASO treatment | After 12X ASO treatment |
|---|---|---|---|---|---|
| PBS | DM200 340 | 0.0325 | 0.0525 | 0.0525 | 0.0575 |
| DM200 344 | 0.0275 | Afib | Afib | Afib | |
| DM200 351 | 0.03 | Afib | Afib | Afib | |
| DM200 366 | 0.0325 | Afib | Afib | Afib | |
| DM200 377 | 0.0325 | 2nd | 0.0425 | 0.05 | |
| DM200 382 | 0.0325 | Afib | 0.0575 | Afib | |
| DM200 385 | 0.03 | 0.085 | 0.075 | Afib | |
| DM200 392 | 0.0325 | 2nd | 0.0375 | 0.04 | |
| DM200 403 | 0.03 | 0.06 | 0.045 | 0.04 | |
| Control-ASO | DM200 350 | 0.0275 | 0.06 | 0.035 | Afib |
| DM200 355 | 0.03 | Afib | 0.03 | 0.03 | |
| DM200 356 | 0.03 | 2nd | 2nd | Afib | |
| DM200 364 | 0.0325 | Afib | Afib | Afib | |
| DM200 367 | 0.0325 | 0.045 | 0.045 | Afib | |
| DM200 387 | 0.03 | 2nd | 2nd | 0.05 | |
| DM200 393 | 0.035 | Afib | Afib | Afib | |
| DM200 394 | 0.03 | 0.05 | 0.0375 | 0.04 | |
| DM200 404 | 0.025 | 0.11 | 0.0475 | 0.055 | |
| ISIS 486178 | DM200 342 | 0.0325 | 0.065 | 0.035 | 0.0325 |
| DM200 345 | 0.03 | 2nd | 0.0375 | 0.0325 | |
| DM200 349 | 0.03 | 0.05 | 0.03 | 0.03 | |
| DM200 376 | 0.0325 | 2nd | 0.035 | 0.035 | |
| DM200 381 | 0.035 | 2nd | 0.0325 | 0.035 | |
| DM200 391 | 0.04 | AFib | 0.04 | 0.0325 | |
| DM200 402 | 0.03 | 3rd | 0.04 | 0.03 |
Afib = atrial fibrillation, 2nd = second-degree heart block, 3rd = third-degree heart block.
The numerical values represent PR intervals in seconds. Normal values are usually between 0.025 and 0.0375 s. Prolonged PR intervals are greater than that.
Please see Supplementary Material, Figure S1 for examples of ECGs.
ISIS 486178 reduces toxic RNA expression in the heart and improves run distance in a mouse model of RNA toxicity
Previously, we showed that induction of toxic RNA expression for 4 weeks made the DM200 mice run a shorter distance as compared to uninduced DM200 mice (24). In the current study, baseline analyses, including treadmill running, were done on all the DM200 mice at 2 months of age prior to doxycycline administration. Analysis of treadmill running showed that the DM200 D+ mice treated with ISIS 486178 for 6 weeks ran further (about 25% more) than DM200s treated with control ASO (P = 0.0067) and 16% further than mice treated with PBS (P = 0.1) (Fig. 3A). By RNA-FISH, we also found that ISIS 486178 reduced the RNA foci in the heart as compared to the DM200 D− mice or the DM200 D+ mice treated with the control ASO (Fig. 3B). Note also that the DM200 D− mice have transgene leakiness in the absence of any doxycycline, and though there are a few RNA foci, this is not associated with conduction abnormalities in these mice (see Table 1). qRT-PCR confirmed a 51% reduction in the toxic RNA level in the ISIS 486178-treated mice as compared to the control ASO-treated group (P = 0.00057) and a 35% reduction as compared to mice treated with PBS (P = 0.01) (Fig. 3C). Interestingly, these treated mice have even less toxic RNA than the DM200 D− mice. This is the first time an ASO targeting DMPK has shown efficacy in reducing toxic RNA levels in the heart.
Figure 3.

ISIS 486178 treatment improves heart function in the RNA toxicity mice. (A) Graph showing percent retained run distance (relative to pre-induction) in DM200 D+ mice after 6 weeks of treatment with ISIS 486178 or control ASO or PBS. Retained run distance is significantly better in the ISIS 486178 as compared to the control ASO-treated group. N = 7–9 mice/group; **P ≤ 0.01; student’s t-test; error bars are mean ± SEM. (B) RNA-FISH detection of RNA foci (red) in the heart from wild-type, DM200 (D−), DM200 (D+) control ASO-treated and DM200 (D+) ISIS 486178-treated mice. No foci were present in wild-type mice. RNA foci were detected in the heart of uninduced mice (DM200 D−) and more in the induced mice (DM200 D+), whereas DM200 D+ treated with ISIS 486178 shows few if any RNA foci. DAPI (blue) was used to stain nuclei. (C) qRT-PCR shows expression of toxic RNAs is reduced significantly in hearts of DM200 D+ mice treated with ISIS 486178 as compared to control ASO-treated mice. N = 5 mice/group; *P ≤ 0.05;***P ≤ 0.001; student’s t-test; error bars are mean ± SEM.
ISIS 486178 rescues mRNA splicing defects in the heart
To determine whether treatment with ASOs would correct the mis-splicing events in the heart caused by the toxic RNA, we analyzed splicing of Cacna1s (exon 29), Atp2a1 (exon 22), Ldb3 (exon 11) and Scn5a (exon 6A), targets which are mis-spliced in individuals with DM1 (31, 49–51). Three of these splicing targets were mis-spliced in the hearts of DM200 D+ mice (Fig. 4). Treatment with ISIS 486178 significantly rescued splicing defects of the target genes Cacna1s (P = 0.0047), Atp2a1 (P = 0.0023) and Ldb3 (P = 0.0073) in the DM200 mouse model (Fig. 4). For Scn5a (exon 6a), the splicing changes were slight, and there was no significant difference between wild-type and DM200 D+ mice; but the treatment with ISIS 486178 did show a statistically significant reduction as compared to the mice treated with the control ASO.
Figure 4.
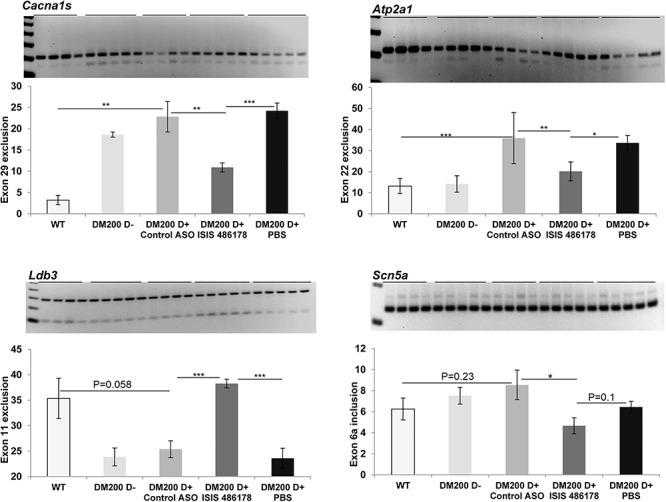
Analysis of RNA splicing defects in the heart of RNA toxicity mice. RT-PCR analysis of several RNA splicing targets, Cacna1s (exon 29), Atp2a1 (exon 22) and Ldb3 (exon 11) in hearts from DM200 D+ mice showing splicing defects of the indicated exon. RT-PCR analysis of Scn5a (exon 6a) shows no significant splicing defects in the DM200 D+ mice. Quantification of the gels show significant splicing defects of the tested targets in the DM200 D+ mice except for Scn5a (exon 6a). ISIS 486178 treatment results in significant rescue of splicing for Cacna1s (exon 29), Atp2a1 (exon 22), Ldb3 (exon 11) and Scn5a (exon 6a) as compared to treatment with control ASO. N = 4–6 mice/groups; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001; student’s t-test; error bars are mean ± S.D.
ISIS 486178 rescues connexin 40 expression defects in the heart
We have previously shown that RNA toxicity affects the expression of genes encoding connexins in the heart (23), where we found decreased levels of connexin 43 (Gja1) and connexin 40 (Gja5). More recently, we demonstrated increased expression of markers of fibrosis (Col1a1) in the hearts of DM200 D+ mice (24). We therefore assessed if the DM200+ mice had changes in expression of Gja1 and Gja5 along with the changes in Col1a1 and if the treatment with ISIS 486178 had any beneficial effects. We found that induction of RNA toxicity resulted in significant decreases in mRNA levels of Gja5 (connexin 40) in the hearts of the DM200 D+ mice (Fig. 5A). Gja5 mRNA levels already showed reductions in the DM200 D− mice to about 50% of wild-type levels, though this was not statistically significant (P = 0.09) or associated with any conduction abnormality. However, Gja5 mRNA levels drastically reduced further to about 20% of wild-type levels in the DM200 D+ mice treated with control ASO or PBS (P = 0.006). Immunofluorescence showed robust connexin 40 expression in the cardiac atria of wild-type and DM200 D− mice (Fig. 5A). But, in DM200 D+ mice treated with either PBS or the control ASO, it showed dramatic loss in connexin 40 expression. Notably, mice treated with ISIS 486178 had normalized expression of connexin 40 in the cardiac atria. This correlated with increased Gja5 mRNA levels to levels similar to those in the DM200 D− mice (P = 0.36). This also correlated with the correction of their cardiac conduction abnormalities, most of which were related to atrial arrhythmias (Table 1).
Figure 5.
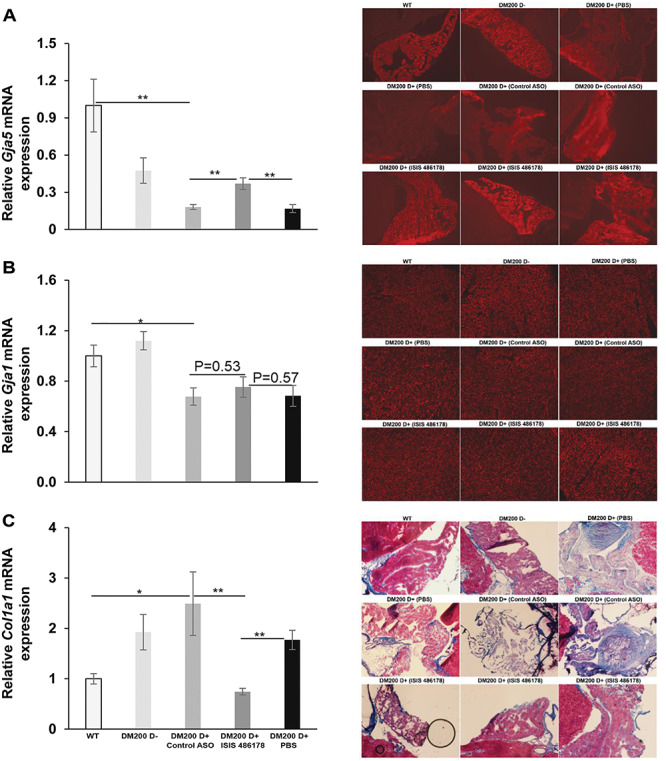
Expression of genes in the hearts of DM200 mice treated with ASO (ISIS 486178). (A) (Left) qRT-PCR analysis shows decreased expression of Gja5 mRNA in the hearts of DM200 D+ mice as compared to wild-type mice and a significant rescue of expression in response to treatment with ISIS 486178. There was no significant difference in the expression of Gja5 mRNA between DM200 D− mice and the ISIS 486178-treated group. N = 4–6 mice/group; **P = 0.01; student’s t-test; error bars are mean ± SEM. (Right) Immunofluorescence for connexin 40 (red) in the atrium shows decreased expression in DM200 D+ mice (PBS as well as control ASO) as compared to DM200 D− mice. ISIS 486178 treatment results in rescue of connexin 40 expression as compared to control ASO or PBS. (B) (Left) qRT-PCR shows decreased expression of Gja1 mRNA in the hearts of DM200 D+ mice treated with PBS or control ASO as compared to wild-type mice. There was no significant change in expression with ISIS 486178 treatment. N = 4–5 mice/group; *P ≤ 0.05; student’s t-test; error bars are mean ± SEM. (Right) Immunofluorescence for connexin 43 (red) shows no visible difference in the ventricular muscle among the groups. (C) (Left) qRT-PCR shows increased expression of Col1a1 mRNA in the hearts of DM200+ D+ mice as compared to wild-type mice and a significant rescue in the expression in response to treatment with ISIS 486178. N = 4–5 mice/group; *P ≤ 0.05, **P ≤ 0.01; student’s t-test; error bars are mean ± SEM. (Right) Gomori trichrome staining of representative mouse atrial sections from WT, DM200 D−, DM200 D+ (PBS), DM200 D+ (control ASO) and DM200 D+ (ISIS 486178) groups showing increased collagen staining in the control ASO- or PBS-treated groups as compared to WT or DM200 D−. DM200 D+ mice treated with ISIS 486178 had visibly less collagen accumulation and more morphologically intact atria and cardiomyocytes.
The levels of Gja1 mRNA were unchanged in the hearts of DM200 D− mice as compared to wild-type mice (Fig. 5B). But, in the DM200 D+ mice treated with PBS or control ASO, the Gja1 mRNA levels reduced to about 68% to 75% of the levels found in the wild-type mice (P = 0.04). Treatment of the DM200 D+ mice with ISIS 486178 did not have any significant effect on the levels of Gja1 mRNA levels (Fig. 5B). Immunofluorescence of the cardiac ventricles, where the majority of Gja1 (connexin 43) is expressed, showed no obvious difference in expression between the different groups of mice (Fig. 5B).
We also evaluated the expression of Col1a1 (the gene for collagen type1, alpha1), a pro-fibrotic marker in the heart (52). In the hearts of DM200 D− mice, the Col1a1 mRNA levels were about twice as high as in wild-type mice (P = 0.03) and increased further to about 2.5-fold in the DM200 D+ mice, similar to our previous observations (24) (Fig. 5C). Treatment of DM200 D+ mice with either PBS or the control ASO had no significant effect on this. However, the ISIS 486178 treatment resulted in significant reductions in Col1a1 mRNA levels (P = 0.002). In fact, the Col1a1 mRNA levels were reduced below those in the DM200 D− mice and were similar to those seen in wild-type mice. Gomori trichrome staining for collagen showed a slight perceptible increase in blue staining (indicative of collagen) in the atria and ventricles of DM200 D− mice as compared to wild-type mice (Fig. 5C, Supplementary Material, Fig. S2). In contrast, in the DM200 D+ mice treated with either PBS or the control ASO, we saw a dramatic increase in expression of collagen in the atria of all these mice (Fig. 5C). In the ventricles of these mice, there seemed to be increased collagen staining as well (Supplementary Material, Fig. S1). Notably, DM200 D+ mice treated with ISIS 486178 did not show this and, instead, had more morphologically intact atria and atrial cardiomyocytes (Fig. 5C). It was difficult to discern if there was a visible reduction in collagen expression in the ventricles of DM200 D+ mice treated with ISIS 486178 as compared to the mice treated with PBS or the control ASO (Supplementary Material, Fig. S2).
Discussion
Myotonic dystrophy type 1 is a multisystemic disorder with skeletal muscle and the heart being two of the most clinically relevant tissues affected in patients. In fact, the combination of myotonia and progressive conduction abnormalities uniquely identifies patients with myotonic dystrophy. Our DM200 inducible model of RNA toxicity associated with expression of the mutant DMPK 3'UTR mRNA is one of the few mouse models to concurrently have both of these defining features of DM1. Using this model, we have shown that these phenotypes are sensitive to the presence of the toxic RNA and that cessation of doxycycline-induced transgene expression results in reversion to normal in mice affected by RNA toxicity (24). Our goal in this study was to use this model to evaluate antisense oligonucleotides that target the DMPK 3'UTR to see if they would be effective as potential therapies.
Antisense oligonucleotides have been used to treat various mouse models of DM1. The most cited is the landmark study by Wheeler et al. (45) in which they used systemic administration of an ASO that targeted the 3'UTR of the HSA mRNA in which an expanded (CUG) tract was expressed. Other studies during that time period used various antisense oligonucleotide chemistries targeting the CUG, injected locally (40, 41, 53, 54). One potential concern with using an ASO that targets only the CUG sequence is that it could have off-target effects. On the flip side, targeting regions outside the CUG tract will most likely not confer allele specificity unless the target is a disease allele-specific variant. However, the expanded CUG tract is the only allele-specific variant that is exclusively associated with DM1. One of the first descriptions of antisense oligonucleotides targeting regions in the DMPK mRNA outside the CUG repeats described pre-clinical evaluation of the ISIS 486178 oligonucleotide in cells, wild-type mice, cynomolgus monkeys and limited evaluation of the effects of this ASO on the levels of the transgene mRNA in the DMSXL mouse model (44). This paper established the safety and molecular efficacy profiles of this ASO and demonstrated that this ASO targeted both the mouse and human DMPK gene. Our data also shows that the ASO is very effective in targeting endogenous Dmpk mRNA levels in the skeletal muscle and heart (Supplementary Material, Fig. S3). Another more recent study was also done to evaluate the same parameters, and it also provided data on the distribution of the ASO in various tissues including the skeletal muscle and heart of rats and monkeys (55). The data from mice is very similar to that published for rats (unpublished data-IONIS). Subsequently this ASO was used in wild-type mice and Dmpk+/− mice to demonstrate that reductions in expression of DMPK had no apparent deleterious effects on cardiac and skeletal muscle function (56). The ISIS 486178 ASO has been evaluated only once in a mouse model of RNA toxicity, namely, the DMSXL mouse model (43). In that study, the investigators administered ISIS 486178 at the same dose we used in the current study and found that the ASO decreased transgene mRNA levels by about 66% in skeletal muscle and about 31% in the heart. There was limited evaluation of phenotypes in that mouse model and no reported assessment of effects on splicing defects. Unfortunately, ASOs that are directed against non-CUG regions of the DMPK mRNA have not been evaluated in a mouse model with robust DM1 phenotypes. So, in this study, we took the opportunity to evaluate the efficacy of such an ASO in a mouse model with robust phenotypes spanning a variety of relevant molecular and clinical readouts. Additionally, no studies have been done to study the efficacy of these ASOs on the effects of RNA toxicity in the heart.
In this study, we found that an ASO targeting a non-CUG sequence within the DMPK 3'UTR (ISIS 486178) significantly reduced RNA foci and mutant DMPK mRNAs in the skeletal muscles of mice with RNA toxicity. RNA foci are a key hallmark of the expression of the mutant DMPK mRNA, as are an increasing number of splicing defects in affected tissues, due to sequestration of RNA-binding proteins (such as MBNL1– MBNL3) by the RNA foci. For example, in DM1, myotonia is attributed to deficiency in sarcolemmal CLC-1 (chloride channel protein 1) due to splicing defects in Clcn1, (e.g. exon 7a inclusion). This concept has been supported by results in mice with MBNL1 deficiency (57). The DM200 mice also have mild but clear myotonia and various splicing defects in skeletal muscle, including a mild increase in Clcn1 exon 7a inclusion (~10–15%). Notably, despite the observed disappearance of RNA foci and MBNL1 aggregates in the skeletal muscles of DM200 mice treated with ISIS 486178, we did not see a significant reversion of the Clcn1 exon7a splicing defect. However, we did find restoration of CLC-1 expression on the sarcolemma of the treated skeletal muscles, and this correlated with an increase in overall Clcn-1 expression. This was surprising. But, it should be noted that a variety of other splicing defects in Clcn-1 (such as inclusion of intron 2) have been reported (47). We developed a new RT-PCR-based assay to assess this. Our assay showed that intron 2 retention is increased in the DM200, in HSA-LR and Mbnl1−/− mice and showed that in the DM200 mice, the ISIS 486178 ASO corrects the splicing defect (Fig. 6). The splicing defects in Clcn-1 are predicted to result in premature stop codons that may make the Clcn-1 mRNA more susceptible to nonsense mediated RNA decay and thus result in decreased CLC-1 (47, 48). Thus, the rescue in total Clcn-1 mRNA levels and concomitant re-expression of CLC-1 at the sarcolemma in the ISIS 486178-treated mice may likely reflect a correction of this deleterious effect of RNA toxicity.
Figure 6.
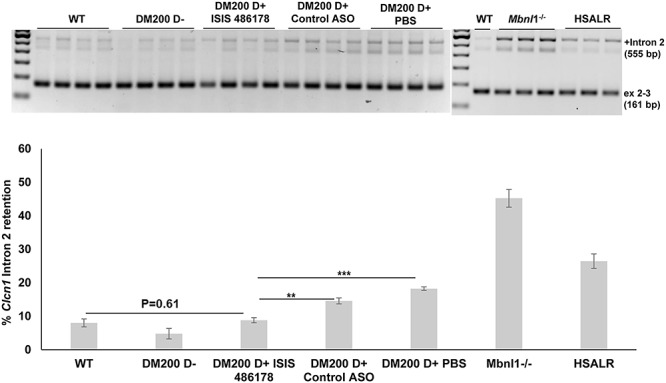
Analysis of Clcn1 intron 2 retention in the skeletal muscle of RNA toxicity mice. (A) RT-PCR analysis of Clcn1 intron 2 retention in the skeletal muscle (quadriceps femoris) shows increased retention in DM200 D+ mice (treated with control ASO or PBS) as compared to wild-type or DM200 D− mice. It also showed increased retention in HSA-LR and Mbnl1−/− mice. Treatment with ISIS 486178 rescues this splicing defect back to wild-type levels. (B) Quantification of results. N = 3–4 mice/group; **P = 0.0025; ***P = 0.000055; student’s t-test; error bars are mean ± SEM.
The DM200 mouse model also provided an unprecedented opportunity to assess the efficacy of systemic ASO therapy in treating cardiac pathologies associated with RNA toxicity in DM1. In patients with DM1, about 30% of the deaths are due to cardiac complications (20, 21, 58). The most characteristic cardiac findings in DM1 are a variety of arrhythmias. The most common are prolongation of the PR interval, atrioventricular conduction blocks and atrial arrhythmias such as atrial fibrillation, flutter and tachycardia. Our DM200 mouse model is one of the few mouse models of DM1 with a robust cardiac phenotype due to RNA toxicity and mirrors the primary arrhythmias seen in DM1 (58). Here, using this mouse model, we found that ISIS 486178 significantly improved a variety of cardiac conduction defects. This was also associated with a better functional outcome, namely, an improved retained run distance in DM200 mice following 6 weeks of treatment, which might be a result of the combined effects on the heart and skeletal muscle. The ISIS 486178 treatment also significantly reduced RNA foci in the heart, and it resulted in an approximately 50% reduction in toxic RNA levels in the heart. This correlated with a rescue of various splicing defects in the heart as well. Our results show that cardiac conduction defects in DM1 can by reversed by using ASOs designed to reduce the level of toxic RNAs in DM1.
Our molecular analyses also provided additional insights into effects on the heart due to RNA toxicity. One aspect of the DM200 mouse model is that it exhibits transgene leakiness even without doxycycline induction. This results in RNA foci and some splicing defects, for example, increased Cacna1s exon 29 exclusion and decreased Ldb3 exon 11 exclusion. Notably, this was not associated with cardiac conduction abnormalities. Other splicing events such as Atp2a1 exon 22 exclusion, or Scn5a exon 6a inclusion, did not show any changes in the DM200 D− mice as compared to wild-type mice. These results are consistent with the idea that splicing events in the heart are variably sensitive to RNA toxicity and ‘free’ MBNL concentrations, as reported in studies using cell-based models and applied to DM1 patient muscle samples (59). Upon induction of transgene induction, there was no significant change as compared to the uninduced mice, in the alternative splicing for the respective exons in Cacna1s, Ldb3 and Scn5a. However, the mice developed cardiac conduction abnormalities. Thus, there was no evident correlation between these splicing events and the cardiac phenotype. In contrast, the Atp2a1 exon 22 exclusion increased significantly with induction of RNA toxicity and correlated with the presence of conduction defects. Notably, all four splicing events showed a significant response to the ISIS 486178 therapeutic, which also reduced the levels of the toxic RNA in the heart, thus demonstrating their sensitivity to the levels of the toxic RNA. Thus, while multiple splicing events may be sensitive to RNA toxicity, only some may correlate with phenotypes such as cardiac conduction abnormalities. The DM200 mice, in conjunction with a therapeutic such as the ISIS 486178, provide an opportunity to dissect these relationships further in future studies.
We had previously demonstrated deleterious effects on connexin expression in the heart in a different mouse model of RNA toxicity (23). Here, we found that the DM200 model also exhibited deleterious effects on connexin expression in the heart (Fig. 5). The connexins are channel proteins that are essential for forming intercellular gap junctions between cardiomyocytes and enabling coordinated electrical conduction in the heart. Connexin 40 expression is limited to the atria and conduction system (His bundle and Purkinje fibers), whereas connexin 43 has a more global expression and is the primary connexin found in ventricular myocytes. Studies using genetically modified mice have shown that deficiencies in connexin 40 result in atrial arrhythmias (such as atrial fibrillation and flutter) and slowed signal propagation through the conduction system (60–63). These are also the prototypical conduction defects in patients with DM1 and in the DM200 mice. They are also present in Mbnl1−/−/Mbnl2+/− compound knockout mice (33). Although atrioventricular conduction defects such as prolongation of the PR interval and varying degrees of heart block are frequent and often cited as the classic cardiac conduction phenotype in DM1, atrial tachyarrhythmias are also common in DM1 patients with a reported frequency of up to 30% in a recent study (18). In fact, atrial arrhythmias are associated with increased mortality and are a strong predictor of sudden death in DM1 (21, 64). Unfortunately, there is little or no information on connexin 40 expression or protein levels in DM1 hearts, as most if not all samples studied are of cardiac ventricles obtained at autopsies. Mouse models such as the DM200 mice provide an opportunity to assess the heart more systematically. Indeed, a recent study of atrial tissue biopsies from human subjects with atrial fibrillation showed clear deficiencies in connexin 40 expression (65). In our study, we too found that Gja5 (connexin 40) mRNA levels in cardiac extracts from DM200 mice with RNA toxicity were drastically reduced. Immunofluorescence showed significant loss of connexin 40 protein expression in the atria of these mice. Notably, treatment with ISIS 486178 restored connexin 40 expression in the atria (Fig. 5A), and this correlated with a beneficial effect on cardiac rhythm in these mice.
Another aspect of cardiac pathology that contributes to cardiovascular morbidity is fibrosis. Often fibrosis is thought of as an end-stage process in response to tissue loss, such as occurs in a myocardial infarction. Historically, early studies of cardiac pathology in DM1 found fibrosis, especially of the sinoatrial node region of the right atrium and the conduction system, along with microscopic patchy interstitial fibrosis in the ventricular myocardium (66, 67). Recent studies have applied techniques such as cardiac magnetic resonance (CMR) imaging and found a high incidence of ventricular cardiac fibrosis in DM1, even in the absence of surface ECG changes (68, 69). In addition, studies in the past year have reported increased incidence of atrial fibrillation in association with CMR detected cardiac fibrosis (70) and interatrial block (18). Interatrial block is an atrial conduction problem associated with atrial fibrosis and atrial fibrillation (71). Moreover, the association of atrial fibrosis as a substrate or sustainer for atrial fibrillation is garnering increased mechanistic and clinical attention (72, 73). Thus, it is notable that we saw increased expression of fibrotic markers and evidence of fibrosis in the hearts of the DM200 mice. The increase in Col1a1 expression was evident even in the DM200 D− mice. Upon induction of RNA toxicity (i.e. DM200 D+), there was a further increase in Col1a1 expression and increased collagen deposition, especially in the atria of affected mice (Fig. 5C). Importantly, treatment with ISIS 486178 significantly reduced the expression of Col1a1 to levels even below the DM00 D− mice and more similar to those found in wild-type mice. Functionally, this was accompanied by a significant reduction in collagen deposition in the heart, especially in the atria of treated mice. As mentioned before, this was associated with a reversal of the cardiac conduction abnormalities.
It is important to note that we collected tissues at the end of 8 weeks of RNA toxicity from all mice. Some of the changes we saw in the mice with RNA toxicity that were treated with PBS or control ASO, for example, the increased fibrosis and loss of connexin 40 in the atria, could be due to loss of viable tissue and replacement by fibrosis, which may not be reversible. Whereas, we started treatment with ISIS 486178 soon after the induction of RNA toxicity (i.e. after 2 weeks). This is consistent with the notion that early treatment of the RNA toxicity may mitigate long-term irreversible damage.
In conclusion, we present here the first study of systemic treatment with an antisense oligonucleotide (ASO) (ISIS 486178) targeted to a non-CUG sequence within the 3'UTR of DMPK that demonstrates benefit against myotonia and cardiac conduction abnormalities, the defining and cardinal phenotypes of DM1. This was associated with key molecular benefits as well, such as restoration of chloride channel expression in the sarcolemma, correction of splicing defects, reduction in RNA foci and redistribution of MBNL1. This study also provided new insights into molecular mechanisms of pathogenesis in the heart such as the deleterious effects on connexin 40 expression in the atria and the early increase in fibrotic activity in the heart. Of note, we demonstrate that the ASO treatment reversed the cardiac conduction abnormalities, and this correlated with restoration of Gja5 (connexin 40) expression and reduction in fibrosis in the heart. Importantly, these improved functional outcomes. Our data also shows, for the first time, that ASOs may be a viable option for treating cardiac pathology in DM1.
Materials and Methods
Experimental mice and subcutaneous injection of ASOs
The DM200 mouse model expresses a GFP-DMPK 3'UTR (CTG) 200 transgene under the control of the human DMPK promoter upon induction with doxycycline (15). The transgenic mice were induced with 0.2% doxycycline in drinking water at the age of 2 months for 2 weeks. Then, these mice were treated with antisense oligonucleotides, ISIS 486178, control ASOs or PBS. These oligonucleotides were dissolved in PBS and administered at a dose of 25 mg/kg by injection subcutaneously (s.c.) twice a week for 6 weeks. All the details about protocols for treadmill running, EMG and ECG are described elsewhere (15, 74, 75). All results are reported as retained function with reference to baseline for each mouse. All animals were used in accordance with protocols approved by the Animal Care and Use Committee at the University of Virginia.
Antisense oligonucleotides
ASOs ISIS 486178 (5'-ACAATAAATACCGAGG-3′; cEt 2′ sugar modification) and control oligo (control: 5′-CCTTCCCTGAAGGTTCCTCC-3′; MOE 2′ sugar modification) were provided by Ionis Pharmaceuticals (43, 44) in a blinded fashion for our studies.
RNA-FISH and immunofluorescence and Gomori trichrome staining
For RNA-FISH, tissue was fixed in 4% paraformaldehyde in 1x PBS and a Cy3-CAG10 (Integrated DNA technologies, IDT) probe used for hybridization. Details about the RNA-FISH protocol are described elsewhere (76). Immunofluorescence was performed as described previously (76). Primary antibodies were anti-CLC-1 (1:100, #CLC11-A, Alpha Diagnostic international, (San Antonio, TX)), anti-MBNL1 (A2764, gift from Dr Charles A. Thornton), anti-connexin 40 (1:100, #36–4900, Invitrogen, Carlsbad, CA) and anti-connexin 43 (1;1000, C6219, Sigma, St. Louis, MO). Secondary antibodies were from Molecular Probes (1; 1000 dilution) (Eugene, OR). Collagen was visualized using Gomori trichrome standard staining protocol [#87020, Thermo Scientific™ Richard-Allan Scientific™ Gomori Trichrome (Blue Collagen), Waltham, MA].
RNA isolation, qRT-PCR assays and splicing analysis
We extracted total RNA from skeletal muscle tissues using protocols as described (37). All RNAs are DNAse treated and confirmed to be free of DNA contamination. QuantiTect Reverse Transcription Kit (Qiagen, Germantown, MD) was used for making cDNA from 1 μg of total RNA. qRT-PCR was done using the Bio-Rad iCycler (Bio-Rad, Hercules, CA) and detected with SYBR Green dye. Data were normalized using an endogenous control (Gapdh), and normalized values were subjected to a 2-ΔΔCt formula to calculate the fold changes between uninduced and induced groups. Primer sequences are given in Tables S1 and S2. All the splicing assays were done in at least five mice or more per group.
Statistical analysis
All the data were analyzed using an unpaired student’s t-test with equal or unequal variance as appropriate. All data are expressed as mean ± standard deviation. *P < 0.05, **P < 0.01, ***P < 0.001 (student’s t-test). P < 0.05 was considered statistically significant unless otherwise specified.
Author Contributions
M.S.M. did experimental design and phenotyping and wrote the manuscript. R.S.Y co-wrote the manuscript and did experimental design and experiments on RNA-FISH, splicing and expression analyses. Q.Y. designed and did phenotyping in concert with M.S.M. Q.Y., M.M. and R.S.Y did splicing and expression analyses. F.R. and C.F.B provided antisense oligonucleotides for the study and helped in the design and implementation of the blinded studies of the ASOs and evaluation and editing of the manuscript.
Supplementary Material
Acknowledgements
C.F.B. is an employee and shareholder in Ionis Pharmaceuticals Inc. and an inventor on patent #WO201502457A3 regarding ISIS 486178 antisense technology for modulation of DMPK expression. F.R. is an employee and shareholder in Ionis Pharmaceuticals Inc.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health (R01AR062189, R01AR071170); generosity of the Stone Circle of Friends.
References
- 1. Day J.W. and Ranum L.P. (2005) RNA pathogenesis of the myotonic dystrophies. Neuromuscul. Disord., 15, 5–16. [DOI] [PubMed] [Google Scholar]
- 2. Mahadevan M., Tsilfidis C., Sabourin L., Shutler G., Amemiya C., Jansen G., Neville C., Narang M., Barcelo J., O'Hoy K. et al. (1992) Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science, 255, 1253–1255. [DOI] [PubMed] [Google Scholar]
- 3. Taneja K.L., McCurrach M., Schalling M., Housman D. and Singer R.H. (1995) Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol., 128, 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Echeverria G.V. and Cooper T.A. (2012) RNA-binding proteins in microsatellite expansion disorders: mediators of RNA toxicity. Brain Res., 1462, 100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Braz S.O., Acquaire J., Gourdon G. and Gomes-Pereira M. (2018) Of mice and men: advances in the understanding of neuromuscular aspects of myotonic dystrophy. Front. Neurol., 9, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim D.H., Langlois M.A., Lee K.B., Riggs A.D., Puymirat J. and Rossi J.J. (2005) HnRNP H inhibits nuclear export of mRNA containing expanded CUG repeats and a distal branch point sequence. Nucleic Acids Res., 33, 3866–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Llamusi B., Bargiela A., Fernandez-Costa J.M., Garcia-Lopez A., Klima R., Feiguin F. and Artero R. (2013) Muscleblind, BSF and TBPH are mislocalized in the muscle sarcomere of a drosophila myotonic dystrophy model. Dis. Model. Mech., 6, 184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ravel-Chapuis A., Belanger G., Yadava R.S., Mahadevan M.S., DesGroseillers L., Cote J. and Jasmin B.J. (2012) The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J. Cell Biol., 196, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paul S., Dansithong W., Kim D., Rossi J., Webster N.J., Comai L. and Reddy S. (2006) Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J., 25, 4271–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pettersson O.J., Aagaard L., Andrejeva D., Thomsen R., Jensen T.G. and Damgaard C.K. (2014) DDX6 regulates sequestered nuclear CUG-expanded DMPK-mRNA in dystrophia myotonica type 1. Nucleic Acids Res., 42, 7186–7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang H., Mankodi A., Swanson M.S., Moxley R.T. and Thornton C.A. (2004) Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet., 13, 3079–3088. [DOI] [PubMed] [Google Scholar]
- 12. Jones K., Wei C., Iakova P., Bugiardini E., Schneider-Gold C., Meola G., Woodgett J., Killian J., Timchenko N.A. and Timchenko L.T. (2012) GSK3beta mediates muscle pathology in myotonic dystrophy. J. Clin. Invest., 122, 4461–4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuyumcu-Martinez N.M., Wang G.S. and Cooper T.A. (2007) Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell, 28, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morriss G.R., Rajapakshe K., Huang S., Coarfa C. and Cooper T.A. (2018) Mechanisms of skeletal muscle wasting in a mouse model for myotonic dystrophy type 1. Hum. Mol. Genet., 27, 2789–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mahadevan M.S., Yadava R.S., Yu Q., Balijepalli S., Frenzel-McCardell C.D., Bourne T.D. and Phillips L.H. (2006) Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat. Genet., 38, 1066–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McNally E.M. and Sparano D. (2011) Mechanisms and management of the heart in myotonic dystrophy. Heart, 97, 1094–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sovari A.A., Bodine C.K. and Farokhi F. (2007) Cardiovascular manifestations of myotonic dystrophy-1. Cardiol. Rev., 15, 191–194. [DOI] [PubMed] [Google Scholar]
- 18. Russo V., Sperlongano S., Gallinoro E., Rago A., Papa A.A., Golino P., Politano L., Nazarian S. and Nigro G. (2019) Prevalence of left ventricular systolic dysfunction in Myotonic dystrophy type 1: a systematic review. J. Card. Fail., S1071–9164(19)30204–0. doi: 10.1016/j.cardfail.2019.07.548. [DOI] [PubMed] [Google Scholar]
- 19. Choudhary P., Nandakumar R., Greig H., Broadhurst P., Dean J., Puranik R., Celermajer D.S. and Hillis G.S. (2016) Structural and electrical cardiac abnormalities are prevalent in asymptomatic adults with myotonic dystrophy. Heart, 102, 1472–1478. [DOI] [PubMed] [Google Scholar]
- 20. Mathieu J., Allard P., Potvin L., Prevost C. and Begin P. (1999) A 10-year study of mortality in a cohort of patients with myotonic dystrophy. Neurology, 52, 1658–1662. [DOI] [PubMed] [Google Scholar]
- 21. Groh W.J., Groh M.R., Saha C., Kincaid J.C., Simmons Z., Ciafaloni E., Pourmand R., Otten R.F., Bhakta D., Nair G.V. et al. (2008) Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N. Engl. J. Med., 358, 2688–2697. [DOI] [PubMed] [Google Scholar]
- 22. Wahbi K. and Furling D. (2019) Cardiovascular manifestations of myotonic dystrophy. Trends Cardiovasc. Med., 30, 232–238. [DOI] [PubMed] [Google Scholar]
- 23. Yadava R.S., Frenzel-McCardell C.D., Yu Q., Srinivasan V., Tucker A.L., Puymirat J., Thornton C.A., Prall O.W., Harvey R.P. and Mahadevan M.S. (2008) RNA toxicity in myotonic muscular dystrophy induces NKX2-5 expression. Nat. Genet., 40, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yadava R.S., Kim Y.K., Mandal M., Mahadevan K., Gladman J.T., Yu Q. and Mahadevan M.S. (2019) MBNL1 overexpression is not sufficient to rescue the phenotypes in a mouse model of RNA toxicity. Hum. Mol. Genet., 28, 2330–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang G.S., Kearney D.L., De Biasi M., Taffet G. and Cooper T.A. (2007) Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J. Clin. Invest., 117, 2802–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang G.S., Kuyumcu-Martinez M.N., Sarma S., Mathur N., Wehrens X.H. and Cooper T.A. (2009) PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J. Clin. Invest., 119, 3797–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chakraborty M., Selma-Soriano E., Magny E., Couso J.P., Perez-Alonso M., Charlet-Berguerand N., Artero R. and Llamusi B. (2015) Pentamidine rescues contractility and rhythmicity in a drosophila model of myotonic dystrophy heart dysfunction. Dis. Model. Mech., 8, 1569–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Algalarrondo V., Wahbi K., Sebag F., Gourdon G., Beldjord C., Azibi K., Balse E., Coulombe A., Fischmeister R., Eymard B. et al. (2015) Abnormal sodium current properties contribute to cardiac electrical and contractile dysfunction in a mouse model of myotonic dystrophy type 1. Neuromuscul. Disord., 25, 308–320. [DOI] [PubMed] [Google Scholar]
- 29. Koshelev M., Sarma S., Price R.E., Wehrens X.H. and Cooper T.A. (2010) Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum. Mol. Genet., 19, 1066–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee K.Y., Li M., Manchanda M., Batra R., Charizanis K., Mohan A., Warren S.A., Chamberlain C.M., Finn D., Hong H. et al. (2013) Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med., 5, 1887–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dixon D.M., Choi J., El-Ghazali A., Park S.Y., Roos K.P., Jordan M.C., Fishbein M.C., Comai L. and Reddy S. (2015) Loss of muscleblind-like 1 results in cardiac pathology and persistence of embryonic splice isoforms. Sci. Rep., 5, 9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chang K.T., Cheng C.F., King P.C., Liu S.Y. and Wang G.S. (2017) CELF1 mediates Connexin 43 mRNA degradation in dilated cardiomyopathy. Circ. Res., 121, 1140–1152. [DOI] [PubMed] [Google Scholar]
- 33. Chou C.C., Chang P.C., Wei Y.C. and Lee K.Y. (2017) Optical mapping approaches on Muscleblind-like compound knockout mice for understanding mechanistic insights into ventricular arrhythmias in Myotonic dystrophy. J. Am. Heart Assoc., 6, e005191. doi: 10.1161/JAHA.116.005191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thornton C.A., Wang E. and Carrell E.M. (2017) Myotonic dystrophy: approach to therapy. Curr. Opin. Genet. Dev., 44, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lopez-Morato M., Brook J.D. and Wojciechowska M. (2018) Small molecules which improve pathogenesis of Myotonic dystrophy type 1. Front. Neurol., 9, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Furling D., Doucet G., Langlois M.A., Timchenko L., Belanger E., Cossette L. and Puymirat J. (2003) Viral vector producing antisense RNA restores myotonic dystrophy myoblast functions. Gene Ther., 10, 795–802. [DOI] [PubMed] [Google Scholar]
- 37. Langlois M.A., Lee N.S., Rossi J.J. and Puymirat J. (2003) Hammerhead ribozyme-mediated destruction of nuclear foci in myotonic dystrophy myoblasts. Mol. Ther., 7, 670–680. [DOI] [PubMed] [Google Scholar]
- 38. Sobczak K., Wheeler T.M., Wang W. and Thornton C.A. (2013) RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol. Ther., 21, 380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gonzalez-Barriga A., Mulders S.A., Giessen J., Hooijer J.D., Bijl S., Kessel I.D., Beers J., Deutekom J.C., Fransen J.A., Wieringa B. et al. (2013) Design and analysis of effects of triplet repeat oligonucleotides in cell models for myotonic dystrophy. Mol. Ther. Nucleic Acids, 2, e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee J.E., Bennett C.F. and Cooper T.A. (2012) RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl. Acad. Sci. U. S. A., 109, 4221–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mulders S.A., Broek W.J., Wheeler T.M., Croes H.J., Kuik-Romeijn P., Kimpe S.J., Furling D., Platenburg G.J., Gourdon G., Thornton C.A. et al. (2009) Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. U. S. A., 106, 13915–13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Francois V., Klein A.F., Beley C., Jollet A., Lemercier C., Garcia L. and Furling D. (2011) Selective silencing of mutated mRNAs in DM1 by using modified hU7-snRNAs. Nat. Struct. Mol. Biol., 18, 85–87. [DOI] [PubMed] [Google Scholar]
- 43. Jauvin D., Chretien J., Pandey S.K., Martineau L., Revillod L., Bassez G., Lachon A., MacLeod A.R., Gourdon G., Wheeler T.M. et al. (2017) Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol Ther Nucleic Acids, 7, 465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pandey S.K., Wheeler T.M., Justice S.L., Kim A., Younis H.S., Gattis D., Jauvin D., Puymirat J., Swayze E.E., Freier S.M. et al. (2015) Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. J. Pharmacol. Exp. Ther., 355, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wheeler T.M., Leger A.J., Pandey S.K., MacLeod A.R., Nakamori M., Cheng S.H., Wentworth B.M., Bennett C.F. and Thornton C.A. (2012) Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature, 488, 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mankodi A., Logigian E., Callahan L., McClain C., White R., Henderson D., Krym M. and Thornton C.A. (2000) Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science, 289, 1769–1773. [DOI] [PubMed] [Google Scholar]
- 47. Charlet B.N., Savkur R.S., Singh G., Philips A.V., Grice E.A. and Cooper T.A. (2002) Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell, 10, 45–53. [DOI] [PubMed] [Google Scholar]
- 48. Mankodi A., Takahashi M.P., Jiang H., Beck C.L., Bowers W.J., Moxley R.T., Cannon S.C. and Thornton C.A. (2002) Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell, 10, 35–44. [DOI] [PubMed] [Google Scholar]
- 49. Kalsotra A., Xiao X., Ward A.J., Castle J.C., Johnson J.M., Burge C.B. and Cooper T.A. (2008) A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. U. S. A., 105, 20333–20338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nakamori M., Sobczak K., Puwanant A., Welle S., Eichinger K., Pandya S., Dekdebrun J., Heatwole C.R., McDermott M.P., Chen T. et al. (2013) Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol., 74, 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Freyermuth F., Rau F., Kokunai Y., Linke T., Sellier C., Nakamori M., Kino Y., Arandel L., Jollet A., Thibault C. et al. (2016) Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nat. Commun., 7, 11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gambini E., Perrucci G.L., Bassetti B., Spaltro G., Campostrini G., Lionetti M.C., Pilozzi A., Martinelli F., Farruggia A., DiFrancesco D. et al. (2018) Preferential myofibroblast differentiation of cardiac mesenchymal progenitor cells in the presence of atrial fibrillation. Transl. Res., 192, 54–67. [DOI] [PubMed] [Google Scholar]
- 53. Wheeler T.M., Sobczak K., Lueck J.D., Osborne R.J., Lin X., Dirksen R.T. and Thornton C.A. (2009) Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science, 325, 336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wojtkowiak-Szlachcic A., Taylor K., Stepniak-Konieczna E., Sznajder L.J., Mykowska A., Sroka J., Thornton C.A. and Sobczak K. (2015) Short antisense-locked nucleic acids (all-LNAs) correct alternative splicing abnormalities in myotonic dystrophy. Nucleic Acids Res., 43, 3318–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ostergaard M.E., Jackson M., Low A., A E.C., R G.L., Peralta R.Q., Yu J., Kinberger G.A., Dan A., Carty R. et al. (2019) Conjugation of hydrophobic moieties enhances potency of antisense oligonucleotides in the muscle of rodents and non-human primates. Nucleic Acids Res., 47, 6045–6058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Carrell S.T., Carrell E.M., Auerbach D., Pandey S.K., Bennett C.F., Dirksen R.T. and Thornton C.A. (2016) Dmpk gene deletion or antisense knockdown does not compromise cardiac or skeletal muscle function in mice. Hum. Mol. Genet., 25, 4328–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kanadia R.N., Shin J., Yuan Y., Beattie S.G., Wheeler T.M., Thornton C.A. and Swanson M.S. (2006) Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. U. S. A., 103, 11748–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Turner C. and Hilton-Jones D. (2014) Myotonic dystrophy: diagnosis, management and new therapies. Curr. Opin. Neurol., 27, 599–606. [DOI] [PubMed] [Google Scholar]
- 59. Wagner S.D., Struck A.J., Gupta R., Farnsworth D.R., Mahady A.E., Eichinger K., Thornton C.A., Wang E.T. and Berglund J.A. (2016) Dose-dependent regulation of alternative splicing by MBNL proteins reveals biomarkers for myotonic dystrophy. PLoS Genet., 12, e1006316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lambiase P.D. and Tinker A. (2015) Connexins in the heart. Cell Tissue Res., 360, 675–684. [DOI] [PubMed] [Google Scholar]
- 61. Jansen J.A., Veen T.A., Bakker J.M. and Rijen H.V. (2010) Cardiac connexins and impulse propagation. J. Mol. Cell. Cardiol., 48, 76–82. [DOI] [PubMed] [Google Scholar]
- 62. Bevilacqua L.M., Simon A.M., Maguire C.T., Gehrmann J., Wakimoto H., Paul D.L. and Berul C.I. (2000) A targeted disruption in connexin40 leads to distinct atrioventricular conduction defects. J. Interv. Card. Electrophysiol., 4, 459–467. [DOI] [PubMed] [Google Scholar]
- 63. Hagendorff A., Schumacher B., Kirchhoff S., Luderitz B. and Willecke K. (1999) Conduction disturbances and increased atrial vulnerability in Connexin40-deficient mice analyzed by transesophageal stimulation. Circulation, 99, 1508–1515. [DOI] [PubMed] [Google Scholar]
- 64. Brembilla-Perrot B., Schwartz J., Huttin O., Frikha Z., Sellal J.M., Sadoul N., Blangy H., Olivier A., Louis S. and Kaminsky P. (2014) Atrial flutter or fibrillation is the most frequent and life-threatening arrhythmia in myotonic dystrophy. Pacing Clin. Electrophysiol., 37, 329–335. [DOI] [PubMed] [Google Scholar]
- 65. Gemel J., Levy A.E., Simon A.R., Bennett K.B., Ai X., Akhter S. and Beyer E.C. (2014) Connexin40 abnormalities and atrial fibrillation in the human heart. J. Mol. Cell. Cardiol., 76, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nguyen H.H., Wolfe J.T. 3rd, Holmes D.R. Jr. and Edwards W.D. (1988) Pathology of the cardiac conduction system in myotonic dystrophy: a study of 12 cases. J. Am. Coll. Cardiol., 11, 662–671. [DOI] [PubMed] [Google Scholar]
- 67. Motta J., Guilleminault C., Billingham M., Barry W. and Mason J. (1979) Cardiac abnormalities in myotonic dystrophy. Electrophysiologic and histopathologic studies. Am. J. Med., 67, 467–473. [DOI] [PubMed] [Google Scholar]
- 68. Petri H., Ahtarovski K.A., Vejlstrup N., Vissing J., Witting N., Kober L. and Bundgaard H. (2014) Myocardial fibrosis in patients with myotonic dystrophy type 1: a cardiovascular magnetic resonance study. J. Cardiovasc. Magn. Reson., 16, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cardona A., Arnold W.D., Kissel J.T., Raman S.V. and Zareba K.M. (2019) Myocardial fibrosis by late gadolinium enhancement cardiovascular magnetic resonance in myotonic muscular dystrophy type 1: highly prevalent but not associated with surface conduction abnormality. J. Cardiovasc. Magn. Reson., 21, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chmielewski L., Bietenbeck M., Patrascu A., Rosch S., Sechtem U., Yilmaz A. and Florian A.R. (2019) Non-invasive evaluation of the relationship between electrical and structural cardiac abnormalities in patients with myotonic dystrophy type 1. Clin. Res. Cardiol., 108, 857–867. [DOI] [PubMed] [Google Scholar]
- 71. Hernandez-Betancor I., Izquierdo-Gomez M.M., Garcia-Niebla J., Laynez-Cerdena I., Garcia-Gonzalez M.J., Barragan-Acea A., Irribarren-Sarria J.L., Jimenez-Rivera J.J. and Lacalzada-Almeida J. (2017) Bayes syndrome and imaging techniques. Curr. Cardiol. Rev., 13, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nattel S. (2017) Molecular and cellular mechanisms of atrial fibrosis in atrial fibrillation. JACC Clin Electrophysiol, 3, 425–435. [DOI] [PubMed] [Google Scholar]
- 73. Hansen B.J., Zhao J. and Fedorov V.V. (2017) Fibrosis and atrial fibrillation: computerized and optical mapping; a view into the human atria at submillimeter resolution. JACC Clin Electrophysiol, 3, 531–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kim Y.K., Mandal M., Yadava R.S., Paillard L. and Mahadevan M.S. (2014) Evaluating the effects of CELF1 deficiency in a mouse model of RNA toxicity. Hum. Mol. Genet., 23, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yadava R.S., Foff E.P., Yu Q., Gladman J.T., Kim Y.K., Bhatt K.S., Thornton C.A., Zheng T.S. and Mahadevan M.S. (2015) TWEAK/Fn14, a pathway and novel therapeutic target in myotonic dystrophy. Hum. Mol. Genet., 24, 2035–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mankodi A., Teng-Umnuay P., Krym M., Henderson D., Swanson M. and Thornton C.A. (2003) Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann. Neurol., 54, 760–768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.