

Abstract
Amyotrophic lateral sclerosis (ALS) and Frontotemporal Degeneration (FTD) are neurodegenerative disorders, related by deterioration of motor and cognitive functions and short survival. Aside from cases with an inherited pathogenic mutation, the causes of the disorders are still largely unknown and no effective treatment currently exists. It has been shown that FTD may coexist with ALS and this overlap occurs at clinical, genetic, and molecular levels. In this work, we review the main pathological aspects of these complex diseases and discuss how the integration of the novel pathogenic molecular insights and the analysis of molecular interaction networks among all the genetic players represents a critical step to shed light on discovering novel therapeutic strategies and possibly tailoring personalized medicine approaches to specific ALS and FTD patients.
Keywords: ALS, Dementia FTLD, FTD, Genetics, Motor neuron disease TDP-43, Stem cell
ALS and FTD: two sides of the same coin?
In principle, Amyotrophic lateral sclerosis (ALS) and Frontotemporal Degeneration (FTD) are two very different neurodegenerative diseases. ALS, whose incidence in Europe is 2.16 per 100000 person-years [1], is a progressive and ultimately fatal disease caused by progressive loss of motor neurons controlling voluntary muscles [2]. On the other hand, FTD, whose incidence in Europe is 3.5 per 100000 person-years [2], is a type of dementia characterized by atrophy of the frontal and temporal lobes, with composite clinical signs ranging from alterations in behavior and language to motor and cognitive dysfunctions [3]. More specifically, there is a general consensus that FTD consists of three distinct clinical syndromes: behavioral variant frontotemporal degeneration (bvFTD), non-fluent variant primary progressive aphasia (nfvPPA) and semantic variant primary progressive aphasia (svPPA) [4].
These pathologies were initially considered two independent clinical entities, because of heterogeneity at clinical and neuropathological levels [5–9]. Indeed, along this line, recent studies have outlined how these two diseases seem to be also characterized by different inflammatory profiles, with predominance of macrophage/microglia activation in ALS and astrocyte malfunctioning in FTD [10–12].
However, it is increasingly recognized that ALS is a multisystem disorder in which other non-motor (and in particular, cognitive and behavioral) impairments can be observed [13, 14], whereas, on the other side, FTD can be associated to signs of motor neuron disease (FTD-MND) [15].
The outlook of ALS and FTD as distinct nosological entities has progressively changed in the last decade due to genetic breakthroughs. Over this time period, more than 50 genes have been associated with ALS and FTD [16–18]. Mutations in at least 44 loci have been linked to ALS and, correspondingly, at least 14 loci have been linked to FTD. At present, the four major ALS-associated genes are the chromosome 9 open reading frame 72 (C9ORF72) [19, 20], Cu-Zn superoxide dismutase 1 (SOD1) [21], TAR DNA-binding protein 43 (TARDBP) [22, 23], and fusion in malignant liposarcoma/translocation in liposarcoma (FUS/TLS) [24, 25], in addition to at least other 40 genes [26]. On the other hand, mutations three main genes (MAPT, GRN and C9ORF72) are found in 60% of familial FTD cases, whereas rare mutations (<5%) have been found in other 11 genes [8, 27, 28].
These findings have contributed to shed light on the major biological processes altered in ALS and FTD/FTLD pathologies (Table 1) [29]. On the other hand, as the genes associated with these diseases were fleshed out, it was apparent that ALS and FTD share pathophysiological processes. In fact, the intersection of the gene sets implicated in the pathogenesis of ALS and FTD shows that at least 11 susceptibility genes are in common between these two disorders (Fig. 1). This genetic observation makes the molecular overlap particularly striking and strengthens the current notion that ALS and FTD share common pathogenic mechanisms.
Table 1.
Main genes and cellular components/processes implicated in the pathogenesis of ALS and FTD
| RNA metabolism | TARDBP, FUS, hnRNPA1, hnRNPA2B1, MATR3, ATXN2, TAF15, SETX, EWSR1, ELP3, ANG |
| Protein trafficking and Proteostasis | C9ORF72, CHMP2B, FIG 4, TBK1, UBQLN2, SQSTM1, SIGMAR1, OPTN, VCP, ALS2, VAPB |
| DNA repair | EWSR1, FUS, SETX, TAF15, TARDBP |
| Mitochondria and Oxidative stress: | SOD1, CHCHD10, C19ORF12 |
| Immune response/Inflammation: | GRN, TREM2, TYROBP |
| Stress granule assembly | ATXN2, C9ORF72, MAPT |
| Glia and Neurons metabolism | GRN, SIGMAR1, SOD1, TREM2, TYROBP |
| Vacuolar transport | C9ORF72, CHMP2B, GRN, TMEM106B, VCP, OPTN, UNC13A |
| Axo-dendritic transport | KIF5A, MAPT, SPG11 |
Fig. 1.

Genetic overlap between ALS and FTD. To this date, more than 50 genes have been associated with ALS and FTD. The Venn diagram summarizes the number of specifically altered genes in each disease and the observed overlap between the two pathologies
No gene is an island by itself
If we analyze more thoroughly the genes implicated in the pathogenesis of ALS and FTD it is interesting to note the intricate network of coexpressions, interactions and pathways interconnecting most of the players (Supplementary Figures 1, 2, 3 and 4). In fact, a prediction of gene functions and connections carried out by using the Genemania online tool [30, 31] suggests that a large number of the susceptibility genes are linked through physical protein-protein interactions (44.57%), and are co-expressed (26.29%), whereas few gene share pathways (7.42%), or show predicted functional interactions based on orthology (6.55%).
Altogether these observations support the hypothesis that such complex diseases as ALS and FTD are likely caused not only by the simple alteration of a single gene and that these pathologies are the result of alteration of multiple genes clustered in the same pathways or in connected pathways, caused by the triggering of a “domino effect” propagating the initial pathological signal to the other physical or functional partners. In this line, TDP-43 and the related TDP-43 proteinopathies are an exemplary case. It is now well established that clinically and neuropathologically, ALS and FTD or its pathological substrate FTLD may be seen as two ends of a spectrum [13, 15, 32] as well as representing a connection with several other neuromuscular diseases [33].
Until recently, the pathological protein/s in these inclusion bodies of ALS and FTLD with ubiquitin-immunoreactive inclusions was thought to be an unknown ubiquitinated protein. Subsequently, TDP-43 was identified as the major component of the cytoplasmic inclusions of both ALS and FTLD neurons and glia [34, 35]. Deeper biochemical analysis of purified neuronal cytoplasmic inclusions has shown that pathologic TDP-43 is not only ubiquitinated, but also hyperphosphorylated, and aggregates as abnormal C-terminal fragments [23]. Phosphorylated TDP-43 intraneuronal inclusions in ALS are found within the motor cortex, brainstem motor nuclei, cranial nerve nuclei V, VII, and X-XII, and in spinal cord motor neurons. It is still unclear whether TDP-43 aggregation might represent a primary event in ALS pathogenesis or rather an epiphenomenon secondary to other pathological processes.
In addition to ALS, TDP-43 proteinopathy now also constitutes 45% of all FTLD molecular pathologies [36] (Table 2). Approximately 50% of all cases contain abnormal tau (namely, tauopathies) and the residual group of about 5% of cases is characterized by abnormal FUS (FUS proteinopathies) [61].
Table 2.
ALS-FTD genotype/phenotype correlations for genes and presence of TDP-43 inclusions
| Gene | Genetic overlap ALS/FTD | TDP inclusions | References |
|---|---|---|---|
| SETX | ALS | Yes | [37] |
| ATXN2 | ALS | Yes | [38] |
| SOD1 | ALS | Yes | [39, 40] |
| VABP | ALS | NR | -- |
| ALS2 | ALS | NR | -- |
| ANG | ALS | Yes | [41] |
| SQSTM1 | ALS | Yes | [42] |
| C21ORF2 | ALS | NR | – |
| MATR3 | ALS | Yes | [43] |
| EWSR1 | ALS | NR | -- |
| TAF15 | ALS | NR | -- |
| HNRPA1 | ALS | Yes | [44, 45] |
| HNRNPA2B1 | ALS | Yes | [44, 45] |
| OPTN | ALS | Yes | [46] |
| TUBA4A | ALS-FTD | NR | [47] |
| TARDBP | ALS-FTD | Yes | [48, 49] |
| C9ORF72 | ALS-FTD | Yes | [50, 51] |
| DCTN1 | ALS-FTD | Yes | [52] |
| TUBA4A | ALS-FTD | NR | [47] |
| TBK1 | ALS-FTD | Yes | [53] |
| CHCHD10 | ALS-FTD | Yes | [54] |
| CCNF | ALS-FTD | Yes | [55] |
| FUS | ALS>FTD | NR | [24, 25] |
| UBQLN2 | ALS>FTD | Yes | [56] |
| SIGMAR1 | ALS>FTD | NR | -- |
| TIA1 | ALS>FTD | NR | -- |
| CHMP2B | FTD>ALS | NR | [56, 57] |
| VCP | FTD>ALS | Yes | [58] |
| GRN | FTD | Yes | [59] |
| MAPT | FTD | NR | -- |
| TMEM106B | FTD | Yes | [60] |
The evidence for mutations linking each gene to the ALS-FTD spectrum is reported (ALS only; FTD only; both ALS-FTD; majority of ALS cases: ALS>FTD; majority of ALS cases: FTD>ALS). NR not reported
Importantly, co-morbidity of TDP-43 proteinopathy seems not to be limited to ALS/FTLD but it has also been detected in several other neurological disorders, such as Alzheimer's disease (about 75% of cases with hippocampal sclerosis, 31% without hippocampal sclerosis, 30% with dementia with Lewy bodies), Parkinson's disease (about 7% and 19% of PD with dementia), dementia Parkinsonism ALS complex of Guam (G-PDC), Pick's disease, corticobasal degeneration and progressive supranuclear palsy (PSP) [62–70]. Furthermore, recent studies reported the altered expression and mislocalization of TDP-43 in brain tissues obtained from a Niemann Pick C (NPC) patient and in NPC cellular models [71].
Disease modifier factors
Genetic mutations are not sufficient to explain the onset and the progression rates of ALS and FTD patients. The common delayed onset of both pathologies [72], and the observation that only specific cellular types are affected suggests that the vulnerability of disease-specific neurons is not induced just by simple genetic factors. Additional environmental, metabolic and genetic factors and modifiers have to be brought into play to explain and increase the complexity of ALS and FTD etiopathogenesis (Fig. 2).
Fig. 2.
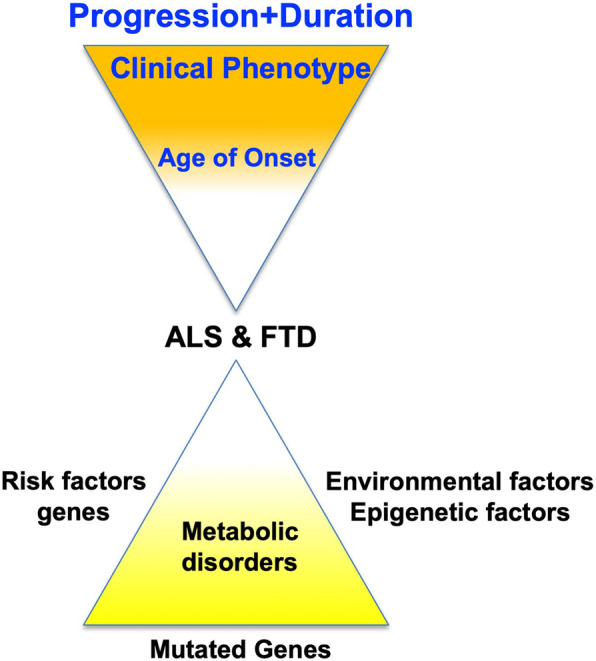
Factors affecting the disease- and clinical- phenotypes of ALS and FTD pathologies. Schematic diagram of the various exogenous and endogenous factors potentially affecting the age at onset, the disease phenotypes, as well as the clinical phenotypes
Regarding the environmental factors, elevated heavy metal levels stand out not only as risk factors for ALS but have also been proposed to correlate with ALS progression [73, 74].
It is interesting to report that different epigenetic mechanisms influencing the expression of C9ORF72, SOD1, GRN, VEGF, and GLT1 have been implicated in the pathogenesis of these disease [75, 76]. While the promoters of SOD1, VEGF, and GLT1 genes have been found to be unmethylated in ALS patients [77, 78], hypermethylation of GRN promoter has been reported in FTD subjects and the observed lower mRNA levels lend support to its correlation with GRN haploinsufficiency and FTD pathogenesis [77, 78]. On the other hand, the role of C9ORF72 gene methylation in ALS and FTD pathogenesis is still unclear. Intriguingly, only 10–30% of c9FTD/ALS subjects present hypermethylation of the C9ORF72 promoter possibly correlated with a decreased C9ORF72 expression.
Although other epigenetic modifications, such as histone and miRNA alterations have been reported in ALS and FTD patients [76], we are still lacking a clear picture of the impact of all the epigenetic findings on the clinical aspects of these pathologies.
Other intriguing lines of research evaluated some metabolic parameters as disease-modifiers that can impact the clinical course in ALS. For example, recent investigations have shown that high-risk cardiovascular profiles such as a high body mass index (BMI) or diabetes mellitus type 2 might be protective for ALS patients and may act by delaying the onset and/or by slowing down clinical progression of the disease [79–85]. Altogether, these finding suggest that caution should be exercised when comorbidities and risk factors are evaluated as prognostic factor in such as complex diseases as ALS and FTD. In particular, a better characterization of the eventual association between the clinicopathological features of the patients and a particular metabolic disorder might be helpful to understand if and how metabolic disorders can influence the subcellular localization, aggregation, and phosphorylation of some of pathogenic proteins, or if the metabolic disorders might rather modulate the toxic effects downstream of these events [81]. In addition, these observations open potentially the way to nutrition, dietary supplements (in particular, antioxidant) and lifestyle interventions as potential strategies for modifying the course of the disease [86–88]. In this line, recent studies have suggested that a high-calorie diet might be an effective treatment for ALS by inducing a hypermetabolic state that counters the increased resting energy expenditure (REE) observed in ALS patients [89–91].
Disease modifier genes
On top of the disease-associated genes, an additional level of complexity is added by the existence of modifier genes. In fact, part of the disease and clinical phenotypes can be ascribed to the action of genes other than those directly implicated in the pathologies (Table 3).
Table 3.
Effect of the genetic modifiers of ALS and FTD
| GRN | SORT1 | rs646776 C-minor allele => decreased GRN plasma expression | [92, 93] |
| GRN | TMEM106B | rs1990622 C-minor allele => lower risk (older age at onset) | [94, 95] |
| C9ORF72 | TMEM106B | rs1990622 C-minor allele => earlier onset | [96] |
| rs3173615 G minor allele => Homozygosity protects from developing FTD but not from developing MND | [97] | ||
| -- | TMEM106B | rs1990622 T-major allele => poorer cognitive performance in FTLD-TDP patients not in ALS patients | [98] |
| -- | TMEM106B | rs1990622 C-minor allele => protective effect on cognitive aging | [99–101] |
In this context, after identification of the first mutations in the GRN gene of FTLD patients, the high clinical variability observed among the carriers of similar GRN mutations led to hypothesize the presence of environmental and/or genetic modifiers. For example, it has been found that each copy of the rs646776 minor C allele, linked to SORT1 expression, is associated with a decreased GRN plasma levels by ∼15% [92, 93].
It has been also shown the existence of a link between GRN and TMEM106B genes. In fact, variation in TMEM106B expression seems to be associated with changes in progranulin expression: in fact, it has been found that TMEM106B rs1990622 C-minor allele is associated with a delay of FTLD onset age (lower risk) for GRN mutation carriers [94, 95]. It has been also shown the existence of a link between GRN and TMEM106B gene and variations in TMEM106B expression seem to be associated with changes in progranulin expression. TMEM106B SNPs has been also reported to be risk factors across the genetic FTLD-TDP spectrum. In addition, the rs1990622 T-major allele has been associated with later age at onset and at death, whereas the homozygosity for rs3173615 G minor allele seems to protects from developing FTD but not from developing MND [96, 97]. To make the puzzle even more challenging, the rs1990622 T-major allele has been also associated with poorer cognitive performance in FTLD-TDP but not in ALS patients [98]. On the other hands, the rs1990622 C-minor allele has been shown to have a protective effect on cognitive aging in MAPT mutation carriers [99–101].
Present drugs
The clinical and molecular heterogeneity of ALS and FTD have represented a significant challenge for the development of effective treatments.
Regarding treatments for FTD, although a lot of symptomatic treatments have been reported so far, it has been found that open-label studies of anticholinesterase medicines and memantine have been negative. In some cases, they may even exacerbate behavioral symptoms [102, 103]. Concerning selective serotonin reuptake inhibitors and antipsychotic therapies these treatments can be helpful in the management of mood and behavioral features in individual patients [102]. Tauopathy has become a target for novel disease-modifying treatments such as Methylene Blue that is a drug in Phase III clinical trials. This compound was found to be effective in arresting age-related cognitive decline of tau-transgenic mice but only if administered at the earliest stages [104]. Regarding ALS, although several new potential drugs are currently being tested in Phase 1 to Phase 3 clinical trials (www.clinicaltrials.gov), there are no effective therapies able to stop the progression of ALS. Riluzole, the only disease-modifying treatment shown to extend life expectancy in patients with ALS, was the first FDA drug approved for clinical use in 1995 [105]. In initial studies, it has been associated with 38,6% reduction in mortality in an efficacy trial [106], and 35% improvement in survival with the 100 mg dose in a dose – ranging trial [107]. Despite being associated with a short survival benefit of 2–3 months equating to a 9% increase in 1-year survival it still represents the most ALS-effective therapy to this date [108]. Although the main neuroprotective activity of Riluzole relies mainly on blocking of the glutamatergic excitotoxicity several other actions have been proposed. These include inactivation of voltage-dependent sodium channels and interference with signal transduction events following interaction of neurotransmitters to excitatory amino acid receptors [109]. Only after 22 years, in 2017, the USA FDA approved the clinical use of the second ALS-specific drug, Edaravone. The Edaravone (MCI-186, also known as Radicava or Radicut) is a neuroprotective drug with free radical scavenger and antioxidant properties [110]. In a double-blind, placebo controlled, Phase 2 study using intravenous Edaravone therapy in ALS patients, Akimoto and colleagues showed a decrease in primary endpoint in the Revised ALS Functional Rating Scale (ALSFRS-R) scores from baseline to 24 weeks after randomization [111]. In the recent Phase 3 study, it has been reported that Edaravone treatment is beneficial in ALS patients even after 6 months of receiving placebo, and efficacy is maintained for up to 1 year [112]. However, both drugs have a relatively small efficacy in delaying the motor function deterioration, and their effectiveness is limited during early stages of the disease [113]. Therefore, new treatments are urgently needed to improve the clinical course of both diseases. Over the years, different approaches are emerging to identify effective therapeutic strategies against ALS and FTD (schematically depicted in Fig. 3).
Fig. 3.

Actual and potential therapeutic approaches to treat ALS and FTD pathologies. This schematic diagram recapitulates the major pharmacological therapeutic approaches that have been tested or are currently approved for treatment of ALS patients (highlighted in red). In addition, it shows the main novel approaches that are currently under development based on recent discoveries in the field of stem-cell/gene therapy and the molecular pathology of these diseases. As shown in this figure, they range from small functional molecules and RNAs against specific protein/RNA-based targets, stem-cell approaches, to transcranial magnetic stimulation (TMS) or transcranial direct current stimulation (tDCS)
Drugs on the way
Arimoclomol
Arimoclomol is a hydroxylamine derivative able to induce heat shock protein expression [114], whose dosing was reported to improve muscle function and survival in different mouse models of motor neuron disease [115]. This compound may have multiple mechanisms of action and several lines of evidence suggest that under non-stressed conditions it may reduce the levels of protein aggregates in motor nerves (a possible cause of ALS). This is achieved by boosting expression of the heat shock proteins Hsp70 and Hsp90 [116]. In addition, Arimoclomol was recently found to induce a HSF1-mediated reduction of the TDP-43 aggregates [117]. Finally, in order to evaluate the efficacy and safety of Arimoclomol, a Phase 3 randomized, double blind, placebo-controlled trial is currently underway (NCT03491462) after a Phase 2 trial suggested a possible therapeutic benefit of this molecule (NCT00706147) [118].
Ceftriaxone
Ceftriaxone is a third generation of cephalosporin antibiotic, with ability to cross the blood brain barrier (BBB) and to exert neuroprotection in different animal models. This activity seems to rely on induction of the astrocytic glutamate transporter EAAT2 expression in humans, and GLT1 glutamate transporter expression in rodents. Apparently, overexpression of glutamate transporters can boost the clearance of synaptic glutamate and protects neurons from glutamate-mediated excitotoxicity, that is thought to be critical in the pathogenesis of ALS [119]. Along this line of evidence, low expression of the glutamate transporter EAAT2 has been reported in ALS animal models as well as in human post-mortem tissue [120]. Unfortunately, this compound did not show efficacy at Phase III clinical trials [121], most likely due to already well-established disease.
Masitinib and cannabinoids
Masitinib is a selective tyrosine kinase inhibitor, with anti-inflammatory activity deriving from its ability to counteract degranulation of the mast cells. In addition, it can also inhibit microglia proliferation and activation [122]. This compound has entered Phase 3 trials for ALS in 2017, as it was observed in a Phase 2 trial that it could be of benefit to ALS patients as an add-on to Riluzole [123]. Regarding anti-inflammatory effects, it has to be noted that cannabinoids have also been proposed to be useful in the treatment of ALS due to their anti-inflammatory, anti-oxidant, and anti-excitotoxic properties [124, 125]. However, a recent meta-analyses of the studies conducted so far on animal models has concluded that more standardized studies should be performed before supporting the treatment of ALS patients with these compounds [126].
Tirasemtiv and structural analogs
Tirasemtiv is a troponin activator with the ability to make troponin more sensitive to calcium and consequently counteract muscle weakness, that is the most common initial symptom in ALS [127]. Tirasemtiv was the subject of a Phase 3 clinical trial program in ALS patients [128, 129] (NCT02496767) but the results were disappointing, as reported in recent congress communications. However, research in this area is still currently evolving with the development of structural analogs of Tirasemtiv such as CK-2066260 [130] and, especially another skeletal muscle activator CK-2127107 [131] called Reldesemtiv has entered Phase 2 trial (NCT03160898) with, hopefully more promising results.
Stem-cell based approaches
Stem cell therapy aimed at counteracting immune dysregulation is another promising novel treatment for several autoimmune diseases (such as rheumatoid arthritis, multiple sclerosis, lupus) [132] as well as for ALS [133]. Initial attempts using neural stem cells in a Phase 2 clinical trial (NCT01730716) did not seem to benefit patients [134]. Therefore, starting from the observation that neurotrophic growth factors seem to extend the survival of motor neurons in ALS [135, 136] a culture-medium based method was used to induce mesenchymal stem cells (MSCs) to secrete neurotrophic growth factors (NTFs) [137].
The intrathecal (i.t.) or intramuscular (i.m.) administration of autologous MSC-NTF cells has been shown to be safe and to provide possible clinical advantages for ALS patients and the potential positive outcomes are supported by other clinical studies [138–140]. However, these results should be further validated by recruiting a larger number of patients and by including a placebo control. Interestingly, it has been suggested that MSCs might modulate the inflammatory responses in ALS patients by increasing the levels of anti-inflammatory cytokines produced by activated regulatory T lymphocytes (Treg) [141, 142]. A Phase 1 study is currently in progress to determine the efficacy of these cells against disease progression [143].
Immunological approaches
Recent studies have shown peculiar differences in the inflammatory responses underlying the pathogenesis of ALS and FTD [11, 144]. These researches pave the way to the approaches on the similarities in the immunological mechanisms of ALS and FTD with other chronic diseases where flogosis plays a critical pathogenetic role [145]. These observations explain current efforts of drug-repositioning with a series of NIH clinical trials aimed at proving the effectiveness on ALS of agents used for the treatment of Rheumatoid arthritis and Multiple sclerosis. However, although some encouraging preliminary reports were observed in animal models with regards to safety and tolerability, no clear improvements for ALS has been found [145].
At the moment, another ALS-specific immunological therapy seemed to be more promising. It was shown, in fact, that peripheral nervous system injury is associated with inhibition of the axonal growth in mammals, through activation of the reticulon 4 receptor (RTN4R, or Nogo receptor) [146]. NogoA is one of the three isoforms of the Nogo protein acting as a neurite outgrowth inhibitor [147]. It is also localized on macrophages and, following Wallerian degeneration, it has a crucial role for the clearance of these cells from the site of injury. NogoA seems to be overexpressed in the skeletal muscle of ALS patients [148]. Therefore, the efficacy of an anti-NogoA antibody (ozanezumab) to slow down ALS progression was tested but no difference in disease progression (ALSFRS-R) or in survival was observed [149].
The therapeutic potential of the gut microbiota
One of the most prominent scientific breakthroughs of the last decade is the demonstration of the link between the gut microbiota and the brain based on the growing number of studies reporting causal effects of the gut microbiota on the brain. Taken together, these studies suggest that the microbiota might influence behavior and play a role in the pathogenesis of several neurological disorders [150–152]. In this context, preclinical studies have demonstrated that there is a difference in the microbiota profile in ALS patients. The results of these studies have suggested that such modifications can alter the gut microbiota brain axis and induce gastrointestinal dysfunction in ALS patients [153, 154]. Notwithstanding the limitation of the studies (in particular, the small panel of human samples and uncertainty whether dysbiosis is primary or secondary to dietary changes in ALS), the role of the gut microbiota in ALS stays on the cutting edge for developing therapies aimed at improving gut dysbiosis as well as the course of the disease using probiotics and prebiotics, both from a prophylactic and therapeutic points of view. Regarding this issue, it is worth to mention a recent study that demonstrates how, in an ALS-prone Sod1 transgenic mouse model, the course of the ALS-like disease can be modulated by administration of specific gut microbial strains. Apparently, administration of Akkermansia muciniphila was associated with improvements of symptoms and prolonged survival whereas Ruminococcus torques and Parabacteroides distasonis were associated with an exacerbation of disease progression. In addition, preliminary analyses of human gut microbiota further supported the hypothesis that Akkermansia might play a role in the progression of human ALS, an exciting discovery [155]. Although more studies are necessary, it is feasible to predict that the management of the gut microbiota might represent a further strategy helpful to prevent or alleviate the symptoms of this neurodegenerative disease.
Therapeutic approaches based on non-invasive brain stimulation
An alternative and innovative approach to pharmacological therapies is provided by non-invasive brain stimulation approaches, represented by transcranial magnetic stimulation (TMS) or transcranial direct current stimulation (tDCS) to slow down clinical progression in patients with ALS. Recent studies have shown limited but promising results on muscle strength, quality of life and neurophysiological measures in ALS patients treated with these options [156, 157]. In the same view, beneficial effect of targeted language training in combination with brain stimulation was demonstrated in agrammatic variant of PPA patients [158].
Therapeutic approaches based on genetic factors
The identification of dominant mutations in TDP-43 and FUS/TLS genes represented the starting point to highlight alterations of common pathogenic pathways in both ALS and FTD mediated by dysfunctions in RNA metabolism [159]. TDP-43 and FUS/TLS have similar structures and are implicated in several steps of mRNA and miRNA processing. On one hand, TDP-43 interacts with over 6000 RNA targets and plays a role during all phases of gene expression. It can act as a transcriptional repressor by direct DNA binding [160] and regulator pre-mRNA splicing, microRNA biogenesis, as well as RNA transport and translation [161]. Conversely, FUS/TLS binds to a single or double-stranded DNA and RNA and promotes or represses transcription and affecting splicing by interacting with RNA polymerase II [162, 163]. Both TDP-43 and FUS/TLS can modify the function of stress granules and regulate synaptic function in neurons, a feature that might represent a crucial link with the pathology [164].
Moreover, a shared characteristic of ALS/FTD is a reactive gliosis [165]. This is an unspecific proliferation of glial cells in injured brain regions characterized by microglial proliferation and astrocytic hypertrophy. Interestingly, gliosis is not the only main feature of the brain regions that show neuronal loss and inclusion pathology. In addition, glia can further promote neurodegeneration by releasing toxic compounds and by decreasing its clearance ability [166].
TDP-43
The lack of proper models of TDP-43 aggregation (that might allow to study the mechanism of formation, and their impact on other cellular components, as well as on cell metabolism) hampers full comprehension of the role of this nuclear factor in the pathogenesis of ALS and FTD [34]. Improving characterization of the mechanisms underlying the activities of these proteins, especially TDP-43, C9ORF72, and FUS/TLS is the main logical approach to better unravel the pathogenesis of ALS and FTLD-TDP. This is currently being done together or in parallel with investigating the relevance of common pathological pathways. For example, perturbation in neurotransmitters has been demonstrated in ALS as well as in FTLD, in particular with glutamate-driven excitotoxicity [160]. This pathway is supposed to be involved in ALS pathophysiology [167] and, more recently, also in FTLD [168, 169].
One still open question in the TDP-43 field has always been to determine whether the pathology caused by this protein was through a mechanism of loss-of-function or gain-of-function (bearing in mind that both mechanisms are not necessarily mutually exclusive and may be acting at the same time or at different stages in disease) [170, 171].
At the loss of function level, aggregates are able to sequester endogenous TDP-43 depleting its nuclear levels and inducing loss of function at the RNA processing level [172]. Importantly, neurodegeneration has recently been observed in selected neuronal populations that are affected early during disease even in the absence of TDP-43 aggregation, an observation suggesting that loss of function may be present even at early stages of disease [173]. No apparent direct cellular toxicity of the aggregates seems to be present beyond the lack of functional TDP-43 although some reports using bacterially-made TDP-43 aggregates have suggested that some degree of toxicity may be present due to alteration of calcium homeostasis [174]. Regarding therapy, therefore, the engineering of protein “disaggregases”, such as Hsp104, that can free and eventually reactivate TDP-43 trapped in the inclusions, could represent a valuable therapy for ALS and FTD patients [175, 176].
As TDP-43 is a ubiquitous protein that plays a general role in cellular and developmental processes of higher organisms, it may be difficult to target this protein in a generalized manner. One possibility, therefore, might be to target regions that are important to trigger the aggregation process, such as the RNA Recognition Motif 1 (RRM-1) that was shown to interact directly with p65-NF-kB [177]. Interestingly, it has been recently shown that antibodies against TDP-43 RRM1, able to disrupt this interaction, can reduce the neuroinflammation and the motor defects in mice that express an ALS-linked TDP-43 mutation [178].
Alternatively, targeting eventual mutations in the sequence of this protein might represent a parallel alternative therapeutic option [179]. To this date, the study of various patient populations has identified more than 50 TDP-43 mutations associated predominantly with ALS [172] and occasionally also in FTLD [180]. Moreover, it is now clear that a significant number of mutations affect other disease-associated proteins such as FUS, hnRNPA1, hnRNPA2/B1, MATR3, and TIA1 [45] with the emerging picture that most of these mutations may lead to altered stress granule (SG) dynamics [181]. Indeed, a growing body of evidence suggests that inhibiting stress granule formation may be a viable therapeutic approach to suppress TDP-43 mediated toxicity [182].
Finally, it may also be possible to target selected TDP-43 RNA processing events that become disrupted by loss of function [183–185] or other functional modifiers of its toxicity such as hnRNP proteins [186], Ataxin-2 [187], matrix metalloproteinase 9 [188], TCERG1 [189], cellular kinases [190], small molecule inhibitors of translational factors [191], and even the TARDBP autoregulatory process at the pre-mRNA level [192].
C9ORF72
The hexanucleotide G4C2, repeat expansion within the C9ORF72 gene has been recently identified as the most frequent genetic cause of both ALS and FTD [19, 20]. The pathogenic hexanucleotide length (>30 repeats) is present in about 10% of all ALS patients [193].
Focal neuronal loss, gliosis and neuronal cytoplasmic TDP-43 inclusions are the characteristic pathological findings in both C9ORF+ ALS and FTLD cases. Among the TDP-43 proteinopathies, the peculiarity of C9ORF72 pathology is that the inclusion bodies also contain dipeptide repeat polymers (DPR) and both loss- and gain- of function (LOF and GOF) of the C9ORF72 protein have been suggested to cause neurodegeneration [194, 195].
Regarding possible functions of C9ORF72, in vitro studies aimed at characterizing the effects of LOF have suggested a role in endosomal and autophagic pathways [196]. In this line, although still to be tested, C9ORF72 haploinsufficiency might eventually be addressed by gene therapy approaches [197].
On the other hand, the finding of both repeat RNA and protein aggregates in post-mortem brain supports the toxic GOF hypothesis [198]. Repeat-containing RNA aggregates, or RNA foci, have been found to trap proteins implicated in RNA splicing, editing, nuclear export, and nuclear function [199]. An alternative mechanism to RNA binding protein sequestration is associated with generation of DPRs by repeat-associated non-ATG translation (RAN), that generate neuronal inclusions with a distribution different from that of the TDP-43 pathology [200]. Small compounds targeting the peculiar G-quadruplex structure of C9ORF72 repeats have successfully used to reduced RNA foci burden and the levels of DPRs in patient iPSC-derived motor and cortical neurons [201]. Likewise to TDP-43 pathology modifiers, the screening for repressors of C9ORF72 toxicity has recently led to identify: 1) the cellular factor DDX3X as capable of inhibiting non-AUG translation of the C9ORF72 dipeptides [202]; 2) several members of the karyopherin family as capable of suppressing dipeptide toxicity [203]; 3) nuclear pore complex component Ref1 [204] and human RanGAP1 [205]. Alternatively, the development of antibodies against selected dipeptides such as GP has been shown capable of inhibiting their cell-to-cell transmission and aggregation [206].
Finally, the use of oligonucleotide-based antisense aimed at decreasing aberrant RNAs expression represents one of the more successful approaches for the treatment of various neurodegenerative disorders [207, 208]. Therefore, it has been suggested that such a strategy might be useful to reduce the levels of mutant C9ORF72 transcripts [209] and successful targeting of mutant C9ORF72 transcripts in different ALS model systems has already been described [210]. Moreover, artificial miRNAs targeting mutant C9ORF72 have been shown to be able to reduce the GOF caused by the repeat-containing transcripts [211]. The therapeutic potential of these approaches, however, still remains to be tested.
FUS
Similarly to TDP-43, FUS is a predominantly nuclear protein and pathological FUS inclusions are mostly localized in the cytosol [212]. Mutations in the FUS/TLS gene account for approximately 4% of fALS (~0.4% of all ALS) [24, 25] and FTLD [213]. Although the phenotype associated with a FUS mutation is variable, most patients predominantly demonstrate loss of lower motor neurons and low survival rate [214]. The mechanisms by which FUS mutations cause ALS and FTD remain controversial and have been linked to a variety of neuronal features, such as dendrite development [215], and alterations in cellular processes, such as paraspeckle formation [216]. Recently, a model has been proposed in which low-complexity domains of FUS drive its reversible assembly into membrane-free, liquid droplet and hydrogen-like structure. Since the inhibition of these fibrillar hydrogel assemblies mitigates neurotoxicity, it has been proposed as a potential therapeutic strategy in early phase ALS and ALS/FTD associated with mutations [217], and approach that could potentially be used also for other RNA binding proteins.
As with TDP-43 and other ALS genes, autophagy enhancement using small molecules has been shown to successfully reduce cytoplasmic FUS levels, to restore RBP homeostasis, and to rescue motor function in vivo [218]. In keeping with this view, it has been shown very recently that a potent HDAC inhibitor, called ACY-738, can cross the blood-brain barrier and improve the motor phenotype and life span of Transgenic FUS+/+ mice [219]. Finally, an early Phase 1 Clinical Trial has been started using the FDA-approved steroid medication Betamethasone (The TRANSLATE Study, NCT03707795) and the recruitment process has been just completed.
GRN
Autosomal dominant mutations in the GRN gene have been implicated in up to 25% of familial FTD cases and these mutations seem to cause the disease due to haploinsufficiency [220, 221]. Mutations in GRN also result in TDP-43 neuropathology in humans, but knockout mice show little pathologically phosphorylated TDP-43 [222], thus indicating that the link between gene defect and pathology is complex. Although the specific functions of GRN have not been fully characterized [223], it is becoming clear that in the neuronal context the expression and function of GRN is an important determinant of proper neurite outgrowth and branching [224]. An obvious therapeutic strategy for carriers of GRN mutations would thus be represented by gene therapy aimed at restoring proper GRN expression levels. This strategy has not yet been tried in humans but recent promising results have shown that using Adeno Associated Vectors (AAV) capable of expressing progranulin in the brain of Grn -/- mice have the ability to improve lysosomal dysfunctions and microglial pathology [225]. Finally, with regards to disease, it is also important to note that TDP-43 loss of function caused by aberrant aggregation can also induce a mis-splicing event in Sortilin1, the neuronal receptor of Progranulin [226, 227]. In this respect, therefore, these recently identified PGRN binding receptors may aid in the development of therapeutics designed to regulate PGRN levels. More recently, it has been announced by Alector company that FDA has granted Fast Track designation to AL101 for the treatment of FTD patients with progranulin gene mutations. AL101 is an anti-sortilin human monoclonal antibody designed with the aim to rescue progranulin haploinsufficiency in the central nervous [228].
Conclusions and perspectives
The clinical and neuropathological heterogeneity of ALS and FTD represents only the tip of the iceberg of these multifaceted diseases and many key issues remain to be fully explained, such as the reason for the selective vulnerability of cell types like specific motor neurons compared to frontal and temporal neurons as well as the influence of exogenous and endogenous modifier factors on the onset and progression of disease. Moreover, there are no appropriate biomarkers capable of accurate diagnosing and predicting disease progression. Nonetheless, it is becoming increasingly clear that the main reason of the complexity of these pathologies arises from the heterogeneity in their etiology. Approximately 90% of patients present sporadic adult-onset forms of unknown etiology. The remaining percentage of patients with genetic forms of the pathologies is characterized by a high degree of genetic heterogeneity both at allele and at locus level [17, 229]. Furthermore, several risk factors and genetic modifiers able to increase susceptibility to the diseases, or to influence the rate of progression have now been identified [17, 230]. In conclusion, the two diseases represent a pathological "continuum" possibly associated with a complex inheritance and influenced by an important interplay between genetic risks and likely environmental factors.
In familial ALS (fALS) many causative gene defects have been described (for an up-to-date list see: http://alsod.iop.kcl.ac.uk/) and in September 2019, FDA released a new guidelines on the elaboration of novel treatments for ALS, providing suggestions for the design of clinical trials and to measure the effectiveness of the potential treatments [231].
At present, the road towards an effective treatment for ALS and FTD lies still in the future. Table 4 summarizes the upstream hurdles to lead the development of novel treatments for ALS and FTD. Recent advances in our better understanding of disease and the genes that are involved in the pathology have considerably improved the outlook of developing innovative therapeutic strategies in the near future. As we have highlighted in this review, and shown in Fig. 3, these therapeutic strategies involve a number of approaches ranging from classic gene therapy to small compounds aimed at modifying aberrant splicing profiles or at reducing selected mRNA expression, as well as to administration of probiotics. Most importantly, the application of other innovative treatments based on antisense oligonucleotides (ASOs) to spinal muscular atrophy and other neurodegenerative diseases provides a solid basis for their utilization also for ALS [232]. Of course, a series of desirable goals need to be accomplished in order to improve the translation of individual genetic information into novel and eventually personalized treatments for ALS and FTD (Table 5). In particular, the development of robust animal models and protocols to minimize eventual off target effects, the optimization of ASOs' delivery across the blood-brain barrier, and the overcoming of the potential concerns with regards to the immunogenicity of the viral vectors as well as to the eventual pre-existing immunity to AAV [233, 234].
Table 4.
Hurdles on the road of developing novel treatments for ALS and FTD
| • Unknown etiology of sporadic cases. | |
| • High degree of heterogeneity at clinical, neuropathological level. | |
| • High degree of genetic heterogeneity both at allele and at locus level. | |
| • Unknown reason(s) for the selective vulnerability of cell types (i.e., specific motor neurons, frontal, and temporal neurons). | |
| • Unknown influence of exogenous factors on the onset and progression of the diseases. | |
| • Unknown influence of endogenous modifier factors on the onset and progression of disease. |
Table 5.
Future directions to improve the translation of individual genetic information into novel and personalized treatments for ALS and FTD
| • Gaining novel insights into molecular mechanisms of ALS and FTD pathophysiology by better integration of Clinical, Neuropathological, Neuroimaging, Next-Generation Sequencing, Proteomics, Pharmacogenetics studies. | |
| • Characterization of common and divergent mechanisms leading to ALS and FTD. | |
| • Revamp of ALS and FTD disease classification system according to the novel genetic and molecular information to identify subgroups of patients that might respond to treatments at a higher (or lower) rate than the population average. | |
| • Identification of reliable biomarkers for diagnosing, monitoring the response to therapy, and predicting disease progression. | |
| • Development of robust animal models and protocols to minimize eventual off target effects. | |
| • Optimization of ASOs' delivery across the blood-brain barrier. | |
| • Decrease/bypass the viral vectors' immunogenicity and the eventual pre-existing immunity to AAV. |
For sure, the more we know about the clinical, neuropathological, genetic and molecular characteristics of patients, the better we will be able to improve ALS and FTD disease classification systems in order to identify subgroups of patients. This will hopefully put researchers on the right path to integrate the novel achievements in everyday clinical practice and the development of personalized approaches to treat specific subsets of patients, based on their particular clinico-genetic “signature”.
Supplementary information
Additional file 1: Supplementary Figure S1. Physical interactions of the genes implicated in the pathogenesis of ALS and FTD. Interactions by binding among the genes shown in Fig. 1 generated by using the online web-portal Genemania (https://http://genemania.org/), gene multiple association network integration algorithm. This analysis shows the complex network of interactions existing among all the genes implicated in the pathologic processes underlying the origin and development of ALS and FTD.
Additional file 2: Supplementary Figure S2. Co-expression of the genes implicated in the pathogenesis of ALS and FTD. Co-expression networks of the genes shown in Fig. 1 generated by using the online web-portal Genemania (https://http://genemania.org/).
Additional file 3: Supplementary Figure S3. Common pathways of the genes implicated in the pathogenesis of ALS and FTD. Shared pathways among the genes shown in Fig. 1 generated by using the online web-portal Genemania (https://http://genemania.org/).
Additional file 4: Supplementary Figure S4. Co-localization of the genes implicated in the pathogenesis of ALS and FTD. Co-localization of the genes shown in Fig. 1 generated by using the online web-portal Genemania (https://http://genemania.org/).
Acknowledgements
This work was supported by: AriSLA Foundation (PathensTDP) and Beneficentia Stiftung – 2019 to EB. All Authors have read the Journal’s authorship statement.
Competing interests
All Authors declare no competing conflict of interest and declare no financial or personal relationship with organizations that could potentially be perceived as influencing the described research.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13024-020-00373-9.
References
- 1.Logroscino G, Traynor BJ, Hardiman O, Chio A, Mitchell D, Swingler RJ, Millul A, Benn E, Beghi E. Eurals Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry. 2010;81(4):385–390. doi: 10.1136/jnnp.2009.183525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mercy L, Hodges JR, Dawson K, Barker RA, Brayne C. Incidence of early-onset dementias in Cambridgeshire, United Kingdom. Neurology. 2008;71(19):1496–9. [DOI] [PubMed]
- 3.Karageorgiou E, Miller BL. Frontotemporal lobar degeneration: a clinical approach. Semin Neurol. 2014;34:189–201. [DOI] [PubMed]
- 4.Olney NT, Spina S, Miller BL. Frontotemporal Dementia. Neurol Clin. 2017;35(2):339–374. doi: 10.1016/j.ncl.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pan XD, Chen XC. Clinic, neuropathology and molecular genetics of frontotemporal dementia: a mini-review. Transl Neurodegener. 2013;2(1):8. doi: 10.1186/2047-9158-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saberi S, Stauffer JE, Schulte DJ, Ravits J. Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurol Clin. 2015;33(4):855–876. doi: 10.1016/j.ncl.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haass C, Neumann M. Frontotemporal dementia: from molecular mechanisms to therapy. J Neurochem. 2016;138(Suppl 1):3–5. doi: 10.1111/jnc.13619. [DOI] [PubMed] [Google Scholar]
- 8.Woollacott IO, Rohrer JD. The clinical spectrum of sporadic and familial forms of frontotemporal dementia. J Neurochem. 2016;138(Suppl 1):6–31. doi: 10.1111/jnc.13654. [DOI] [PubMed] [Google Scholar]
- 9.Tard C, Defebvre L, Moreau C, Devos D, Danel-Brunaud V. Clinical features of amyotrophic lateral sclerosis and their prognostic value. Rev Neurol (Paris) 2017;173(5):263–272. doi: 10.1016/j.neurol.2017.03.029. [DOI] [PubMed] [Google Scholar]
- 10.Peteri UK, Niukkanen M, Castren ML. Astrocytes in Neuropathologies Affecting the Frontal Cortex. Front Cell Neurosci. 2019;13:44. doi: 10.3389/fncel.2019.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oeckl P, Weydt P, Steinacker P, Anderl-Straub S, Nordin F, Volk AE, Diehl-Schmid J, Andersen PM, Kornhuber J, Danek A, et al. Different neuroinflammatory profile in amyotrophic lateral sclerosis and frontotemporal dementia is linked to the clinical phase. J Neurol Neurosurg Psychiatry. 2019;90(1):4–10. doi: 10.1136/jnnp-2018-318868. [DOI] [PubMed] [Google Scholar]
- 12.McCauley ME, Baloh RH. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019;137(5):715–730. doi: 10.1007/s00401-018-1933-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strong MJ, Grace GM, Freedman M, Lomen-Hoerth C, Woolley S, Goldstein LH, Murphy J, Shoesmith C, Rosenfeld J, Leigh PN, et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10(3):131–146. doi: 10.1080/17482960802654364. [DOI] [PubMed] [Google Scholar]
- 14.Grossman M. Amyotrophic lateral sclerosis - a multisystem neurodegenerative disorder. Nat Rev Neurol. 2019;15(1):5–6. doi: 10.1038/s41582-018-0103-y. [DOI] [PubMed] [Google Scholar]
- 15.Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL, 3rd, Schneider JA, Grinberg LT, Halliday G, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114(1):5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front Neurosci. 2019;13:1310. doi: 10.3389/fnins.2019.01310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pottier C, Ravenscroft TA, Sanchez-Contreras M, Rademakers R. Genetics of FTLD: overview and what else we can expect from genetic studies. J Neurochem. 2016;138(Suppl 1):32–53. doi: 10.1111/jnc.13622. [DOI] [PubMed] [Google Scholar]
- 18.Alsultan AA, Waller R, Heath PR, Kirby J. The genetics of amyotrophic lateral sclerosis: current insights. Degener Neurol Neuromuscul Dis. 2016;6:49–64. doi: 10.2147/DNND.S84956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 22.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 23.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 24.Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 25.Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Volk AE, Weishaupt JH, Andersen PM, Ludolph AC, Kubisch C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med Genet. 2018;30(2):252–258. doi: 10.1007/s11825-018-0185-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sirkis DW, Geier EG, Bonham LW, Karch CM, Yokoyama JS. Recent advances in the genetics of frontotemporal dementia. Curr Genet Med Rep. 2019;7(1):41–52. doi: 10.1007/s40142-019-0160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olszewska DA, Lonergan R, Fallon EM, Lynch T. Genetics of Frontotemporal Dementia. Curr Neurol Neurosci Rep. 2016;16(12):107. doi: 10.1007/s11910-016-0707-9. [DOI] [PubMed] [Google Scholar]
- 29.Solomon DA, Mitchell JC, Salcher-Konrad MT, Vance CA, Mizielinska S. Review: Modelling the pathology and behaviour of frontotemporal dementia. Neuropathol Appl Neurobiol. 2019;45(1):58–80. doi: 10.1111/nan.12536. [DOI] [PubMed] [Google Scholar]
- 30.Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(Web Server issue):W214–W220. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zuberi K, Franz M, Rodriguez H, Montojo J, Lopes CT, Bader GD, Morris Q. GeneMANIA prediction server 2013 update. Nucleic Acids Res. 2013;41(Web Server issue):W115–W122. doi: 10.1093/nar/gkt533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liscic RM, Breljak D. Molecular basis of amyotrophic lateral sclerosis. Prog Neuro-Psychopharmacol Biol Psychiatry. 2011;35(2):370–372. doi: 10.1016/j.pnpbp.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 33.Ng AS, Rademakers R, Miller BL. Frontotemporal dementia: a bridge between dementia and neuromuscular disease. Ann N Y Acad Sci. 2015;1338:71–93. doi: 10.1111/nyas.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liscic RM. Molecular basis of ALS and FTD: implications for translational studies. Arh Hig Rada Toksikol. 2015;66(4):285–290. doi: 10.1515/aiht-2015-66-2679. [DOI] [PubMed] [Google Scholar]
- 35.Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ. ALS and FTLD: two faces of TDP-43 proteinopathy. Eur J Neurol. 2008;15(8):772–780. doi: 10.1111/j.1468-1331.2008.02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hergesheimer RC, Chami AA, de Assis DR, Vourc'h P, Andres CR, Corcia P, Lanznaster D, Blasco H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: a resolution in sight? Brain. 2019;142(5):1176–1194. doi: 10.1093/brain/awz078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bennett CL, Dastidar SG, Ling SC, Malik B, Ashe T, Wadhwa M, Miller DB, Lee C, Mitchell MB, van Es MA, et al. Senataxin mutations elicit motor neuron degeneration phenotypes and yield TDP-43 mislocalization in ALS4 mice and human patients. Acta Neuropathol. 2018;136(3):425–443. doi: 10.1007/s00401-018-1852-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466(7310):1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sumi H, Kato S, Mochimaru Y, Fujimura H, Etoh M, Sakoda S. Nuclear TAR DNA binding protein 43 expression in spinal cord neurons correlates with the clinical course in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2009;68(1):37–47. doi: 10.1097/NEN.0b013e3181919cb5. [DOI] [PubMed] [Google Scholar]
- 40.Jeon GS, Shim YM, Lee DY, Kim JS, Kang M, Ahn SH, Shin JY, Geum D, Hong YH, Sung JJ. Pathological Modification of TDP-43 in Amyotrophic Lateral Sclerosis with SOD1 Mutations. Mol Neurobiol. 2019;56(3):2007–2021. doi: 10.1007/s12035-018-1218-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kirby J, Highley JR, Cox L, Goodall EF, Hewitt C, Hartley JA, Hollinger HC, Fox M, Ince PG, McDermott CJ, Shaw PJ. Lack of unique neuropathology in amyotrophic lateral sclerosis associated with p.K54E angiogenin (ANG) mutation. Neuropathol Appl Neurobiol. 2013;39(5):562–571. doi: 10.1111/nan.12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Zee J, Van Langenhove T, Kovacs GG, Dillen L, Deschamps W, Engelborghs S, Matej R, Vandenbulcke M, Sieben A, Dermaut B, et al. Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol. 2014;128(3):397–410. doi: 10.1007/s00401-014-1298-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, Pliner HA, Abramzon Y, Marangi G, Winborn BJ, et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci. 2014;17(5):664–666. doi: 10.1038/nn.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495(7442):467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Purice MD, Taylor JP. Linking hnRNP Function to ALS and FTD Pathology. Front Neurosci. 2018;12:326. doi: 10.3389/fnins.2018.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kamada M, Izumi Y, Ayaki T, Nakamura M, Kagawa S, Kudo E, Sako W, Maruyama H, Nishida Y, Kawakami H, et al. Clinicopathologic features of autosomal recessive amyotrophic lateral sclerosis associated with optineurin mutation. Neuropathology. 2014;34(1):64–70. doi: 10.1111/neup.12051. [DOI] [PubMed] [Google Scholar]
- 47.Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, Kenna KP, Scotter EL, Kost J, Keagle P, Miller JW, et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron. 2014;84(2):324–331. doi: 10.1016/j.neuron.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 49.Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7(5):409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Solomon DA, Stepto A, Au WH, Adachi Y, Diaper DC, Hall R, Rekhi A, Boudi A, Tziortzouda P, Lee YB, et al. A feedback loop between dipeptide-repeat protein, TDP-43 and karyopherin-alpha mediates C9orf72-related neurodegeneration. Brain. 2018;141(10):2908–2924. doi: 10.1093/brain/awy241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee SM, Asress S, Hales CM, Gearing M, Vizcarra JC, Fournier CN, Gutman DA, Chin LS, Li L, Glass JD. TDP-43 cytoplasmic inclusion formation is disrupted in C9orf72-associated amyotrophic lateral sclerosis/frontotemporal lobar degeneration. Brain Commun. 2019;1(1):fcz014. doi: 10.1093/braincomms/fcz014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wider C, Dickson DW, Stoessl AJ, Tsuboi Y, Chapon F, Gutmann L, Lechevalier B, Calne DB, Personett DA, Hulihan M, et al. Pallidonigral TDP-43 pathology in Perry syndrome. Parkinsonism Relat Disord. 2009;15(4):281–286. doi: 10.1016/j.parkreldis.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gijselinck I, Van Mossevelde S, van der Zee J, Sieben A, Philtjens S, Heeman B, Engelborghs S, Vandenbulcke M, De Baets G, Baumer V, et al. Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology. 2015;85(24):2116–2125. doi: 10.1212/WNL.0000000000002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woo JA, Liu T, Trotter C, Fang CC, De Narvaez E, LePochat P, Maslar D, Bukhari A, Zhao X, Deonarine A, et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat Commun. 2017;8(1):15558. doi: 10.1038/ncomms15558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Williams KL, Topp S, Yang S, Smith B, Fifita JA, Warraich ST, Zhang KY, Farrawell N, Vance C, Hu X, et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat Commun. 2016;7(1):11253. doi: 10.1038/ncomms11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holm IE, Englund E, Mackenzie IR, Johannsen P, Isaacs AM. A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J Neuropathol Exp Neurol. 2007;66(10):884–891. doi: 10.1097/nen.0b013e3181567f02. [DOI] [PubMed] [Google Scholar]
- 58.Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Taylor JP, Kretzschmar HA, Kimonis VE, Forman MS. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 2007;66(2):152–157. doi: 10.1097/nen.0b013e31803020b9. [DOI] [PubMed] [Google Scholar]
- 59.Mackenzie IR. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007;114(1):49–54. doi: 10.1007/s00401-007-0223-8. [DOI] [PubMed] [Google Scholar]
- 60.Jun MH, Han JH, Lee YK, Jang DJ, Kaang BK, Lee JA. TMEM106B, a frontotemporal lobar dementia (FTLD) modifier, associates with FTD-3-linked CHMP2B, a complex of ESCRT-III. Mol Brain. 2015;8(1):85. doi: 10.1186/s13041-015-0177-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neumann M, Mackenzie IRA. Review: Neuropathology of non-tau frontotemporal lobar degeneration. Neuropathol Appl Neurobiol. 2019;45(1):19–40. doi: 10.1111/nan.12526. [DOI] [PubMed] [Google Scholar]
- 62.Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61(5):435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hasegawa M, Arai T, Akiyama H, Nonaka T, Mori H, Hashimoto T, Yamazaki M, Oyanagi K. Book TDP-43 is deposited in the Guam parkinsonism-dementia complex brains. - PubMed - NCBI (Editor ed.^eds.) 2007. TDP-43 is deposited in the Guam parkinsonism-dementia complex brains. - PubMed - NCBI; pp. 1386–1394. [DOI] [PubMed] [Google Scholar]
- 64.Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K, Togo T, Katsuse O, Uchikado H, Furukawa Y, et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer's disease and dementia with Lewy bodies. Brain Res. 2007;1184:284–294. doi: 10.1016/j.brainres.2007.09.048. [DOI] [PubMed] [Google Scholar]
- 65.Kalmar B, Lu CH, Greensmith L. Book The role of heat shock proteins in Amyotrophic Lateral Sclerosis: The therapeutic potential of Arimoclomol (Editor ed.^eds.) 2014. The role of heat shock proteins in Amyotrophic Lateral Sclerosis: The therapeutic potential of Arimoclomol; pp. 40–54. [DOI] [PubMed] [Google Scholar]
- 66.Koga S, Sanchez-Contreras M, Josephs KA, Uitti RJ, Graff-Radford N, van Gerpen JA, Cheshire WP, Wszolek ZK, Rademakers R, Dickson DW. Distribution and characteristics of transactive response DNA binding protein 43 kDa pathology in progressive supranuclear palsy. Mov Disord. 2017;32(2):246–255. doi: 10.1002/mds.26809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin WL, Dickson DW. Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol. 2008;116(2):205–213. doi: 10.1007/s00401-008-0408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, Arnold SE, Siderowf A, Grossman M, Leverenz JB, et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007;114(3):221–229. doi: 10.1007/s00401-007-0261-2. [DOI] [PubMed] [Google Scholar]
- 69.Uryu K, Nakashima-Yasuda H, Forman MS, Kwong LK, Clark CM, Grossman M, Miller BL, Kretzschmar HA, Lee VM, Trojanowski JQ, Neumann M. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol. 2008;67(6):555–564. doi: 10.1097/NEN.0b013e31817713b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dugger BN, Dickson DW. Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol. 2017;9(7):a028035. [DOI] [PMC free article] [PubMed]
- 71.Dardis A, Zampieri S, Canterini S, Newell KL, Stuani C, Murrell JR, Ghetti B, Fiorenza MT, Bembi B, Buratti E. Altered localization and functionality of TAR DNA Binding Protein 43 (TDP-43) in niemann- pick disease type C. Acta Neuropathol Commun. 2016;4(1):52. doi: 10.1186/s40478-016-0325-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferrari R, Kapogiannis D, Huey ED, Momeni P. FTD and ALS: a tale of two diseases. Curr Alzheimer Res. 2011;8(3):273–294. doi: 10.2174/156720511795563700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Benedetti S, Lucchini G, Del Bo C, Deon V, Marocchi A, Penco S, Lunetta C, Gianazza E, Bonomi F, Iametti S. Blood trace metals in a sporadic amyotrophic lateral sclerosis geographical cluster. Biometals. 2017;30(3):355–365. doi: 10.1007/s10534-017-0011-4. [DOI] [PubMed] [Google Scholar]
- 74.Cicero CE, Mostile G, Vasta R, Rapisarda V, Signorelli SS, Ferrante M, Zappia M, Nicoletti A. Metals and neurodegenerative diseases. A systematic review. Environ Res. 2017;159:82–94. doi: 10.1016/j.envres.2017.07.048. [DOI] [PubMed] [Google Scholar]
- 75.Jimenez-Pacheco A, Franco JM, Lopez S, Gomez-Zumaquero JM, Magdalena Leal-Lasarte M, Caballero-Hernandez DE, Cejudo-Guillen M, Pozo D. Epigenetic Mechanisms of Gene Regulation in Amyotrophic Lateral Sclerosis. Adv Exp Med Biol. 2017;978:255–275. doi: 10.1007/978-3-319-53889-1_14. [DOI] [PubMed] [Google Scholar]
- 76.Belzil VV, Katzman RB, Petrucelli L. ALS and FTD: an epigenetic perspective. Acta Neuropathol. 2016;132(4):487–502. doi: 10.1007/s00401-016-1587-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oates N, Pamphlett R. An epigenetic analysis of SOD1 and VEGF in ALS. Amyotroph Lateral Scler. 2007;8(2):83–86. doi: 10.1080/17482960601149160. [DOI] [PubMed] [Google Scholar]
- 78.Yang Y, Gozen O, Vidensky S, Robinson MB, Rothstein JD. Epigenetic regulation of neuron-dependent induction of astroglial synaptic protein GLT1. Glia. 2010;58(3):277–286. doi: 10.1002/glia.20922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kioumourtzoglou MA, Rotem RS, Seals RM, Gredal O, Hansen J, Weisskopf MG. Diabetes Mellitus, Obesity, and Diagnosis of Amyotrophic Lateral Sclerosis: A Population-Based Study. JAMA Neurol. 2015;72(8):905–911. doi: 10.1001/jamaneurol.2015.0910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Manera U, D’Ovidio F, D’Errico A, Carnà P, Calvo A, Costa G, Chiò A. The role of pre-morbid diabetes on developing ALS: a population-based study (P6.462) Neurology. 2018;90(15 Supplement):P6.462. doi: 10.1111/ene.13465. [DOI] [PubMed] [Google Scholar]
- 81.Jawaid A, Khan R, Polymenidou M, Schulz PE. Disease-modifying effects of metabolic perturbations in ALS/FTLD. Mol Neurodegener. 2018;13(1):63. doi: 10.1186/s13024-018-0294-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jawaid A, Paganoni S, Hauser C, Schulz PE. Trials of antidiabetic drugs in amyotrophic lateral sclerosis: proceed with caution? Neurodegener Dis. 2014;13(4):205–208. doi: 10.1159/000353158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jawaid A, Poon M, Strutt AM, Rice LK, McDowell EJ, Salamone AR, Qureshi SU, Simpson E, Appel SH, York MK, Schulz PE. Does apolipoprotein E genotype modify the clinical expression of ALS? Eur J Neurol. 2011;18(4):618–624. doi: 10.1111/j.1468-1331.2010.03225.x. [DOI] [PubMed] [Google Scholar]
- 84.Jawaid A, Salamone AR, Strutt AM, Murthy SB, Wheaton M, McDowell EJ, Simpson E, Appel SH, York MK, Schulz PE. ALS disease onset may occur later in patients with pre-morbid diabetes mellitus. Eur J Neurol. 2010;17(5):733–739. doi: 10.1111/j.1468-1331.2009.02923.x. [DOI] [PubMed] [Google Scholar]
- 85.Jawaid A, Murthy SB, Wilson AM, Qureshi SU, Amro MJ, Wheaton M, Simpson E, Harati Y, Strutt AM, York MK, Schulz PE. A decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS. Amyotroph Lateral Scler. 2010;11(6):542–548. doi: 10.3109/17482968.2010.482592. [DOI] [PubMed] [Google Scholar]
- 86.Rosenfeld J, Ellis A. Nutrition and dietary supplements in motor neuron disease. Phys Med Rehabil Clin N Am. 2008;19(3):573–589. doi: 10.1016/j.pmr.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nieves JW, Gennings C, Factor-Litvak P, Hupf J, Singleton J, Sharf V, Oskarsson B, Fernandes Filho JA, Sorenson EJ, D'Amico E, et al. Association Between Dietary Intake and Function in Amyotrophic Lateral Sclerosis. JAMA Neurol. 2016;73(12):1425–1432. doi: 10.1001/jamaneurol.2016.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang LP, Fan DS. Diets for Patients with Amyotrophic Lateral Sclerosis: Pay Attention to Nutritional Intervention. Chin Med J. 2017;130(15):1765–1767. doi: 10.4103/0366-6999.211549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Al-Chalabi A. High-calorie diets in amyotrophic lateral sclerosis. Lancet. 2014;383(9934):2028–2030. doi: 10.1016/S0140-6736(14)60270-1. [DOI] [PubMed] [Google Scholar]
- 90.Bouteloup C, Desport JC, Clavelou P, Guy N, Derumeaux-Burel H, Ferrier A, Couratier P. Hypermetabolism in ALS patients: an early and persistent phenomenon. J Neurol. 2009;256(8):1236–1242. doi: 10.1007/s00415-009-5100-z. [DOI] [PubMed] [Google Scholar]
- 91.Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci U S A. 2004;101(30):11159–11164. doi: 10.1073/pnas.0402026101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hu F, Padukkavidana T, Vaegter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68(4):654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carrasquillo MM, Nicholson AM, Finch N, Gibbs JR, Baker M, Rutherford NJ, Hunter TA, DeJesus-Hernandez M, Bisceglio GD, Mackenzie IR, et al. Genome-wide screen identifies rs646776 near sortilin as a regulator of progranulin levels in human plasma. Am J Hum Genet. 2010;87(6):890–897. doi: 10.1016/j.ajhg.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, Crook R, Hunter T, Ghidoni R, Benussi L, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76(5):467–474. doi: 10.1212/WNL.0b013e31820a0e3b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cruchaga C, Graff C, Chiang HH, Wang J, Hinrichs AL, Spiegel N, Bertelsen S, Mayo K, Norton JB, Morris JC, Goate A. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol. 2011;68(5):581–586. doi: 10.1001/archneurol.2010.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC, Al-Sarraj S, Neumann M, Gelpi E, Ghetti B, et al. TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 2014;127(3):407–418. doi: 10.1007/s00401-013-1239-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.van Blitterswijk M, Mullen B, Wojtas A, Heckman MG, Diehl NN, Baker MC, DeJesus-Hernandez M, Brown PH, Murray ME, Hsiung GY, et al. Genetic modifiers in carriers of repeat expansions in the C9ORF72 gene. Mol Neurodegener. 2014;9:38. doi: 10.1186/1750-1326-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vass R, Ashbridge E, Geser F, Hu WT, Grossman M, Clay-Falcone D, Elman L, McCluskey L, Lee VM, Van Deerlin VM, et al. Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol. 2011;121(3):373–380. doi: 10.1007/s00401-010-0782-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rhinn H, Abeliovich A. Differential Aging Analysis in Human Cerebral Cortex Identifies Variants in TMEM106B and GRN that Regulate Aging Phenotypes. Cell Syst. 2017;4(4):404–415. doi: 10.1016/j.cels.2017.02.009. [DOI] [PubMed] [Google Scholar]
- 100.White CC, Yang HS, Yu L, Chibnik LB, Dawe RJ, Yang J, Klein HU, Felsky D, Ramos-Miguel A, Arfanakis K, et al. Identification of genes associated with dissociation of cognitive performance and neuropathological burden: Multistep analysis of genetic, epigenetic, and transcriptional data. PLoS Med. 2017;14(4):e1002287. doi: 10.1371/journal.pmed.1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Katsumata Y, Nelson PT, Ellingson SR, Fardo DW. Gene-based association study of genes linked to hippocampal sclerosis of aging neuropathology: GRN, TMEM106B, ABCC9, and KCNMB2. Neurobiol Aging. 2017;53:193 e117–193 e125. doi: 10.1016/j.neurobiolaging.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Devenney E, Vucic S, Hodges JR, Kiernan MC. Motor neuron disease-frontotemporal dementia: a clinical continuum. Expert Rev Neurother. 2015;15(5):509–522. doi: 10.1586/14737175.2015.1034108. [DOI] [PubMed] [Google Scholar]
- 103.Boxer AL, Gold M, Huey E, Gao FB, Burton EA, Chow T, Kao A, Leavitt BR, Lamb B, Grether M, et al. Frontotemporal degeneration, the next therapeutic frontier: molecules and animal models for frontotemporal degeneration drug development. Alzheimers Dement. 2013;9(2):176–188. doi: 10.1016/j.jalz.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hochgrafe K, Sydow A, Matenia D, Cadinu D, Konen S, Petrova O, Pickhardt M, Goll P, Morellini F, Mandelkow E, Mandelkow EM. Preventive methylene blue treatment preserves cognition in mice expressing full-length pro-aggregant human Tau. Acta Neuropathol Commun. 2015;3:25. doi: 10.1186/s40478-015-0204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Miller RG, Rosenberg JA, Gelinas DF, Mitsumoto H, Newman D, Sufit R, Borasio GD, Bradley WG, Bromberg MB, Brooks BR, et al. Practice parameter: the care of the patient with amyotrophic lateral sclerosis (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology: ALS Practice Parameters Task Force. Neurology. 1999;52(7):1311–1323. doi: 10.1212/wnl.52.7.1311. [DOI] [PubMed] [Google Scholar]
- 106.Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- 107.Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996;347(9013):1425–1431. doi: 10.1016/s0140-6736(96)91680-3. [DOI] [PubMed] [Google Scholar]
- 108.Dharmadasa T, Kiernan MC. Riluzole, disease stage and survival in ALS. Lancet Neurol. 2018;17(5):385–386. doi: 10.1016/S1474-4422(18)30091-7. [DOI] [PubMed] [Google Scholar]
- 109.Cheah BC, Vucic S, Krishnan AV, Kiernan MC. Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr Med Chem. 2010;17(18):1942–1199. doi: 10.2174/092986710791163939. [DOI] [PubMed] [Google Scholar]
- 110.Takei K, Watanabe K, Yuki S, Akimoto M, Sakata T, Palumbo J. Edaravone and its clinical development for amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(sup1):5–10. doi: 10.1080/21678421.2017.1353101. [DOI] [PubMed] [Google Scholar]
- 111.Writing G, Edaravone ALSSG. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16(7):505–512. doi: 10.1016/S1474-4422(17)30115-1. [DOI] [PubMed] [Google Scholar]
- 112.Shefner J, Heiman-Patterson T, Pioro EP, Wiedau-Pazos M, Liu S, Zhang J, Agnese W, Apple S. Long-term edaravone efficacy in amyotrophic lateral sclerosis: Post-hoc analyses of Study 19 (MCI186-19) Muscle Nerve. 2020;61(2):218–221. doi: 10.1002/mus.26740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jaiswal MK. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med Res Rev. 2019;39(2):733–748. doi: 10.1002/med.21528. [DOI] [PubMed] [Google Scholar]
- 114.Kalmar B, Lu CH, Greensmith L. The role of heat shock proteins in Amyotrophic Lateral Sclerosis: The therapeutic potential of Arimoclomol. Pharmacol Ther. 2014;141(1):40–54. doi: 10.1016/j.pharmthera.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 115.Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10(4):402–405. doi: 10.1038/nm1021. [DOI] [PubMed] [Google Scholar]
- 116.Scotter EL, Chen HJ, Shaw CE. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics. 2015;12(2):352–363. doi: 10.1007/s13311-015-0338-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang P, Wander CM, Yuan CX, Bereman MS, Cohen TJ. Acetylation-induced TDP-43 pathology is suppressed by an HSF1-dependent chaperone program. Nat Commun. 2017;8(1):82. doi: 10.1038/s41467-017-00088-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Benatar M, Wuu J, Andersen PM, Atassi N, David W, Cudkowicz M, Schoenfeld D. Randomized, double-blind, placebo-controlled trial of arimoclomol in rapidly progressive SOD1 ALS. Neurology. 2018;90(7):e565–e574. doi: 10.1212/WNL.0000000000004960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jagadapillai R, Mellen NM, Sachleben LR, Jr, Gozal E. Ceftriaxone preserves glutamate transporters and prevents intermittent hypoxia-induced vulnerability to brain excitotoxic injury. PLoS One. 2014;9(7):e100230. doi: 10.1371/journal.pone.0100230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7(11):616–630. doi: 10.1038/nrneurol.2011.152. [DOI] [PubMed] [Google Scholar]
- 121.Cudkowicz ME, Titus S, Kearney M, Yu H, Sherman A, Schoenfeld D, Hayden D, Shui A, Brooks B, Conwit R, et al. Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: a multi-stage, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(11):1083–1091. doi: 10.1016/S1474-4422(14)70222-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hammam K, Saez-Ayala M, Rebuffet E, Gros L, Lopez S, Hajem B, Humbert M, Baudelet E, Audebert S, Betzi S, et al. Dual protein kinase and nucleoside kinase modulators for rationally designed polypharmacology. Nat Commun. 2017;8(1):1420. doi: 10.1038/s41467-017-01582-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mora JS, Genge A, Chio A, Estol CJ, Chaverri D, Hernandez M, Marin S, Mascias J, Rodriguez GE, Povedano M, et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized clinical trial. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(1-2):5–14. doi: 10.1080/21678421.2019.1632346. [DOI] [PubMed] [Google Scholar]
- 124.Bilsland LG, Greensmith L. The endocannabinoid system in amyotrophic lateral sclerosis. Curr Pharm Des. 2008;14(23):2306–2316. doi: 10.2174/138161208785740081. [DOI] [PubMed] [Google Scholar]
- 125.Giacoppo S, Mazzon E. Can cannabinoids be a potential therapeutic tool in amyotrophic lateral sclerosis? Neural Regen Res. 2016;11(12):1896–1899. doi: 10.4103/1673-5374.197125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Urbi B, Owusu MA, Hughes I, Katz M, Broadley S, Sabet A. Effects of cannabinoids in Amyotrophic Lateral Sclerosis (ALS) murine models: a systematic review and meta-analysis. J Neurochem. 2019;149(2):284–297. doi: 10.1111/jnc.14639. [DOI] [PubMed] [Google Scholar]
- 127.Romano M, Buratti E. Tirasemtiv (CK-2017357) Drugs Future. 2013;38(3):157–162. [Google Scholar]
- 128.Shefner JM, Cudkowicz ME, Hardiman O, Cockcroft BM, Lee JH, Malik FI, Meng L, Rudnicki SA, Wolff AA, Andrews JA, Group V-AS A phase III trial of tirasemtiv as a potential treatment for amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2019;0(0):1–11. [Google Scholar]
- 129.Andrews JA, Cudkowicz ME, Hardiman O, Meng L, Bian A, Lee J, Wolff AA, Malik FI, Shefner JM. VITALITY-ALS, a phase III trial of tirasemtiv, a selective fast skeletal muscle troponin activator, as a potential treatment for patients with amyotrophic lateral sclerosis: study design and baseline characteristics. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19(3-4):259–266. doi: 10.1080/21678421.2018.1426770. [DOI] [PubMed] [Google Scholar]
- 130.Hwee DT, Cheng AJ, Hartman JJ, Hinken AC, Lee K, Durham N, Russell AJ, Malik FI, Westerblad H, Jasper JR. The Ca(2+) sensitizer CK-2066260 increases myofibrillar Ca(2+) sensitivity and submaximal force selectively in fast skeletal muscle. J Physiol. 2017;595(5):1657–1670. doi: 10.1113/JP273248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Andrews JA, Miller TM, Vijayakumar V, Stoltz R, James JK, Meng L, Wolff AA, Malik FI. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve. 2018;57(5):729–734. doi: 10.1002/mus.26017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mazzini L, Ferrero I, Luparello V, Rustichelli D, Gunetti M, Mareschi K, Testa L, Stecco A, Tarletti R, Miglioretti M, et al. Mesenchymal stem cell transplantation in amyotrophic lateral sclerosis: A Phase I clinical trial. Exp Neurol. 2010;223(1):229–237. doi: 10.1016/j.expneurol.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 133.Beers DR, Appel SH. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol. 2019;18(2):211–220. doi: 10.1016/S1474-4422(18)30394-6. [DOI] [PubMed] [Google Scholar]
- 134.Glass JD, Hertzberg VS, Boulis NM, Riley J, Federici T, Polak M, Bordeau J, Fournier C, Johe K, Hazel T, et al. Transplantation of spinal cord-derived neural stem cells for ALS: Analysis of phase 1 and 2 trials. Neurology. 2016;87(4):392–400. doi: 10.1212/WNL.0000000000002889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gouel F, Rolland AS, Devedjian JC, Burnouf T, Devos D. Past and future of neurotrophic growth factors therapies in ALS: from single neurotrophic growth factor to stem cells and human platelet lysates. Front Neurol. 2019;10(JUL):835. [DOI] [PMC free article] [PubMed]
- 136.Henriques A, Pitzer C, Schneider A. Book Neurotrophic growth factors for the treatment of amyotrophic lateral sclerosis: Where do we stand? (Editor ed.^eds.) 2010. Neurotrophic growth factors for the treatment of amyotrophic lateral sclerosis: Where do we stand? [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Petrou P, Gothelf Y, Argov Z, Gotkine M, Levy YS, Kassis I, Vaknin-Dembinsky A, Ben-Hur T, Offen D, Abramsky O, et al. Safety and Clinical Effects of Mesenchymal Stem Cells Secreting Neurotrophic Factor Transplantation in Patients With Amyotrophic Lateral Sclerosis: Results of Phase 1/2 and 2a Clinical Trials. JAMA Neurol. 2016;73(3):337–344. doi: 10.1001/jamaneurol.2015.4321. [DOI] [PubMed] [Google Scholar]
- 138.Oh KW, Moon C, Kim HY, Oh SI, Park J, Lee JH, Chang IY, Kim KS, Kim SH. Phase I trial of repeated intrathecal autologous bone marrow-derived mesenchymal stromal cells in amyotrophic lateral sclerosis. Stem Cells Transl Med. 2015;4(6):590–597. doi: 10.5966/sctm.2014-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Oh KW, Noh MY, Kwon MS, Kim HY, Oh SI, Park J, Kim HJ, Ki CS, Kim SH. Repeated Intrathecal Mesenchymal Stem Cells for Amyotrophic Lateral Sclerosis. Ann Neurol. 2018;84(3):361–373. doi: 10.1002/ana.25302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Staff NP, Madigan NN, Morris J, Jentoft M, Sorenson EJ, Butler G, Gastineau D, Dietz A, Windebank AJ. Safety of intrathecal autologous adipose-derived mesenchymal stromal cells in patients with ALS. Neurology. 2016;87(21):2230–2234. doi: 10.1212/WNL.0000000000003359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE, Zhao W, Moore DH, Powell SZ, Appel SH. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med. 2013;5(1):64–79. doi: 10.1002/emmm.201201544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sheean RK, McKay FC, Cretney E, Bye CR, Perera ND, Tomas D, Weston RA, Scheller KJ, Djouma E, Menon P, et al. Association of Regulatory T-Cell Expansion With Progression of Amyotrophic Lateral Sclerosis: A Study of Humans and a Transgenic Mouse Model. JAMA Neurol. 2018;75(6):681–689. doi: 10.1001/jamaneurol.2018.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Thonhoff JR, Beers DR, Zhao W, Pleitez M, Simpson EP, Berry JD, Cudkowicz ME, Appel SH. Expanded autologous regulatory T-lymphocyte infusions in ALS: A phase I, first-in-human study. Neurol Neuroimmunol Neuroinflamm. 2018;5(4):e465. doi: 10.1212/NXI.0000000000000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vucic S. Differences in inflammatory profiles between ALS and FTD. J Neurol Neurosurg Psychiatry. 2019;90(1):1. doi: 10.1136/jnnp-2018-319377. [DOI] [PubMed] [Google Scholar]
- 145.Khalid SI, Ampie L, Kelly R, Ladha SS, Dardis C. Immune Modulation in the Treatment of Amyotrophic Lateral Sclerosis: A Review of Clinical Trials. Front Neurol. 2017;8:486. doi: 10.3389/fneur.2017.00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.David S, Fry EJ, Lopez-Vales R. Novel roles for Nogo receptor in inflammation and disease. Trends Neurosci. 2008;31(5):221–226. doi: 10.1016/j.tins.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 147.Pernet V, Schwab ME. The role of Nogo-A in axonal plasticity, regrowth and repair. Cell Tissue Res. 2012;349(1):97–104. doi: 10.1007/s00441-012-1432-6. [DOI] [PubMed] [Google Scholar]
- 148.Berges A, Bullman J, Bates S, Krull D, Williams N, Chen C. Ozanezumab dose selection for amyotrophic lateral sclerosis by pharmacokinetic-pharmacodynamic modelling of immunohistochemistry data from patient muscle biopsies. PLoS One. 2015;10(2):e0117355. doi: 10.1371/journal.pone.0117355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Meininger V, Genge A, van den Berg LH, Robberecht W, Ludolph A, Chio A, Kim SH, Leigh PN, Kiernan MC, Shefner JM, et al. Safety and efficacy of ozanezumab in patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2017;16(3):208–216. doi: 10.1016/S1474-4422(16)30399-4. [DOI] [PubMed] [Google Scholar]
- 150.Quigley EMM. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr Neurol Neurosci Rep. 2017;17(12):94. doi: 10.1007/s11910-017-0802-6. [DOI] [PubMed] [Google Scholar]
- 151.Dinan TG, Cryan JF. The Microbiome-Gut-Brain Axis in Health and Disease. Gastroenterol Clin N Am. 2017;46(1):77–89. doi: 10.1016/j.gtc.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 152.Wang HX, Wang YP. Gut Microbiota-brain Axis. Chin Med J. 2016;129(19):2373–2380. doi: 10.4103/0366-6999.190667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mazzini L, Mogna L, De Marchi F, Amoruso A, Pane M, Aloisio I, Cionci NB, Gaggia F, Lucenti A, Bersano E, et al. Potential Role of Gut Microbiota in ALS Pathogenesis and Possible Novel Therapeutic Strategies. J Clin Gastroenterol. 2018;52(Suppl 1):S68–S70. doi: 10.1097/MCG.0000000000001042. [DOI] [PubMed] [Google Scholar]
- 154.McCombe PA, Henderson RD, Lee A, Lee JD, Woodruff TM, Restuadi R, McRae A, Wray NR, Ngo S, Steyn FJ. Gut microbiota in ALS: possible role in pathogenesis? Expert Rev Neurother. 2019;19(9):785–805. doi: 10.1080/14737175.2019.1623026. [DOI] [PubMed] [Google Scholar]
- 155.Blacher E, Bashiardes S, Shapiro H, Rothschild D, Mor U, Dori-Bachash M, Kleimeyer C, Moresi C, Harnik Y, Zur M, et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature. 2019;572(7770):474–480. doi: 10.1038/s41586-019-1443-5. [DOI] [PubMed] [Google Scholar]
- 156.Benussi A, Alberici A, Cotelli MS, Dell'Era V, Cantoni V, Bonetta E, Manenti R, Filosto M, Morini R, Datta A, et al. Cortico-spinal tDCS in ALS: A randomized, double-blind, sham-controlled trial. Brain Stimul. 2019;12(5):1332–1334. doi: 10.1016/j.brs.2019.06.011. [DOI] [PubMed] [Google Scholar]
- 157.Di Lazzaro V, Pellegrino G, Ranieri F, Florio L, Musumeci G, Caulo M, Ferretti A, Capone F. Effects of repetitive TMS of the motor cortex on disease progression and on glutamate and GABA levels in ALS: A proof of principle study. Brain Stimul. 2017;10(5):1003–1005. doi: 10.1016/j.brs.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 158.Cotelli M, Manenti R, Petesi M, Brambilla M, Cosseddu M, Zanetti O, Miniussi C, Padovani A, Borroni B. Treatment of primary progressive aphasias by transcranial direct current stimulation combined with language training. J Alzheimers Dis. 2014;39(4):799–808. doi: 10.3233/JAD-131427. [DOI] [PubMed] [Google Scholar]
- 159.Taylor JP, Brown RH., Jr Cleveland DW Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front Mol Neurosci. 2019;12:25. doi: 10.3389/fnmol.2019.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Butti Z, Patten SA. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front Genet. 2018;9:712. doi: 10.3389/fgene.2018.00712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tan AY, Manley JL. TLS/FUS: a protein in cancer and ALS. Cell Cycle. 2012;11(18):3349–3350. doi: 10.4161/cc.21875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tan AY, Riley TR, Coady T, Bussemaker HJ, Manley JL. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc Natl Acad Sci U S A. 2012;109(16):6030–6035. doi: 10.1073/pnas.1203028109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lenzi J, De Santis R, de Turris V, Morlando M, Laneve P, Calvo A, Caliendo V, Chio A, Rosa A, Bozzoni I. ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell-derived motoneurons. Dis Model Mech. 2015;8(7):755–766. doi: 10.1242/dmm.020099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Radford RA, Morsch M, Rayner SL, Cole NJ, Pountney DL, Chung RS. The established and emerging roles of astrocytes and microglia in amyotrophic lateral sclerosis and frontotemporal dementia. Front Cell Neurosci. 2015;9:414. doi: 10.3389/fncel.2015.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rostalski H, Leskela S, Huber N, Katisko K, Cajanus A, Solje E, Marttinen M, Natunen T, Remes AM, Hiltunen M, Haapasalo A. Astrocytes and Microglia as Potential Contributors to the Pathogenesis of C9orf72 Repeat Expansion-Associated FTLD and ALS. Front Neurosci. 2019;13:486. doi: 10.3389/fnins.2019.00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rowland LP. Ameliorating amyotrophic lateral sclerosis. N Engl J Med. 2010;362(10):953–954. doi: 10.1056/NEJMcibr0912229. [DOI] [PubMed] [Google Scholar]
- 168.Benussi A, Alberici A, Buratti E, Ghidoni R, Gardoni F, Di Luca M, Padovani A, Borroni B. Toward a Glutamate Hypothesis of Frontotemporal Dementia. Front Neurosci. 2019;13:304. doi: 10.3389/fnins.2019.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Borroni B, Stanic J, Verpelli C, Mellone M, Bonomi E, Alberici A, Bernasconi P, Culotta L, Zianni E, Archetti S, et al. Anti-AMPA GluA3 antibodies in Frontotemporal dementia: a new molecular target. Sci Rep. 2017;7(1):6723. doi: 10.1038/s41598-017-06117-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. 2011;13(1):38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Halliday G, Bigio EH, Cairns NJ, Neumann M, Mackenzie IR, Mann DM. Mechanisms of disease in frontotemporal lobar degeneration: gain of function versus loss of function effects. Acta Neuropathol. 2012;124(3):373–382. doi: 10.1007/s00401-012-1030-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Buratti E. Functional Significance of TDP-43 Mutations in Disease. Adv Genet. 2015;91:1–53. doi: 10.1016/bs.adgen.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 173.Nana AL, Sidhu M, Gaus SE, Hwang JL, Li L, Park Y, Kim EJ, Pasquini L, Allen IE, Rankin KP, et al. Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol. 2019;137(1):27–46. doi: 10.1007/s00401-018-1942-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Cascella R, Capitini C, Fani G, Dobson CM, Cecchi C, Chiti F. Quantification of the Relative Contributions of Loss-of-function and Gain-of-function Mechanisms in TAR DNA-binding Protein 43 (TDP-43) Proteinopathies. J Biol Chem. 2016;291(37):19437–19448. doi: 10.1074/jbc.M116.737726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Torrente MP, Chuang E, Noll MM, Jackrel ME, Go MS, Shorter J. Mechanistic Insights into Hsp104 Potentiation. J Biol Chem. 2016;291(10):5101–5115. doi: 10.1074/jbc.M115.707976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tariq A, Lin J, Noll MM, Torrente MP, Mack KL, Murillo OH, Jackrel ME, Shorter J. Potentiating Hsp104 activity via phosphomimetic mutations in the middle domain. FEMS Yeast Res. 2018;18(5):foy042. doi: 10.1093/femsyr/foy042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Swarup V, Phaneuf D, Dupre N, Petri S, Strong M, Kriz J, Julien JP. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J Exp Med. 2011;208(12):2429–2447. doi: 10.1084/jem.20111313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pozzi S, Thammisetty SS, Codron P, Rahimian R, Plourde KV, Soucy G, Bareil C, Phaneuf D, Kriz J, Gravel C, Julien JP. Virus-mediated delivery of antibody targeting TAR DNA-binding protein-43 mitigates associated neuropathology. J Clin Invest. 2019;129(4):1581–1595. doi: 10.1172/JCI123931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nishimura AL, Shum C, Scotter EL, Abdelgany A, Sardone V, Wright J, Lee YB, Chen HJ, Bilican B, Carrasco M, et al. Allele-specific knockdown of ALS-associated mutant TDP-43 in neural stem cells derived from induced pluripotent stem cells. PLoS One. 2014;9(3):e91269. doi: 10.1371/journal.pone.0091269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Borroni B, Bonvicini C, Alberici A, Buratti E, Agosti C, Archetti S, Papetti A, Stuani C, Di Luca M, Gennarelli M, Padovani A. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum Mutat. 2009;30(11):E974–E983. doi: 10.1002/humu.21100. [DOI] [PubMed] [Google Scholar]
- 181.Khalfallah Y, Kuta R, Grasmuck C, Prat A, Durham HD, Vande VC. TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci Rep. 2018;8(1):7551. doi: 10.1038/s41598-018-25767-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chen Y, Cohen TJ. Aggregation of the nucleic acid-binding protein TDP-43 occurs via distinct routes that are coordinated with stress granule formation. J Biol Chem. 2019;294(10):3696–3706. doi: 10.1074/jbc.RA118.006351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.De Conti L, Akinyi MV, Mendoza-Maldonado R, Romano M, Baralle M, Buratti E. TDP-43 affects splicing profiles and isoform production of genes involved in the apoptotic and mitotic cellular pathways. Nucleic Acids Res. 2015;43(18):8990–9005. doi: 10.1093/nar/gkv814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019;22(2):167–179. doi: 10.1038/s41593-018-0300-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Melamed Z, Lopez-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019;22(2):180–190. doi: 10.1038/s41593-018-0293-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Appocher C, Mohagheghi F, Cappelli S, Stuani C, Romano M, Feiguin F, Buratti E. Major hnRNP proteins act as general TDP-43 functional modifiers both in Drosophila and human neuronal cells. Nucleic Acids Res. 2017;45(13):8026–8045. doi: 10.1093/nar/gkx477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, Messing J, Kim HJ, Soriano A, Auburger G, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544(7650):367–371. doi: 10.1038/nature22038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Spiller KJ, Khan T, Dominique MA, Restrepo CR, Cotton-Samuel D, Levitan M, Jafar-Nejad P, Zhang B, Soriano A, Rigo F, et al. Reduction of matrix metalloproteinase 9 (MMP-9) protects motor neurons from TDP-43-triggered death in rNLS8 mice. Neurobiol Dis. 2019;124:133–140. doi: 10.1016/j.nbd.2018.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Pons M, Prieto S, Miguel L, Frebourg T, Campion D, Sune C, Lecourtois M. Identification of TCERG1 as a new genetic modulator of TDP-43 production in Drosophila. Acta Neuropathol Commun. 2018;6(1):138. doi: 10.1186/s40478-018-0639-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Liachko NF, McMillan PJ, Strovas TJ, Loomis E, Greenup L, Murrell JR, Ghetti B, Raskind MA, Montine TJ, Bird TD, et al. The tau tubulin kinases TTBK1/2 promote accumulation of pathological TDP-43. PLoS Genet. 2014;10(12):e1004803. doi: 10.1371/journal.pgen.1004803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, Lee VM, Finkbeiner S, Gitler AD, Bonini NM. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46(2):152–160. doi: 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.White MA, Kim E, Duffy A, Adalbert R, Phillips BU, Peters OM, Stephenson J, Yang S, Massenzio F, Lin Z, et al. TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat Neurosci. 2018;21(4):552–563. doi: 10.1038/s41593-018-0113-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Fratta P, Polke JM, Newcombe J, Mizielinska S, Lashley T, Poulter M, Beck J, Preza E, Devoy A, Sidle K, et al. Screening a UK amyotrophic lateral sclerosis cohort provides evidence of multiple origins of the C9orf72 expansion. Neurobiol Aging. 2015;36(1):546 e541–546 e547. doi: 10.1016/j.neurobiolaging.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Mizielinska S, Isaacs AM. C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia: gain or loss of function? Curr Opin Neurol. 2014;27(5):515–523. doi: 10.1097/WCO.0000000000000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Rohrer JD, Isaacs AM, Mizielinska S, Mead S, Lashley T, Wray S, Sidle K, Fratta P, Orrell RW, Hardy J, et al. C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol. 2015;14(3):291–301. doi: 10.1016/S1474-4422(14)70233-9. [DOI] [PubMed] [Google Scholar]
- 196.Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, Hendricks E, Linares GR, Wang Y, Son EY, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018;24(3):313–325. doi: 10.1038/nm.4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Mis MSC, Brajkovic S, Tafuri F, Bresolin N, Comi GP, Corti S. Development of Therapeutics for C9ORF72 ALS/FTD-Related Disorders. Mol Neurobiol. 2017;54(6):4466–4476. doi: 10.1007/s12035-016-9993-0. [DOI] [PubMed] [Google Scholar]
- 198.Balendra R, Isaacs AM. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol. 2018;14(9):544–558. doi: 10.1038/s41582-018-0047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Conlon EG, Manley JL. RNA-binding proteins in neurodegeneration: mechanisms in aggregate. Genes Dev. 2017;31(15):1509–1528. doi: 10.1101/gad.304055.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A. 2011;108(1):260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Simone R, Balendra R, Moens TG, Preza E, Wilson KM, Heslegrave A, Woodling NS, Niccoli T, Gilbert-Jaramillo J, Abdelkarim S, et al. G-quadruplex-binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo. EMBO Mol Med. 2018;10(1):22–31. doi: 10.15252/emmm.201707850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Cheng W, Wang S, Zhang Z, Morgens DW, Hayes LR, Lee S, Portz B, Xie Y, Nguyen BV, Haney MS, et al. CRISPR-Cas9 Screens Identify the RNA Helicase DDX3X as a Repressor of C9ORF72 (GGGGCC)n Repeat-Associated Non-AUG Translation. Neuron. 2019;104(5):885–898. doi: 10.1016/j.neuron.2019.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW, 3rd, Sun S, Herdy JR, Bieri G, et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015;18(9):1226–1229. doi: 10.1038/nn.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525(7567):129–133. doi: 10.1038/nature14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525(7567):56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zhou Q, Lehmer C, Michaelsen M, Mori K, Alterauge D, Baumjohann D, Schludi MH, Greiling J, Farny D, Flatley A, et al. Antibodies inhibit transmission and aggregation of C9orf72 poly-GA dipeptide repeat proteins. EMBO Mol Med. 2017;9(5):687–702. doi: 10.15252/emmm.201607054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Bennett CF, Krainer AR, Cleveland DW. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu Rev Neurosci. 2019;42:385–406. doi: 10.1146/annurev-neuro-070918-050501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.St Martin JL, Wang L, Kaprielian Z. Toxicity in ALS: TDP-43 modifiers and C9orf72. Neurosci Lett. 2020;716:134621. doi: 10.1016/j.neulet.2019.134621. [DOI] [PubMed] [Google Scholar]
- 209.Riboldi G, Zanetta C, Ranieri M, Nizzardo M, Simone C, Magri F, Bresolin N, Comi GP, Corti S. Antisense oligonucleotide therapy for the treatment of C9ORF72 ALS/FTD diseases. Mol Neurobiol. 2014;50(3):721–732. doi: 10.1007/s12035-014-8724-7. [DOI] [PubMed] [Google Scholar]
- 210.Martier R, Liefhebber JM, Garcia-Osta A, Miniarikova J, Cuadrado-Tejedor M, Espelosin M, Ursua S, Petry H, van Deventer SJ, Evers MM, Konstantinova P. Targeting RNA-Mediated Toxicity in C9orf72 ALS and/or FTD by RNAi-Based Gene Therapy. Mol Ther Nucleic Acids. 2019;16:26–37. doi: 10.1016/j.omtn.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Martier R, Liefhebber JM, Miniarikova J, van der Zon T, Snapper J, Kolder I, Petry H, van Deventer SJ, Evers MM, Konstantinova P. Artificial MicroRNAs Targeting C9orf72 Can Reduce Accumulation of Intra-nuclear Transcripts in ALS and FTD Patients. Mol Ther Nucleic Acids. 2019;14:593–608. doi: 10.1016/j.omtn.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136(6):1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Van Langenhove T, van der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, Gijselinck I, Van den Broeck M, Mattheijssens M, Peeters K, De Deyn PP, et al. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology. 2010;74(5):366–371. doi: 10.1212/WNL.0b013e3181ccc732. [DOI] [PubMed] [Google Scholar]
- 214.Naumann M, Peikert K, Gunther R, van der Kooi AJ, Aronica E, Hubers A, Danel V, Corcia P, Pan-Montojo F, Cirak S, et al. Phenotypes and malignancy risk of different FUS mutations in genetic amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2019;6(12):2384–2394. doi: 10.1002/acn3.50930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Shang Y, Huang EJ. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 1647;2016:65–78. doi: 10.1016/j.brainres.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.An H, Skelt L, Notaro A, Highley JR, Fox AH, La Bella V, Buchman VL, Shelkovnikova TA. ALS-linked FUS mutations confer loss and gain of function in the nucleus by promoting excessive formation of dysfunctional paraspeckles. Acta Neuropathol Commun. 2019;7(1):7. doi: 10.1186/s40478-019-0658-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Murakami T, Qamar S, Lin JQ, Schierle GS, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FT, Michel CH, et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron. 2015;88(4):678–690. doi: 10.1016/j.neuron.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Marrone L, Drexler HCA, Wang J, Tripathi P, Distler T, Heisterkamp P, Anderson EN, Kour S, Moraiti A, Maharana S, et al. FUS pathology in ALS is linked to alterations in multiple ALS-associated proteins and rescued by drugs stimulating autophagy. Acta Neuropathol. 2019;138(1):67–84. doi: 10.1007/s00401-019-01998-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Rossaert E, Pollari E, Jaspers T, Van Helleputte L, Jarpe M, Van Damme P, De Bock K, Moisse M, Van Den Bosch L. Restoration of histone acetylation ameliorates disease and metabolic abnormalities in a FUS mouse model. Acta Neuropathol Commun. 2019;7(1):107. doi: 10.1186/s40478-019-0750-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Guven G, Lohmann E, Bras J, Gibbs JR, Gurvit H, Bilgic B, Hanagasi H, Rizzu P, Heutink P, Emre M, et al. Mutation Frequency of the Major Frontotemporal Dementia Genes, MAPT, GRN and C9ORF72 in a Turkish Cohort of Dementia Patients. PLoS One. 2016;11(9):e0162592. doi: 10.1371/journal.pone.0162592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Rainero I, Rubino E, Michelerio A, D'Agata F, Gentile S, Pinessi L. Recent advances in the molecular genetics of frontotemporal lobar degeneration. Funct Neurol. 2017;32(1):7–16. doi: 10.11138/FNeur/2017.32.1.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wils H, Kleinberger G, Pereson S, Janssens J, Capell A, Van Dam D, Cuijt I, Joris G, De Deyn PP, Haass C, et al. Cellular ageing, increased mortality and FTLD-TDP-associated neuropathology in progranulin knockout mice. J Pathol. 2012;228(1):67–76. doi: 10.1002/path.4043. [DOI] [PubMed] [Google Scholar]
- 223.Gass J, Prudencio M, Stetler C, Petrucelli L. Progranulin: an emerging target for FTLD therapies. Brain Res. 2012;1462:118–128. doi: 10.1016/j.brainres.2012.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Gass J, Lee WC, Cook C, Finch N, Stetler C, Jansen-West K, Lewis J, Link CD, Rademakers R, Nykjaer A, Petrucelli L. Progranulin regulates neuronal outgrowth independent of sortilin. Mol Neurodegener. 2012;7:33. doi: 10.1186/1750-1326-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Arrant AE, Onyilo VC, Unger DE, Roberson ED. Progranulin Gene Therapy Improves Lysosomal Dysfunction and Microglial Pathology Associated with Frontotemporal Dementia and Neuronal Ceroid Lipofuscinosis. J Neurosci. 2018;38(9):2341–2358. doi: 10.1523/JNEUROSCI.3081-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Prudencio M, Jansen-West KR, Lee WC, Gendron TF, Zhang YJ, Xu YF, Gass J, Stuani C, Stetler C, Rademakers R, et al. Misregulation of human sortilin splicing leads to the generation of a nonfunctional progranulin receptor. Proc Natl Acad Sci U S A. 2012;109(52):21510–21515. doi: 10.1073/pnas.1211577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Mohagheghi F, Prudencio M, Stuani C, Cook C, Jansen-West K, Dickson DW, Petrucelli L, Buratti E. TDP-43 functions within a network of hnRNP proteins to inhibit the production of a truncated human SORT1 receptor. Hum Mol Genet. 2016;25(3):534–545. doi: 10.1093/hmg/ddv491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Boxer AL, Gold M, Feldman H, Boeve BF, Dickinson SL, Fillit H, Ho C, Paul R, Pearlman R, Sutherland M, et al. New directions in clinical trials for frontotemporal lobar degeneration: Methods and outcome measures. Alzheimers Dement. 2020;16(1):131–143. doi: 10.1016/j.jalz.2019.06.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017;88(7):540–549. doi: 10.1136/jnnp-2016-315018. [DOI] [PubMed] [Google Scholar]
- 230.Yanagi KS, Wu Z, Amaya J, Chapkis N, Duffy AM, Hajdarovic KH, Held A, Mathur AD, Russo K, Ryan VH, et al. Meta-analysis of Genetic Modifiers Reveals Candidate Dysregulated Pathways in Amyotrophic Lateral Sclerosis. Neuroscience. 2019;396:A3–A20. doi: 10.1016/j.neuroscience.2018.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.FDA . Book Amyotrophic Lateral Sclerosis: Developing Drugs for Treatment Guidance for Industry (Editor ed.^eds.) 2019. Amyotrophic Lateral Sclerosis: Developing Drugs for Treatment Guidance for Industry. [Google Scholar]
- 232.Klim JR, Vance C, Scotter EL. Antisense oligonucleotide therapies for Amyotrophic Lateral Sclerosis: Existing and emerging targets. Int J Biochem Cell Biol. 2019;110:149–153. doi: 10.1016/j.biocel.2019.03.009. [DOI] [PubMed] [Google Scholar]
- 233.Colella P, Ronzitti G, Mingozzi F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. In: Book Emerging Issues in AAV-Mediated In Vivo Gene Therapy (Editor ed.^eds.), vol. 8: Cell Press; 2018. p. 87–104. City. [DOI] [PMC free article] [PubMed]
- 234.Schoch KM, Miller TM. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron. 2017;94(1097-4199 (Electronic)):1056–1070. doi: 10.1016/j.neuron.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary Figure S1. Physical interactions of the genes implicated in the pathogenesis of ALS and FTD. Interactions by binding among the genes shown in Fig. 1 generated by using the online web-portal Genemania (https://http://genemania.org/), gene multiple association network integration algorithm. This analysis shows the complex network of interactions existing among all the genes implicated in the pathologic processes underlying the origin and development of ALS and FTD.
Additional file 2: Supplementary Figure S2. Co-expression of the genes implicated in the pathogenesis of ALS and FTD. Co-expression networks of the genes shown in Fig. 1 generated by using the online web-portal Genemania (https://http://genemania.org/).
Additional file 3: Supplementary Figure S3. Common pathways of the genes implicated in the pathogenesis of ALS and FTD. Shared pathways among the genes shown in Fig. 1 generated by using the online web-portal Genemania (https://http://genemania.org/).
Additional file 4: Supplementary Figure S4. Co-localization of the genes implicated in the pathogenesis of ALS and FTD. Co-localization of the genes shown in Fig. 1 generated by using the online web-portal Genemania (https://http://genemania.org/).