

Abstract
Neuronal proteostasis is maintained by the dynamic integration of different processes that regulate the synthesis, folding, quality control, and localization of proteins. The endoplasmic reticulum (ER) serves as a fundamental pillar of the proteostasis network, and is emerging as a key compartment to sustain normal brain function. The unfolded protein response (UPR), the main mechanism that copes with ER stress, plays a central role in the quality control of many ion channels and receptors, in addition to crosstalk with signaling pathways that regulate connectivity, synapse formation, and neuronal plasticity. We provide here an overview of recent advances in the involvement of the UPR in maintaining neuronal proteostasis, and discuss its emerging role in brain development, neuronal physiology, and behavior, as well as the implications for neurodegenerative diseases involving cognitive decline.
Regulation of Protein Synthesis in Neuronal Communication
The ability of neurons to modulate the strength of their connections underlies the storage of new information, which is essential for survival and central to adaptive behavior. Two major forms of activity-dependent changes (see Glossary) in synaptic function have been described in the brain: long-term potentiation (LTP) and long-term depression (LTD), which refer to long-lasting increases and decreases in the efficacy of synaptic connection, respectively [1]. Both forms of synaptic plasticity require tightly regulated synthesis of key proteins and robust quality control mechanisms [2].
Regulation of protein synthesis in neurons is key to maintaining proteostasis [3]. During LTP, high-frequency stimulation leads to the synthesis of new proteins required either for the formation of new synapses or for strengthening of existing ones. Conversely, during LTD a different subset of proteins is synthesized which promote the weakening of pre-existing synapses. Protein synthesis is required not only for bidirectional control of activity-dependent changes in synaptic function [2] but also for other changes in the brain [2,4].
To ensure that ‘plasticity-related proteins’ are functional, each step in their synthesis must be tightly regulated. These steps, which are crucial for normal neuronal physiology and function, include mRNA translation, protein folding, protein maturation and secretion from the ER, and trafficking to the final destination [5]. Not surprisingly, alterations in proteostasis have been associated with a variety of cognitive disorders [2] and neurodegenerative diseases [6]. While various molecular mechanisms regulate neuronal proteostasis, in this review we focus on the emerging role of the unfolded protein response (UPR) pathway - a key homeostatic mechanism that is responsible for buffering cellular stress caused by an overabundance of misfolded proteins in the ER. We also overview the roles of the UPR in brain development, neuronal function, and disease. Moreover, we specifically highlight areas in which further research will be necessary to better understand the involvement of the UPR in neuronal function, and discuss potential therapeutic approaches for cognitive disorders in which neuronal ER proteostasis is dysfunctional.
Control of ER Proteostasis: Signaling Mechanisms and Function
Proteostasis requires the dynamic coordination of all processes underlying the maintenance of a functional proteome and prevention of abnormal aggregation (Figure 1) [3]. The ER is the largest intracellular organelle that serves as a signaling platform to mediate the synthesis and folding of ~30% of the proteome in eukaryotic cells, including all plasma membrane channels and receptors important for synaptic function. Dozens of chaperones, foldases, cofactors, and processing enzymes are expressed in the ER to assist protein folding, maturation, quality control, and degradation. Protein folding in the ER has a relatively high degree of failure, where ~10% of the total cargo does not reach its final destination, and are degraded through the ER-associated degradation (ERAD) pathway. In fact, many proteins, particularly those with multiple transmembrane regions, are likely to spontaneously misfold and aggregate during maturation, and only a small fraction overcome ER protein quality control mechanisms [3].
Figure 1. The Proteostasis Network.
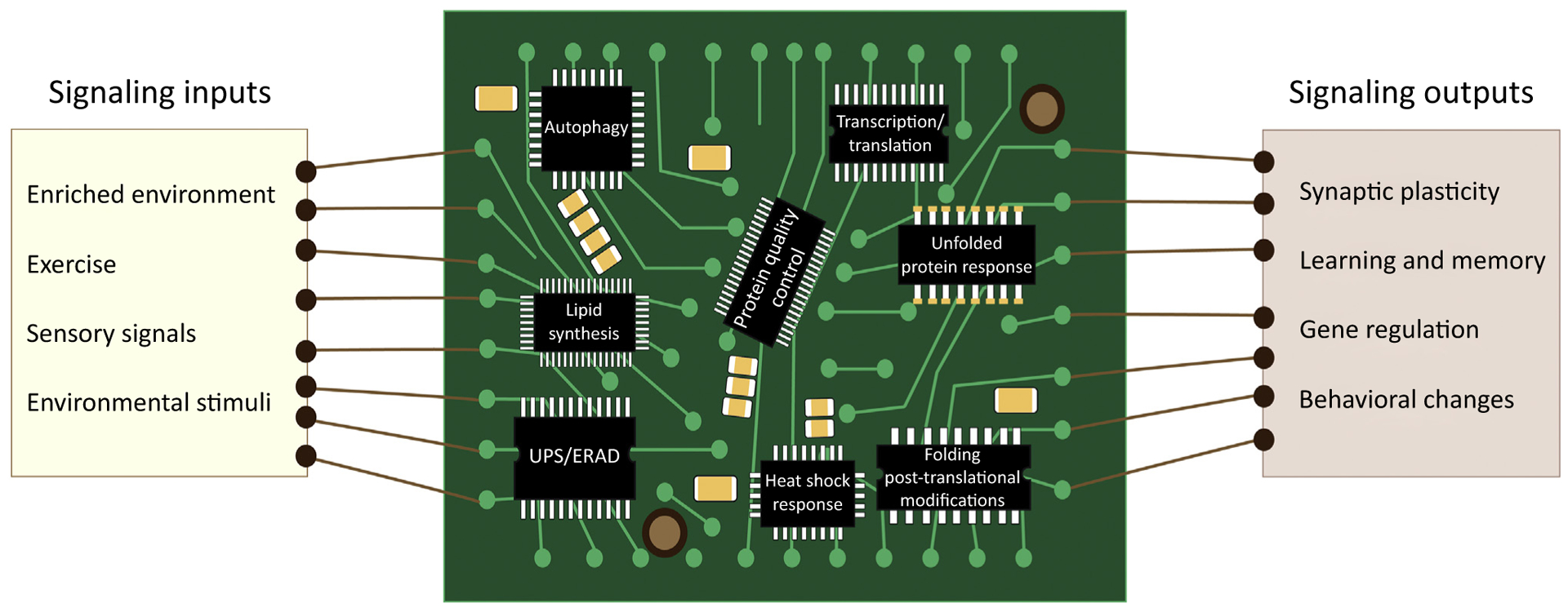
Several signaling inputs such as environmental enrichment, exercise, and various sensory signals induce changes in the brain proteostasis network, where the healthy brain adapts to these proteostatic perturbations, and facilitate resulting changes in gene expression that induce synaptic plasticity, learning, memory, and behavioral changes. The proteostasis network includes the autophagy pathway, the endoplasmic reticulum (ER)-associated degradation machinery (ERAD), the unfolded protein response (UPR), the heat shock response (HSR), the ubiquitin-proteasome system (UPS), and mechanisms to improve protein quality control, transcription, translation, folding, post-translational modifications, lipid synthesis, chaperones, and foldases to re-establish homeostasis and maintain neuronal cell function.
Proteostasis imbalance in the ER (known as ER stress) engages the UPR, an interconnected signaling network that transduces information about the protein folding status from the ER lumen to the cytosol and nucleus to initiate a stress response that restores cellular homeostasis [7]. The UPR is initiated following the activation of three major type I ER transmembrane proteins known as inositol-requiring enzyme 1α (here termed IRE1) and 1β, PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6) α and β [7]. Induction of the UPR leads to upregulation of target genes involved in protein folding, ERAD, autophagy, amino acid metabolism, and vesicular trafficking, among others [8]. Collectively, UPR signaling results in the inhibition of global protein synthesis, increase in the degradation of misfolded proteins, and improved folding efficiency, thereby reducing the overall burden of misfolded proteins. If the UPR is unable to restore cellular proteostasis, apoptotic pathways are triggered and damaged cells are eliminated (Figure 2) [9].
Figure 2. The Endoplasmic Reticulum (ER) Proteostasis Network.
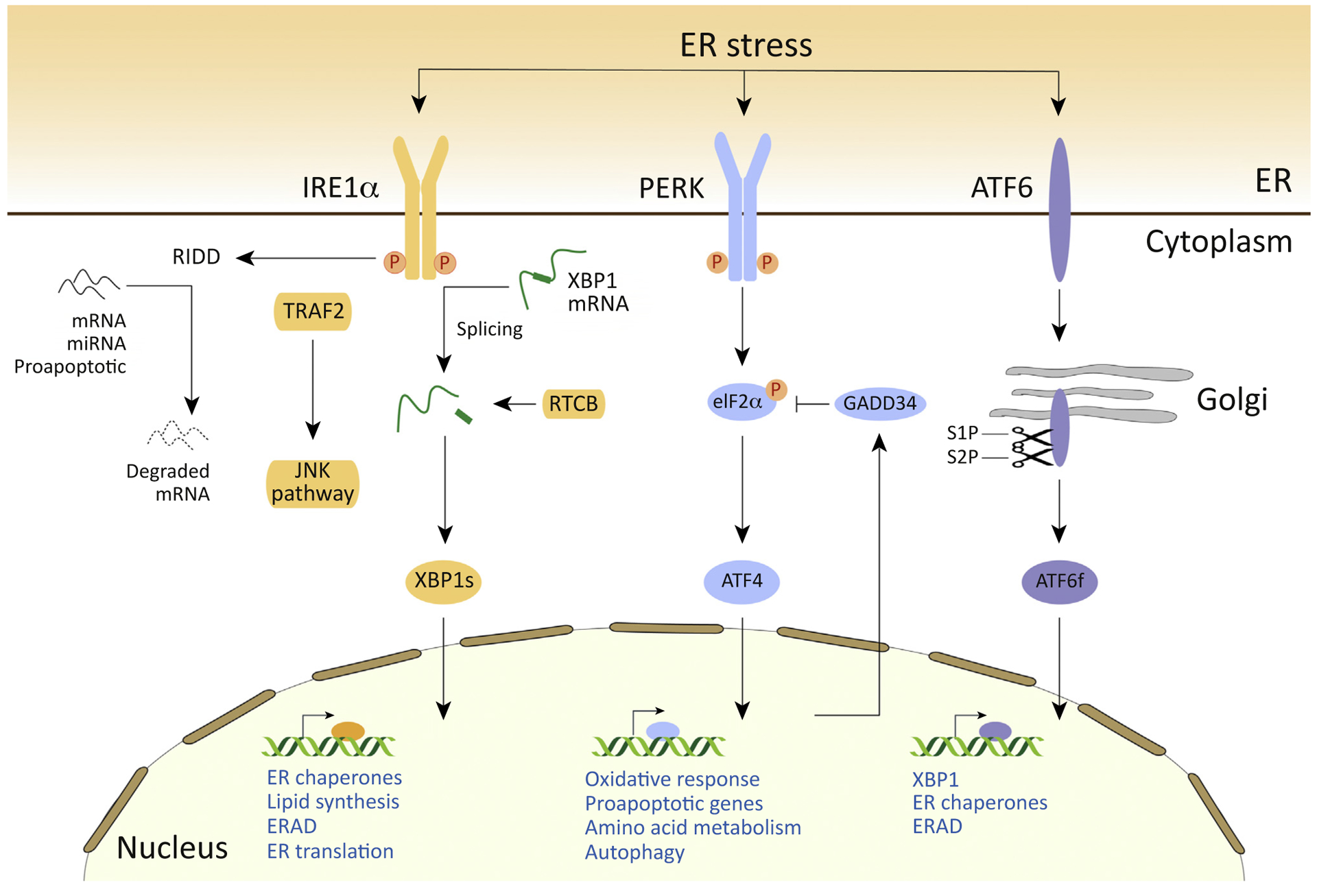
The Unfolded protein response (UPR): misfolded protein accumulation in the ER activates the UPR sensors inositol-requiring enzyme 1α (IRE1), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6). Upon ER stress, ATF6 is transported to the Golgi apparatus, where it is cleaved by site 1 protease (S1P) and S2P, releasing the cytosolic ATF6 fragment (ATF6f) which operates as a transcription factor. ATF6f induces genes required for ER-associated protein degradation (ERAD) and regulates X-box binding protein 1 (XBP1) mRNA levels. ER stress also activates PERK, which phosphorylates (P) eukaryotic translation initiation factor 2α (eIF2α), which in turn inhibits global protein translation, with the exception of some mRNAs including ATF4. ATF4 induces the expression of ER chaperones, genes related to autophagy, redox control, and amino acid metabolism. ATF4 also controls genes related to apoptosis, including C/EBP-homologous protein (CHOP). Active IRE1 generates the splicing of mRNA encoding XBP1 in a reaction that is completed by the RTCB ligase, leading to the expression of an active transcription factor. Spliced XBP1 (XBP1s) upregulates ER chaperones, genes involved in the ERAD pathway, and lipid synthesis. In addition, IRE1 is associated with tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2) and induces activation of c-Jun N-terminal kinase (JNK), apoptosis signal-regulating kinase (ASK1), and nuclear factor κB (NF-κB), thereby modulating autophagy and apoptosis. IRE1 endoRNase activity also induces a process known as regulated IRE1-dependent mRNA decay (RIDD) that affects different pathways, including those involved in lipid biosynthesis and apoptosis.
How do proteins such as IRE1, ATF6, and PERK sense the presence of misfolded proteins in the ER lumen? The ER chaperone binding immunoglobulin protein (BiP/Grp78) is proposed to modulate this process. Under basal conditions, BiP interacts with the luminal domain of PERK and IRE1, maintaining the transmembrane proteins in an inactive monomeric state, whereas its association with ATF6 masks a translocation signal to the Golgi apparatus. Under ER stress BiP is recruited by misfolded proteins, thereby releasing UPR transducers to allow their activation [10]. In addition, IRE1 may directly bind misfolded proteins to engage downstream signaling events [11].
IRE1
IRE1 activation is characterized by its dimerization and trans-autophosphorylation [7]. In mammalian cells, active IRE1 splices out a 26 nt intron from mRNA encoding X-box binding protein 1 (XBP1), shifting the reading frame and resulting in the rapid expression of a stable and active transcription factor, XBP1s [7], which upregulates the expression of a broad spectrum of target genes to restore global proteostasis [12,13]. In addition, IRE1 controls the stability of different types of RNAs through direct degradation by a process called ‘regulated IRE1-dependent decay’ (RIDD) [14]. RIDD contributes to reducing stress levels by degrading ER-localized mRNAs, but also modulates processes such as inflammation, cell migration, and apoptosis [14,15]. Moreover, IRE1 interacts with adapter proteins to mediate signaling crosstalk with stress pathways including cJUN amino-terminal kinase (JNK) and nuclear factor κB (NF-κB) [16]. Overall, IRE1 can be viewed as a rheostat that integrates information about the intensity and duration of the stress stimuli in a highly regulated manner, constituting a signaling platform referred to as the UPRosome [17].
ATF6
Under ER stress, ATF6 is transported to the Golgi apparatus, where it is cleaved by site 1 protease (S1P) and S2P, releasing the cytosolic ATF6 fragment (ATF6f). ATF6f is a transcription factor that enhances the transcription of XBP1 [18] and induces the expression of genes required for ERAD [19]. In addition, XBP1s and ATF6f heterodimerize and drive a specific transcriptional program [20].
PERK
The ER kinase PERK oligomerizes in response to ER stress, and phosphorylates the α-subunit of eukaryotic initiation factor 2 (eIF2α) at serine 51 [7]. Phosphorylation of eIF2α inhibits global translation while upregulating the translation of specific mRNAs containing upstream open reading frames (uORFs) in their 5′ untranslated regions. One such mRNA, Atf4, encodes a transcription factor that upregulates the expression of ER chaperones and genes involved in redox control and amino acid metabolism [11,21]. ATF4 also controls the expression of genes related to apoptosis, including C/EBP-homologous protein (CHOP, also known as GADD153) and GADD34 [22]. GADD34, whose expression is induced by ER stress, is a cofactor of a phosphatase complex that dephosphorylates eIF2α [23], representing an important feedforward loop to shut down PERK signaling.
Several studies have investigated the contribution of the UPR to organ physiology, indicating an essential role in sustaining the function of highly secretory cells, energy metabolism, and inflammation [24]. In the context of the central nervous system (CNS), most studies have focused on defining the impact of ER stress and the UPR to neurodegenerative diseases (reviewed elsewhere; e.g., [6]); however, little is known about the possible activity of the UPR in neurophysiology and glial cells. Interestingly, studies in models of spinal cord injury [25] and multiple sclerosis [26] have shown that oligodendrocytes undergo damaging ER stress more readily, consistent with their basal stress due to a high rate of myelin synthesis. Overall, the UPR represents a major mechanism to overcome ER stress and maintain cell functionality in physiology and disease.
The UPR and the Physiology of the Nervous System
Recent studies suggest that the UPR has important physiological functions in the CNS. As a regulator of the secretory pathway, components of the UPR have been shown to mediate the maturation and expression of different synaptic proteins, thereby impacting on brain development. Moreover, UPR signaling components may have a relevant role in neuronal plasticity that is independent of their classical functions in the ER stress pathway. Finally, recent reports have uncovered crosstalk between components of the UPR and canonical pathways involved in neuronal plasticity and behavior. In this section we discuss key evidence supporting an emerging role of the UPR in the physiology and function of the CNS at basal levels.
Brain Development
The UPR is activated during brain development in mice, C. elegans, and flies (reviewed in [27,28]). It was recently reported that PERK expression is important in the regulation of neurogenesis, as well as for the generation of intermediate progenitors and projection neurons in different cortical layers, thereby impacting on overall brain architecture [29]. Moreover, this study uncovered that PERK signaling favors direct neurogenesis, and disruption of this pathway results in microcephaly [29]. In agreement, mutant mice lacking the essential ER chaperone BiP also display disorganized cerebral cortex and cerebellar lamination, and die after birth [30]. Mutations in filamin A are the underlying cause of periventricular heterotopia, a disease condition driven by altered neuronal migration during brain development [31]. We recently reported a physical interaction between IRE1 and Filamin A [32]. Importantly, targeting IRE1 expression in the developing brain cortex phenocopy Filamin A deficiency, associated with alterations in actin cytoskeleton dynamics and cell movement [31,32]. Interestingly, another study also indicated that PERK interacts with Filamin A [33], however the possible contribution of this protein complex to brain cortex development was not defined.
Although potential contributions of ATF6 in brain development have not yet been deeply studied, analysis of gene expression patterns suggests that it is activated during development [27,34]. PERK and IRE1 have been found to be involved in neuronal differentiation of mouse stem cells in vitro [35], and pharmacological induction of ER stress activates neuronal differentiation in stem cells [35]. An inverse correlation between ATF4 levels and neurogenesis was reported that was possibly linked with cell-cycle control [36]. Another study defined the function of mesencephalic astrocyte-derived neurotrophic factor (MANF), an ER-resident protein, in brain development. Genetic ablation of MANF expression resulted in retarded neuronal migration and impaired neurite outgrowth during cortical development, and these were associated with modulation of the UPR [37]. These results suggest an important role of the UPR in brain development.
Neuronal Differentiation, Connectivity, and Gene Expression Control
Dendritic spine formation and the insertion of ionotropic transmembrane receptors at synapses are some of the processes that underlie synaptic plasticity [38,39]. Studies in C. elegans demonstrated the involvement of the IRE1/XBP1 pathway in the transport of glutamate receptors subunits GLR-1, GLR-2, and GLR-5 to the plasma membrane [40]. Mutant worms lacking Ire1 display impaired dendritic morphogenesis, specifically in neurons with complex dendritic arbors, whereas axons and neurons with fewer dendrites were not affected [41]. Studies in primary neuron cultures show that induction of ER stress results in aberrant neuronal differentiation and an attenuation of dendritic outgrowth [42], whereas primary neurons from mice lacking XBP1 show decreased axonal length and dendritic growth in neurons stimulated with brain-derived neurotrophic factor (BDNF) [43]. Overall these data suggest that IRE1 signaling regulates neuronal differentiation and possibly synapse formation (Figure 3).
Figure 3. Novel Outputs of the Unfolded Protein Response (UPR) Related to Learning and Memory Processes.
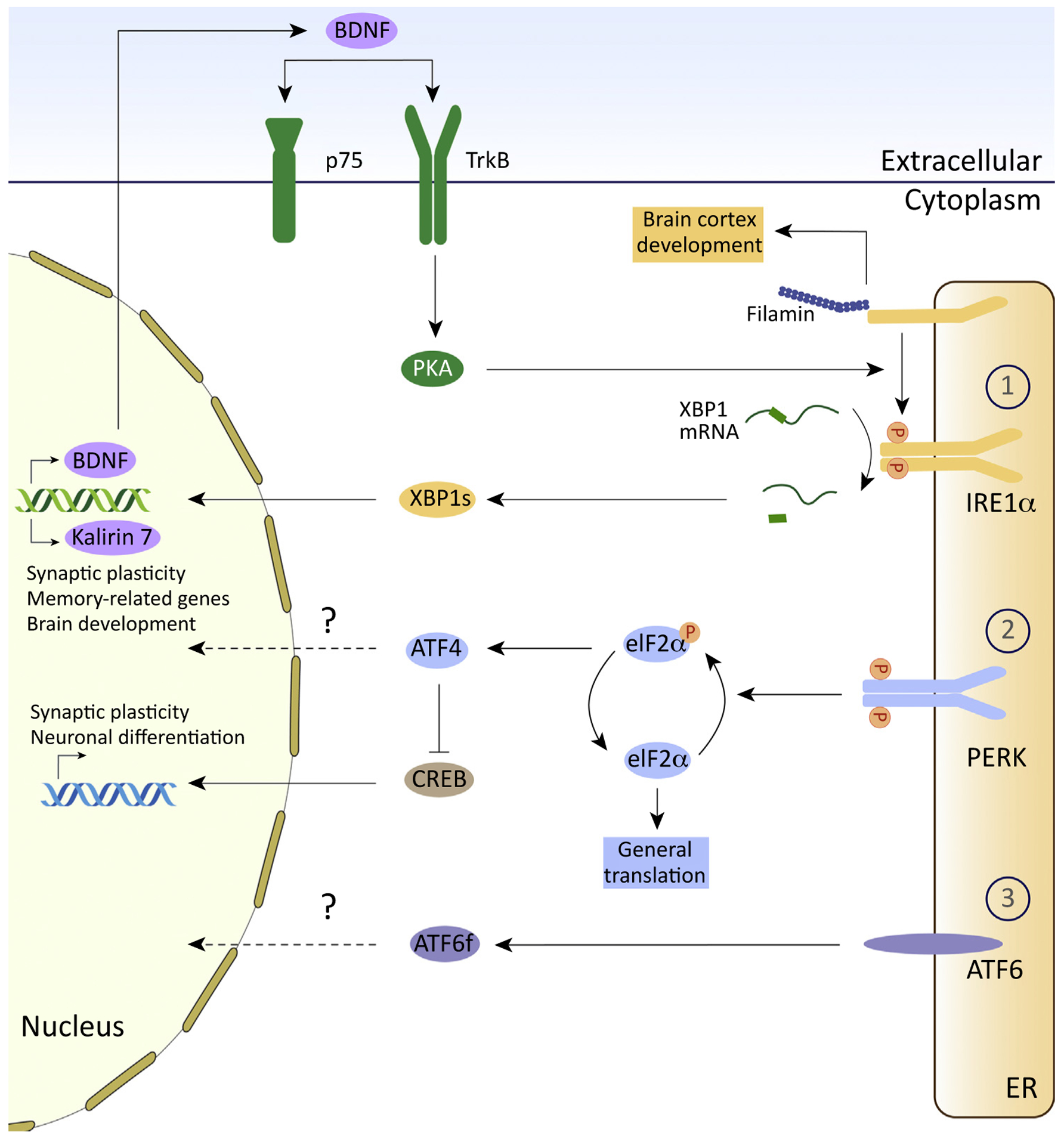
Endoplasmic reticulum (ER) stress (right) leads to activation of three branches of the UPR. (1) The inositol-requiring enzyme 1α (IRE1a) branch, (2) the PERK pathway, and (3) the ATF6 pathway. (1) Activation of IRE1a via dimerization and autophosphorylation leads to splicing of X-box binding protein 1 (XBP1) mRNA, which is then locally translated within dendrites and transported into the nucleus. Once in the nucleus, XBP1s activates the transcription of genes encoding brain-derived neurotrophic factor (BDNF), kalirin 7, and other functions involved in neuronal plasticity. BDNF can then localize extracellularly where it activates the TrkB receptor, resulting in activation of PKA and IRE1α/XBP1 signaling. Thus, a positive feedback loop maintains the activity of IRE1α/XBP1s pathway and promotes the expression of target genes involved in synaptic plasticity and memory. IRE1α also physically interacts with filamin A that regulates the development of the cerebral cortex. (2) Protein kinase R (PKR)-like ER kinase (PERK) is activated during ER stress. PERK phosphorylates (P) the α-subunit of eukaryotic translation initiation factor 2 (eIF2α) which plays a key role in learning and memory. Phosphorylation of eIF2α leads to global attenuation of translation and selective expression of ATF4. Increased expression of ATF4 represses cAMP-responsive element-binding protein (CREB) and inhibits the expression of genes involved in synaptic plasticity and memory. (3) ER stress can also activate the ATF6 pathway, but whether ATF6 signaling in the brain is involved in synaptic plasticity and memory processes is not known.
The function of XBP1s in the brain may be related to its ability to regulate distinct sets of target genes involved in neuronal physiology. Gene expression analysis of XBP1-deficient neurons revealed its role in establishing gene expression programs downstream of BDNF that are associated with the control of inhibitory neurotransmitter GABA, including genes encoding calbindin 1, somatostatin, and neuropeptide Y [44]. In the brain XBP1s may have additional effects because it directly transactivates BDNF expression in neurons [45]. BDNF is a master regulator of several physiological processes in the adult brain that impacts dendritic branching, synaptic plasticity, and LTP - three hallmarks of memory formation and normal cognitive processes [46]. Accordingly, XBP1s belongs to the ATF/CREB superfamily of transcription factors [45] which are known to regulate gene expression control downstream of BDNF receptors. A recent report identified the activation of PKA as one of the signaling events engaging IRE1 downstream of the BDNF receptor [47]. These studies suggested the occurrence of a feedback loop where BDNF signaling triggers the activation of IRE1, which in turn upregulates BDNF through XBP1s.
XBP1s has additional target genes that are relevant for neurophysiology. For example, XBP1s transactivate the promoter of the gene encoding karilin 7 [48], a factor with a central role in spine formation/maintenance, synaptic function, and behavior [48,49]. We also reported that XBP1 regulates the levels of Kinesin17 (Kif17) [45], a motor protein involved in the trafficking of NMDA receptors to the plasma membrane with essential roles to neuronal plasticity [50]. The ER folding network has been also linked to the maintenance of neuronal connectivity at the level of neuromuscular junctions. When essential ER chaperones are genetically ablated, the structure and function of the neuromuscular junction are drastically altered, impacting on motor control [51–54]. Moreover, XBP1s expression in peripheral nerves accelerates axon regeneration and connectivity after damage [55]. Taken together, these findings suggest that IRE1 signaling contributes to neuronal differentiation and function through the control of the expression of neuron-specific genes unrelated to ER stress (Figure 3).
Synaptic Plasticity and Memory Storage
Several exogenous inputs, such as physical exercise and environmental enrichment, are known to improve learning behavior, correlating with the upregulation of XBP1s [56–58]. Behavioral stress in rats induces splicing of Xbp1 mRNA in the hippocampus [59], whereas depolarization in human motoneurons can activate IRE1/XBP1 signaling [60]. The UPR, and more specifically eIF2α phosphorylation, has been extensively studied in synaptic plasticity. Mice with reduced eIF2α phosphorylation show enhanced LTP but impaired protein synthesis-dependent LTD. By contrast, chemical/genetic or pharmacological induction of eIF2α phosphorylation elicits LTD, but blocks protein synthesis-mediated LTP [61–63]. Thus, eIF2α phosphorylation bidirectionally regulates the two major forms of synaptic plasticity in the brain. More importantly, reducing eIF2α phosphorylation - by replacing the eIF2α phosphorylation site serine 51 with alanine (eIF2αS/A mice) or by genetically deleting the eIF2α kinases GCN2, PKR, or PERK - enhances long-term memory formation [61,64–67]. Unlike eIF2αS/A mice or mice lacking GCN2 or PKR, deletion of PERK in the forebrain causes repetitive and perseverant behaviors [68]. Thus, it is possible that PERK regulates perseverant behaviors by other targets in an eIF2α phosphorylation-independent fashion.
The role of ATF4 (also called CREB2) - a downstream target of eIF2α - in learning and memory formation remains somewhat controversial [61,64,68–70]. ATF4 negatively regulates CREB-driven gene expression [70,71]. Deleting GCN2 or diminishing the levels of eIF2α phosphorylation reduces ATF4 expression and promotes memory formation, associated with changes in synaptic function [61,64]. Similarly, PERK deficiency in the prefrontal cortex decreases ATF4 expression in the insular cortex, associated with enhanced learning and memory [66]. Accordingly, blocking ATF4 expression with a dominant negative in the forebrain enhances CREB-driven gene expression and facilitates LTP and the subsequent formation of long-term memory [70]. Interestingly, inhibition of the ATF4 ortholog ApCREB2 in Aplysia, a CREB inhibitor, enhances a form of long-term synaptic function called long-term facilitation [71]. By contrast, a recent report shows that downregulation of ATF4 using short hairpin RNA (shRNA) in the hippocampus blocks LTP and spatial memory [69]. These discrepancies may be explained by the fact that some strategies to target ATF4 fail to completely abrogate its function in the brain. Using mice in which ATF4 could be conditionally deleted [72] in specific brain regions (or neuronal types) might help to clarify the role of ATF4 in memory formation.
The physiological role of IRE1/XBP1 signaling in the nervous system remained elusive until recently. The development of a conditional mouse model for XBP1 in the brain [73] allowed the uncovering of a previously unanticipated function for XBP1s in memory formation and synaptic plasticity (Figure 3) [45]. This unexpected involvement of XBP1 in cognition was mapped to the regulation of BDNF levels in the hippocampus, thereby impacting on synaptic plasticity. Moreover, the basal performance of mice in learning and memory tasks was improved using gain-of-function studies in XBP1s transgenic mice or through the local expression of XBP1s in the hippocampus of adult animals using gene therapy [45]. Importantly, heterozygous XBP1 mice did not exhibit a strong phenotype in several behavioral tests [74], suggesting that a full deficiency is necessary to produce behavioral deficits. In summary, recent evidence place the activity of IRE1/XBP1 and eIF2α/ATF4 signaling branches as relevant regulators of neuronal plasticity and behavior.
Neuronal UPR and Energy Control
Recent evidence also suggests that the neuronal UPR may influence global organismal physiology. For instance, a study in the context of obesity revealed that XBP1 deficiency in the hypothalamus augments leptin resistance by blocking leptin receptor signaling in mice [75]. In addition, expression of XBP1 in pro-opiomelanocortin (POMC) neurons of the hypothalamus impacts on the control of whole-body metabolism, a discovery that expanded the implications of neuronal UPR in global physiology [76]. Notably, the effects of expressing XBP1s in POMC neurons were mapped to the activation of the UPR in peripheral organs in a cell-nonautono-mous manner that fine-tunes liver physiology [76]. However, another report suggested that IRE1 deficiency in POMC neurons leads to a resistance to high-fat-induced obesity as well as insulin resistance [77]. These animals developed higher energy expenditure, correlating with increased thermogenesis in brown adipose tissue. Targeting IRE1 in POMC neurons also resulted in increased levels of α-melanocyte-stimulating hormone in the hypothalamus [77]. In sharp contrast, a recent report suggested a completely opposite phenotype of IRE1 deficiency in POMC neurons [78].
ER proteostasis control may also affect other relevant functions of the nervous system that relate to general physiological processes. For example, treatment with the eIF2α phosphatase inhibitor salubrinal, which increases phosphorylation of eIF2α and prolongs ER stress, modified sleep behavior and hypothalamic activity [79]. In addition, the regulation of the circadian clock by light is associated with the establishment of XBP1s-dependent gene expression patterns in the pineal gland [80]. Thus, neuronal UPR may contribute to integrating global animal physiology through central control.
Overall, all these studies suggest a new concept where the UPR has emerging functions in the nervous system beyond ER stress and disease, impacting on the regulation and function of the CNS at basal levels.
Proteostasis Alterations and Synaptic Dysfunction in Disease
Aging is a major risk factor for the development of neurodegenerative diseases, and disrupted proteostasis control has been described as one of the hallmarks of aging, highlighting ER stress as a relevant factor [81,82]. The causal role of ER stress and the UPR in neurodegenerative disease has been extensively studied by analyzing its impact on abnormal protein aggregation and neuronal death, as reviewed elsewhere [6,83,84]. In this section we focus only on studies linking the activity of the ER proteostasis network to synaptic function in the context of brain diseases.
ER stress in Alzheimer’s disease (AD) has been proposed to repress synthesis of synaptic proteins via increased phosphorylation of eIF2α [83]. Deleting PERK or GCN2 decreases eIF2α phosphorylation in AD mice and restores synaptic plasticity and memory formation [68,85]. Moreover, an increase in PKR phosphorylation has been reported in brain samples from AD mice [86], as well as in cerebrospinal fluid (CSF) samples from human patients [87]. Functional studies demonstrated that deletion of PKR restores the cognitive decline associated with AD in mice [88,89]. Traumatic brain injury also increases eIF2α phosphorylation in the brain [90]. Of note, administration of a small-molecule ISRIB (integrated stress response inhibitor), which blocks the downstream translational events mediated by eIF2α phosphorylation [91,92], prevents cognitive decline in traumatic brain injury [93], but not in AD [94]. ISRIB administration was also shown to improve basal learning and memory in mice [91].
Sustained eIF2α phosphorylation is also involved in prion-mediated neurodegeneration [95]. Indeed, repression of the eIF2α phosphorylation translational program using the PERK inhibitor GSK2606414 or treatment with ISRIB delayed disease progression after prion infection [96,97]. Mutations in EIF2AK3, the gene encoding PERK, have been suggested to be risk factor for developing tauopathies [98]. Notably, administration of PERK inhibitors to a model of frontotemporal dementia induced by Tau overexpression provided strong neuroprotection [99]. We also reported that administration of GSK2606414 to a Parkinson’s disease (PD) model protects dopaminergic neurons against degeneration and improves motor performance involving increased expression of synaptic proteins and improved dopamine levels [100]. The PERK-eIF2α pathway seems to be the main UPR pathway involved in prion replication and pathogenesis because ablation of XBP1 expression in the brain had no effect on prion-mediated degeneration [73].
Genetic studies, human tissue analyses, and work in animal models also implicate the IRE1/XBP1 pathway in AD [101]. Genetic studies in the Chinese population, for instance, indicated that the - 116C/G polymorphism in the XBP1 promoter, which is proposed to reduce XBP1 levels [102], is associated with a higher likelihood of developing AD [103]. Analysis of human AD tissue at different Braak stages indicated that IRE1 phosphorylation-a measure of its activation - is directly correlated with the severity of the disease [104]. In addition, IRE1 is hyperactivated in AD patients in most neurons containing neurofibrillary tangles [104]. In mouse models of AD, we provided functional studies using brain-specific deletion of IRE1 and demonstrated a strong neuroprotective effect mediated by decreasing amyloid plaques, in addition to ameliorated synaptic and cognitive dysfunction [104]. Another report showed that the overexpression of XBP1s in the hippocampus of AD mice improves synaptic function, spine density, LTP, and alleviates long term potentiation, and deficits in spatial memory [48]. Taken together, these results suggest that different outputs of the IRE1 pathway may have opposite effects on AD progression and amyloid β metabolism (Figure 3). Thus, altered UPR signaling underlies, at least in part, the memory and synaptic deficits associated with neurodegeneration.
Mutations in UPR regulatory elements have also been associated with psychiatric disorders [105]. The - 116C/G polymorphism in the XBP1 promoter was initially discovered as a risk factor for developing bipolar disorder [102], and was subsequently implicated in schizophrenia [106]. Another study, conducted in a Japanese population, implicated the - 116C/G polymorphism in personality alterations [107]. In addition, this polymorphism is associated with differential responses to lithium treatment in patients with bipolar disorder [108]. However, in populations of European descent, the - 116C/G polymorphism did not correlate with the incidence of psychiatric disorders. The PERK and ATF4 pathways have been also implicated in schizophrenia because their expression is reduced in frontal cortex of patients [68]. Interestingly, ISRIB administration to a model of neuropsychiatric disorders (CACNA1C-deficient mice) reversed the social deficits and elevated anxiety-like behaviors [109]. Finally, another report indicated that the adverse effects of post-traumatic stress disorder on the learning and memory capacity of an animal model are prevented by the administration of the PERK antagonist GSK2606414 [110].
Other studies in disease models also reinforce the idea that ER proteostasis is key to sustain neuronal connectivity and function. Rare genetic variants in PDI and ERp57 also increase the risk of developing amyotrophic lateral sclerosis (ALS) [111–113] by altering neuronal connectivity at the level of neuromuscular junctions, as demonstrated by functional studies [54]. Genetic mutations in ATF6 have been associated with achromatopsia, a disorder characterized by color blindness, photophobia, nystagmus, and severely reduced visual acuity [114,115]. Taken together, these findings suggest a causal link between ER proteostasis and functional neuronal impairment, including in sensorimotor systems.
Overall, these studies added a new layer of complexity to the involvement of UPR in neuro-degeneration, suggesting that this pathway regulates multiple processes including (i) mitigation of ER stress and reduction of protein aggregation through canonical proteostasis effectors; (ii) cell death, by activating terminal apoptotic pathways; and (iii) modulation of synaptic function and connectivity.
Concluding Remarks and Future Perspectives
Much of the focus in research regarding the role of UPR in brain function has been placed on models of pathology [84,116]. However, recent evidence suggests that the ER proteostasis network is fundamental in maintaining neuronal physiology at the level of synaptic function, connectivity, and brain development [117,118], opening interesting directions for future research (see Outstanding Questions). An emerging concept suggests that UPR components also operate as signaling elements that crosstalk with relevant pathways involved in neuronal plasticity [119]. Studies in animals with disrupted brain expression of XBP1 have revealed selective changes in gene expression that differ from previous characterizations in other tissues [45]. How XBP1s drives specific gene expression patterns in the brain is not known. We speculate that this phenomenon may involve the formation of heterodimers with neuron-specific transcription factors and post-translational modifications as reported in other systems [119]. Recent reports show that key components of the UPR may also have ER stress-independent functions, as reported in the context of aging, cell differentiation, metabolic control, angiogenesis, and inflammation [16,119]. In addition, based on the known role of the UPR in sustaining the function of specialized secretory cells, we speculate that cells such as oligodendrocytes and Schwann cells, which produce high rates of myelin, may specifically depend on the activity of the UPR for optimal function.
Outstanding Questions.
How are distinct components of the UPR activated during CNS development? Are they involved in determining cell fate and cell differentiation?
Is the UPR necessary for inducing neurogenesis?
Does UPR activation dictate neuro-immune signaling?
How is the UPR activated in different cell types in the nervous system? How does this relate to neuronal stimulation and behavioral inputs?
The UPR propagates from the CNS to the whole organism through cell-non-autonomous mechanisms. Which molecules mediate this propagation? How many physiological processes are affected? Do these signals also propagate through neuronal circuits?
A better understanding of the UPR in pathology and normal brain function will require full dissection of the pathway in a cell type-specific manner. Of note, ablating dopamine transporter expression or treatment of dopaminergic neurons with dopamine results in UPR activation [120–122]. In addition, the inhibition of XBP1 expression in the substantia nigra in mice results in spontaneous increase in ER stress [123], suggesting that the UPR has a relevant function in sustaining the function of dopaminergic neurons at resting conditions. Importantly, studies in other organs also suggest that the UPR may impact on immune and inflammatory responses [124–126]. It remains to be determined whether the activity of the UPR in glial cells contributes to basal brain function and to disease stages.
From a therapeutic standpoint, it would be interesting to determine whether some of the promising genetic manipulations that block or activate UPR components in animal models can be recapitulated using pharmacological approaches (Table 1), and, if so, to determine their translational potential in human patients. Another important question is whether complete blockade of only one branch of the UPR (typically using overexpression or knockout animals) is more efficient in reverting pathology than partial modulation of all three branches. The use of a combination of inhibitors, or heterozygous mice for all three UPR branches, in models of disease will help to address this question.
Table 1.
Summary of Inhibitors of the PERK Pathway and Their Impact on CNS Function
| Drug | Molecular target/effect | Readout | Model/disease | Effect on the CNS | Refs. |
|---|---|---|---|---|---|
| GSK2606414 | PERK kinase domain/inhibitor | Inhibition of eIF2α phosphorylation | Mouse/prion disease | Increased neuroprotection | [97] |
| Mouse/frontotemporal dementia | Decreased neuronal loss and brain atrophy | [99] | |||
| Mouse/PD | Protection of nigral dopaminergic neurons | [100] | |||
| Rat/PTSDa model | Reversion of learning and memory defiicits | [110] | |||
| Guanabenz | Inhibition of GADD34/PP1 complex | Repression of translation, decreased protein misfolding overload | Mouse/ALS | Increased survival of mSOD1 model mice | [6] |
| Integrated stress response inhibitor (ISRIB) | Guanine nucleotide exchange factor eIF2B/inhibitor | Decreased ATF4 expression | Mouse/prion disease | Increased survival and decreased neuronal loss in the hippocampus | [96] |
| Mouse/wild type | Enhanced cognitive function | [91] | |||
| Mouse/traumatic brain injury | Reversed cognitive deficits | [93] | |||
| Mouse/AD | No effect | [94] | |||
| Mouse/neuropsychiatric disorder | Reversed social deficits | [109] | |||
| Salubrinal | eIF2α phosphatase complexes/inhibitor | Repression of translation, decreased protein misfolding overload | Mouse/prion disease | Decreased survival and increased neurotoxicity | [95] |
| Mouse/ALS | Decreased axon pathology and denervation | [6] | |||
| Rat and mouse/PD | Increased survival and decreased neurodegeneration | [6] | |||
| Sephin 1 | PPP1R15A eIF2α phosphatase/inhibitor | Repression of translation, decreased protein misfolding overload | Mouse/ALS | Decreased motoneuron degeneration | [6] |
PTSD, post-traumatic stress disorder.
Finally, the UPR is emerging as a key player in modulating the global capacity of the organism to cope with stress by integrating the systemic response at the whole-animal level that may originate from a neuronal compartment. For instance, recent studies in C. elegans, flies, and mice indicate that the neuronal UPR, and especially the IRE1/XBP1 pathway, is involved in maintaining homeostasis of peripheral organs and tissues through a cell-nonautonomous manner [76,127–129]. Precise mechanisms of global proteostasis control and the neuronal circuits mediating the propagation of UPR signaling between cells remain to be determined. Given that ER stress responses could have specific, and sometimes opposite, effects on neuronal function as well as on survival in disease states, understanding their normal physiological function will be crucial for predicting and mitigating possible side effects of targeting these pathways as we move forward with new approaches to treat brain diseases.
Highlights.
ER proteostasis is essential for maintaining neuronal physiology.
UPR signaling is involved in brain development, synaptic plasticity, and memory formation.
Aberrations in UPR signaling alter memory formation and behavior.
UPR activity in the central nervous system controls global physiology through cell-nonautonomous mechanisms.
Targeting the UPR in the context of neurodegenerative diseases may restore perturbations in proteostasis and neuronal function through various mechanisms.
Acknowledgments
This work was funded by US Office of Naval Research-Global (ONR-G) grant N62909-16-1-2003, Millennium Institute P09-015-F, FONDEF ID16I10223, US Air Force Office of Scientific Research FA9550-16-1-0384, CONICYT-Brazil 441921/2016-7, and a Leading House for the Latin American Region seed money grant (C.H.). We also thank FONDECYT 3150637 (GM). In addition, we are grateful for the support from FONDEF D11E1007, Muscular Dystrophy and FONDECYT 1180186 (C.H.), and for grants from the National Institutes of Health to M.C.M. (NIMH 096816, NINDS 076708, and the Sammons Foundation).
Glossary
- Activating transcription factor 6 (ATF6)
a transmembrane protein in the endoplasmic reticulum (ER) that senses unfolded proteins, and upon activation, acts as a transcription factor to facilitate the expression of ‘stress response’ proteins
- Activity-dependent changes
structural and functional plasticity of neuronal connections resulting from their activation
- Binding immunoglobulin protein (BiP)
an ER chaperone that facilitates the correct folding of newly synthesized proteins
- ER-associated protein degradation (ERAD)
a mechanism for degrading misfolded proteins in the ER via their ubiquitination and degradation by the proteasome
- ER stress
the accumulation of misfolded/unfolded proteins in the ER, resulting in activation of UPR stress sensors to adapt to stress or trigger apoptosis of irreversibly damaged cells
- Inositol-requiring enzyme (IRE1)
a conserved transmembrane protein in the ER that senses unfolded proteins and activates a signaling cascade to upregulate the expression of ‘stress response’ proteins
- Long-term depression (LTD)
a long-lasting decrease in synaptic strength resulting from low-frequency electrical stimulation or chemical activation of particular receptors
- Long-term potentiation (LTP)
a long-lasting (hours to days) increase in synaptic strength resulting from a brief high-frequency brief stimulus (for instance, 1 s stimulation at 100 Hertz)
- Neurodegenerative disease
a disease characterized by loss of neuronal structure and function that ultimately leads to neuronal cell death
- PKR-like ER kinase (PERK)
a transmembrane kinase in the ER that is activated in response to unfolded proteins. PERK phosphorylates eIF2α to attenuate global translation
- Proteostasis
a combination of the words protein and homeostasis. Refers to the concept of integrated biological pathways within cells that control the biogenesis, folding, trafficking, and degradation of proteins present within and outside the cell
- Spines
cytoskeletal protrusions on the dendrites that form synapses and receive input from other neurons
- Synapse
the junction between two neurons through which signal propagates from one neuron to the next
- Synaptic plasticity
the ability of synapses to strengthen or weaken over time in response to alterationsin their activity.
- Trafficking
the process by which synthesized proteins (and other cargos) are modified and packaged into vesicles for distribution to their target location
- Unfolded protein response (UPR)
a signal transduction pathway activated in response to the accumulation of unfolded or misfolded proteins in the ER lumen. The UPR facilitates the mitigation of protein folding stress or the elimination of non-functional cells by apoptosis
- X-box binding protein 1 (XBP1)
a transcription factor that is activated by IRE1-mediated splicing during ER stress. Spliced XBP1 facilitates the expression of ‘stress response’ proteins
References
- 1.Neves G et al. (2008) Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat. Rev. Neurosci 9, 65–75 [DOI] [PubMed] [Google Scholar]
- 2.Buffington SA et al. (2014) Translational control in synaptic plasticity and cognitive dysfunction. Annu. Rev. Neurosci 37, 17–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balch WE et al. (2008) Adapting proteostasis for disease intervention. Science 319, 916–919 [DOI] [PubMed] [Google Scholar]
- 4.Costa-Mattioli M et al. (2009) Translational control of long-lasting synaptic plasticity and memory. Neuron 61, 10–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sonenberg N and Hinnebusch AG (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hetz C and Saxena S (2017) ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol 13, 477–491 [DOI] [PubMed] [Google Scholar]
- 7.Walter P and Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 8.Wang M and Kaufman RJ (2016) Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326–335 [DOI] [PubMed] [Google Scholar]
- 9.Urra H et al. (2013) When ER stress reaches a dead end. Biochim. Biophys. Acta 1833, 3507–3517 [DOI] [PubMed] [Google Scholar]
- 10.Hetz C and Papa FR (2018) The unfolded protein response and cell fate control. Mol. Cell 69, 1–13 [DOI] [PubMed] [Google Scholar]
- 11.Hetz C et al. (2011) The unfolded protein response: integrating stress signals through the stress sensor IRE1α. Physiol. Rev 91, 1219–1243 [DOI] [PubMed] [Google Scholar]
- 12.Lee A-H et al. (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol 23, 7448–7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Acosta-Alvear D et al. (2007) XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 27, 53–66 [DOI] [PubMed] [Google Scholar]
- 14.Maurel M et al. (2014) Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci 39, 245–254 [DOI] [PubMed] [Google Scholar]
- 15.Moore K and Hollien J (2015) Ire1-mediated decay in mammalian cells relies on mRNA sequence, structure, and translational status. Mol. Biol. Cell 26, 2873–2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hetz C et al. (2015) Proteostasis control by the unfolded protein response. Nat. Cell Biol 17, 829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hetz C and Glimcher LH (2009) Fine-tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol. Cell 35, 551–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshida H et al. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto K et al. (2007) Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 13, 365–376 [DOI] [PubMed] [Google Scholar]
- 20.Shoulders MD et al. (2013) Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 3, 1279–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang M and Kaufman RJ (2014) The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 14, 581–597 [DOI] [PubMed] [Google Scholar]
- 22.Tabas I and Ron D (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol 13, 184–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsaytler P and Bertolotti A (2013) Exploiting the selectivity of protein phosphatase 1 for pharmacological intervention. FEBSJ. 280, 766–770 [DOI] [PubMed] [Google Scholar]
- 24.Hotamisligil GS (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valenzuela V et al. (2016) Injury to the nervous system: a look into the ER. Brain Res. 1648, 617–625 [DOI] [PubMed] [Google Scholar]
- 26.Clayton BL and Popko B (2016) Endoplasmic reticulum stress and the unfolded protein response in disorders of myeli-nating glia. Brain Res. 1648, 594–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Godin JD et al. (2016) Emerging roles for the unfolded protein response in the developing nervous system. Trends Neurosci. 39, 394–404 [DOI] [PubMed] [Google Scholar]
- 28.Murao N and Nishitoh H (2017) Role of the unfolded protein response in the development of central nervous system. J. Biochem 162, 155–162 [DOI] [PubMed] [Google Scholar]
- 29.Laguesse S et al. (2015) A dynamic unfolded protein response contributes to the control of cortical neurogenesis. Dev. Cell 35, 553–567 [DOI] [PubMed] [Google Scholar]
- 30.Mimura N et al. (2008) Altered quality control in the endoplasmic reticulum causes cortical dysplasia in knock-in mice expressing a mutant BiP. Mol. Cell. Biol 28, 293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarkisian MR et al. (2008) Trouble making the first move: interpreting arrested neuronal migration in the cerebral cortex. Trends Neurosci. 31, 54–61 [DOI] [PubMed] [Google Scholar]
- 32.Urra H et al. IRE1α governs cytoskeleton remodeling and cell migration through a direct interaction with lamin A. Nat. Cell Biol (in press). [DOI] [PubMed] [Google Scholar]
- 33.van Vliet AR et al. (2017) The ER stress sensor PERK coordinates ER-plasma membrane contact site formation through interaction with filamin-A and F-actin remodeling. Mol. Cell 65, 885–899 [DOI] [PubMed] [Google Scholar]
- 34.Salzberg Y et al. (2017) Reduced insulin/insulin-like growth factor receptor signaling mitigates defective dendrite morphogenesis in mutants of the ER stress sensor IRE-1. PLoS Genet. 13, e1006579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho YM et al. (2009) Induction of unfolded protein response during neuronal induction of rat bone marrow stromal cells and mouse embryonic stem cells. Exp. Mol. Med 41, 440–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frank CL et al. (2010) Control of activating transcription factor 4 (ATF4) persistence by multisite phosphorylation impacts cell cycle progression and neurogenesis. J. Biol. Chem 285, 33324–33337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tseng KY et al. (2017) MANF is essential for neurite extension and neuronal migration in the developing cortex. eNeuro 4, e0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Segal M (2005) Dendritic spines and long-term plasticity. Nat. Rev. Neurosci 6, 277–284 [DOI] [PubMed] [Google Scholar]
- 39.Ramirez OA and Couve A (2011) The endoplasmic reticulum and protein trafficking in dendrites and axons. Trends Cell Biol. 21, 219–227 [DOI] [PubMed] [Google Scholar]
- 40.Shim J et al. (2004) The unfolded protein response regulates glutamate receptor export from the endoplasmic reticulum. Mol. Biol. Cell 15, 4818–4828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei X et al. (2015) The unfolded protein response is required for dendrite morphogenesis. eLife 4, e06963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawada K et al. (2014) Aberrant neuronal differentiation and inhibition of dendrite outgrowth resulting from endoplasmic reticulum stress. J. Neurosci. Res 92, 1122–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayashi A et al. (2007) The role of brain-derived neurotrophic factor (BDNF)-induced XBP1 splicing during brain development. J. Biol. Chem 282, 34525–34534 [DOI] [PubMed] [Google Scholar]
- 44.Hayashi A et al. (2008) Attenuated BDNF-induced upregulation of GABAergic markers in neurons lacking Xbp1. Biochem. Biophys. Res. Commun 376, 758–763 [DOI] [PubMed] [Google Scholar]
- 45.Martinez G et al. (2016) Regulation of memory formation by the transcription factor XBP1. Cell Rep. 14, 1382–1394 [DOI] [PubMed] [Google Scholar]
- 46.Minichiello L (2009) TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci 10, 850–860 [DOI] [PubMed] [Google Scholar]
- 47.Saito A et al. (2018) Neuronal activity-dependent local activation of dendritic unfolded protein response promotes expression of brain-derived neurotrophic factor in cell soma. J. Neurochem 144, 35–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cisse M et al. (2016) The transcription factor XBP1s restores hippocampal synaptic plasticity and memory by control of the kalirin-7 pathway in Alzheimer model. Mol. Psychiatry 22, 1562–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma XM et al. (2008) Kalirin-7 is required for synaptic structure and function. J. Neurosci 28, 12368–12382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yin X et al. (2011) Molecular motor KIF17 is fundamental for memory and learning via differential support of synaptic NR2A/2B levels. Neuron 70, 310–325 [DOI] [PubMed] [Google Scholar]
- 51.Bernard-Marissal N et al. (2012) Reduced calreticulin levels link endoplasmic reticulum stress and Fas-triggered cell death in motoneurons vulnerable to ALS. J. Neurosci 32, 4901–4912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Filezac de L’Etang A et al. (2015) Marinesco-Sjögren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nat. Neurosci 18, 227–338 [DOI] [PubMed] [Google Scholar]
- 53.Kraus A et al. (2010) Calnexin deficiency leads to dysmyelination. J. Biol. Chem 285, 18928–18938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woehlbier U et al. (2016) ALS-linked protein disulfide isomerase variants cause motor dysfunction. EMBO J. 35, 845–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Onate M et al. (2016) Activation of the unfolded protein response promotes axonal regeneration after peripheral nerve injury. Sci. Rep 6, 21709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rampon C et al. (2000) Effects of environmental enrichment on gene expression in the brain. Proc. Natl. Acad. Sci. U. S. A 97, 12880–12884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park-York M et al. (2012) Cage food location alters energy balance and endoplasmic reticulum stress in the brain of mice. Physiol. Behav 106, 158–163 [DOI] [PubMed] [Google Scholar]
- 58.Kim Y et al. (2010) Three weeks voluntary running wheel exercise increases endoplasmic reticulum stress in the brain of mice. Brain Res. 1317, 13–23 [DOI] [PubMed] [Google Scholar]
- 59.Toda H et al. (2006) Behavioral stress and activated serotonergic neurotransmission induce XBP-1 splicing in the rat brain. Brain Res. 1112, 26–32 [DOI] [PubMed] [Google Scholar]
- 60.Kiskinis E et al. (2014) Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14, 781–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Costa-Mattioli M et al. (2007) eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 129, 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Di Prisco GV et al. (2014) Translational control of mGluR-dependent long-term depression and object-place learning by eIF2alpha. Nat. Neurosci 17, 1073–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang Z et al. (2010) eIF2alpha phosphorylation-dependent translation in CA1 pyramidal cells impairs hippocampal memory consolidation without affecting general translation. J. Neurosci 30, 2582–2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Costa-Mattioli M et al. (2005) Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature 436, 1166–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu PJ et al. (2011) Suppression of PKR promotes network excitability and enhanced cognition by interferon-gammamediated disinhibition. Cell 147, 1384–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ounallah-Saad H et al. (2014) Genetic or pharmacological reduction of PERK enhances cortical-dependent taste learning. J. Neurosci 34, 14624–14632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zimmermann HR et al. (2018) Genetic removal of eIF2alpha kinase PERK in mice enables hippocampal L-LTP independent of mTORC1 activity. J. Neurochem Published online January 16, 2018. 10.1111/jnc.14306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trinh MA et al. (2012) Brain-specific disruption of the eIF2alpha kinase PERK decreases ATF4 expression and impairs behavioral flexibility. Cell Rep. 1, 676–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pasini S et al. (2015) Specific downregulation of hippocampal ATF4 reveals a necessary role in synaptic plasticity and memory. Cell Rep. 11, 183–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen A et al. (2003) Inducible enhancement of memory storage and synaptic plasticity in transgenic mice expressing an inhibitor of ATF4 (CREB-2) and C/EBP proteins. Neuron 39, 655–669 [DOI] [PubMed] [Google Scholar]
- 71.Bartsch D et al. (1995) Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell 83, 979–992 [DOI] [PubMed] [Google Scholar]
- 72.Ebert SM et al. (2012) Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J. Biol. Chem 287, 27290–27301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hetz C et al. (2008) Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc. Natl. Acad. Sci. U. S. A 105, 757–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takata A et al. (2010) Behavioral and gene expression analyses in heterozygous XBP1 knockout mice: possible contribution of chromosome 11qA1 locus to prepulse inhibition. Neurosci. Res 68, 250–255 [DOI] [PubMed] [Google Scholar]
- 75.Ozcan L et al. (2009) Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 9, 35–51 [DOI] [PubMed] [Google Scholar]
- 76.Williams KW et al. (2014) Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab. 20, 471–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xiao Y et al. (2016) Knockout of inositol-requiring enzyme 1alpha in pro-opiomelanocortin neurons decreases fat mass via increasing energy expenditure. Open Biol. 6, 160131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yao T et al. (2017) Ire1alpha in Pomc neurons is required for thermogenesis and glycemia. Diabetes 66, 663–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Methippara M et al. (2012) Salubrinal, an endoplasmic reticulum stress blocker, modulates sleep homeostasis and activation of sleep- and wake-regulatory neurons. Neuroscience 209, 108–118 [DOI] [PubMed] [Google Scholar]
- 80.Hatori M et al. (2011) Light-dependent and circadian clock-regulated activation of sterol regulatory element-binding protein, X-box-binding protein 1, and heat shock factor pathways. Proc. Natl. Acad. Sci. U. S. A 108, 4864–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lopez-Otin C et al. (2013) The hallmarks of aging. Cell 153, 1194–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martinez G et al. (2017) Endoplasmic reticulum proteostasis impairment in aging. Aging Cell 16, 615–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Freeman OJ and Mallucci GR (2016) The UPR and synaptic dysfunction in neurodegeneration. Brain Res. 1648, 530–537 [DOI] [PubMed] [Google Scholar]
- 84.Scheper W and Hoozemans JJ (2015) The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. 130, 315–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ma T et al. (2013) Suppression of eIF2alpha kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat.Neurosci 16, 1299–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Peel AL and Bredesen DE (2003) Activation of the cell stress kinase PKR in Alzheimer’s disease and human amyloid precursor protein transgenic mice. Neurobiol. Dis 14, 52–62 [DOI] [PubMed] [Google Scholar]
- 87.Dumurgier J et al. (2013) Cerebrospinal fluid PKR level predicts cognitive decline in Alzheimer’s disease. PLoS One 8, e53587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Segev Y et al. (2015) PKR inhibition rescues memory deficit and ATF4 overexpression in ApoE epsilon4 human replacement mice. J. Neurosci 35, 12986–12993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lourenco MV et al. (2013) TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 18, 831–843 [DOI] [PubMed] [Google Scholar]
- 90.Dash PK et al. (2015) Inhibition of eukaryotic Initiation factor 2 alpha phosphatase reduces tissue damage and improves learning and memory after experimental traumatic brain injury. J. Neurotrauma 32, 1608–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sidrauski C et al. (2013) Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife 2, e00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tsai JC et al. (2018) Structure of the nucleotide exchange factor eIF2B reveals mechanism of memory-enhancing molecule. Science 359, eaaq0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chou A et al. (2017) Inhibition of the integrated stress response reverses cognitive deficits after traumatic brain injury. Proc. Natl. Acad. Sci. U. S. A 114, E6420–E6426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Briggs DI et al. (2017) Role of endoplasmic reticulum stress in learning and memory impairment and Alzheimer’s disease-like neuropathology in the PS19 and APPSwe mouse models of tauopathy and amyloidosis. eNeuro 4, e0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Moreno JA et al. (2012) Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 485, 507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Halliday M et al. (2015) Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 6, e1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moreno JA et al. (2013) Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med 5, 206ra138. [DOI] [PubMed] [Google Scholar]
- 98.Hoglinger GU et al. (2011) Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet 43, 699–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Radford H et al. (2015) PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 130, 633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mercado G et al. (2018) Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson’s disease. Neurobiol. Dis 112, 136–148 [DOI] [PubMed] [Google Scholar]
- 101.Gerakis Y and Hetz C (2018) Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 285, 995–1011 [DOI] [PubMed] [Google Scholar]
- 102.Kakiuchi C et al. (2003) Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nat. Genet 35, 171–175 [DOI] [PubMed] [Google Scholar]
- 103.Liu SY et al. (2013) Polymorphism - 116C/G of human X-box-binding protein 1 promoter is associated with risk of Alzheimer’s disease. CNS Neurosci. Ther 19, 229–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Duran-Aniotz C et al. (2017) IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. 134, 489–506 [DOI] [PubMed] [Google Scholar]
- 105.Cheng D et al. (2014) The - 116C/G polymorphism in XBP1 gene is associated with psychiatric illness in Asian population: a meta-analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet 165B, 665–672 [DOI] [PubMed] [Google Scholar]
- 106.Kakiuchi C et al. (2004) Association of the XBP1 - 116C/G polymorphism with schizophrenia in the Japanese population. Psychiatry Clin. Neurosci 58, 438–440 [DOI] [PubMed] [Google Scholar]
- 107.Kusumi I et al. (2005) Relationship between XBP1 genotype and personality traits assessed by TCI and NEO-FFI. Neurosci. Lett 391, 7–10 [DOI] [PubMed] [Google Scholar]
- 108.Kakiuchi C and Kato T (2005) Lithium response and - 116C/G polymorphism of XBP1 in Japanese patients with bipolar disorder. Int. J. Neuropsychopharmacol 8, 631–632 [DOI] [PubMed] [Google Scholar]
- 109.Kabir ZD et al. (2017) Rescue of impaired sociability and anxiety-like behavior in adult cacna1c-deficient mice by pharmacologically targeting eIF2alpha. Mol. Psychiatry 22, 1096–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wen L et al. (2017) PERK signalling pathway mediates single prolonged stress-induced dysfunction of medial prefrontal cortex neurons. Apoptosis 22, 753–768 [DOI] [PubMed] [Google Scholar]
- 111.Gonzalez-Perez P et al. (2015) Identification of rare protein disulfide isomerase gene variants in amyotrophic lateral sclerosis patients. Gene 566, 158–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yang Q and Guo ZB (2016) Polymorphisms in protein disulfide isomerase are associated with sporadic amyotrophic lateral sclerosis in the Chinese Han population. Int. J. Neurosci 126, 607–611 [DOI] [PubMed] [Google Scholar]
- 113.Kwok CT et al. (2013) Association studies indicate that protein disulfide isomerase is a risk factor in amyotrophic lateral sclerosis. Free Radic. Biol. Med 58, 81–86 [DOI] [PubMed] [Google Scholar]
- 114.Kohl S et al. (2015) Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat. Genet 47, 757–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ansar M et al. (2015) Mutation of ATF6 causes autosomal recessive achromatopsia. Hum. Genet 134, 941–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hetz C and Mollereau B (2014) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci 15, 1–19 [DOI] [PubMed] [Google Scholar]
- 117.Arimura N and Kaibuchi K (2007) Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat. Rev. Neurosci 8, 194–205 [DOI] [PubMed] [Google Scholar]
- 118.Cajigas IJ et al. (2010) Protein homeostasis and synaptic plasticity. EMBO J. 29, 2746–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol 13, 89–102 [DOI] [PubMed] [Google Scholar]
- 120.Tinsley RB et al. (2009) Dopamine D2 receptor knockout mice develop features of Parkinson disease. Ann. Neurol 66, 472–484 [DOI] [PubMed] [Google Scholar]
- 121.Jayanthi S et al. (2009) Methamphetamine induces dopamine D1 receptor-dependent endoplasmic reticulum stress-related molecular events in the rat striatum. PLoS One 4, e6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dukes AA et al. (2008) Changes in endoplasmic reticulum stress proteins and aldolase A in cells exposed to dopamine. J. Neurochem 106, 333–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Valdes P et al. (2014) Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. U. S. A 111, 6804–6809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Martinon F et al. (2010) TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol 11, 411–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Osorio F et al. (2014) The unfolded-protein-response sensor IRE-1alpha regulates the function of CD8alpha+ dendritic cells. Nat. Immunol 15, 248–257 [DOI] [PubMed] [Google Scholar]
- 126.Bettigole SE and Glimcher LH (2015) Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol 33, 107–138 [DOI] [PubMed] [Google Scholar]
- 127.Taylor RC and Dillin A (2013) XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153, 1435–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang L et al. (2015) PERK limits Drosophila lifespan by promoting intestinal stem cell proliferation in response to ER stress. PLoS Genet. 11, e1005220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schinzel R and Dillin A (2015) Endocrine aspects of organelle stress - cell non-autonomous signaling of mitochondria and the ER. Curr. Opin. Cell Biol 33, 102–110 [DOI] [PubMed] [Google Scholar]