

Abstract
Dominant mutations in the mitochondrial paralogs coiled-helix-coiled-helix (CHCHD) domain 2 (C2) and CHCHD10 (C10) were recently identified as causing Parkinson’s disease and amyotrophic lateral sclerosis/frontotemporal dementia/myopathy, respectively. The mechanism by which they disrupt mitochondrial cristae, however, has been uncertain. Using the first C2/C10 double knockout (DKO) mice, we report that C10 pathogenesis and the normal function of C2/C10 are intimately linked. Similar to patients with C10 mutations, we found that C2/C10 DKO mice have disrupted mitochondrial cristae, because of cleavage of the mitochondrial-shaping protein long form of OPA1 (L-OPA1) by the stress-induced peptidase OMA1. OMA1 was found to be activated similarly in affected tissues of mutant C10 knock-in (KI) mice, demonstrating that L-OPA1 cleavage is a novel mechanism for cristae abnormalities because of both C10 mutation and C2/C10 loss. Using OMA1 activation as a functional assay, we found that C2 and C10 are partially functionally redundant, and some but not all disease-causing mutations have retained activity. Finally, C2/C10 DKO mice partially phenocopied mutant C10 KI mice with the development of cardiomyopathy and activation of the integrated mitochondrial integrated stress response in affected tissues, tying mutant C10 pathogenesis to C2/C10 function.
Introduction
Autosomal dominant mutations in the mitochondrial paralogs coiled-helix-coiled-helix (CHCH) domain 2 (C2) and CHCHD10 (C10) were recently shown to cause, respectively, Parkinson’s disease (PD) and a spectrum of neuromuscular disorders that includes myopathy and amyotrophic lateral sclerosis (ALS) [1–4]. Most mutations with proven pathogenicity in C2/C10 cluster in a common hydrophobic α-helix N-terminal to the CHCH domain, consisting of dual CX9C motifs. These include the C10 S59L substitution, which was originally identified in a family with ALS, frontotemporal dementia (FTD) and myopathy, and substitution of the neighboring residue, G58R, identified in a family with myopathy [1,2].
C2 and C10 are co-expressed in relevant cellular subtypes, such as substantia nigral neurons, pyramidal neurons and myocytes, where they directly interact within a ~200 kDa complex in the mitochondrial intermembrane space [5–7]. Although their precise function remains unknown, cristae abnormalities have been observed in muscle tissue and cell lines from patients with C10 mutations, as well as in Drosophila lacking the C2/C10 ortholog [1,2,8]. To explain cristae abnormalities in C2/C10 patients, C10 and later C2 were proposed to be structural components of the mitochondrial contact site and cristae organizing system (MICOS) complex [2,9,10]. However, recent reports have challenged this model, suggesting that C2/C10 mutation or loss may lead to cristae abnormalities by another as of yet unidentified mechanism [5–7].
Cristae are infoldings of the inner mitochondrial membrane and can be considered the fundamental bioenergetic unit within mitochondria. Studded with respiratory complexes and rimmed with dimers of the F1Fo ATP synthase, they increase surface area for oxidative phosphorylation [11]. Cristae are shaped by at least three protein complexes: the MICOS complex, bending the cristae membrane at its junction with the boundary membrane; the GTPase OPA1, which mediates inner membrane fusion and supports cristae structure by bridging apposing membranes in the cristae fold; and ATP synthase dimers, which bend the membrane at the cristae edge [12–19].
OPA1, in particular, is highly regulated by mitochondrial bioenergetics and proteostatic stress to dynamically shape the inner membrane in response to changing conditions within the mitochondrial network. The peptidase OMA1 is activated by mitochondrial stressors to cleave the active long form of OPA1 (L-OPA1) from its membrane anchor, leading to mitochondrial fragmentation and alterations in cristae structure [20,21]. L-OPA1 processing by OMA1 also occurs upon loss of quality control proteases, such as YME1, AFG3L2 and SPG7, as well as inner membrane scaffolding proteins of the SPFH family, namely, prohibitins and SLP-2, which organize quality control proteases within inner membrane microdomains [20,22–25]. Thus, OMA1 provides an escape mechanism for cristae with failure of protein quality control, separating dysfunctional mitochondrial units from the network for degradation by autophagy [26].
Very recently, mice null for C10 and knock-in (KI) for the pathogenic S59L mutation have been reported [5,27,28]. Whereas C10−/− mice have a normal life span, C10S59L/+ KI mice develop a cardiomyopathy by 23 weeks, which is uniformly fatal at 13 months, supporting a gain-of-function mechanism of pathogenesis. Anderson et al. [27], in particular, observed C10 levels increase and co-aggregate with C2 in affected tissues, leading them to propose a toxic aggregation model of pathogenesis, in which C2/C10 toxic aggregates trigger the integrated mitochondrial integrated stress response (mt-ISR) and tissue degeneration. However, it remains unclear in this aggregate model whether the pathogenesis of mutant C10 relates to C2/C10 physiological function.
Here, we evaluate the first C2/C10 DKO mouse model with two novel findings. First, we identify OPA1 cleavage by the stress-induced peptidase OMA1 to be at least partially responsible for cristae abnormalities in absence of C2/C10. We further show that OMA1 activation is a key event in mutant C10 pathogenesis in vivo. Thus, we establish for the first time OPA1 cleavage as a potential mechanism for cristae abnormalities in patients with C10 mutations and C2/C10 models. Second, we observe that C2/C10 DKO mice, unlike C10 KO mice, partially phenocopy the recently reported mutant C10 model, including activation of the mt-ISR and development of cardiomyopathy. This suggests that C10 pathogenesis and C2/C10 physiological function are intimately linked. We propose that C2/C10 levels are required in a narrow range of expression to maintain normal L-OPA1/short form of OPA1 (S-OPA1) balance, as either too little or too much wild-type (WT) C2 or C10 actives OMA1. Mutations in C10 may lead to OMA1 activation by a dominant negative action (e.g. through co-aggregation with WT C2/C10) or by lowering the concentration at which mutant C10 expression triggers OMA1 activation.
Results
C2/C10 double knockout leads to cristae abnormalities because of increased L-OPA1 processing by the stress-induced protease OMA1 in cell culture
To investigate the physiological role of C2 and C10 in mammalian primary cells, we generated C2 and C10 single knockout (KO) and double knockout (DKO) mice (Fig. 1A). Whereas cristae structure was normal for WT and C10 single KO cell lines by transmission electron microscopy (TEM) (Supplementary Material, Fig. S1A), primary fibroblasts from DKO mice exhibited abnormalities in mitochondrial ultrastructure, including convoluted cristae, which were sometimes circular and detached from boundary membrane, as well as a decrease in cristae number (8.232 vs. 16.43 cristae/μm2, P < 0.0001) (Supplementary Material, Fig. S1A and Fig. 1B–D). Cristae number was also mildly decreased for C10 single KO cells compared with WT (14.87 vs. 16.43 cristae/μm2, P = 0.030) (Fig. 1B–D). By confocal microscopy, mitochondrial area was also found to be slightly reduced in C2/C10 DKO compared with two WT primary fibroblasts (Supplementary Material, Fig. S1B).
Figure 1.
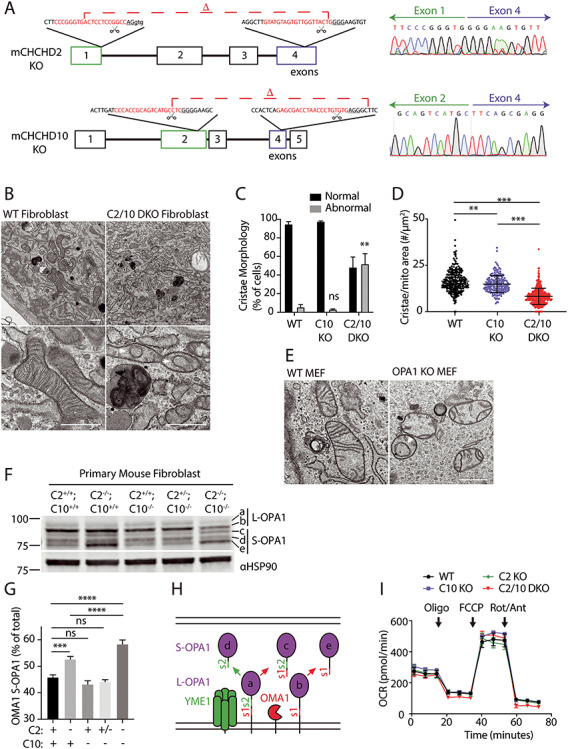
Cristae abnormalities in C2/C10 DKO primary fibroblasts associated with OPA1 processing by OMA1 activation. (A) C2 and C10 KO mice were generated by CRIPSR/Cas9 genomic editing with CRISPR cut sites (scissor icon) and guide RNA sequence (red text) and photospacer adjacent motif (PAM) sequence (underlined) indicated. Sanger sequencing across the deletions verifies loss of the intervening exons. (B–D) Analysis of TEM images from WT and C2/C10 DKO fibroblast cells. Representative images are shown in (B). Arrowhead indicates detached cristae and arrow points to convoluted cristae. (C) Quantification of abnormal mitochondria. (D) The number of cristae per mitochondrion area (μm2) was calculated. For (C) and (D) ≥267 mitochondria were quantified per condition from ≥21 cells. Two independent WT cell lines, three independent C10 KO cell lines (two litter matched to DKO and one to WT) and two independent C2/C10 DKO cell lines were evaluated. Data from cell lines were pooled by genotype. A total of three biological replicates were performed per genotype on two occasions. Standard error of the mean in depicted in the graphs. **P < 0.01, ***P < 0.001 and ****P < 0.0001 after correction for multiple comparisons. (E) Representative TEM images of transformed WT and OPA1 KO MEF cells. Arrowhead indicates detached cristae. (F and G) Immunoblot from lysates of WT, C2−/−, C10−/−, C2+/−; C10−/− and C2−/−; C10−/− (DKO) primary mouse fibroblasts. Immunoblot is representative of three biological replicates. HSP90 serves as the loading control. OPA1 isoforms (a–e) depicted in (H) are indicated. Standard error of the mean in depicted in the graphs. Ns indicates values are not significant, ***P < 0.001 and ***P < 0.0001 after correction for multiple comparisons. (H) Model depicts processing of the two membrane-bound L-OPA1 at protease sites s1 and s2 into the three S-OPA1 by the proteases OMA1 (at the s1 site) and YME1 (at the s2 site). Red arrows depict cleavage by OMA1 at the s1 site to generate isoform c from a and e from b. The green arrow depicts cleavage at s2 by YME1 to generate d from a. (I) OCR of WT, C2−/−, C10−/−, C2−/−; C10−/− (DKO) primary fibroblasts measured by the Seahorse instrument. Injections of oligomycin (Oligo), carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) and rotenone (Rot)/antimycin (Ant) are indicated. Plot is representative of at least six biological replicates performed on at least two occasions. Scale bars in all images = 600 nm.
As the cristae abnormalities in C2/C10 DKO fibroblasts resembled those with OPA1 KO (Fig. 1E), we assessed OPA1 processing by immunoblotting (Fig. 1F–H). There are five OPA1 isoforms visible by immunoblotting corresponding to the membrane-bound L-OPA1 forms (a and b), and their proteolytic cleavage products, the S-OPA1 forms (c–e), generated by YME1 (d) or OMA1 cleavage (c and e), respectively (Fig. 1H) [20,21]. In C2/C10 DKO fibroblasts, L-OPA1 forms decreased and OMA1-dependent S-OPA1 forms (c and e) increased, consistent with OMA1 cleavage (Fig. 1F and G). Notably, L-OPA1 processing in C2 single KO cells was between WT and C2/C10 DKO cells, and normal in C10 KO cells, consistent with partial functional redundancy of the paralogs.
Protein levels of the MICOS subunits, Mic19 and Mic27 were also mildly reduced in C2/C10 DKO fibroblasts compared with WT fibroblasts, similar to what we observed previously in HEK293 C2/C10 DKO cells [6] (Supplementary Material, Fig. S1C). Mic60 levels were also decreased, although the trend was not significant. A recent report found that OMA1 can cleave Mic19 on activation, and so we considered whether this could be the mechanism for the observed decrease in MICOS subunits in C2/C10 DKO cells [29]. However, we did not detect cleaved Mic19 in HEK293 C2/C10 DKO cells, suggesting that this mechanism of crosstalk between OMA1 and the MICOS complex likely does not account for the decrease in MICOS subunits (Supplementary Material, Fig. S2A). In contrast to levels of L-OPA1 and MICOS subunits, DRP1 and Mitofusin-2 levels were similar in all cell lines (Supplementary Material, Fig. S1C and D).
As OMA1 is activated in response to bioenergetic collapse, we investigated cellular respiration in DKO cells by oximetry. Consistent with our prior observations in HEK293 cells, basal and maximal oxygen consumption was comparable for DKO and WT primary fibroblasts under growth conditions in which L-OPA1 processing was disrupted (Fig. 1I) [6]. Mitochondrial membrane potential measured by tetramethylrhodamine methyl ester (TMRM) fluorescence was also similar for litter matched C10 KO and C2/10 DKO fibroblasts (Supplementary Material, Fig. S1C). Membrane potential in C2/C10 single KO and DKO was reduced when compared with WT lines from unrelated mice (Supplementary Material, Fig. S1C). Together these findings argue against OPA1 instability in C2/10 DKO cells resulting from disrupted mitochondrial bioenergetics.
To assess whether OPA1 processing was altered also in human cells, we evaluated the OMA1/OPA1 axis in HEK293 DKO cells that we described previously [6]. Similar to DKO mouse fibroblasts, cristae number was significantly reduced in HEK293 DKO cells (Fig. 2A and B). Consistently, L-OPA1 was found to be processed in a pattern reflective of OMA1 activation (Fig. 2C). OMA1 levels were likewise decreased, as expected given that OMA1 is degraded shortly after its activation [21]. Similar to primary fibroblasts, we previously found only a mild oxygen consumption deficit in HEK293 DKO cells compared with WT cells [6], and basal tetramethylrhodamine ethyl ester (TMRE) fluorescence was slightly higher for DKO cells compared with WT cells, indicating increased membrane potential (Supplementary Material, Fig. S2C). TMRE fluorescence decreased in response to carbonyl cyanide m-chlorophenyl hydrazone (CCCP) and increased in response to oligomycin in both WT and DKO cells, consistent with TMRE reflecting membrane potential in the unquenched mode. To test whether a moderate reduction of oxidative phosphorylation (OXPHOS) is sufficient to activate OMA1 in HEK293 cells, we inhibited mitochondrial translation in HEK293 cells with 200 μg/ml chloramphenicol for 5–7 days. This caused a 35% decrease in membrane potential and a 78% decrease in expression of the complex IV subunit, COX2 (Supplementary Material, Fig. S2D and E). By comparison, COX2 levels were 37% lower in untreated HEK293 C2/C10 DKO cells compared with WT cells (Supplementary Material, Fig. S2E). However, even the large reduction in COX2 expression with chloramphenicol treatment was insufficient to trigger L-OPA1 processing by OMA1. Together, these data demonstrate that L-OPA1 processing in C2/C10 DKO cells is not driven by a bioenergetic deficit or the observed mild reduction in OXPHOS subunits.
Figure 2.
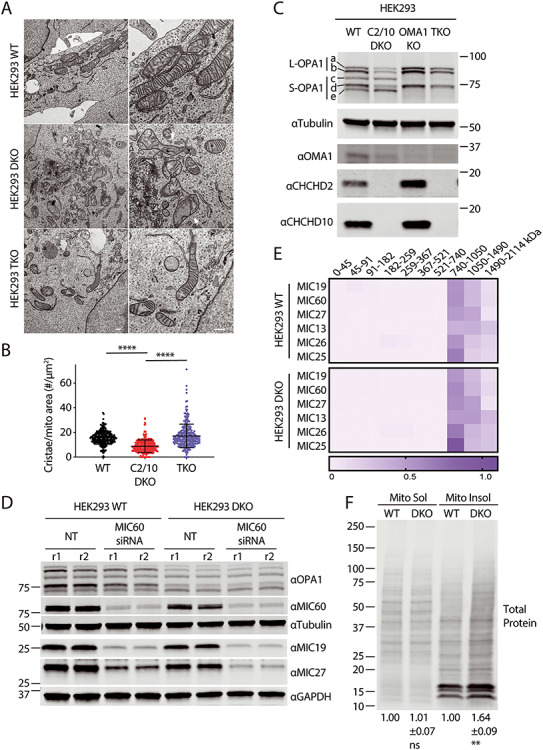
Cristae abnormalities in C2/C10 DKO cells are due at least in part to OMA1 activation. (A) Representative TEM images of WT, C2/C10 DKO and TKO HEK293 cells. Scale bar = 800 nm. Arrows indicate abnormal mitochondria. (B) The number of cristae per mitochondrion area (μm2) was calculated. For each condition ≥275 mitochondria were measured from ≥22 cells in three biological replicates performed on two occasions. ****P < 0.0001 after correction for multiple comparisons. Error bars represent standard error of the mean. (C) Immunoblot from lysates of WT, C2/C10 DKO, OMA1 KO and TKO HEK293 cells. Tubulin served as the loading control. Blot is representative of at least three biological replicates performed on at least two occasions. (D) Immunoblot of WT and C2/C10 DKO HEK293 cells following knockdown of MIC60 or left untransfected not treated (NT). Immunoblot is representative of three biological replicates obtained on two different occasions. Lysates from two replicates (r1 and r2) are shown. (E) Heat map depicts relative abundance of MICOS complex subunits in stable isotope labelling of amino acids in cell culture (SILAC) labeled WT and DKO HEK293 mitochondria, solubilized in 1% digitonin and separated by BN-PAGE prior to detection by quantitative mass spectrometry. Columns in the heat map represent gel slices at the indicated position. Rows indicate proteins detected. Data represent two biological replicates performed on one occasion. (F) Total protein was stained from soluble and insoluble mitochondrial fractions of HEK293 WT and C2/C10 DKO separated on SDS-PAGE gels and transferred to a membrane. Relative intensity of total protein in soluble or insoluble HEK293 DKO fractions relative to WT was quantified for N = 3 biological replicates performed on at least three occasions. Mean±standard error of the mean is represented on the bottom of the membrane. Values were normalized to WT samples.
Next, to test formally whether OMA1 is responsible for L-OPA1 cleavage in the absence of C2/C10, we generated OMA1 KO and C2/C10/OMA1 triple KO (TKO) HEK293 cell lines (Fig. 2A–C). Loss of OMA1 in HEK293 cells led to a mild decrease in total OPA1 (68.1% of WT levels, P = 0.0006) and CHCHD2 levels were slightly increased (128% of WT levels, P = 0.0099) (Supplementary Material, Fig. S3A). Crucially, in TKO cells L-OPA1 isoforms (a and b) were stabilized against cleavage to isoforms c and e, whereas basal processing of L-OPA1 isoform a to d by the protease YME1 was unaffected (Fig. 2C). Additionally, cristae density was partially restored to C2/C10-deficient cells following KO of OMA1 (Fig. 2B). Together, these findings demonstrate that cristae abnormalities in the absence of C2 and C10 are caused at least in part by OMA1 cleavage of L-OPA1.
We assessed the specificity of activated OMA1 to L-OPA1 in HEK293 DKO cells, by examining another substrate of activated OMA1, PGAM5. PGAM5 processing was similar in WT and DKO cells both under basal conditions and following mitochondrial uncoupling with CCCP, suggesting that OMA1 activation with C2/C10 DKO may preferentially affect L-OPA1 processing (Supplementary Material, Fig. S3B) [30]. Consistent with our previously reported observations, C2 and C10 increased in response to CCCP treatment in cell culture (Supplementary Material, Fig. S3B) [6]. These findings suggest that OMA1 activation in response to C2/C10 loss may be relatively specific for L-OPA1.
As the MICOS complex is reported to interact with OPA1, and we observe a mild decrease in some MICOS subunits in the absence of C2/C10 [9,10,31,32], we tested whether the reduction in MICOS subunits could account for the observed OMA1 activation and L-OPA1 cleavage. As was found in primary fibroblasts and as we reported previously in HEK293 cells [6], levels of MICOS subunits, MIC19, MIC27 and MIC60, were mildly reduced in HEK293 DKO cells compared with WT cells (77.4% for MIC19, 69.8% for MIC27 and 80.1% for MIC60, Fig. 2D and Supplementary Material, Fig. S3C). As expected, depletion of a central subunit of the MICOS complex, MIC60, further destabilized MIC19 and MIC27, components of the respective MICOS subcomplexes in both DKO and WT cells (Fig. 2D). OPA1 processing, in contrast, was unaffected under these conditions, demonstrating that MICOS complex instability alone cannot account for OPA1 cleavage by OMA1.
We additionally assessed the relative abundance of the MICOS and other complexes previously found to affect OMA1 activation and OPA1 cleavage using LC-MS/MS-based complexomics in WT and DKO HEK293 cells [24,25]. Detected subunits of the SPY, Prohibitin and MICOS complexes were of similar abundance in WT and DKO cells (Fig. 2E, Supplementary Material, Fig. S3D and Table S1).
Loss of mitochondrial proteostasis is known to activate OMA1. To assess whether there is a general loss in mitochondrial proteostasis in in the absence of C2/C10, we fractionated mitochondria from WT and DKO cells into soluble and insoluble fractions. Consistent with a general loss in mitochondrial proteostasis, HEK293 cells lacking C2/C10 had a 64% increase in total protein in the insoluble mitochondrial fraction (Fig. 2F). This association supports the idea that OMA1 may be activated by loss of mitochondrial proteostasis in C2/C10 DKO cells.
We next assessed for interactions among C2, OPA1, OMA1 and proteins forming the SPY complex involved in quality control proteostasis, SLP-2, PARL and YME1 [25]. As reported, previously, C10 robustly co-immunoprecipitates with C2-Flag (Supplementary Material, Fig. S4A and B). In contrast, OPA1 and OMA1 failed to co-immunoprecipitate with C2 under the same conditions. Interactions between C2 and PARL and C10 and YME1 of the SPY complex have been identified, previously, in LC-MS/MS co-immunoprecipitation experiments [25,33]. We were able to confirm an interaction between C2-Flag and YME1. However, we were not able to detect interactions between C2-Flag and PARL or C2-Flag and SLP-2 under the conditions used (Supplementary Material, Fig. S4A and B). Similarly, whereas PARL-Flag pulled down endogenous SLP-2, it failed to pull down endogenous C2 or C10. Together, these findings could be consistent with weak transient interactions with the SPY complex (and in particular with YME1) but argue against strong physical interactions between C2 and OMA1, the SPY complex, or OPA1.
Given that YME1 loss activates OMA1, and C2 interacts with YME1, we assessed whether decreased YME1 activity might be mediating the effect of C2/C10 DKO on OMA1. As expected, loss of either YME1 in HeLa cells or C2/C10 in HEK293 cells led to OMA1 activation, with a decrease in L-OPA1, an increase in OMA1 cleaved S-OPA1 (bands c and e), and a decrease in OMA1 levels (Supplementary Material, Fig. S4C) [22]. Activation of OMA1 appeared to be even stronger in C2/C10 DKO cells than YME1 KO cells, although this may also reflect a difference in cell type. Whereas YME1 KO led to the expected increase in its substrates TIM23 and PRELID [34,35], loss of C2/C10 resulted in slightly reduced levels of these YME1 substrates (Supplementary Material, Fig. S4C and D). This suggests that YME1 activity may be slightly increased in C2/C10 DKO cells. Taken together, these results suggest that decreased YME1 activity does not account for OMA1 activation in C2/C10 DKO cells.
Exogenous C2 and C10 and some (but not all) pathogenic mutants rescue OMA1 processing in C2/C10 DKO cells
We next addressed whether OMA1 activation in C2/C10 DKO cells can be rescued by exogenous expression of either C2 or C10, as would be expected if the paralogs are functionally redundant. Consistent with functional redundancy between C2 and C10, stable expression of either C2 or C10 reduced OMA1-specific S-OPA1 cleavage products in HEK293 cells lacking endogenous C2 and C10 (Fig. 3A and B).
Figure 3.
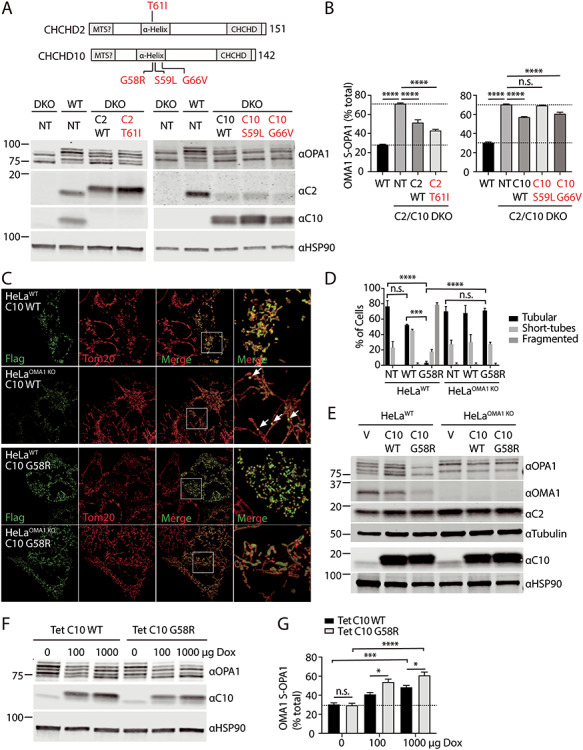
Mitochondrial fragmentation induced by C10 mutant depends on OMA1 processing of OPA1. (A and B) HEK293WT and HEK293C2/C10 DKO were not transduced (NT) or stably transduced with C10 or C2 containing the disease-causing mutations indicated. Lysates were separated on SDS-PAGE gels and OPA1 processing was assessed by immunoblotting. (B) OMA1 activity was measured as the abundance of OMA1 cleaved S-OPA1 isoforms (c and e) divided by the sum of all five isoforms in three biological replicates performed on at least two occasions. N = 3 biological replicates performed on at least two occasions. Error bars represent standard error of the mean. ****P < 0.0001 after correction for multiple comparisons. (C) Representative Airyscan confocal images of WT and OMA1 KO HeLa cells transiently transfected with C10 WT-Flag (C10 WT) or C10 G58R-Flag (C10 G58R). Anti-Flag immunostaining in green and anti-Tom20 in red. (D) Quantification of mitochondrial morphology depicted in (A) for >150 cells in three biological replicates per condition. Standard error of the mean in depicted in the graphs. ***P < 0.001 and ****P < 0.0001 after correction for multiple comparisons. Untransfected cells (NT) were identified by the absence of Flag staining in each of the samples. Counts were pooled as they did not differ among the samples. (E) Immunoblot from lysates of WT and OMA1 KO HeLa cells transduced with vector (V), C10 WT (C10 WT) or C10 G58R (C10 G58R). Tubulin and HSP90 serve as the loading controls in the respective blots. Immunoblot is representative of two biological replicates. (F and G) HeLaWT cells stably expressing tetracycline-inducible C10 WT and C10 G58R constructs were treated with 100 or 1000 μg/ml doxycycline for 48 h and whole-cell lysates were immunoblotted for OPA1, C10 and HSP90, which served as a loading control. OMA1-cleaved OPA1 was quantified as percent of the OMA1 specific isoforms (c and e) divided by the sum of all five isoforms. Experiment was performed in three biological replicates on two occasions. *P < 0.05, ***P < 0.001 and ****P < 0.0001 after correction for multiple comparisons. Error bars represent standard error of the mean. Ns, not significant.
We next examined whether disease-causing mutations in C2 or C10 disrupted their activity, using OMA1 activation as a functional readout. We generated HEK293 DKO lines that were stably expressing mutations previously demonstrated to cause familial PD (C2 T61I) or neuromuscular disorders (C10 S59L or G66V) [2–4] (Fig. 3A and B). We also attempted to generate a stable cell line expressing the C10 G58R mutation at a similar level but failed because of toxicity, which was investigated further below (data not shown). The C2 T61I and C10 G66V mutants rescued L-OPA1 processing, similar to their WT counterparts (Fig. 3A and B). In contrast, the C10 S59L mutant did not suppress OMA1 activation, suggesting that the S59L mutation disrupts C10 activity (Fig. 3A and B). This analysis demonstrated that some but not all pathogenic mutations retain activity and is consistent with a dominant negative or toxic gain-of-function mechanism of disease for the dominant mutations.
Mitochondrial fragmentation by C10 mutant depends on L-OPA1 cleavage by OMA1
Having identified a functional interaction between C2/C10 and OMA1/OPA1, we next asked whether OMA1 activation is responsible for mitochondrial fragmentation that we and others observed previously following overexpression of the pathogenic C10 mutation G58R in human cells [1,6]. Overexpression of C10 G58R caused severe mitochondrial fragmentation on a WT background, as was expected (intermediate or fragmented mitochondria in 96.7% of C10 G58R cells vs. 23.3% of untransfected cells, P < 0.0001) (Fig. 3C and D). Notably, this fragmentation was blocked by OMA1 KO (intermediate or fragmented in 29.67% C10 G58R expressing HeLaOMA1 KO cells vs. 28.67% of untransfected cells, P > 0.9999). Overexpression of WT C10 also resulted in a trend toward increased mitochondrial fragmentation, although this trend did not reach statistical significance (Fig. 3C and D). These findings demonstrated that C10 G58R overexpression causes mitochondrial fragmentation by activating OMA1.
The G58R substitution lies within a conserved hydrophobic α-helix of C10 and contributes to a GXXXGXXXG motif that often mediates protein–protein interactions [6,36] (Fig. 3A, schematic at top). The G58R substitution, which introduces a positive charge into the predicted hydrophobic glycine cleft of the GXXXGXXXG helix, is likely to disrupt the α-helix and/or its interactions. To test this idea further, we deleted the α-helix of C10 (ΔαH). Similar to C10 G58R, C10 (ΔαH) induced the fragmentation of mitochondria in an OMA1-dependent manner (Supplementary Material, Fig. S5A and B). These findings are consistent with the notion that the C10 G58R mutation disrupts the α-helix of C10 to induce OMA1/OPA1-dependent mitochondrial fragmentation.
Forced C2 and C10 expression activates OMA1, and C10 mutant activates OMA1 at a lower concentration than C10 WT
Consistent with the observed OMA1-dependent mitochondrial fragmentation with C10 G58R overexpression, C10 G58R strongly stimulated OMA1-dependent processing of L-OPA1 to S-OPA1 (Fig. 3E, lanes 1–3). Unexpectedly, overexpression of WT C10 also induced L-OPA1 processing by OMA1 (Fig. 3E, lanes 4–6). To more directly compare C10 WT and C10 G58R expression in HeLa cells, we established tetracycline-inducible cell lines. Treatment with 100 ng/ml doxycycline-induced expression of C10 WT and C10 G58R to 9.5 and 7.2 times endogenous levels, respectively (P = 0.90 for WT vs. G58R comparison). In total, 1000 ng/ml doxycycline further increased expression of C10 WT and C10 G58R to 12.31 and 11.12 times endogenous levels, respectively (P = 0.99 for WT vs. G58R comparison). A dose-responsive increase in OMA1 activation was observed with both WT C10 and C10 G58R overexpression; however, OMA1 activation was significantly greater for C10 G58R at each level of expression (Fig. 3F and G). Thus, the C10 G58R mutation leads to greater OMA1 activation at a similar concentration as C10 WT.
Intense C2/C10 foci accumulate on blocking L-OPA1 cleavage by OMA1
We observed that WT C10-Flag formed foci within OMA1 KO cells, rather than the typical smooth appearance throughout the mitochondrial network of overexpressed C10 protein by light microscopy (Fig. 3C, arrows in right panels). To investigate whether the pattern of endogenous C2 and C10 distribution also changes in the absence of OMA1, we used antisera specific for these proteins in OMA1 KO HeLa cells (Fig. 4A–C and Supplementary Material, Fig. S5C and D). We previously reported that endogenous C2 and C10 form foci in the intermembrane space that extend into the mitochondrial cristae in WT HeLa cells [6]. Strikingly, in the absence of OMA1, C2 and C10 foci exhibited substantially increased intensity (Fig. 4A–C and Supplementary Material, Fig. 5C and D). As the pattern was similar for both C2 and C10, we focused on C2 in subsequent analyses. High-intensity foci were present in the majority of OMA1 KO cells and absent in OMA1 KO cells re-expressing WT but not protease-dead OMA1 (Fig. 4A and B). The appearance of these foci did not appear to be driven by a global change in bioenergetics, as membrane potential in OMA1 KO HeLa cells was similar to WT HeLa cells (Supplementary Material, Fig. S5E). Thus, the pattern of C2 and C10 distribution is greatly altered in the absence of OMA1 with intensification of C2/C10 foci in the mitochondrial network.
Figure 4.
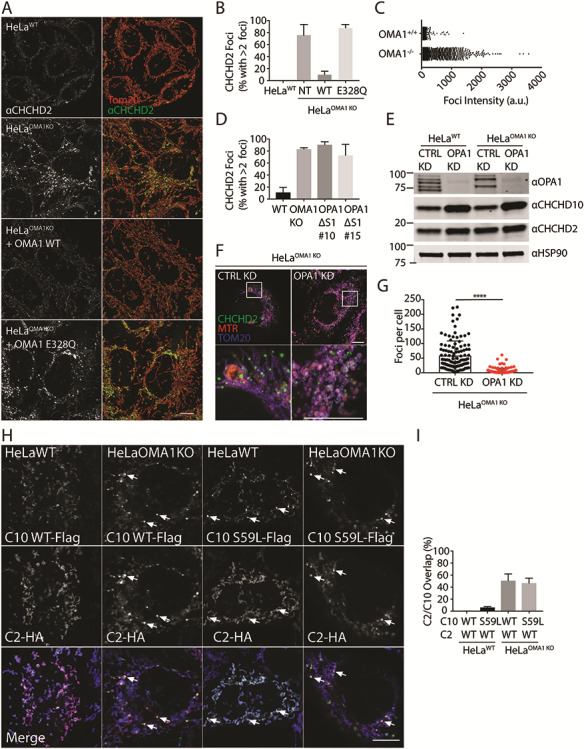
Altered C2/C10 localization and processing in the absence of OMA1. (A) Airyscan confocal images from of WT and OMA1 KO HeLa cells immunostained for Tom20 (red) and C2 (green), where indicated OMA1 KO HeLa cells stably expressed OMA1 WT or the protease-dead OMA1 E328Q mutation. Scale bar = 20 μm. (B) Quantification of cells with >2 intense foci in (A). More than 150 cells were scored in three biological replicates. (C) Graph depicts intensities of individual foci from representative WT and OMA1 KO HeLa cells in (A) and (B). Error bars represent standard error of the mean. (D) Quantification of cells with >2 intense C2 foci in WT or two independent OPA1Δs1 HeLa cell lines (#10 and #15). More than 150 cells were scored in three biological replicates for each condition. Error bars represent standard error of the mean. (E) Immunoblot of WT and OMA1 KO HeLa cells treated with control or OPA1 siRNA. HSP90 served as the loading control. Immunoblot is representative of three biological replicates. (F) Representative Airyscan confocal images of OMA1 KO HeLa cells treated with control or OPA1 siRNA and stained for C2 (green), Mitotracker Red (MTR) (red), and Tom20 (blue). (G) Quantification of intense C2 foci in OMA1 KO Hela cells following treatment with either control or OPA1 siRNA. More than 150 cells were scored in three biological replicates for each condition. ****P < 0.0001. (H) Representative confocal images of WT or OMA1 KO HeLa cells transiently co-transfected with CHCHD2-hemagglutinin (HA) and either CHCHD10(WT)-Flag or CHCHD10(S59L)-Flag, immunostained for Flag and HA. (I) Quantification of (H) for CHCHD10 foci that overlap with CHCHD2 foci. More than 20 cells in each condition were analyzed from N = 3 biological replicates. Scale bars in all images = 10 μm. All confocal images were acquired as a z-stack and represented as a maximum projection image.
Given OPA1 processing is altered in OMA1 KO cells, we next investigated the dependence of high-intensity C2 foci on OPA1 and its processing. To assess whether C2 foci formation depends on OPA1 processing by OMA1, we used genomic editing to delete the canonical OMA1 cleavage site within the endogenous OPA1 locus (HeLaOPA1Δs1). This blocked formation of the canonical OMA1-cleaved products c and e—although residual degradation of OPA1 was observed following mitochondrial depolarization by CCCP, possibly because of limited cleavage by OMA1 at a non-canonical site (Supplementary Material, Fig. S5F). Importantly, C2 foci were observed also in HeLaOPA1Δs1 cells, indicating that partially blocking cleavage of OPA1 by OMA1 leads to formation of high-intensity C2 foci (Fig. 4D and Supplementary Material, Fig. S5G). To further assess the dependence of C2/C10 foci formation on OPA1, we depleted OPA1 using small interfering RNA (siRNA) (Fig. 4E). Interestingly, C10 levels increased following OPA1 knockdown (KD) in HeLa cells (302 ± 75% in HeLaWT and 325 ± 85% in HeLaOMA1KO, P = 0.0018). Despite an increase in total C2 protein levels, C2 foci were found to be significantly decreased following OPA1 KD in OMA1 KO cells (Fig. 4F and G). Together, these findings demonstrate that C2 foci formation is a consequence of L-OPA1 stabilization.
The C2/C10 foci resembled those that we previously observed to form spontaneously with transient expression of the C10 mutant S59L [6]. To further explore the relationship between these foci, we co-expressed C2(WT)-HA with C10(WT)-Flag or C10(S59L)-Flag. Only C10(S59L)-Flag formed foci in WT HeLa cells; notably, however, it failed to recruit C2(WT)-HA into the foci (Fig. 4H and I). In contrast, both C10(S59L)-Flag and C10(WT)-Flag formed foci in similar numbers in HeLa OMA1 KO cells. The majority of these foci also contained C2-HA. Although not conclusive, we hypothesize these intense foci may represent stalled intermediates that can form either from failure of C10(S59L) to disperse, because of increased hydrophobicity in the N-terminal α-helix, or from blocked OPA1 cleavage in the absence of OMA1.
A pool of C2/C10 is degraded following activation of OMA1 by mitochondrial stressors
We and others previously demonstrated that C2 and C10 exhibit short half-lives in cultured cells [5,6]. To test whether OMA1 or another intermembrane space-facing protease such as YME1 or PARL might be the protease responsible for basal C2/C10 degradation, we assessed C2/C10 levels in KO HeLa cell lines both at steady state and following inhibition of translation with cycloheximide (CHX). CHX treatment times were chosen based on our prior work [6]. Similar to our findings in HEK293 cells, we found that C2/C10 steady-state levels are slightly increased in HeLa cells following loss of OMA1 when normalized for loading (Supplementary Material, Fig. S5H). C2/C10 protein levels were slightly increased following KO of PARL and slightly decreased following loss of YME1 (Supplementary Material, Fig. S6A and B). However, C2/C10 half-lives were similar to WT cells following KO of OMA1, YME1 or PARL, demonstrating that these proteases are not solely responsible for basal turnover of C2/C10 (Fig. 5A and Supplementary Material, Fig. S6A and B) [25]. Thus, another protease or combination of proteases may be responsible for basal turnover of C2/C10.
Figure 5.
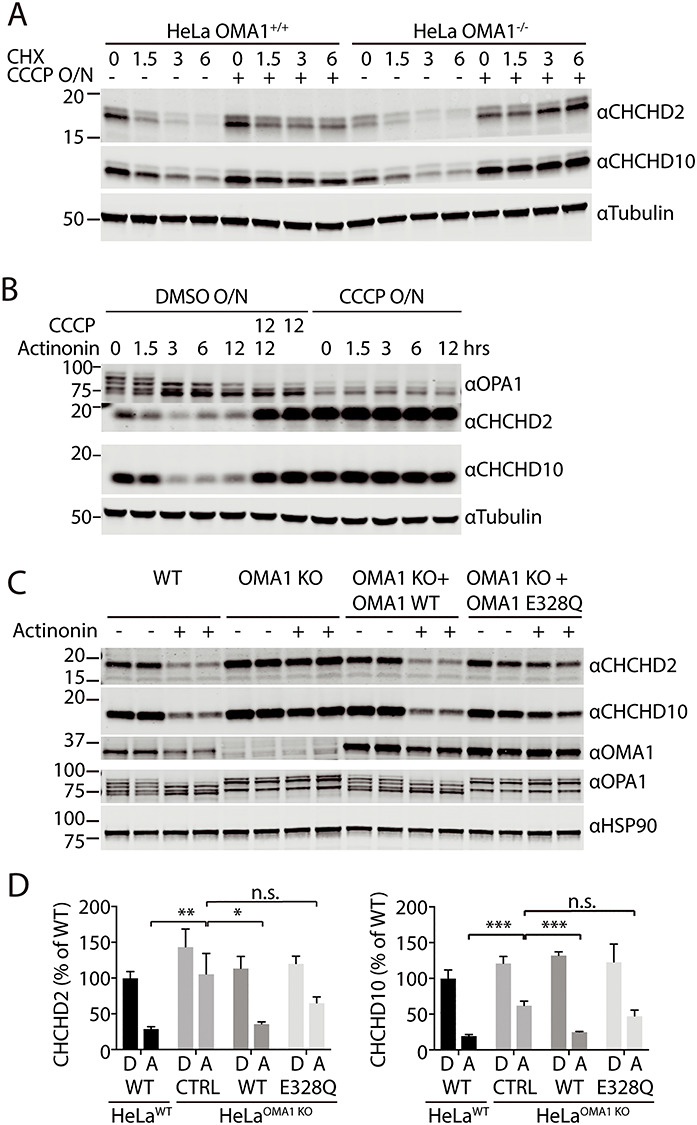
C2/C10 are degraded by OMA1 activated by mitochondrial stress. (A) Immunoblot of OMA1 KO and WT HeLa cells treated with dimethyl sulfoxide (DMSO) or 10 μm CCCP overnight (O/N) and then treated with 100 μm CHX for the indicated number of hours. GAPDH or tubulin served as controls where indicated. (B) Immunoblot of WT HeLa cells treated with DMSO or 10 μm CCCP O/N, followed by the addition of actinonin, CCCP or 125 μm actinonin + 10 μm CCCP for the indicated number of hours. Tubulin served as a loading control. N = 3 biological replicates per sample. (C) Immunoblot of WT and OMA1 KO HeLa cell lines, which stably express OMA1 WT or OMA1 E328Q mutation where indicated, treated with either vehicle or 125 μm actinonin for 3 h. HSP90 served as the loading control. (D) Quantification of C2 and C10 levels from (C). N = 3 biological replicates per sample. The levels of C2 of each cell type following DMSO (D) or actinonin (A) treatment are normalized to those from WT HeLa with DMSO treatment. Three biological replicates were quantified. Error bars represent standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001.
We next tested cells subject to mitochondrial stressors known to activate OMA1. Actinonin treatment for 3 h has been previously shown to accumulate aberrant mitochondrial DNA (mtDNA)-encoded polypeptides in the inner membrane and potently activate OMA1 prior to collapse of the mitochondrial membrane potential [37]. Unexpectedly, we found that C2/C10 levels are reduced following 3 h treatment with actinonin (Fig. 5B–D). This reduction was dependent on OMA1, as it was partially blocked in OMA1 KO cells. Consistently, degradation was restored in OMA1 KO cells stably re-expressing WT but not catalytically inactive OMA1. The block in degradation in OMA1 KO cells was more robust for C2 than C10 but was observed for both. The direction of the effect is the same for C2 and C10, which suggests that the general mechanism of regulation is likely similar for C2 and C10; however, there appear to be quantitative differences in their regulation following mitochondrial stress. Together, these findings suggest that OMA1 is dispensable for basal turnover of C2/C10, but, when activated by inner membrane proteostatic stress, degrades C2/C10.
OMA1 is also potently activated by mitochondrial uncoupling with drugs such as CCCP [20,21]. As we reported previously and in contrast to treatment with actinonin, C2/C10 levels increased in response to loss of membrane potential [6]. This increase in C2/C10 abundance with uncoupling was independent of OMA1, as it occurred in both WT and OMA1 KO HeLa cells (Fig. 5A and B). Co-treatment with CCCP and actinonin resulted in accumulation of C2/C10 similar to treatment with CCCP alone (Fig. 5B). Thus, although C2/C10 is degraded following OMA1 activation in the presence of a mitochondrial membrane potential, the pool of C2/C10 that accumulates with loss of membrane potential is protected from OMA1 and other proteases.
Together, these findings suggest that OMA1 degrades C2/C10 upon activation but is not responsible for their basal turnover. This also implies that the formation of C2/C10 intense foci in the absence of OMA1 is not a direct consequence of failed C2/C10 degradation by OMA1, but rather reflects loss of OMA1 activity against another substrate (e.g. L-OPA1).
In summary, these findings suggest that the functional interaction between C2/C10 and OPA1/OMA1 is bidirectional. C2/C10 loss or C10 mutation leads to OMA1 activation and OPA1 processing. Conversely, OMA1 loss blocks the stress-induced degradation of C2/C10; and C2/C10 collect in intense foci in OMA1-deficient mitochondria.
Pathogenic C10 mutation activates OPA1 cleavage by OMA1 in vivo
Having identified a functional interaction between pathogenic C10 mutants and OMA1 activation in cell culture, we next asked whether mutant C10 activates OMA1 in vivo.
To model the pathogenic S59L mutation in human C10, we generated a transgenic mouse with the equivalent substitution in mouse C10 (S55L in mouse; but, for clarity, numbering corresponding to human C10 is used throughout) (Fig. 6A). Consistent with two recent reports on independent lines [27,28], we found that C10S59L/+ mice developed cardiomyopathy by early adulthood (17–42 weeks). Percent ejection fraction (%EF) in C10S59L/+ animals was significantly lower than WT littermates by echocardiography (39.6% vs. 59.7%, P = 0.003) (Fig. 6B–D and Supplementary Material, Table S2). Similarly, peak left ventricular outflow tract (LVOT) and pulmonary artery (PA) velocities were significantly reduced, indicating, respectively, left and right ventricular dysfunction (624.1 vs. 1204 mm/s, P < 0.0001 and 424.5 vs. 686.5 mm/s, P < 0.0001, respectively). Additionally, the anterior and posterior left ventricular walls were significantly thickened in diastole by echocardiography (Supplementary Material, Fig. S7A and Table S2). Vacuole formation in cardiac tissue and fibrosis were apparent on histology with hematoxylin and eosin (H&E) and Masson trichrome staining, respectively (Fig. 6E). There was also an increase in apoptotic nuclei detected in C10S59L/+ KI mice compared to control mice detected by the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNNEL) assay (Supplementary Material, Fig. S7B). Primary fibroblasts from WT and C10S59L/+ KI mice were similarly susceptible to cytochrome c release in response to H2O2, suggesting that tissue context is important for apoptotic cell death because of mutant C10 (Supplementary Material, Fig. S7C).
Figure 6.
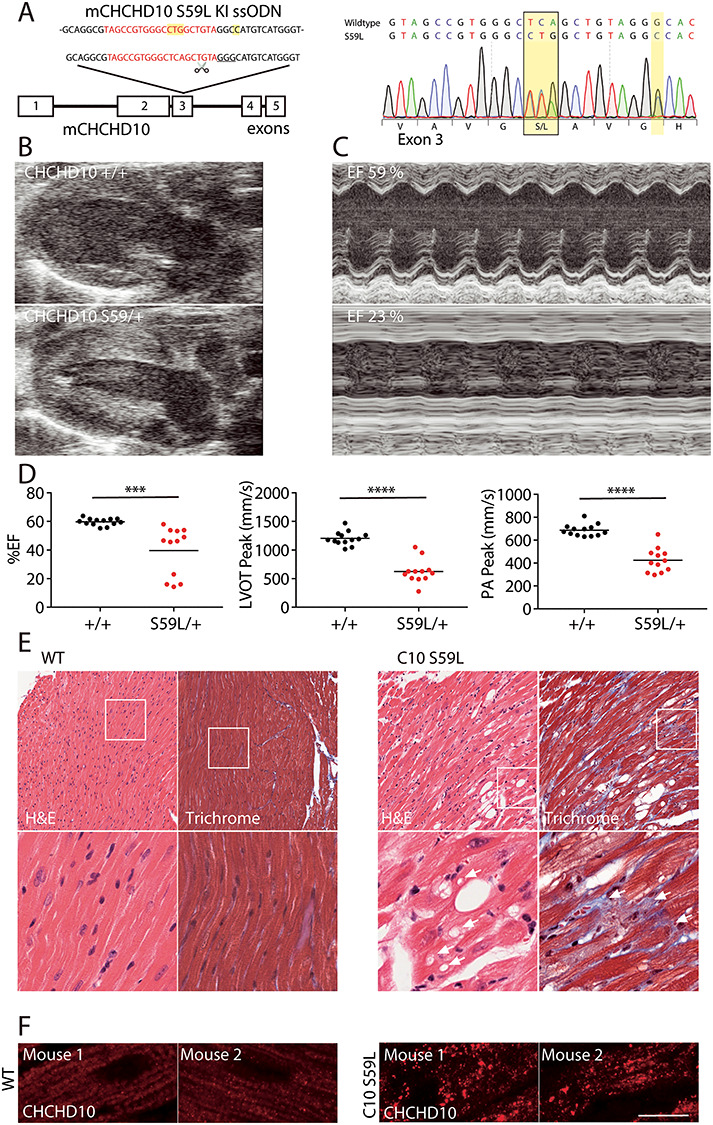
C10 S59L/+ KI mice develop cardiomyopathy. (A) C10S59L/+ KI mice were generated using CRIPSR/Cas9 genomic editing. The targeted CRISPR cut site (scissors), guide RNA sequence (red) and PAM sequence (underlined) are indicated. The donor single-stranded donor oligonucleotides (ssODN) used for repair is shown above. Sanger sequencing demonstrates the TCA → CTG codon change, as well as a silent GGG → GGC mutation at G58 introduced to disrupt the PAM sequence. (B) Representative echocardiogram in long axis view from WT and C10S59L/+ mice demonstrating thickening of the ventricular wall in C10S59L/+ mice. (C) Motion (M)-mode view from the short axis of echocardiogram, demonstrating decreased left ventricular contractility of the C10S59L/+ heart. (D) Graph representing %EF, LVOT velocities and PA velocities of litter-matched WT (N = 13) and C10S59L/+ (N = 12) mice between the ages of 17–42 weeks. Velocities were measured by pulsed-wave Doppler echocardiography. ***P < 0.001 and ****P < 0.0001. (E) Representative images of H&E stain (left) and Masson trichrome stain (right) from adult WT and C10S59L/+ mice. Arrows indicate vacuoles. Images are representative of at least three animals per genotype evaluated. (F) Airyscan confocal images of C10 immunostaining of heart slices from WT and C10S59L/+ mice. Scale bar = 10 μm. All confocal images were acquired as a z-stack and represented as a maximum projection image. Images are representative of at least two animals per genotype evaluated.
Additionally, we found C10 distribution to be greatly altered in cardiac tissue from C10S59L/+ KI mice, forming aggregates, similar to what was reported previously [27] (Fig. 6F). Unlike the foci that formed in cell culture with transient overexpression of C10 S59L, however, C10 aggregates in cardiac tissue did not co-localize with mitochondria (Figs 4H and 6F and Supplementary Material, Fig. S7D). Together, these findings confirmed development of hypertrophic cardiomyopathy in C10S59L/+ KI mice by early adulthood, previously reported in two independent mouse models [27,28].
Examining cardiac tissue lysates from C10S59L/+ mice, we observed a substantial elevation in C2 and C10, consistent with a recent report in vivo and our prior observation that C2 and C10 increase in response to mitochondrial distress in cell culture (Fig. 7A) [6,27]. Interestingly, C2 and C10 protein levels decreased slightly with aging in WT mice. methylenetetrahydrofolate dehydrogenase (MTHFD)2 and MTHFD1L, which are sensitive markers of the mt-ISR in muscle [38,39], also progressively increased from younger (9–13 weeks) to older (22–37 weeks) C10S59L/+ animals (Fig. 7A). Consistent with activation of the mt-ISR in C10S59L/+ mice, expression of transcription factors reported to mediate the response in cardiac tissue, ATF4, ATF5, CHOP and Myc [39], was significantly elevated relative to younger WT mice (Fig. 7B). Altogether, in the younger C10S59L/+ versus younger WT comparison, altered expression reached gene-level significance for 15 of 22 pre-specified mt-ISR-associated genes, recently identified to be differentially regulated genes (DEGs) in the heart of an independent C10 S59L/+ mouse line (Supplementary Material, Fig. S8 and Table S3) [27].
Figure 7.
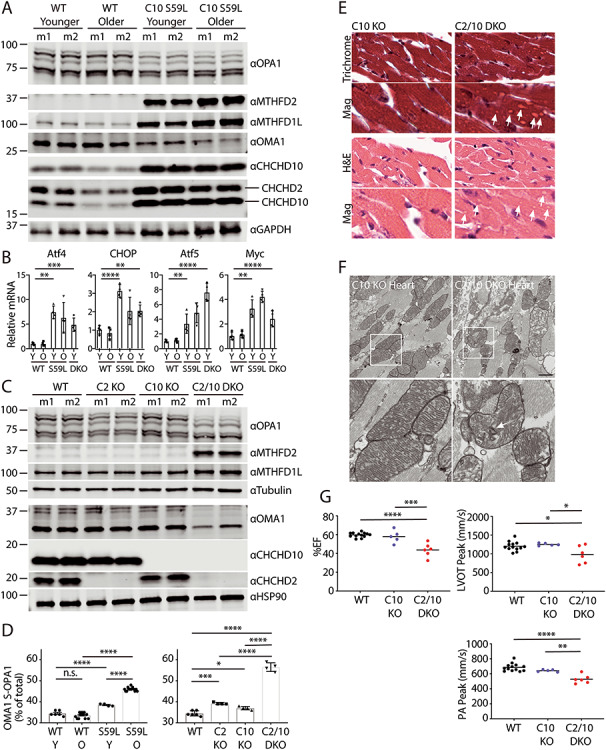
OMA1 activation coincident with cardiomyopathy and mt-ISR phenocopied in C2/C10 DKO and C10S59L/+ KI mice. (A) Representative immunoblot of heart lysates from younger (9–13 weeks) and older (36–37 weeks) WT and C10S59L/+ KI mice. Lysates from two different mice (m1 and m2) are shown for each condition. (B) Relative transcript levels of Atf4, CHOP, Atf5 and Myc in heart extracts of younger WT, C10S59L/+ KI and C2/C10 DKO mice (9–13 weeks) and older WT and C10S59L/+ KI (22–37 weeks) mice measured by microarray and normalized to average of younger WT mice. N = 4 for each group. Error bars represent standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.001 after correction for multiple comparisons among groups for each gene. Array wide corrected P-values are shown in Supplementary Material, Table S3. (C) Representative immunoblot of heart lysates from WT, C2 KO, C10 KO and C2/C10 DKO mice (age 9–13 weeks). Lysates from two different mice (m1 and m2) are shown for each condition. (D) Quantification of OMA1-cleaved S-OPA1 relative to total OPA1 from heart lysates as in (A and C). Note: WT comparison group is same as in the right and left graphs. Error bars represent standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.001 after correction for multiple comparisons. (E) Representative images of H&E stain and Masson trichrome stain from sibling-matched C10 KO and C2/C10 DKO mice (9–13 weeks). Arrows indicate vacuoles. (F) Representative TEM images of C10 KO and C2/C10 DKO hearts. Arrow indicates abnormal cristae. (G) Graph representing echography measurements of %EF and LVOT and PA peak velocities measured for sibling-matched C10 KO and C2/C10 DKO mice and WT mice. WT group in all graphs is reproduced from Figure 6D. C10 KO and C2/C10 DKO mice were 29–56 weeks old at the time of echocardiography. At least five animals per genotype were assessed. Error bars represent standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 after correction for multiple comparisons.
We next assessed OPA1 processing in C10S59L/+ mice. Notably, in the heart, L-OPA1 (isoforms a and b) was excessively processed to OMA1 cleavage products (isoforms c and e) in younger C10S59L/+ mice and to an even greater extent in older C10S59L/+ mice (Fig. 7A and D). OMA1 levels were likewise decreased in C10S59L/+ hearts, as expected given that OMA1 is degraded shortly after its activation [21]. Sampling lysates from the atria and ventricles, separately, demonstrated that the myocardium is similarly affected throughout (Supplementary Material, Fig. S9A and B). OMA1 activation was absent in the brain and liver but present in the muscle (Supplementary Material, Fig. S10A). C2 and C10 protein levels were also normal in the liver and brain but slightly elevated in skeletal muscle; thus, OMA1 activation correlates with elevated C2 and C10 levels in vivo. Notably, OMA1 was not activated in either C10S59L/+ or C10S59L/S59L primary fibroblast lines, in contrast to C2/C10 DKO cell lines, suggesting that the pathogenic effect of mutant C10 may require its high expression in the heart (Supplementary Material, Fig. S10B). Together, these findings demonstrate that the pathogenic C10 mutation activates OPA1 cleavage by OMA1 in vivo, possibly following its accumulation with C2 in response to an inciting mitochondrial stress.
C2/C10 DKO activates OPA1 cleavage by OMA1 and mt-ISR in vivo
Guided by parallel results in DKO fibroblasts and C10S59L/+ mutant hearts, we assayed OPA1 processing in lysates from younger C2/C10 single KO and DKO mice (9–13 weeks). Whereas processing of OPA1 to OMA1-cleaved forms was mildly increased in hearts from C2/C10 single KO mice, loss of both C2 and C10 had a synergistic effect on L-OPA1 cleavage (Fig. 7C and D; Fig. 1F and G). OMA1 levels were likewise decreased in DKO hearts, as expected given that OMA1 is degraded shortly after its activation. OMA1 cleavage of L-OPA1 was also apparent in liver, brain and skeletal muscle at this age, albeit to a lesser extent (Supplementary Material, Fig. S10A and B). Comparing OMA1 activation in the younger C10S59L model with the younger KO models showed that OMA1 activation in C10S59L to be intermediate between C10 single KO and C2/C10 DKO (Fig. 7D). Additionally, whereas OMA1 activation was observed in all tissues examined in C2/C10 DKO mice, it was only observed in skeletal muscle and heart of C10 S59L mice, coincident with the elevated C2 and C10 levels in those tissues. Together, these findings demonstrated that OMA1 is activated by C2 and C10 loss in vivo, phenocopying OMA1 activation by C10S59L/+.
It was recently reported that C10 KO does not activate the mt-ISR in the mouse heart, in contrast to C10S59L/+ KI. Consistently, we did not see an increase in MTHFD2 or MTHFD1L in hearts of either C10 or C2 single KO mice (Fig. 7C). In contrast, DKO of C2 and C10 led to a substantial elevation in MTHFD2 and a more modest elevation in MTFHD1L protein levels. MTHFD2 was similarly activated in skeletal muscle and brain (Supplementary Material, Fig. S11A and B). Consistently, expression of transcription factors associated with the mt-ISR (namely, ATF4, ATF5 and Myc) was significantly elevated in younger DKO mice compared with younger WT mice with CHOP reaching nominal significance (Fig. 7B and Supplementary Material, Table S3). Altogether, 6/22 genes in a pre-specified set of mt-ISR-associated genes previously identified as DEGs in the C10S59L/+ KI model reached gene-level significance in the overall DKO versus WT comparison (Fig. 7B and Supplementary Material, Fig. S8). With the exception of ATF5 expression, generally the mt-ISR was less robust in C2/C10 DKO mice compared with C10S59L/+ KI mice examined at a similar age. Comparison of total DEGs between DKO versus WT and C10S59L/+ versus WT also underscored the similarity in overall transcriptional response and the greater severity in the younger C10 S59L/+ mice compared with younger DKO mice. The C10 S59L/+ versus WT comparison revealed 1386 DEGs, whereas the C2/C10 DKO versus WT comparison produced 548 DEGs, 290 (52.9%) of which were shared. Thus, a similar transcriptional response occurs in both C2/C10 DKO and C10S59L/+ KI hearts but involves a more extensive set of genes in C10S59L/+ KI hearts.
As we observed early biochemical changes in the heart, we next assessed our younger DKO mice for early signs of cardiomyopathy. Although fibrosis was not apparent by 13 weeks, numerous vacuoles similar to those seen in C10S59L/+ KI were observed in tissues from DKO mice with both H&E and Masson trichrome staining (Fig. 7E). These changes were not apparent in single C10 KO littermates. In an exploratory analysis, there was also a slight increase in apoptotic nuclei detected in C2/C10 DKO mice by the TUNEL assay compared with their C10 KO littermates (Supplementary Material, Fig. S7B). This trend did not reach significance but was underpowered as limited samples were available for analysis. Primary fibroblasts from WT and C2/C10 DKO mice were similarly susceptible to cytochrome c release in response to H2O2 treatment, suggesting that tissue context is important for apoptotic cell death because of loss of C2/C10 (Supplementary Material, Fig. S7C). Consistent with TEM findings in C10 KO and C2/C10 DKO primary fibroblasts and L-OPA1 cleavage patterns in the heart, TEM cristae structure was abnormal in the majority of mitochondria from a C2/C10 DKO heart but normal in most mitochondria from a litter-matched C10 KO heart [96/116 (82.8%) abnormal vs. 11/143 (7.6%) abnormal] (Fig. 7F). To assess for cardiomyopathy, we evaluated our oldest cohorts of C2/C10 DKO and C10 KO littermates by echocardiography. By 29–56 weeks, significantly decreased %EF was apparent in DKO mice when compared with both their C10 KO littermates and unrelated WT mice of the same strain (43.70% vs. 59.96% and 43.70% vs. 59.68%, P < 0.0001 and P = 0.0002, respectively) (Fig. 7G and Supplementary Material, Table S2). Similarly, LVOT peak velocity and PA peak velocity were decreased in DKO mice compared with their C10 KO littermates and unrelated WT mice, consistent with left and right ventricular dysfunction, respectively (Fig. 7G and Supplementary Material, Table S2). Overall, cardiac dysfunction in C2/C10 DKO mice was milder than in unrelated C10S59L/+ KI mice. Whereas all C10S59L/+ KI mice had succumbed to cardiomyopathy by 14 months, the two oldest C2/C10 DKO mice in our colony were still alive at 14 months. Thus, similar to C10S59L/+ KI mice, C2/C10 DKO mice develop cardiomyopathy, but to a milder degree. Together, these findings demonstrate that C2/C10 DKO partially phenocopies C10S59L/+ KI mice in vivo with the development of cardiomyopathy and activation of mt-ISR and OMA1 in affected tissues.
Discussion
In this study, we identified a functional interaction between C2/C10 and stress-induced processing of L-OPA1 by OMA1 that is shared by C2/C10 loss of function and mutant C10 gain-of-function. This suggests a model in which the normal function of C2/C10 may be related to the mechanism of C10 mutant pathogenesis (Fig. 8). Together, our findings suggest that WT C2 and C10 are required within a narrow range of expression to maintain normal L-OPA1/S-OPA1 balance, as either too little or too much WT C2 or C10 actives OMA1 (Fig. 8A). This may account for dramatic changes in C2/C10 protein expression levels in response to, for example, bioenergetic stress [6].
Figure 8.
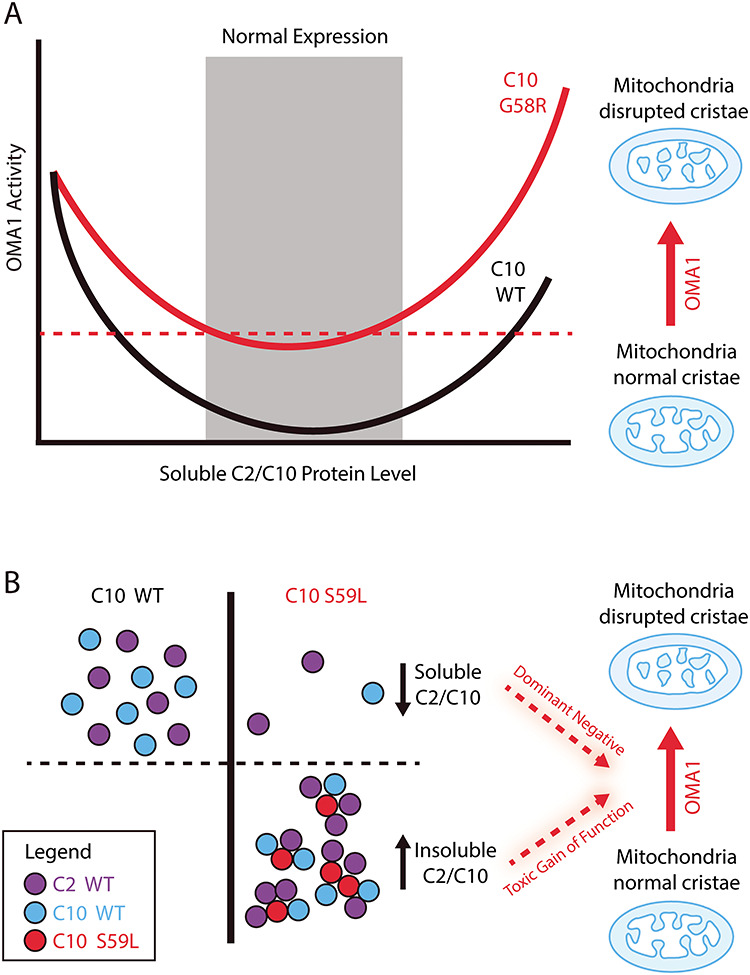
Model of OMA1 activation because of loss of C2/C10 and mutant C10. (A) WT C2/C10 are required in a narrow range of expression to prevent L-OPA1 processing by the stress-induced peptidase OMA1. Too low or too high expression of WT C2 or C10 activates OMA1. Activated OMA1 disrupts mitochondrial cristae by cleaving L-OPA1. Some C10 mutations such as G58R appear to lower the threshold concentration for activation of OMA1 by C10. (B) Aggregation-prone mutants of C2 or C10, such as C10 S59L, may activate OMA1 by a different mechanism. Aggregation-prone C10 S59L may co-aggregate with soluble C2 and C10, pulling them into an insoluble fraction. In this setting, OMA1 may be activated by decreased soluble C2/C10, thereby mimicking C2/C10 DKO in a dominant negative mechanism. Alternatively (or in addition), increased insoluble C2/C10 may activate OMA1 by a toxic gain-of-function mechanism.
In this context, we propose that mutant C2 or C10 may cause aberrant OMA1 activation by one of two mechanisms. With the first mechanism, the mutation may cause C2 or C10 to activate OMA1 at a lower concentration than its WT counterpart (Fig. 8A). If a given cell has a concentration of mutant C2 or C10 above that threshold, then OMA1 may be activated at the physiological level of C2 and C10 in the absence of aggregation. We suggest that this is the mechanism by which the myopathy-causing mutation C10 G58R leads to OMA1 activation (Fig. 8A). This is based on the observations that: (1) C10 G58R is not prone to insolubility [6] and (2) C10 G58R activates OMA1 at a lower concentration than C10 WT in cellular models (Fig. 3C–G). We observed similar behavior in C10 with the middle α-helical region deleted, suggesting that the G58R substitution may induce a large structural change that leads to OMA1 activation at a lower concentration than WT C10.
In a second mechanism, mutation in C2 or C10 may render the protein prone to aggregate with WT C2 and C10, leading to increased total C2/C10 and decreased soluble C2/C10 (Fig. 8B). In this scenario, OMA1 may be activated because of decreased soluble C2/C10 (phenocopying C2/C10 DKO by a dominant negative mechanism), increased insoluble C2/C10 (triggering activation of OMA1 by a toxic gain-of-function mechanism) or a combination. We propose that this is the mechanism by which the ALS/FTD/myopathy-causing mutation C10 S59L activates OMA1 based on the observations that: (1) C10 S59L forms aggregates in cells and affected tissues, (2) C10 S59L leads to decreased solubility of C2 and C10 in affected tissues and (3) increased tissue C2/C10 levels coincides with OMA1 activation, both of which are observed in heart and muscle but not liver and brain [27,40]. Notably, the PD-causing mutation C2 T61I is also prone to insolubility in cell culture and in autopsy tissue, and the SMAJ-causing mutation C10 G66V is prone to insolubility in cell culture, suggesting that this mechanism may also be relevant to the pathogenesis of these disorders [6,41,42].
Examining primary cells from the first C2/C10 DKO mouse, we found cristae to be decreased and occasionally circular and detached from the boundary membrane, phenocopying cristae abnormalities observed in fibroblasts and skeletal muscle from patients with C10 mutations (S59L and G58R, respectively) [1,2]. These findings are also consistent with swirled cristae recently reported in Drosophila muscle lacking the predominant C2/C10 ortholog in that tissue, CG5010 [8]. As an explanation for cristae abnormalities, we identified L-OPA1 cleavage by activated OMA1. This was observed in HEK293 cells, primary fibroblasts and heart and skeletal muscle in vivo. OMA1 activation was independent of the mild decrease in MICOS subunits observed in C2/C10 DKO cells, as disruption of the MICOS complex with MIC60 KD was insufficient to activate OMA1. However, we did not rule out the possibility that the observed decrease in MICOS subunits with C2/C10 loss may also contribute to the cristae abnormalities. We extended these findings to mutant C10, identifying OMA1 processing of OPA1 as a key event in affected organs, such as the heart and skeletal muscle. Thus, we identify L-OPA1 cleavage by OMA1 as a novel mechanism for cristae abnormalities resulting from dominant C10 mutation or C2/C10 loss.
C2 and C10 are 58% identical and have a single ortholog in yeast and Drosophila, suggesting that they are paralogs produced by gene duplication [43]. They have also been shown to directly interact [5–7]. Whereas these observations imply that C2 and C10 may be functionally redundant, our results provide the strongest evidence that this is indeed the case. Cardiomyopathy with strong activation of OMA1 and the mt-ISR was observed in C2/C10 DKO but not single C2 or C10 KO mice. In cell culture, loss of C2 as well as C10 had a synergistic effect on L-OPA1 processing, and enforced expression of either C2 or C10 in C2/C10 DKO cells partially rescued L-OPA1 processing. Together, these findings demonstrate that C2 and C10 are partially functionally redundant paralogs.
We found that L-OPA1 processing also provided a useful functional assay for C2 and C10 activity more generally. Notably, the ALS/FTD/myopathy-causing mutation C10 S59L failed to rescue C2/C10 processing, suggesting that it does not retain function. This was not due solely to its propensity for insolubility, as two other disease-causing mutations C2 T61I and C10 G66V, which are more insoluble in cell culture, retained function [6]. Together with the observation that the C10 G58R mutation strongly activates OMA1 at a low concentration, these observations suggests that mutations in C2/C10 may form at least three distinct classes, namely, those with (1) decreased solubility and retained function (C2 T61I and C10 G66V), (2) decreased solubility and loss of function (C10 S59L) and (3) retained solubility and dominant OMA1 activation (C10 G58R). We speculate that these differences in C2/C10 mutant behavior may account for phenotypic differences clinically, as the mutant protein interacts with distinct cellular environments, including myocytes (with C10 G58R and C10 S59L), lower motor neurons (with C10 S59L and C10 G66V), peripheral nerves (C10 G66V), upper motor neurons (C10 S59L), cortical neurons (C10 S59L) and dopaminergic neurons (C2 T61I) [1–4,44].
The precise trigger for OMA1 activation in the absence of C2 and C10 is not clear at present. However, there are at least three plausible models: (1) C2/C10 loss or mutation causes bioenergetic stress that indirectly activates OMA1, (2) C2/C10 loss or mutation causes proteostatic stress that indirectly activates OMA1 and (3) C2/C10 directly modulates L-OPA1 processing by OMA1.
Our data seem to rule out the first model, that is, OMA1 is activated indirectly because of bioenergetic collapse. Under growth conditions in which OMA1 was activated in C2/C10 DKO cells, we found that oxygen consumption and membrane potential were minimally affected. Additionally, inducing a moderate bioenergetic stress in HEK293 cells (by inhibiting mtDNA translation with chloramphenicol) resulted in a reduced membrane potential and a more severe reduction in COX2 expression compared with HEK293 C2/C10 DKO cells but failed to activate OMA1. Together, these data suggest that loss of C2/C10 does not cause bioenergetic stress substantial enough to activate OMA1 on its own.
The second model, that loss of C2/C10 triggers OMA1 activation by increasing proteostatic stress, is intriguing and has some circumstantial evidence in support of it. First, we observe that insoluble protein increases in the mitochondrial fraction following C2/C10 loss. Additionally, previous studies have identified interactions between either C2 or C10 and the proteases YME1 and PARL, which are components of the intermembrane space-facing SPY complex that also contains OMA1 [25,33]. We were able to replicate an interaction between C2 and YME1 in this study (although interactions with PARL and SLP-2 were not observed under our study conditions). These findings are at least consistent with a transient interaction between C2/C10 and components of this complex of quality control proteases and a possible role for C2/C10 in maintaining proteostasis. Notably, other twin CX9C proteins in the intermembrane space have chaperoning functions in the intermembrane space mediated by the CX9C motif, including Mdm35/TRIAP1, CHCHD4/Mia40 and Cox11 [34,45]. Together, these findings suggest that it is at least plausible that C2/C10 may activate OMA1 through decreased proteostasis perhaps because of loss of a chaperoning function. Direct evidence for such a function, however, is not clear at present and will require further work.
Finally, these are some circumstantial evidence in favor of the third model, i.e. C2/C10 directly modulates processing of L-OPA1 by OMA1. First, we found that both low and high levels of C2/C10 activate OPA1 processing by OMA1. This suggests that the proteins are required in a narrow range of expression to prevent OMA1 activation. Additionally, we found that C2/C10 distribution dramatically changes in the absence of OMA1, and C2/C10 protein levels increase in response to OPA1 or OMA1 reduction. This suggests a bidirectional functional interaction between C2/C10 and OPA1/OMA1. Finally, we find that C2/C10 are possible substrates of activated OMA1, suggesting that OMA1 and C2/C10 may be in close proximity. Against this model, we failed to detect direct physical interactions between C2 and OPA1 or OMA1 stable enough for immunocapture, and at least the predominant pool of C2/C10 appears in complexes that are of distinct in size from the predominant OPA1- and OMA1-containing complexes by blue native polyacrylamide gel electrophoresis (BN-PAGE) [7,15,25]. However, this does not rule out more transient interactions between C2/C10 and either OPA1 or OMA1. Thus, while the direct model has circumstantial evidence in favor of it, a direct link between C2/C10 and OPA1/OMA1 has not been identified, and, if present, will require further work to establish.
Identifying OMA1 activation as a key event in C10 mutant pathogenesis also nominates OMA1 as a potential therapeutic target for C2/C10-related disorders. Although OMA1 is required for OPA1 processing in response to mitochondrial stress, it is not essential for mammalian life [46]. The OMA1 KO mouse is viable, and loss-of-function mutations occur in humans at close to the rate predicted by chance, including two ‘KO’ adult individuals with homozygous frameshift mutations (observed SNV/expected SNV = 0.78 (0.55–1.13); gnomAD v.2.1.1 database) [47]. Indeed, in the setting of a high proteostatic load, such as that resulting from loss of YME1 or the scaffolding protein prohibitin-2, its excessive activation may be detrimental, leading to heart failure and neurodegeneration, respectively [48,49].
In this scenario, OMA1 cleavage of OPA1 and the resulting cristae abnormalities may be responsible for the observed cardiomyopathy, multiple mtDNA deletions and mt-ISR activation following C2/C10 loss or dominant mutation. L-OPA1 is known to be necessary for mtDNA stability, raising the possibility that OPA1 cleavage by OMA1 may drive mtDNA instability in patients and the C10 S59L/+ mouse [2,28]. Similarly, OMA1 activation was recently suggested to cause mt-ISR activation, by cleaving the inner mitochondrial membrane protein DELE1 [50,51]. DELE1 subsequently retro-translocates to the cytosol and activates the eIF2α kinase heme-regulated eIF2? kinase (HRI), which in turn, activates ATF-4. It is interesting to speculate that this may be the mechanism for mt-ISR activation in response to C2/C10 loss and dominant C10 mutation, given the strong OMA1 activation observed in this context. However, we cannot rule out the alternative explanation that a mitochondrial stress resulting from C2/C10 loss or dominant mutation may independently cause OMA1 activation and drive cardiomyopathy, mtDNA mutations and the mt-ISR. In this case, OMA1 activation would be a biomarker of C2/C10-related toxicity but would not cause the subsequent tissue degeneration. Future studies of mutant C10 (and C2) on an OMA1 KO background will be important for evaluating OMA1 as a therapeutic target, as well as determining which aspects of the mutant phenotype (e.g. cardiomyopathy, mt-ISR activation, multiple mtDNA deletion, cytosolic TDP-43 accumulation) are dependent on L-OPA1 processing by OMA1.
Materials and Methods
Generation of transgenic mice
C2−/−, C10−/− and C10S59L/+ transgenic lines were produced using clustered regularly interspaced short palindromic repeats (CRISPR)/caspase 9 (Cas9) endonuclease-mediated genome editing on the C57Bl6J background. CRISPR guide RNAs targeted exon 2 and 4 in mC10 and exon 1 and 4 in mC2 to generate breaks in DNA. Animals were screened for large deletions lacking the aforementioned exons by Sanger sequencing. C10 KI mice were generated with a guide RNA targeting near the S55 position (equivalent of S59 in human C10) in mC10 co-injected with Cas9 and an ssODN containing a TCA → CTG substitution at the S55L codon in the C57Bl6J background. All animal studies were approved by the Animal Care Use Committee at the National Institute of Neurological Disorders and Stroke (NINDS) intramural program. Both genders were used in all studies.
Cell culture
WT, C2, C10 and DKO fibroblast cells were generated from 1-day-old newborn pups. Pups were removed from litter and placed in Dulbecco’s modified Eagle medium (DMEM) containing 1% L-glutamine, 15% fetal bovine serum (FBS), MEM non-essential amino acid solution (MEM-NEAA), 1% sodium pyruvate, ×1 penicillin/streptomycin (pen/strep), ×1 gentamicin and ×1 amphotericin B. Sterile scalpels were used to cut the skin from the animals into thin slices around 1–2 mm squares. The tissues were transferred to a 1% gelatin-coated plate containing the dissection media and covered with sterile cover slips. High-glucose DMEM with sodium pyruvate and supplemented with 10% FBS and pen/strep was used for all other tissue culture and for primary fibroblasts after passage 1.
OPA1-null mouse embryonic fibroblasts (MEFs) were obtained from the American Type Culture Collection (ATCC CRL-2995) and transformed MEF WT cells with SV40 were obtained as a kind gift from the laboratory of David Chan (Caltech). Generation of the HEK293 C2/C10 DKO cell line was described previously [6]. HeLaPARL KO, HeLaOMA1 KO, HeLaOMA1 KO stably expressing OMA1 WT or OMA1 E328Q and matched HeLaWT cell lines were a kind from the Richard Youle (the National Institutes of Health) and have been described previously [52]. HeLaYME1 KO and matched HeLaWT cell lines were a kind gift from the Thomas Langer (Cologne) and have been described previously [53].
HEK293 OMA1 KO and HEK293 C2/C10/OMA1 TKO lines were produced by CRISPR/Cas9, using the pSpCas9(BB)-2A-GFP (PX458) plasmid with the guide sequences 5′-CACCGAGTGCAATCGAGACGTCCCG-3′ and 5′-CACCGAGATCGCACACGCAGTCCTG-3′ targeting exons 4 and 5, respectively. KO was verified by immunoblotting for OMA1 and characteristic change in OPA1 processing. The HeLaOPA1Δs1 was produced using the pSpCas9(BB)-2A-GFP (PX458) plasmid with the guide sequence 5′-CACCGCGTTTAGAGCAACAGATCG-3′ and the ssODN sequence: 5′-gggttgcatatttatctttaagGTTCTCCGGAAGATCGTGGATCTGAAAGTGACAAGCATTTTAGAAAGg-3′. pSpCas9(BB)-2A-GFP (PX458) was a gift from the Feng Zhang (Addgene plasmid# 48138; http://n2t.net/addgene:48138; RRID:Addgene_48138). HeLa cells stably expressing C2-His6 or C2-Flag were generated by lentiviral transduction, using the pCIG3 vector containing the C2-His6 or C2-Flag sequence. pCIG3 (pCMV-IRES-GFP version 3) was a gift from the Felicia Goodrum (Addgene plasmid# 78264; http://n2t.net/addgene:78264; RRID:Addgene_78264). The fibroblasts were incubated at 37°C with 5% CO2 and 5% O2. All other cell lines were incubated at 37°C with 5% CO2 and 21% O2.
Antibodies
The following antibodies were used: anti-FLAG M2 (Sigma, cat# F1804-1MG), anti-Tom20 F-10 (Santa Cruz, cat# sc-17764), anti-C2 C-term (Sigma, cat# HPA027407), anti-cytochrome c 6H2.B4 (BD Pharmingen, cat# 556432), anti-OPA1 (BD Bioscience, cat# 612606), anti-β-Tubulin (Sigma, cat# T8328), OMA1 (ProteinTech, cat# 17116-1-AP), anti-C2 (ProteinTech, cat# 19424-1-AP), anti-C2 (ProteinTech, cat# 6602-1-Ig), anti-C10 (Sigma, cat# HPA003440), anti-MIC60 (ProteinTech, cat# 430909), anti-MIC19 (Sigma, cat# HPA042935), anti-MIC27 AntI-MIC27 (Sigma, cat# HPA000612), anti-GAPDH (ProteinTech, cat# 60004), anti-HSP90 (ProteinTech, cat# 13171-1-AP), anti-MTHFD2 (ProteinTech, cat# 12270-1-AP) and anti-MTHFD1L (ProteinTech, cat# 16113-1-AP).
TEM
Mouse fibroblasts were fixed with 4% glutaraldehyde (Electron Microscopy Services) in electron microscopy (EM) buffer (0.1 N sodium cacodylate at pH 7.4 with 2 mM calcium chloride) for 30 min at room temperature and then at 4oC for at least 24 h. Samples were washed with buffer and treated with 1% osmium tetroxide in 0.1 N cacodylate buffer at pH 7.4 for 1 h on ice, washed and en bloc stained with 0.25–1% uranyl acetate in 0.1 N acetate buffer at pH 5.0 overnight at 4°C, dehydrated with a graded series of ethanol washes and finally embedded in epoxy resins. Ultrathin sections (70 nm) were stained with lead citrate and imaged with a JEOL 1200 EXII TEM. Images were evaluated and scored by a blinded experimenter. Mitochondria were scored as abnormal if they displayed cristae that were ring shaped and detached from the boundary membrane, had a total course of the cristae turned >90°, and/or there was extreme variability in mitochondrial diameter in which the region with the thinnest caliber was less than half of the region with the greatest caliber. Cristae were only scored when the outer and boundary inner membrane surrounding the cristae were sharply in view.
Oxygen consumption rate
Seahorse Extracellular Flux Analyzer XF (Aglient) was used to measure the oxygen consumption rates (OCR) of mouse primary fibroblasts. Fibroblast cells were seeded at 20 000 cells/well in XF96 cell culture microplates coated with 1% gelatin and incubated for 24 h at 37°C with 5% CO2 in DMEM containing glucose. An hour before the assay, the cells were placed in DMEM lacking bicarbonate and culture plates were moved to a 37°C incubator with atmospheric CO2. The assay measures OCR at basal levels, after addition of 1 μm oligomycin, 2 μm FCCP and 0.5 μm of rotenone and antimycin. Data are represented as means ± standard error of at least six biological replicates, normalized by protein concentration as determined by the bichinchoninic acid (BCA) assay.
Mass spectrometry
HEK293WT cells were labeled to >95% incorporation of 12C6,15N2 L-Lysine and 13C6,15N4 L-Arginine (Cambridge Isotope Laboratories) in SILAC DMEM media lacking L-Lysine and L-Arginine (Thermo Fischer). Mitochondria were isolated from both heavy-labeled HEK293WT and light-labeled HEK293DKO cells. Protein concentration was determined by BCA assay and heavy and light mitochondria were mixed 1:1. The mixed mitochondria were solubilized in 1% digitonin and complexes were separated on a BN-PAGE gel with bovine heart mitochondria used as a molecular weight standard. Lanes were cut into 10 gel slices and digested with trypsin. Extracted peptides were desalted and used for LC-MS/MS data acquisition on an Orbitrap Luminos mass spectrometer (Thermo Fisher Scientific) coupled with a 3000 Ultimate high-pressure liquid chromatography instrument (Thermo Fisher Scientific). Peptides were separated on an ES802 column (Thermo Fisher Scientific) with mobile phase B increasing from 2 to 27% over 60 min. The LC-MS/MS data were acquired in data-dependent mode. The resolution of the survey scan (300–1600 m/z) was set at 60k at m/z 400 with a target value of 10 × 106 ions. Collision-induced dissociation was performed on the top 10 most abundant precursor ions with an isolation window of 2.0 Da. Database search and heavy/light (H/L) ratio calculation were performed using MaxQuant against Sprot Human database [54]. Oxidation (M), Label: 13C(6)15N(2) (K) and Label: 13C(6)15N(4) (R) were included as variable modifications in the database search. Identified protein groups tagged as reverse, contaminants or identified only by modified peptide were filtered out using the MaxQuant companion program, Perseus. For the HEK293WT condition intensities were normalized for each protein group across the gel slices by dividing protein intensities in each gel slice by the sum of the intensities from all of the gel slices. Intensities for the HEK293DKO values were calculated by multiplying the relative WT value for each protein group in each gel slice by the light/heavy (L/H) ratio quantified for each slice. When a ratio was missing the WT value was multiplied by 1.
Transfection, immunocytochemistry and confocal microscopy
Cells were plated on eight-well chambered slides (Ibidi) at a seeding density of 25 000 cells/well overnight, where indicated cells were transfected with plasmids using FugeneHD (Promega). C10 (del ΔH) was produced by cloning a gene block (IDT) comprising a human codon optimized C10 Δ(45–69) into a YFP-N1 vector (Clontech) digested with NotI and BamHI to replace the YFP insert, using HiFi NEB-Builder (NEB). Other plasmids were cloned as described previously [6]. For siRNA KD experiments, cells were plated on day 1, transfected with siRNA on day 2, transfected again on day 4 and fixed on day 6. OPA1 was targeted using SMARTpool: On-TARGETplus (Dharmacon, cat# L-005273-00-0005) and Mic60/IMMT was targeted using siGENOME Human IMMT (10989) siRNA—SMARTpool (Dharmacon, cat# M-019832-01-0005), using RNAiMAX (Thermo Fischer). Cells were fixed for 10 min with 4% paraformaldehyde (Electron Microscopy Services) in 1× phosphate-buffered saline (PBS). Cells were then permeabilized with 0.25% Triton X-100, blocked with 1% bovine serum albumin (BSA) for at least 30 min and then incubated in primary antibody for 2 h to overnight in PBS at 4°C. Cells were washed with PBS three times and then incubated at room temperature for 1 h with Alexa Fluor goat secondary antibodies (488, 555, 594 or 647) at 1:1000 dilution in 1% BSA. Confocal microscopy was performed on Fluoview3000 (Olympus) or Airy LSM 880 (Zeiss). The antibodies used for immunocytochemistry were anti-FLAG M2 (Sigma cat# F1804-1MG), anti-Tom20 F-10 (Santa Cruz, cat# sc-17764), anti-C2 C-term (Sigma, cat# HPA027407), anti-HA (3F10) rat monoclonal (Sigma, 11867423001) and anti-cytochrome c 6H2.B4 (BD Pharmingen, cat# 556432).
Fragmentation of the mitochondria in HeLa cells stained with Mitotracker Red (Thermo Fischer) or immunostained for cytochrome c or Tom20 was scored to fall in into one of three categories: tubular, short-tubes and fragmented mitochondrial network. Cells with and without transfection were determined through anti-Flag M2 (Sigma) immunocytochemistry staining. To determine number of cells with >2 foci, HeLa cells stained with Tom20 (Santa Cruz) and C2 (Sigma) were imaged with confocal microscopy. The experimenter counted the number of cells in each condition that contained >2 foci visible by wide-field fluorescent microscopy using a ×60 oil objective. To determine C2 foci count and intensity in HeLa cells, Imaris software was used to identify and calculate fluorescent intensity from z-stack confocal images. The ‘Spots’ detection tool in Imaris was used to identify foci from C2 (Sigma) immunostaining. For co-localization of C10 and C2 foci, z-stack confocal images were analyzed in Imaris. Then, the total number of C10 foci that co-localized with any C2 focus was calculated. Two foci are considered co-localized if their centers were within 0.01 mm.
Echocardiography
Mice were lightly anesthetized with isoflurane during examinations and placed in the supine position over a heated platform with electrocardiography leads and a rectal temperature probe. Heart images were acquired using the Vevo2100 ultrasound system (VisualSonics, Toronto, Canada) with a 30 MHz ultrasound probe (VisualSonics, MS-400 transducer). Measurements were made from standard 2D and M-mode images from the parasternal long axis and midpapillary short axis views of the left ventricle.
Histology analysis
Hearts were harvested from mice anesthetized with isoflurane. Harvested hearts were cut into three segments with a razor blade. The apex and base were snap frozen and a midsection was fixed in 4% paraformaldehyde (Electron Microscopy Services) for 24 h at 4oC, washed with PBS, dehydrated and embedded in paraffin (through HistoServ), sliced into 4 μm sections and stained with H&E and Masson trichrome (through HistoServ). Sections were imaged with a wide-field microscope (Zeiss).
For immunohistochemistry, deparaffinized cardiac sections were treated in blocking buffer (5% goat serum in 0.3% Triton X-100, 0.02% NaN2). The sections were then incubated in primary antibody anti-C10 (1:1000, Sigma-Aldrich, HPA003440) and anti-Tom20 (Santa Cruz sc-17764 1:100 dilution) in PBS overnight at 4oC. After washes in PBS, the sections were incubated with secondary antibodies (Alexa Fluor-labeled goat antibodies) in blocking buffer for 1 h at room temperature. After immunostaining, the tissue was mounted with mounting agent (KPL 71-00-16) and imaged with constant microscopy settings on a Fluoview3000 confocal microscope (Olympus) with a ×40 silicon oil immersion lens. The antibodies used are anti-Tom20 F-10 (Santa Cruz, cat# sc-17764) and anti-C10 (Sigma, cat# HPA003440).
Immunoblotting
Lysates from HEK293, HeLa and fibroblast cells were processed as described previously [6]. Mouse heart and skeletal muscle were lysed in radioimmunoprecipitation assay buffer and a buffer containing 20 mm Tris pH 7.8, 137 mm NaCl, 2.7 mm KCl, 1 mm MgCl2, 1% Triton X-100, 10% glycerol, 1 mm ethylenediamine tetraacectic acid and 1 mm dithiothreitol, respectively with 1% proteinase inhibitor. Lysates were sonicated by a Vibra-Cell Ultrasonic Disruptor for 15 s, four times at an output level of 20. Protein concentration was determined by the BCA assay. Lysates were separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and analyzed by immunoblotting. OMA1-produced S-OPA1 cleavage products were calculated by measuring the maximum intensity of each of the five bands in a linescan of the optical density of the five OPA1 bands on the blot using the software program FIJI. After subtracting background intensity, the peaks of c and e bands were summed and divided by the sum of the five bands (a–e) to obtain percentage of OMA1-generated S-OPA1 from total OPA1.
RNA expression studies
For each for the following groups, RNA was extracted from hearts of four mice: WT younger, WT older, C10S59L/+ younger, C10S59L/+ older and C2/C10 DKO mice. RNA expression was measured using the Clariom_S_Mouse microarray (Affymetrix). Data were analyzed using the Transcriptome Analysis Console Software (version 4.0) on the default settings (Affymetrix). Specifically, data were summarized using the Gene-Level–signal space transformation - robust multiple-array average normalization method (SST-RMA) model. Differentially expressed genes (DEGs) were required to have gene-level fold change ≤2 or >2 and gene-level P < 0.05 measured using the ebayes analysis of variance (ANOVA) method. Additionally, expression levels of a pre-specified list of 24 individual genes consisting of C2, myc and 22 genes previously identified as mt-ISR-related DEGs in an independent C10S59L/+ mouse line were extracted for each sample and analyzed nominally using one-way ANOVA with Sidak’s multiple comparison test applied for each gene.
Statistical analysis
For all statistical analyses with two samples, Student’s t-tests (two-tailed) were performed in Excel (Microsoft) or Prism (Graphpad). For analyses comparing >2 samples one-way ANOVA with Sidak’s multiple comparison test was performed in Prism (Graphpad). Statistical analysis for RNA expression studies is discussed above. The animal and cellular experiments were performed as exploratory studies and explicit power analysis was not performed prior to experimentation. For initial animal studies of C10S59L/+ KI mice groups of echocardiography groups, N > 12 were chosen based on review of the literature and prior experience. Based on the large effect size observed for the C10S59L/+ KI mice and availability of animals old enough for analysis groups of five–six litter-matched C2/C10 DKO and C10 KO mice were tested by echocardiography. For RNA expression studies, groups of four were used based on review of the literature and prior experience. For all cellular studies, with the exception of electron microscopy, at least three replicates on two occasions were performed. For electron microscopy studies of cells, three biological replicates were performed with the following exceptions: one biological replicate was performed for electron microscopy analysis of mouse heart tissue and one replicate for analysis of OPA1 KO mouse fibroblasts. No randomization was used in the animal experiments as a treatment was not being tested. Experiments were performed unblinded.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Authors thank Dragan Maric, PhD, and the NINDS Flow Cytometry Core Facility for technical assistance with FACS experiments, Virginia Crocker and the NINDS EM Facility for technical assistance with TEM, Carolyn Smith, PhD, and the NINDS Light Microscopy Facility for technical assistance with confocal microscopy, Abdel Elkahloun, PhD, and the NHGRI/DIR Microarray Core for technical assistance with RNA expression studies, Yan Li at the NINDS Protein/peptide Sequencing Facility for the SILAC-based proteomics analysis, and Richard Youle, PhD, for critical reading of the manuscript and insightful comments.
Conflict of Interest Statement. None declared.
Funding
The National Institutes of Health (the Intramural Research Program of the NINDS).
References
- 1. Ajroud-Driss S., Fecto F., Ajroud K., Lalani I., Calvo S.E., Mootha V.K., Deng H.-X., Siddique N., Tahmoush A.J., Heiman-Patterson T.D. and Siddique T. (2015) Mutation in the novel nuclear-encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy. Neurogenetics, 16, 1–9. doi: 10.1007/s10048-014-0421-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bannwarth S., Ait-El-Mkadem S., Chaussenot A., Genin E.C., Lacas-Gervais S., Fragaki K., Berg-Alonso L., Kageyama Y., Serre V., Moore D.G. et al. (2014) A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain J. Neurol., 137, 2329–2345. doi: 10.1093/brain/awu138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Funayama M., Ohe K., Amo T., Furuya N., Yamaguchi J., Saiki S., Li Y., Ogaki K., Ando M., Yoshino H. et al. (2015) CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: a genome-wide linkage and sequencing study. Lancet Neurol., 14, 274–282. doi: 10.1016/S1474-4422(14)70266-2. [DOI] [PubMed] [Google Scholar]
- 4. Penttilä S., Jokela M., Bouquin H., Saukkonen A.M., Toivanen J. and Udd B. (2015) Late onset spinal motor neuronopathy is caused by mutation in CHCHD10. Ann. Neurol., 77, 163–172. doi: 10.1002/ana.24319. [DOI] [PubMed] [Google Scholar]
- 5. Burstein S.R., Valsecchi F., Kawamata H., Bourens M., Zeng R., Zuberi A., Milner T.A., Cloonan S.M., Lutz C., Barrientos A. and Manfredi G. (2018) In vitro and in vivo studies of the ALS-FTLD protein CHCHD10 reveal novel mitochondrial topology and protein interactions. Hum. Mol. Genet., 27, 160–177. doi: 10.1093/hmg/ddx397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang X., Wu B.P., Nguyen D., Liu Y.-T., Marani M., Hench J., Bénit P., Kozjak-Pavlovic V., Rustin P., Frank S. and Narendra D.P. (2018) CHCHD2 accumulates in distressed mitochondria and facilitates oligomerization of CHCHD10. Hum. Mol. Genet., 27, 3881–3900. doi: 10.1093/hmg/ddy270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Straub I.R., Janer A., Weraarpachai W., Zinman L., Robertson J., Rogaeva E. and Shoubridge E.A. (2018) Loss of CHCHD10-CHCHD2 complexes required for respiration underlies the pathogenicity of a CHCHD10 mutation in ALS. Hum. Mol. Genet., 27, 178–189. doi: 10.1093/hmg/ddx393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meng H., Yamashita C., Shiba-Fukushima K., Inoshita T., Funayama M., Sato S., Hatta T., Natsume T., Umitsu M., Takagi J., Imai Y. and Hattori N. (2017) Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun., 8, 15500. doi: 10.1038/ncomms15500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Genin E.C., Plutino M., Bannwarth S., Villa E., Cisneros-Barroso E., Roy M., Ortega-Vila B., Fragaki K., Lespinasse F., Pinero-Martos E. et al. (2016) CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med., 8, 58–72. doi: 10.15252/emmm.201505496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou W., Ma D., Sun A.X., Tran H.-D., Ma D.-L., Singh B.K., Zhou J., Zhang J., Wang D., Zhao Y. et al. (2019) PD-linked CHCHD2 mutations impair CHCHD10 and MICOS complex leading to mitochondria dysfunction. Hum. Mol. Genet., 28, 1100–1116. doi: 10.1093/hmg/ddy413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davies K.M., Strauss M., Daum B., Kief J.H., Osiewacz H.D., Rycovska A., Zickermann V. and Kühlbrandt W. (2011) Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc. Natl. Acad. Sci., 108, 14121–14126. doi: 10.1073/pnas.1103621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ban T., Ishihara T., Kohno H., Saita S., Ichimura A., Maenaka K., Oka T., Mihara K. and Ishihara N. (2017) Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol., 19, 856–863. doi: 10.1038/ncb3560. [DOI] [PubMed] [Google Scholar]
- 13. Davies K.M., Anselmi C., Wittig I., Faraldo-Gómez J.D. and Kühlbrandt W. (2012) Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc. Natl. Acad. Sci., 109, 13602–13607. doi: 10.1073/pnas.1204593109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dudkina N.V., Sunderhaus S., Braun H.-P. and Boekema E.J. (2006) Characterization of dimeric ATP synthase and cristae membrane ultrastructure from Saccharomyces and Polytomella mitochondria. FEBS Lett., 580, 3427–3432. doi: 10.1016/j.febslet.2006.04.097. [DOI] [PubMed] [Google Scholar]
- 15. Frezza C., Cipolat S., Martins de Brito O., Micaroni M., Beznoussenko G.V., Rudka T., Bartoli D., Polishuck R.S., Danial N.N., De Strooper and Scorrano L. (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell, 126, 177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 16. Harner M., Körner C., Walther D., Mokranjac D., Kaesmacher J., Welsch U., Griffith J., Mann M., Reggiori F. and Neupert W. (2011) The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J., 30, 4356–4370. doi: 10.1038/emboj.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hoppins S., Collins S.R., Cassidy-Stone A., Hummel E., DeVay R.M., Lackner L.L., Westermann B., Schuldiner M., Weissman J.S. and Nunnari J. (2011) A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J. Cell Biol., 195, 323–340. doi: 10.1083/jcb.201107053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paumard P., Vaillier J., Coulary B., Schaeffer J., Soubannier V., Mueller D.M., Brèthes D., di Rago and Velours J. (2002) The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J., 21, 221–230. doi: 10.1093/emboj/21.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. von der Malsburg, Müller J.M., Bohnert M., Oeljeklaus S., Kwiatkowska P., Becker T., Loniewska-Lwowska A., Wiese S., Rao S., Milenkovic D. et al. (2011) Dual role of Mitofilin in mitochondrial membrane organization and protein biogenesis. Dev. Cell, 21, 694–707. doi: 10.1016/j.devcel.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 20. Ehses S., Raschke I., Mancuso G., Bernacchia A., Geimer S., Tondera D., Martinou J.-C., Westermann B., Rugarli E.I. and Langer T. (2009) Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol., 187, 1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Head B., Griparic L., Amiri M., Gandre-Babbe S. and van der Bliek (2009) Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol., 187, 959–966. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anand R., Wai T., Baker M.J., Kladt N., Schauss A.C., Rugarli E. and Langer T. (2014) The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol., 204, 919–929. doi: 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Magri S., Fracasso V., Plumari M., Alfei E., Ghezzi D., Gellera C., Rusmini P., Poletti A., Bella D.D., Elia A.E., Pantaleoni C. and Taroni F. (2018) Concurrent AFG3L2 and SPG7 mutations associated with syndromic parkinsonism and optic atrophy with aberrant OPA1 processing and mitochondrial network fragmentation. Hum. Mutat., 39, 2060–2071. doi: 10.1002/humu.23658. [DOI] [PubMed] [Google Scholar]
- 24. Merkwirth C., Dargazanli S., Tatsuta T., Geimer S., Löwer B., Wunderlich F.T., von Kleist-Retzow, Waisman A., Westermann B. and Langer T. (2008) Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev., 22, 476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wai T., Saita S., Nolte H., Müller S., König T., Richter-Dennerlein R., Sprenger H.-G., Madrenas J., Mühlmeister M., Brandt U., Krüger M. and Langer T. (2016) The membrane scaffold SLP2 anchors a proteolytic hub in mitochondria containing PARL and the i-AAA protease YME1L. EMBO Rep., 17, 1844–1856. doi: 10.15252/embr.201642698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. MacVicar T.D.B. and Lane J.D. (2014) Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. J. Cell Sci., 127, 2313–2325. doi: 10.1242/jcs.144337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Anderson C.J., Bredvik K., Burstein S.R., Davis C., Meadows S.M., Dash J., Case L., Milner T.A., Kawamata H., Zuberi A. et al. (2019) ALS/FTD mutant CHCHD10 mice reveal a tissue-specific toxic gain-of-function and mitochondrial stress response. Acta Neuropathol. (Berlin), 138, 103–121. doi: 10.1007/s00401-019-01989-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Genin E.C., Madji Hounoum B., Bannwarth S., Fragaki K., Lacas-Gervais S., Mauri-Crouzet A., Lespinasse F., Neveu J., Ropert B., Augé G. et al. (2019) Mitochondrial defect in muscle precedes neuromuscular junction degeneration and motor neuron death in CHCHD10S59L/+ mouse. Acta Neuropathol. (Berlin), 138, 123–145. doi: 10.1007/s00401-019-01988-z. [DOI] [PubMed] [Google Scholar]
- 29. Tang J., Zhang K., Dong J., Yan C., Hu C., Ji H., Chen L., Chen S., Zhao H. and Song Z. (2020) Sam50-Mic19-Mic60 axis determines mitochondrial cristae architecture by mediating mitochondrial outer and inner membrane contact. Cell Death Differ., 27, 146–160. doi: 10.1038/s41418-019-0345-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sekine S., Kanamaru Y., Koike M., Nishihara A., Okada M., Kinoshita H., Kamiyama M., Maruyama J., Uchiyama Y., Ishihara N., Takeda K. and Ichijo H. (2012) Rhomboid protease PARL mediates the mitochondrial membrane potential loss-induced cleavage of PGAM5. J. Biol. Chem., 287, 34635–34645. doi: 10.1074/jbc.M112.357509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Darshi M., Mendiola V.L., Mackey M.R., Murphy A.N., Koller A., Perkins G.A., Ellisman M.H. and Taylor S.S. (2011) ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function. J. Biol. Chem., 286, 2918–2932. doi: 10.1074/jbc.M110.171975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ding C., Wu Z., Huang L., Wang Y., Xue J., Chen S., Deng Z., Wang L., Song Z. and Chen S. (2015) Mitofilin and CHCHD6 physically interact with Sam50 to sustain cristae structure. Sci. Rep., 5, 16064. doi: 10.1038/srep16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Floyd B.J., Wilkerson E.M., Veling M.T., Minogue C.E., Xia C., Beebe E.T., Wrobel R.L., Cho H., Kremer L.S., Alston C.L. et al. (2016) Mitochondrial protein interaction mapping identifies regulators of respiratory chain function. Mol. Cell, 63, 621–632. doi: 10.1016/j.molcel.2016.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Potting C., Tatsuta T., König T., Haag M., Wai T., Aaltonen M.J. and Langer T. (2013) TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab., 18, 287–295. doi: 10.1016/j.cmet.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 35. Rainbolt T.K., Atanassova N., Genereux J.C. and Wiseman R.L. (2013) Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation. Cell Metab., 18, 908–919. doi: 10.1016/j.cmet.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Russ W.P. and Engelman D.M. (2000) The GxxxG motif: a framework for transmembrane helix-helix association. J. Mol. Biol., 296, 911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 37. Richter U., Lahtinen T., Marttinen P., Suomi F. and Battersby B.J. (2015) Quality control of mitochondrial protein synthesis is required for membrane integrity and cell fitness. J. Cell Biol., 211, 373–389. doi: 10.1083/jcb.201504062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Khan N.A., Nikkanen J., Yatsuga S., Jackson C., Wang L., Pradhan S., Kivelä R., Pessia A., Velagapudi V. and Suomalainen A. (2017) mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab., 26, 419–428.e5. doi: 10.1016/j.cmet.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 39. Kühl I., Miranda M., Atanassov I., Kuznetsova I., Hinze Y., Mourier A., Filipovska A. and Larsson N.-G. (2017) Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. eLife, 6, e30952. doi: 10.7554/eLife.30952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang X., Wu B.P., Nguyen D., Liu Y.-T., Marani M., Hench J., Bénit P., Kozjak-Pavlovic V., Rustin P., Frank S. and Narendra D.P. (2019) CHCHD2 accumulates in distressed mitochondria and facilitates oligomerization of CHCHD10. Hum. Mol. Genet., 28, 349. doi: 10.1093/hmg/ddy340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cornelissen T., Spinazzi M., Martin S., Imberechts D., Vangheluwe P., Bird M., Strooper B. and Vandenberghe W. (2020) CHCHD2 harboring the Parkinson’s disease-linked T61I mutation precipitates inside mitochondria and induces precipitation of wild-type CHCHD2. Hum. Mol. Genet.. doi: 10.1093/hmg/ddaa028/5739952. [DOI] [PubMed] [Google Scholar]
- 42. Ikeda A., Nishioka K., Meng H., Takanashi M., Hasegawa I., Inoshita T., Shiba-Fukushima K., Li Y., Yoshino H., Mori A. et al. (2019) Mutations in CHCHD2 cause α-synuclein aggregation. Hum. Mol. Genet., 28, 3895–3911. doi: 10.1093/hmg/ddz241. [DOI] [PubMed] [Google Scholar]
- 43. Cavallaro G. (2010) Genome -wide analysis of eukaryotic twin CX 9 C proteins. Mol. BioSyst., 6, 2459–2470. doi: 10.1039/C0MB00058B. [DOI] [PubMed] [Google Scholar]
- 44. Auranen M., Ylikallio E., Shcherbii M., Paetau A., Kiuru-Enari S., Toppila J.P. and Tyynismaa H. (2015) CHCHD10 variant p.(Gly66Val) causes axonal Charcot-Marie-tooth disease. Neurol. Genet., 1, e1. doi: 10.1212/NXG.0000000000000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Longen S., Bien M., Bihlmaier K., Kloeppel C., Kauff F., Hammermeister M., Westermann B., Herrmann J.M. and Riemer J. (2009) Systematic analysis of the twin Cx9C protein family. J. Mol. Biol., 393, 356–368. doi: 10.1016/j.jmb.2009.08.041. [DOI] [PubMed] [Google Scholar]
- 46. Quirós P.M., Ramsay A.J., Sala D., Fernández-Vizarra E., Rodríguez F., Peinado J.R., Fernández-García M.S., Vega J.A., Enríquez J.A., Zorzano A. and López-Otín C. (2012) Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J., 31, 2117–2133. doi: 10.1038/emboj.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P. et al. (2019) Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv, 531210. doi: 10.1101/531210. [DOI] [Google Scholar]
- 48. Korwitz A., Merkwirth C., Richter-Dennerlein R., Tröder S.E., Sprenger H.-G., Quirós P.M., López-Otín C., Rugarli E.I. and Langer T. (2016) Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria. J. Cell Biol., 212, 157–166. doi: 10.1083/jcb.201507022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wai T., García-Prieto J., Baker M.J., Merkwirth C., Benit P., Rustin P., Rupérez F.J., Barbas C., Ibañez B. and Langer T. (2015) Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science, 350, aad0116. doi: 10.1126/science.aad0116. [DOI] [PubMed] [Google Scholar]
- 50. Fessler E., Eckl E.-M., Schmitt S., Mancilla I.A., Meyer-Bender M.F., Hanf M., Philippou-Massier J., Krebs S., Zischka H. and Jae L.T. (2020) A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature. doi: 10.1038/s41586-020-2076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guo X., Aviles G., Liu Y., Tian R., Unger B.A., Lin Y.-H.T., Wiita A.P., Xu K., Correia M.A. and Kampmann M. (2020) Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature. doi: 10.1038/s41586-020-2078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sekine S., Wang C., Sideris D.P., Bunker E., Zhang Z. and Youle R.J. (2019) Reciprocal roles of Tom7 and OMA1 during mitochondrial import and activation of PINK1. Mol. Cell, 73, 1028–1043.e5. doi: 10.1016/j.molcel.2019.01.002. [DOI] [PubMed] [Google Scholar]
- 53. Hartmann B., Wai T., Hu H., MacVicar T., Musante L., Fischer-Zirnsak B., Stenzel W., Gräf R., van den Heuvel, Ropers H.-H. et al. (2016) Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation. eLife, 5. doi: 10.7554/eLife.16078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tyanova S., Temu T. and Cox J. (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc., 11, 2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.