

Abstract
Secondary joint damage is the process by which a single injury can lead to detrimental changes in adjacent tissue structures, typically through the spread of inflammatory responses. We recently developed an in vitro model of secondary joint damage using a murine rotator cuff explant system, in which injuries to muscle and bone cause massive cell death in otherwise uninjured tendon. The purpose of the present study was to test the ability cytokine-targeted and broad-spectrum therapeutics to prevent cell death and tissue degeneration associated with secondary joint damage. We treated injured bone-tendon-muscle explants with either interleukin-1 receptor antagonist, etanercept, or dexamethasone (DEX) for up to 7 days in culture. Only the low-dose DEX treatment was able to prevent cell death and tissue degeneration. We then identified a critical window between 24–72 hours following injury for maximal benefit of DEX treatment through timed administration experiments. Finally, we performed two tendon-only explant studies to identify mechanistic effects on tendon health. Interestingly, DEX did not prevent cell death and degeneration in a model of cytokine-induced damage, suggesting other targets of DEX activity. Future studies will aim to identify factors in joint inflammation that may be targeted by DEX treatment, as well as to investigate novel delivery strategies.
Keywords: dexamethasone, inflammation, tendon, explant, cytokines
Statement of Clinical Significance:
Overall, this work demonstrates beneficial effects of dexamethasone administration on preventing tenocyte death and extracellular matrix degeneration in an explant model of secondary joint damage, supporting the clinical use of low-dose glucocorticoids for short-term treatment of joint inflammation.
Introduction:
Tendon degeneration or tendinopathy is a chronic disorder marked by a substantial increase in tissue cellularity, a progressive disruption and disorganization of the extracellular matrix, and a loss of mechanical function. Together, these changes weaken the tendon, leading to painful and debilitating acute tendon tears. While this end-stage degeneration is extremely common in the clinic, particularly with the elderly population, the pathogenesis is still unclear. The initiating factors, as well as disease progression, are multifactorial and complex, although overuse exercise is frequently implicated as a major risk factor.1 Historically the role of inflammation in tendinopathy has been somewhat controversial, but recent studies finding evidence of multiple relevant cell types (neutrophils, macrophages, T cells, etc.) and the presence of inflammatory signaling in pathological human samples2–4 have shifted perspective.
One scenario where inflammation has been well established to play a significant role is the process of secondary joint damage, where acute injury to one tissue can influence the health of adjacent tissues within the joint. The release of cytokines into the joint from the injured tissue can cause cell death, extracellular matrix degeneration, and aberrant tissue signaling. This mechanism is thought to be a key participant in the progression of post-traumatic osteoarthritis in the knee, where there is a clear relationship between an anterior cruciate ligament (ACL) and/or meniscus tear and subsequent cartilage damage.5,6 Recent studies have been able to mimic the disease progression of post-traumatic osteoarthritis through in vitro and in vivo models leading to significant advancements in our understanding of inflammation-induced cartilage degeneration.7,8
Secondary joint damage has been implicated in rotator cuff injury and disease as well. In particular, several recent studies have shown damage to the biceps tendon and the glenoid articular cartilage in the weeks following an acute rotator cuff tear in rodent models.9–11 Furthermore, high levels of pro-inflammatory cytokines (interleukin-1 (IL-1), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α)) have been measured in human and animal models of acute tendon injury as well as in tendon disease. Concentrations have been reported as much as a 4000-fold increase compared to uninjured controls, with levels measured between 1–20 ng/ml depending on the cytokine.12–15 The presence of these cytokines has been shown to induce matrix degeneration through increased expression of matrix metalloproteinases,16,17 to promote non-tenogenic morphology and signaling,18 and to induce apoptosis.19,20 However, many of these studies have been performed in vitro under 2D cell-alone culture conditions and, therefore, it is still unclear how the native tissue environment of tenocytes and tendon progenitor cells would influence cell response to inflammatory cytokines since we know that the matrix environment plays a critical role in influencing gene expression, particularly metabolic regulation, proliferation, differentiation, and protein synthesis21–24
We recently developed an in vitro model of secondary joint damage using a murine rotator cuff explant system, which allows us to explore the response of intact tendons to physiologically relevant inflammation without disrupting cell-matrix interactions. This model system contains uninjured supraspinatus tendon connected to pieces of bone and muscle in a three-dimensional explant model. In our previous study, we reported that acute trauma to the bone and muscle induces a remarkable release of pro-inflammatory cytokines from both tissues, and that there was a rapid loss of tenocyte viability in the first 48 hours following this injury.25 The levels of pro-inflammatory cytokines produced from this muscle/bone injury are consistent with those previously measured in human synovial fluid immediately following various joint injuries.26–28 We also demonstrated that exposing otherwise healthy flexor explants (tendon-only cultures) to conditioned medium collected from this injury model, or to individual pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α), resulted in similar degenerative changes, indicating the role of inflammatory mediators in inducing tissue damage and degeneration.25
The purpose of the present study was to determine the efficacy of selected therapeutics to preventing inflammation-induced cell death and tissue degeneration caused by secondary joint damage. Specifically, we explored the ability of IL-1 receptor antagonist (‘RA’) and etanercept (‘EN’) to target and prevent detrimental effects causes by IL-1β and TNF-α, respectively. We also tested the efficacy of a low-dose broad spectrum treatment with dexamethasone (‘DEX’), which has shown benefits for native tendon and other connective tissues.7,29,30 After determining that only DEX treatment was capable of preventing cell death and mitigating tissue damage in our secondary joint damage model, we then asked, (1) how long a treatment was necessary to maintain therapeutic benefits, and (2) how long could administration be delayed and still achieve success. Finally, we sought to understand the specific targeting of DEX treatment on joint inflammation through a series of control experiments.
Methods:
Tendon Harvest and Culture
All tendon explants for this study were harvested from 126 C57BL/6J male mice at 4 months of age directly following sacrifice per approved animal use protocol (MIT CAC #0618-061-21). We used two explant culture models: a 10-mm intrasynovial segment of the flexor digitorum longus tendon (‘FDL’), and a rotator cuff bone-tendon-muscle (‘BTM’) explant, as previously described.25 Control BTM and FDL explant data in this manuscript were previously gathered for initial studies25 and are reproduced here for direct comparison with new treatment groups. All explants were washed in sterile 1x phosphate-buffered saline (PBS) for up to 45 minutes and then placed directly into culture medium. Base culture medium consisted of low glucose Dulbecco’s Modified Eagle Media (1 g/L (Corning Life Sciences, Tewksbury, MA)) supplemented with 10% fetal bovine serum (GE Healthcare Life Sciences, Pittsburgh, PA), 100 units/ml penicillin G, 100 μg/ml streptomycin, and 0.25μg/ml amphotericin B (Sigma). Medium was changed every 2 days in culture for up to 7 days. For FDL groups, conditioned medium (‘CM’) and 3-cytokine medium (‘3C’) replaced fresh tendon medium on the 1st medium change, at day 2 in culture. CM consisted of a 1:1 mixture of BTM medium (taken after 24 hours of culture) mixed with fresh medium; CM was previously measured to contain 10ng/mL IL-6, and 10pg/mL TNF-α using a commercially available multiplex ELISA kit.25 To create 3C medium, 10 ng/ml IL-1β, 10 ng/ml IL-6, and 10 pg/ml TNF-α were added to fresh culture medium.
Therapeutics were added to cultures at the same time that cytokine exposure commenced (day 0 for BTM cultures, day 2 for FDL cultures). For the present study, we used 100nM dexamethasone (PeproTech Inc., Rocky Hill, NJ),7,30 0.25 μg/mL etanercept (Immunex Corp., Seattle, WA),31 and 100ng/mL IL-1RA (PeproTech).31,32 Pilot studies were performed to determine the dose-dependence of preventing cell death with various treatments (Supplemental Figs. S1–S3). We performed several experiments investigating the dependence of therapeutic benefit on the timing of DEX administration. All explants were assessed for changes in viability, metabolism, proliferation, biosynthesis, and extracellular matrix content at days 1, 3, 5, and 7.
Explant Viability
Explants (n=4–6/group/time) were assessed for tenocyte viability using fluorescent probes, as described previously.7,25 Whole explants were incubated for 5 minutes in 1x PBS containing fluorescein diacetate (FDA; 4 mg/ml, Sigma) and propidium iodide (1 mg/ml, Sigma) for viable and non-viable cells, respectively. Tendons only (no muscle and bone in BTM cultures) were then imaged using confocal laser scanning microscope at 10x magnification, and composites were created from confocal z-stacks by averaging maximum intensity. On average, stacks of confocal images penetrated approximately 230μm and 150μm into the depth of the BTM and FDL explants, respectively. Viability was quantified using Fiji33 by counting the numbers of viable and non-viable cells via selection of fluorescent maxima under a defined threshold for each region of interest. Viability is expressed as the ratio of viable cells to total cells, expressed as a percentage, within the region of interest.
Metabolism
Metabolism of the entire explant (including bone, tendon and muscle for BTM explants) was assessed during culture via oxidation-reduction conversion of the weakly fluorescent blue dye resazurin to the highly red fluorescent resorufin during a specified culture period.34 Samples (n=5/group/time) were incubated for 3 hours with medium containing 10% v/v resazurin (25 mM, Fisher Scientific, Hampton, NH) diluted in 1x PBS. Following incubation, fluorescence intensity was measured at 530/590nm, then normalized to daily control wells that contained resazurin media with no explants. For BTM cultures, it is important to note that this metabolic assay is representative of the entire explant including bone, tendon and muscle as opposed to all other assays which were specific to tendon.
Tenocyte Biosynthesis and Proliferation, and Analyses of Tendon Biochemical Constituents
Synthesis of sulfated glycosaminoglycans (sGAG) and cell proliferation were measured by incorporation of 35S-sulfate (20 μCi/mL, Perkin-Elmer, Norwalk, CT) and 3H-thymidine (1 μCi/mL), respectively. Following 24-hour incubation, tendons only (n=5/group/time) were harvested from the explant culture, washed in 1x PBS and digested with proteinase K (5mg/ml) (Roche, Indianapolis, MN). Radiolabel incorporation was measured via liquid scintillation counting, while sGAG content was measured using dimethyl methylene blue (DMMB).35 Samples were then hydrolyzed, dried, resuspended and assayed for total collagen content using the hydroxyproline (OHP) assay.36 Double-stranded DNA (dsDNA) content was also measured using the PicoGreen dye-binding assay.37
All data presented in this manuscript are unnormalized, raw data. For both explant cultures, tendons are measured to approximately the same size prior to digestion (~2.5mm for BTM tendons and ~10mm for FDL explants); therefore, we do not believe there is large variation in tissue size. Explant weights were not measured due to inconsistencies in measuring the very low weights of BTM tendons. Finally, since measured dsDNA accounts for DNA from both live and dead cells in tissue samples, we did not feel that normalization to DNA was appropriate for live-cell parameters, such as biosynthesis. However, we recognize the importance of analyzing properties on a per-cell basis, and therefore all data normalized to DNA content are included as Supplemental Data. Furthermore, conclusions discussed in this study reflective raw and DNA-normalized data.
Statistical Comparisons
Based on our study hypotheses, all BTM treatment groups were compared only to the BTM control group. Additional FDL models (+CM, +3C) were compared to the FDL control group. All FDL treatment groups were compared to their appropriate FDL control group (e.g., +CM or +3C), and to the FDL control group to investigate the ability of treatment to rescue negative effects. Statistical comparisons were made between experimental and control groups using two-way ANOVAs with effects for time in culture, treatment and the interaction. Bonferroni-corrected post-hoc t-tests were then used to identify differences within each time point where appropriate, with significance set at *p<0.025 and a trend set at #p<0.05.
Results:
In this injury model, BTM explants exhibit a reduction in tenocyte viability following the first 24 hours in culture, leading to a complete loss of viability by 5 days in culture (Fig.1A).25 Here, we first investigated whether cytokine inhibitors (EN and RA) or a broad-spectrum glucocorticoid (DEX) could ameliorate this loss of tenocyte viability in BTM cultures. RA treatment did not significantly alter viability compared to BTM controls (Supplemental Fig. S4). While EN showed promising results in the first 3 days of culture, viability was still reduced by day 5 similar to controls (Supplemental Fig. S4). Only low-dose DEX administration was capable of reducing the loss of viability in BTM explants (Fig. 1D,E). DEX treatment maintained stable viability levels of 50%, significantly different from BTM controls by day 5 in culture.
Figure 1.
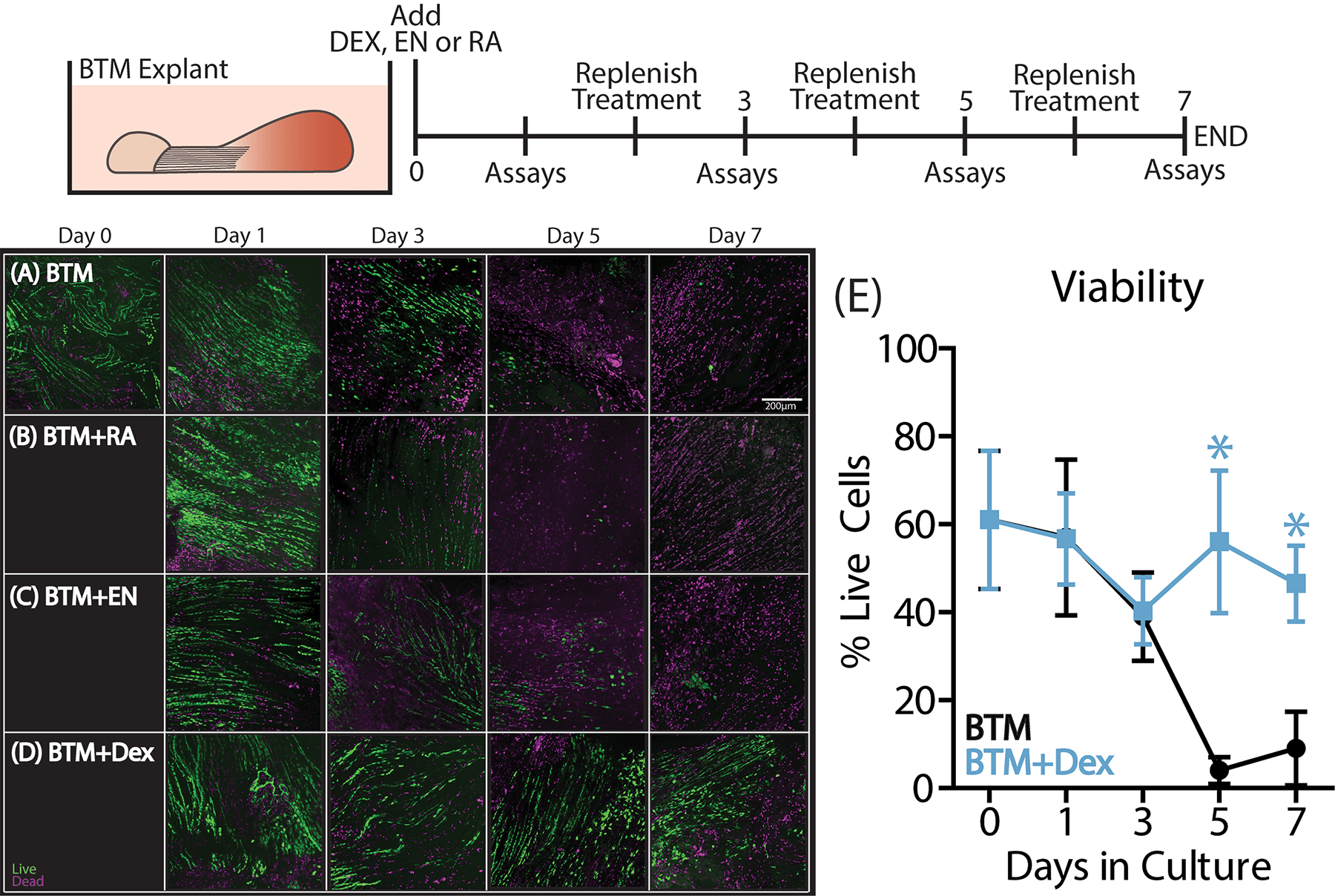
BTM explants were harvested from 4-month old mice and placed into culture with RA (n= 4–6/timepoint), EN (n=4–6/timepoint) or DEX treatment (n=8–10/timepoint) for the duration of the 7-day culture period. Explant viability is reduced in (A) control BTM cultures between days 1 and 5, after which point viability was less than 20%. Neither (B) RA nor (C) EN treatment were capable of preventing this loss in viability. However, (D) low-dose DEX treatment was able to maintain ~50% viability for the duration of the culture period with significant differences between the BTM+DEX and BTM groups at days 5 and 7. Data are presented as mean ± 95% confidence interval with significance represented by stars (*).
While treatment with RA and EN did reduce explant metabolism as well as loss of sGAG from tendon over time in culture (Supplemental Fig. S4), the inability of either treatment to reduce the loss of tenocyte viability prevented them from being considered as a viable treatment options. Since low-dose DEX was able to prevent this injury-induced tenocyte death, the effects of DEX on other measures of tendon health are presented in detail here (Fig. 2, Supplemental Fig. S5). BTM samples incubated with DEX had reduced overall explant metabolism over the course of culture, most notably at days 1, 5, and 7 in culture (Fig. 2A). DEX also caused reduced tenocyte proliferation and sGAG biosynthesis after just 24 hours in incubation (Fig. 2D,E). However, this did not result in any differences in dsDNA, sGAG, or total collagen content (Fig. 2B,C,F).
Figure 2.
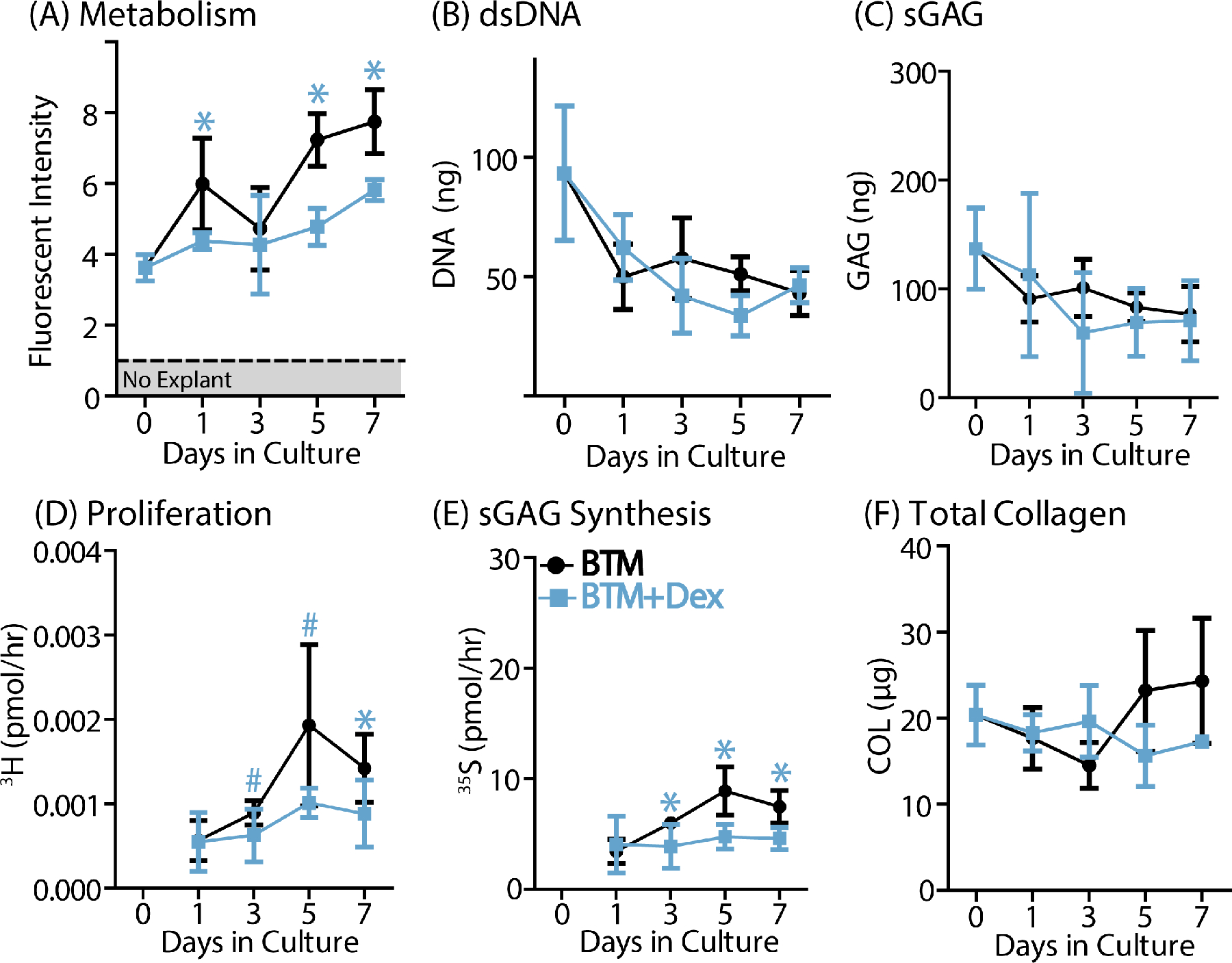
DEX treatment resulted in reduced (A) metabolic activity, (D) cell proliferation, and (E) sulfated GAG synthesis over the course of the culture period. There were no significant differences in (B) DNA, (C) sGAG, or (F) total collagen content. Data are presented as mean ± 95% confidence interval where statistical significance is denoted by stars (*) at each time point (n=5/group/timepoint).
We then investigated the duration of DEX treatment needed for therapeutic benefit through a timed removal experiment (Fig. 3). DEX was added to all groups at the beginning of the culture period and selectively removed at either 1, 2, 3, or 7 days in culture upon medium change. When DEX was removed after 24 hours of treatment (Fig. 3B,E), viability was reduced at day 3 compared to that in control BTM explants. Removal of DEX treatment after 48 hours (Fig. 3C,E) and 72 hours (Fig. 3D,E) resulted in increased viability at day 5 compared to BTM controls but there were no differences by day 7, with less than 20% tenocyte viability in each group. Sustained DEX treatment for a full 7 days was the only course of action that resulted in complete prevention of the cell death caused by secondary joint damage.
Figure 3.
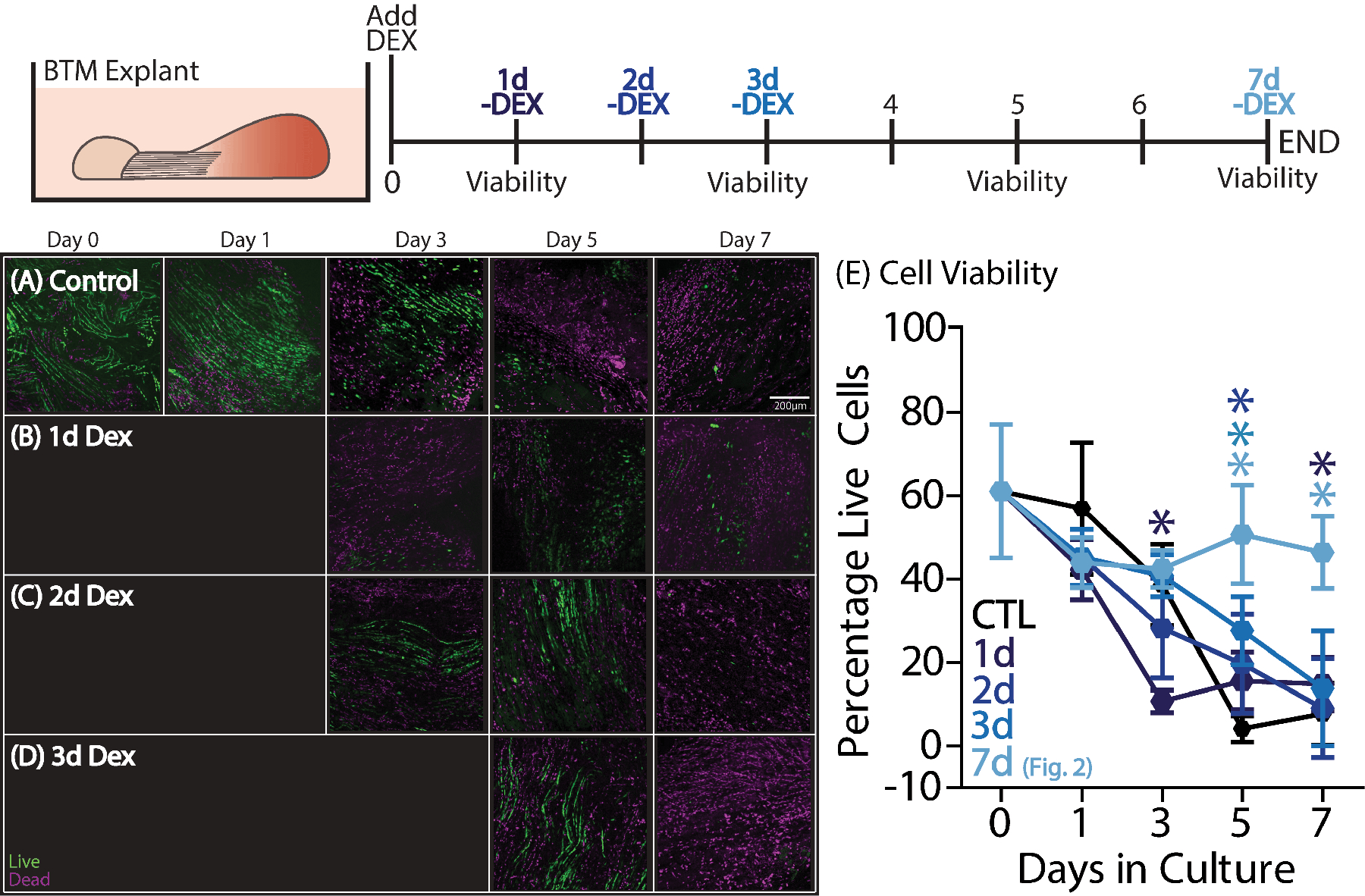
To investigate how long DEX treatment was required to mitigate secondary tendon damage, BTM explants were incubated with DEX for the first 1, 2, 3, or 7 days in the culture period (n=7–10/group/timepoint). (B) One day of DEX treatment significantly reduced viability at day 3 but was improved by day 7 compared to (A) control BTM. (C) Two and (D) three days of treatment improved viability only at day 5 compared to controls. Data are presented as mean ± 95% confidence interval where significant differences compared to the BTM control group are represented by stars (*).
Seeking to determine how long after injury initiation of DEX treatment could exert beneficial effects, we performed a delayed administration experiment by adding DEX to cultures at the time of injury (0 hours) or at 24, 48, or 72 hours after the injury (Fig. 4). Administering DEX at 24 hours (Fig. 4B,E) after the initial injury resulted in sustained benefit throughout the culture period with no differences between the 24h and 0h groups and significant improvements over the no-DEX control group at days 5 and 7 (Fig. 4A,E). Administration at 48 hours after injury (Fig. 4C) resulted in increased viability at days 3 and 5 compared to the control BTM, but no significant palliative effect by day 7. Finally, administration at 72 hours after injury (Fig. 4C–E) resulted in increased viability at day 5 only, but no significant difference by day 7 when compared to control BTM.
Figure 4.
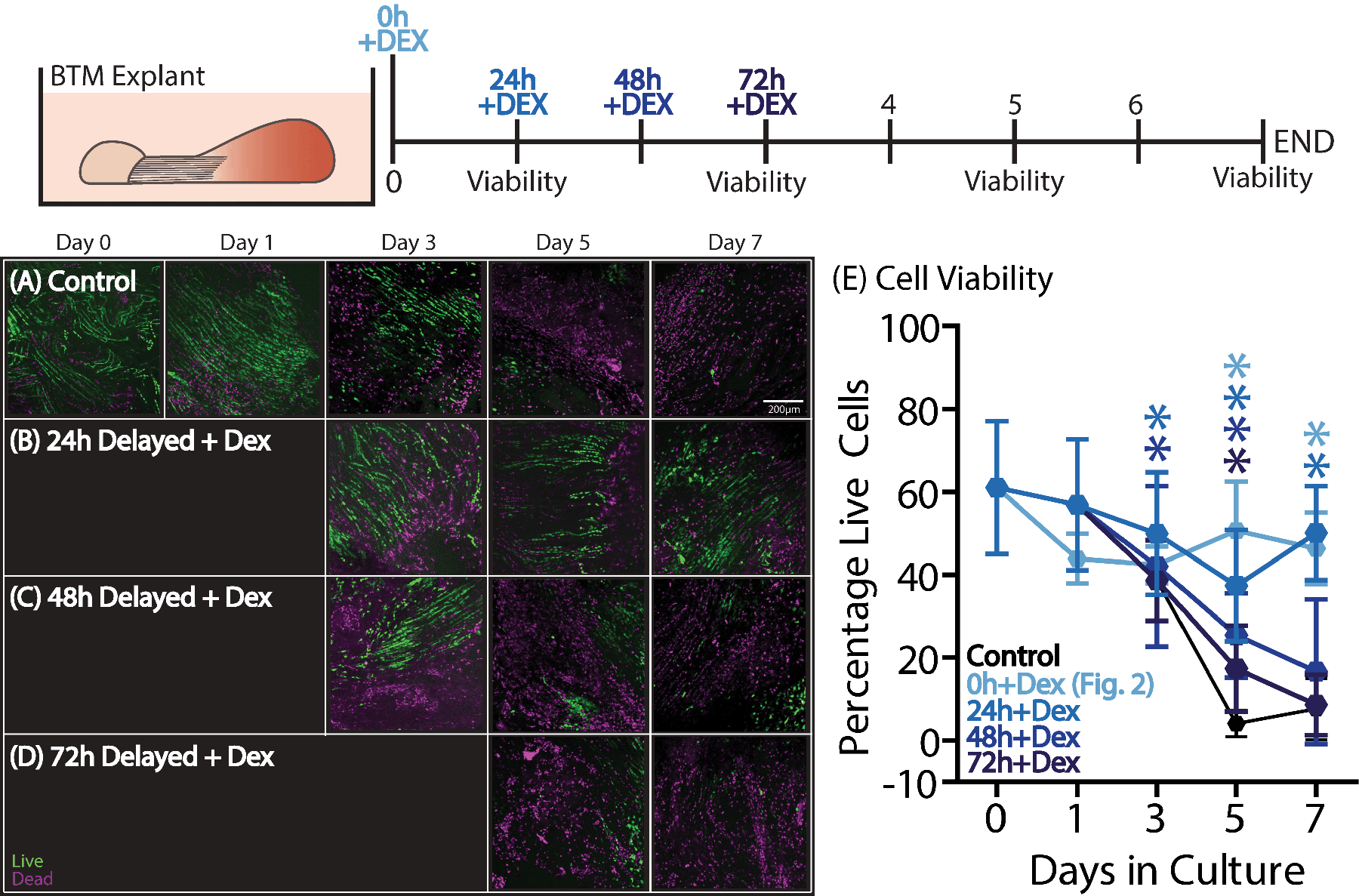
To investigate how long DEX administration could be delayed, DEX treatment was added at 0, 24, 48, or 72 hours after the initial injury (n=7–10/group/timepoint). (B) A 24-hour delay improved viability at days 3–7 compared to (A) control BTM. (C) A 48-hour delayed administration resulted in improved viability at days 3 and 5, but there were no differences by day 7. Finally, (D) a 72-hour delayed administration was only different from control BTM at day 5. Data are presented as mean ± 95% confidence interval where significant differences compared to the BTM control group are represented by stars (*).
To further confirm that DEX treatment was targeting effects in tendon (rather than bone/muscle), flexor digitorum longus tendon (‘FDL’) explants were incubated in conditioned medium (‘CM’) obtained from BTM cultures to replicate BTM injury in the absence of muscle and bone (Fig. 5). We then added each of the three treatments (RA, EN or DEX) to FDL+CM samples to evaluate efficacy of each treatment and to validate BTM results. Control FDL explants maintained approximately 75% viability for the duration of the 7-day culture period, as previously shown and reproduced here (Fig. 5A).25 When incubated with CM, FDL explants demonstrate a marked loss of viability following the first 24 hours in culture, similar to control BTM explants (Fig. 5B). Neither EN or RA treatment were capable of preventing cell death in FDL+CM explants (Supplemental Fig. S6), although RA did exhibit several changes similar to DEX treatment in metabolic activity, proliferation and biosynthesis. EN has very little effect on FDL+CM explants, similar to BTM explants.
Figure 5.
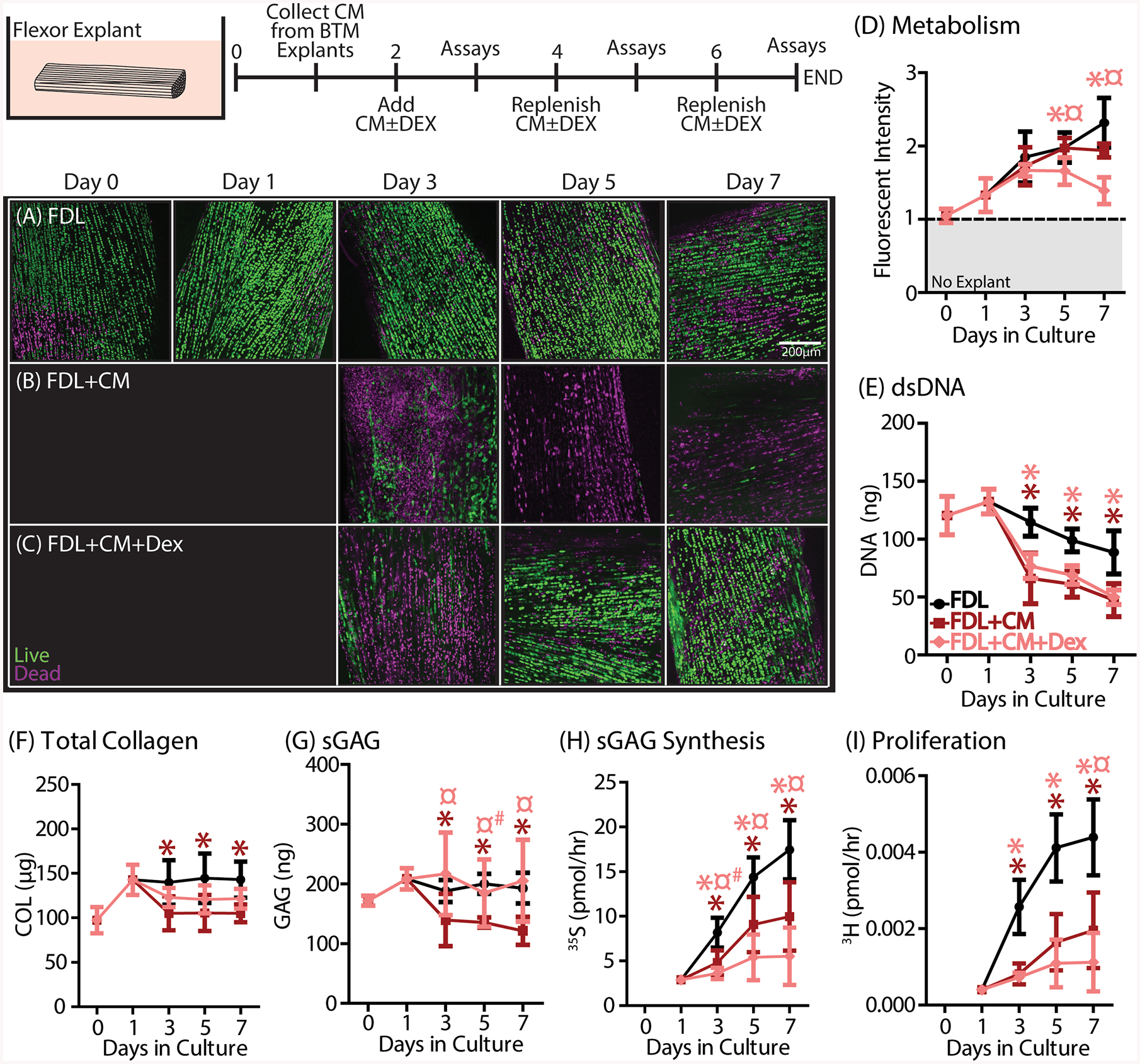
Flexor explants were incubated with CM with or without DEX from day
2–7 to simulate BTM injury and treated injury in the absence of living
muscle and bone (n=5/group/timepoint). (A) Otherwise healthy FDL explants
( ) remain viable for the
entire 7-day culture period. (B) FDL+CM explants have reduced viability after 24
hours in CM that persists through day 7. (C) FDL+CM+DEX explants demonstrated
rescued viability compared to the FDL+CM group. While there were no differences
in (D) metabolism, (E) DNA, (F) total collagen, (G) sGAG, (H) sGAG synthesis,
and (I) proliferation were all reduced in FDL+CM explants (
) remain viable for the
entire 7-day culture period. (B) FDL+CM explants have reduced viability after 24
hours in CM that persists through day 7. (C) FDL+CM+DEX explants demonstrated
rescued viability compared to the FDL+CM group. While there were no differences
in (D) metabolism, (E) DNA, (F) total collagen, (G) sGAG, (H) sGAG synthesis,
and (I) proliferation were all reduced in FDL+CM explants ( ). DEX treatment (
). DEX treatment ( ) further reduced metabolic activity,
sGAG synthesis, and proliferation but rescued matrix loss in total collagen and
sGAG. Data are presented as mean ± 95% confidence interval where
significant differences compared to the FDL control group are represented by
stars (*) and compared to the FDL+CM group are represented by the scarab symbol
(¤). Trends are additionally denoted by a hashtag symbol (#).
) further reduced metabolic activity,
sGAG synthesis, and proliferation but rescued matrix loss in total collagen and
sGAG. Data are presented as mean ± 95% confidence interval where
significant differences compared to the FDL control group are represented by
stars (*) and compared to the FDL+CM group are represented by the scarab symbol
(¤). Trends are additionally denoted by a hashtag symbol (#).
In contrast, treatment with DEX was successful in preventing cell death and loss of matrix content (Fig. 5C). There is an initial loss in viability at day 3, but at day 5 and beyond there is no difference between the FDL+CM+Dex group and the FDL control group (Supplemental Fig. S7A), which are both significantly different from the FDL+CM group. With CM alone, there were no differences in explant metabolism (Fig. 5D). However, these explants did have reduced dsDNA content and demonstrated loss of sGAG and collagen after just 24 hours in culture (starting at day 3) that persisted throughout the culture period (Fig. 5E–G, Supplemental Fig. S8A–D). In addition, FDL+CM groups showed reduced proliferation and biosynthesis (Fig.5H,I). Treatment with DEX resulted in further reduced explant metabolism, proliferation and sGAG biosynthesis when compared to the FDL+CM group (Fig. 5D,H,I). Furthermore, while there was no effect on dsDNA content (Fig. 5E), DEX treatment did result in a partial or complete rescue of sGAG and total collagen loss (Fig. 5F,G).
To examine whether DEX treatment was specifically targeting cytokine-induced damage, FDL explants were incubated in a three-cytokine medium (‘3C’= IL-1 + TNF-α + IL-6) to replicate effects of those constituents of cytokine-induced effects (Fig. 6). When incubated with 3C, FDL+3C explants also exhibit a loss in cell viability (Fig. 6B). However, this loss in viability is not nearly as dramatic as in FDL+CM or in BTM control explants, and it is not significantly different from healthy FDL explants until day 7 in culture (Supplemental Fig. S7B). Treatment with DEX has no effect on cell viability when compared to the FDL+3C group, with both groups exhibiting marked cell death at 7 days of culture (Fig. 6C). Furthermore, DEX treatment had no effect on any of the changes associated with the 3C-induced injury. Metabolism, dsDNA content, sGAG content, cell proliferation, and sGAG biosynthesis were all significantly reduced in FDL+3C explants (Fig. 6D–I, Supplemental Fig. S8). Treatment with DEX did not result in any significant changes between the FDL+3C and FDL+3C+DEX groups.
Figure 6.
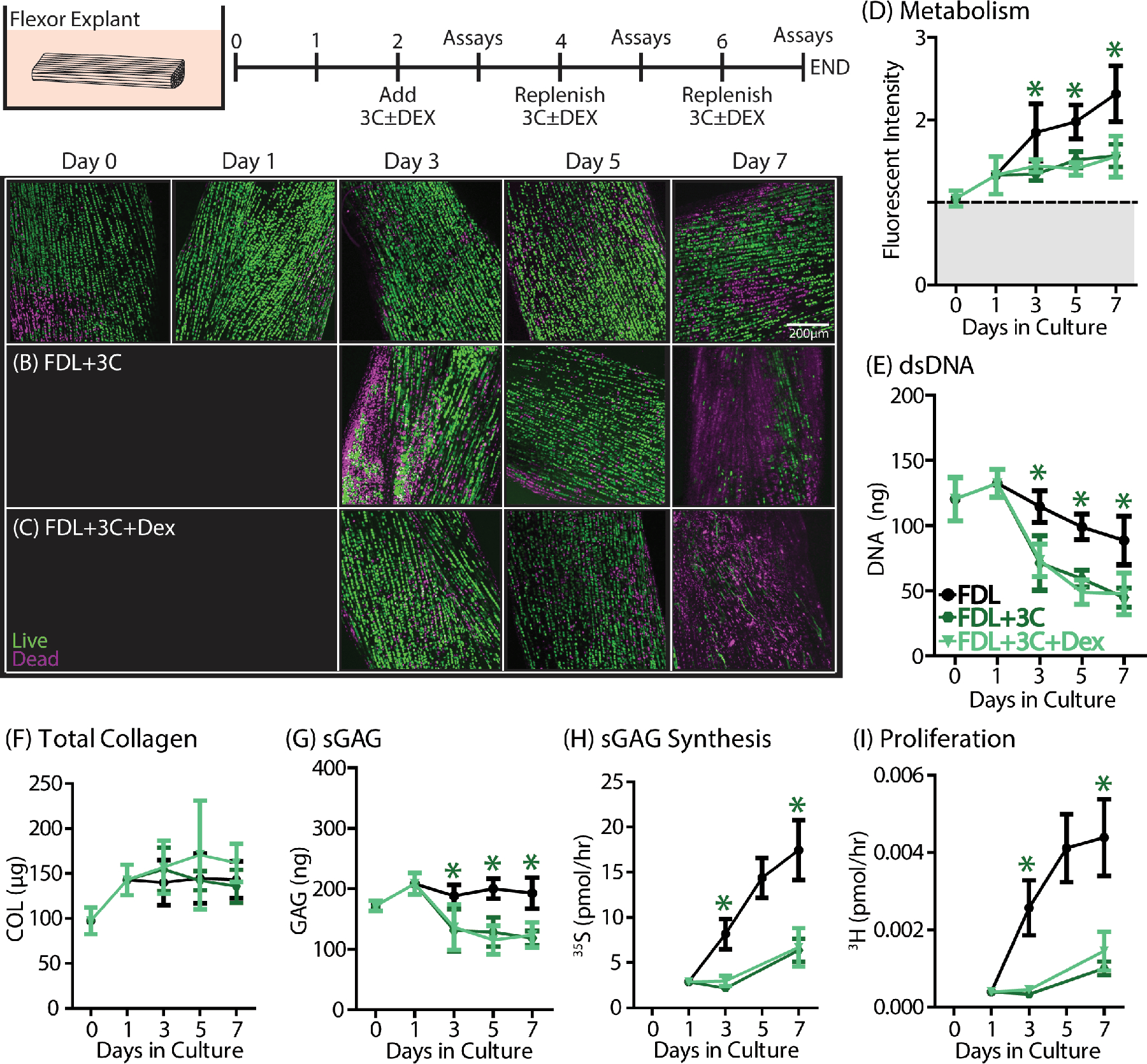
Flexor explants were incubated in 3C medium with or without DEX from day
2–7 to determine whether DEX acted specifically on cytokine-induced
effects (n=5/group/timepoint). (A) Otherwise healthy FDL explants
( ) remain viable for the
entire 7-day culture period. (B) FDL+3C explants incubated in 3C medium have
reduced viability only at day 7 compared to control. (C) FDL+3C+DEX explants
were no different from the FDL+3C group. (D) Metabolism, (E) DNA, (G) sGAG, (H)
sGAG synthesis, and (I) proliferation were all reduced in FDL+3C explants
(
) remain viable for the
entire 7-day culture period. (B) FDL+3C explants incubated in 3C medium have
reduced viability only at day 7 compared to control. (C) FDL+3C+DEX explants
were no different from the FDL+3C group. (D) Metabolism, (E) DNA, (G) sGAG, (H)
sGAG synthesis, and (I) proliferation were all reduced in FDL+3C explants
( ). There were no
differences in (F) total collagen content. DEX treatment (
). There were no
differences in (F) total collagen content. DEX treatment ( ) had no effect on FDL+3C explants. Data
are presented as mean ± 95% confidence interval where significant
differences compared to the FDL control group are represented by stars (*) and
compared to the FDL+3C group are represented by scarabs (¤). Trends are
additionally denoted by a hashtag symbol (#).
) had no effect on FDL+3C explants. Data
are presented as mean ± 95% confidence interval where significant
differences compared to the FDL control group are represented by stars (*) and
compared to the FDL+3C group are represented by scarabs (¤). Trends are
additionally denoted by a hashtag symbol (#).
Discussion:
In multiple tendon explant studies presented here, we found that administration of DEX was beneficial towards mitigating detrimental effects of inflammation induced by secondary joint damage, specifically by injuries to muscle and bone. There has been much debate concerning the use of glucocorticoids for musculoskeletal injuries, with active ongoing research in multiple organisms and disease models, as well as basic studies at the cell and tissue level. Recent reports have shown benefit for DEX in preventing cytokine-induced damage in cartilage explants.30,38,39 In contrast, there are many studies suggesting detrimental effects of glucocorticoid treatment in tendons.40–42 However, most of this work has focused on 2D tenocyte or tendon stem cell culture; at least one in vivo healing study appears to demonstrate a benefit of DEX treatment on healing tendons.43 We believe that much of the debate on glucocorticoid use centers around the dose and frequency of administration, as well as the injury state of the tissue. Our work addresses several of these concerns, and for the first time explores the potential benefit of a low-dose DEX treatment to prevent tendon damage in a 3D explant culture system.
While our studies showed beneficial effects of DEX treatment in preventing inflammation-induced tissue damage, several other measures of cell health did appear to be altered in agreement with other studies. In particular, DEX treatment consistently reduced metabolic activity, matrix biosynthesis and proliferative capacity. Previous monolayer cell cultures studies have discouraged the use of DEX treatment, citing negative effects of cytotoxicity, proliferative and migratory capacity, and differentiation potential.44,45 In the case of injury, where there are changes in cell activity associated with inflammatory and repair processes, the reduction in activity caused by DEX may have other benefits such as reducing matrix degeneration by inhibiting production of MMPs46,47 and reducing cell death. However, this may not be beneficial for all scenarios. One could speculate that DEX treatment of normal tissues in the absence of injury could result in the inability of tissues to perform important basic functions, such as repair of daily matrix damage or adequate control of matrix production. In the case of sustained DEX treatment over time, this could result in chronic degeneration.
Several recent studies have suggested that treatment with DEX in 2D cell culture resulted in increased cellular senescence, particularly with increased and sustained DEX doses.48,49 Senescent cells are growth-arrested cells that exhibit reduced activity, induction of anti-apoptotic genes and enhanced secretion of pro-inflammatory factors through the senescence-associated secretory phenotype.50 While the reduction in certain aspects of cell activity with DEX treatment in the present study does appear to agree with the onset of cellular quiescence, or perhaps senescence, this has not yet been confirmed in our culture system and will be a focus of future work. This finding would have a number of implications regarding clinical use of DEX, especially for an increasingly aged clinical population where the accumulation and spread of senescent cells is already implicated as a potentially dangerous mechanism of chronic tissue degeneration.
Remarkably, the timing of DEX administration did have a substantial impact on its therapeutic benefit in preventing cell death. Although shorter periods of delivery did have some benefit by the end of the culture period, DEX treatment was most beneficial when given sustained for the entire culture period. Furthermore, determining the window of treatment for maximal benefit was more complex than expected. Treatment immediately at the time of injury and continued during the first 24-hours after injury actually appeared to be detrimental, evidenced by slightly reduced viability in all groups receiving DEX during that time compared to the control and 24-hour delayed group. However, delayed treatment starting beyond the 48-hour mark was unable to replicate the benefit of early DEX treatment with all groups had reduced viability compared to the 24-hour delayed and 7-day sustained groups. This could be explained by the temporal inflammatory response occurring in the culture, where the maximal levels of inflammation are occurring at day 3 in the 7-day culture period.25 If DEX is added before this period and rapidly reduces cell activity, protective responses to subsequent inflammatory insults may also be diminished. However, if DEX is added too late, the damage may already be done and treatment will have limited benefit. Future studies will need to optimize beneficial effects of DEX by further investigating dosing and timing strategies, as well as exploring local drug delivery techniques for targeted therapy.
We also found that while DEX treatment was beneficial in preventing inflammation-induced changes in BTM and FDL+CM injury models, it had virtually no effect on inflammatory cytokine-induced damage via the FDL+3C model. This suggested that DEX is not directly targeting tissue damage induced by these three cytokines alone, contrary to our initial hypotheses and to other studies in cartilage injury models induced by selected cytokine treatment.7 Since DEX was beneficial in our clinically-relevant joint damage model, it points to additional targets including other cytokines, chemokines or soluble factors present in BTM explants (or in CM) that are relevant to joint injury and responsive to DEX treatment. Our previous studies demonstrated that in addition to pro-inflammatory cytokines, several cytokines associated with anti-inflammatory functions were also measured experimentally in the conditioned medium, such as IL-10 and IFN-γ, which could be working in concert with DEX treatment.25 Furthermore, our original studies also measured high levels of the chemokine CXCL1, which is responsible for neutrophil recruitment, and there are many other potential chemokines that were not measured and could interact with DEX treatment. Future studies will focus on fully characterizing the spent medium from our model of secondary joint damage in an attempt to determine the mechanism for therapeutic benefit by DEX, as well as to explore other biomarkers and targets that can be validated with animal and human studies.
While our model is valid for understanding initial changes associated with acute injury, it is inherently limited by the lack of the circulating system that would be found in vivo. Metabolic and inflammatory changes in the synovial fluid are dynamic and time-sensitive in vivo, but our system does not currently have circulation with the exception of medium. Therefore, the concentrations of cytokines that tissues are exposed to are continuously elevated and static. Furthermore, there is no infiltration of other cell types which are capable of interacting with and altering inflammatory responses. Both of these factors would likely reduce the overall inflammatory response in vivo and therefore responses in our system may be super-physiologic and on an accelerated timescale. Furthermore, we did not control for undefined levels of cytokines present in fetal bovine serum added to all media in the present study, which could influence inflammatory responses. However, our model system does allow us to understand the local damage responses in isolation and allows for the addition of different cellular and molecular stimuli in a controlled fashion, providing some benefits over in vivo work.
Furthermore, all of the aforementioned studies were performed under stress-deprived conditions. Mechanical loading is critically important to tenocyte health, particularly in maintaining a delicate balance between matrix catabolism and anabolism.51 Loss of mechanical tension has been reported to decrease tendon phenotype and promote inflammation in vitro using tissue-engineered tendon constructs and tendon explant models as well as in vivo in animal models.52,53 Previous studies from our group as well as previous model systems have shown that stress deprivation, or the lack of mechanical stimulus, may induce degenerative changes by itself.25,54 However, our studies indicate that matrix degeneration, apoptosis, and loss in mechanical function due to stress deprivation does not occur in flexor (FDL) explants during the first seven days of the culture period.25 Our system is designed to detect early changes associated with cellular responses to biochemical or mechanical insult. Future studies will be extended to investigate long-term changes, such as matrix degeneration and/or altered mechanical function. In addition, we are currently working to repeat these studies in conditions of physiological static and cyclic tensile loading to investigate interactions between inflammation-induced and mechanically-induced damage. We hypothesize based on preliminary results from a small pilot study that while mechanical stimulus may reduce activity associated with stress deprivation (increase in proliferation, metabolism, and matrix biosynthesis over the culture period), returning explant cultures closer to in vivo baseline values, these explants will still exhibit inflammation-induced damage that is effectively treated with low-dose DEX administration.
In conclusion, this study is the first to identify that, in an intact explant model, low-dose DEX treatment may be beneficial for mitigating tendon damage stemming from inflammation. Given the prevalence and benefits of corticosteroid use for pain relief, this work provides much needed support to the use of DEX in the clinic as a potential disease modifying drug for the treatment of musculoskeletal injuries. At the present, our data support the use of low-dose short-term administration of DEX for preventing tendon damage following acute musculoskeletal injuries. However, there are still questions that need to be answered regarding the appropriate delivery strategy for administration, as well as further exploration into how DEX treatment may be influencing long-term cell behavior. Furthermore, this study only investigated responses to inflammation in male explants; future studies will also aim to detect any sex differences that may exist, which is particularly important if we aim to study therapeutics in the future. Using these novel in vitro models, future studies will seek to understand the progression of initial musculoskeletal injury to end-stage degenerative joint disease and the role of inflammation in this process.
Supplementary Material
Acknowledgements:
This study was supported by funding from the NIH/NIA (F32‐AG052284, K99‐AG063896). The authors would like to thank Han-Hwa Hung for expertise and assistance.
References:
- 1.Soslowsky LJ, Thomopoulos S, Tun S, et al. 2000. Neer Award 1999. Overuse activity injures the supraspinatus tendon in an animal model: a histologic and biomechanical study. J. Shoulder Elb. Surg. Am. Shoulder Elb. Surg. Al 9(2):79–84. [PubMed] [Google Scholar]
- 2.Dean BJF, Gettings P, Dakin SG, Carr AJ. 2016. Are inflammatory cells increased in painful human tendinopathy? A systematic review. Br J Sports Med 50(4):216–220. [DOI] [PubMed] [Google Scholar]
- 3.Millar NL, Hueber AJ, Reilly JH, et al. 2010. Inflammation is present in early human tendinopathy. Am. J. Sports Med 38(10):2085–2091. [DOI] [PubMed] [Google Scholar]
- 4.Millar NL, Murrell GAC, McInnes IB. 2017. Inflammatory mechanisms in tendinopathy - towards translation. Nat. Rev. Rheumatol 13(2):110–122. [DOI] [PubMed] [Google Scholar]
- 5.Carbone A, Rodeo S. 2017. Review of current understanding of post-traumatic osteoarthritis resulting from sports injuries. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 35(3):397–405. [DOI] [PubMed] [Google Scholar]
- 6.Dare D, Rodeo S. 2014. Mechanisms of post-traumatic osteoarthritis after ACL injury. Curr. Rheumatol. Rep 16(10):448. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Wang Y, Chubinskaya S, et al. 2015. Effects of insulin-like growth factor-1 and dexamethasone on cytokine-challenged cartilage: relevance to post-traumatic osteoarthritis. Osteoarthritis Cartilage 23(2):266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu YCS, Evans CH, Grodzinsky AJ. 2011. Effects of short-term glucocorticoid treatment on changes in cartilage matrix degradation and chondrocyte gene expression induced by mechanical injury and inflammatory cytokines. Arthritis Res. Ther 13(5):R142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reuther KE, Sarver JJ, Schultz SM, et al. 2012. Glenoid cartilage mechanical properties decrease after rotator cuff tears in a rat model. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 30(9):1435–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas SJ, Reuther KE, Tucker JJ, et al. 2014. Biceps detachment decreases joint damage in a rotator cuff tear rat model. Clin. Orthop 472(8):2404–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kramer EJ, Bodendorfer BM, Laron D, et al. 2013. Evaluation of cartilage degeneration in a rat model of rotator cuff tear arthropathy. J. Shoulder Elbow Surg 22(12):1702–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ackermann PW, Domeij-Arverud E, Leclerc P, et al. 2013. Anti-inflammatory cytokine profile in early human tendon repair. Knee Surg. Sports Traumatol. Arthrosc. Off. J. ESSKA 21(8):1801–1806. [DOI] [PubMed] [Google Scholar]
- 13.Fabiś J, Szemraj J, Strek M, et al. 2014. Is resection of the tendon edge necessary to enhance the healing process? An evaluation of the expression of collagen type I, IL-1β, IFN-γ, IL-4, and IL-13 in the distal 1 cm of a torn supraspinatus tendon: part II. J. Shoulder Elbow Surg 23(12):1779–1785. [DOI] [PubMed] [Google Scholar]
- 14.Manning CN, Havlioglu N, Knutsen E, et al. 2014. The early inflammatory response after flexor tendon healing: a gene expression and histological analysis. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 32(5):645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaida JE, Bagge J, Purdam C, et al. 2012. Evidence of the TNF-α system in the human Achilles tendon: expression of TNF-α and TNF receptor at both protein and mRNA levels in the tenocytes. Cells Tissues Organs 196(4):339–352. [DOI] [PubMed] [Google Scholar]
- 16.Thampatty BP, Li H, Im H-J, Wang JH-C. 2007. EP4 receptor regulates collagen type-I, MMP-1, and MMP-3 gene expression in human tendon fibroblasts in response to IL-1 beta treatment. Gene 386(1–2):154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsuzaki M, Guyton G, Garrett W, et al. 2003. IL-1 beta induces COX2, MMP-1, −3 and −13, ADAMTS-4, IL-1 beta and IL-6 in human tendon cells. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 21(2):256–264. [DOI] [PubMed] [Google Scholar]
- 18.John T, Lodka D, Kohl B, et al. 2010. Effect of pro-inflammatory and immunoregulatory cytokines on human tenocytes. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 28(8):1071–1077. [DOI] [PubMed] [Google Scholar]
- 19.Machner A, Baier A, Wille A, et al. 2003. Higher susceptibility to Fas ligand induced apoptosis and altered modulation of cell death by tumor necrosis factor-α in periarticular tenocytes from patients with knee joint osteoarthritis. Arthritis Res. Ther 5(5):R253–R261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandberg O, Eliasson P, Andersson T, et al. 2012. Etanercept does not impair healing in rat models of tendon or metaphyseal bone injury. Acta Orthop. 83(3):305–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gargotti M, Lopez-Gonzalez U, Byrne HJ, Casey A. 2018. Comparative studies of cellular viability levels on 2D and 3D in vitro culture matrices. Cytotechnology 70(1):261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wiesner C, Le-Cabec V, El Azzouzi K, et al. 2014. Podosomes in space: Macrophage migration and matrix degradation in 2D and 3D settings. Cell Adhes. Migr 8(3):179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonnier F, Keating ME, Wróbel TP, et al. 2015. Cell viability assessment using the Alamar blue assay: A comparison of 2D and 3D cell culture models. Toxicol. In Vitro 29(1):124–131. [DOI] [PubMed] [Google Scholar]
- 24.Wen JH, Vincent LG, Fuhrmann A, et al. 2014. Interplay of matrix stiffness and protein tethering in stem cell differentiation. Nat. Mater 13(10):979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Connizzo BK, Grodzinsky AJ. 2018. Release of pro-inflammatory cytokines from muscle and bone causes tenocyte death in a novel rotator cuff in vitro explant culture model. Connect. Tissue Res :1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ko J-Y, Wang F-S, Huang H-Y, et al. 2008. Increased IL-1beta expression and myofibroblast recruitment in subacromial bursa is associated with rotator cuff lesions with shoulder stiffness. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 26(8):1090–1097. [DOI] [PubMed] [Google Scholar]
- 27.Bigoni M, Sacerdote P, Turati M, et al. 2013. Acute and late changes in intraarticular cytokine levels following anterior cruciate ligament injury. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 31(2):315–321. [DOI] [PubMed] [Google Scholar]
- 28.Cameron M, Buchgraber A, Passler H, et al. 1997. The natural history of the anterior cruciate ligament-deficient knee. Changes in synovial fluid cytokine and keratan sulfate concentrations. Am. J. Sports Med 25(6):751–754. [DOI] [PubMed] [Google Scholar]
- 29.Mousavizadeh R, Backman L, McCormack RG, Scott A. 2015. Dexamethasone decreases substance P expression in human tendon cells: an in vitro study. Rheumatol. Oxf. Engl 54(2):318–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grodzinsky AJ, Wang Y, Kakar S, et al. 2017. Intra-articular dexamethasone to inhibit the development of post-traumatic osteoarthritis. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 35(3):406–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patwari P, Lin SN, Kurz B, et al. 2009. Potent inhibition of cartilage biosynthesis by coincubation with joint capsule through an IL-1-independent pathway. Scand. J. Med. Sci. Sports 19(4):528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gouze J-N, Gouze E, Palmer GD, et al. 2003. A comparative study of the inhibitory effects of interleukin-1 receptor antagonist following administration as a recombinant protein or by gene transfer. Arthritis Res. Ther 5(5):R301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schindelin J, Arganda-Carreras I, Frise E, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9(7):676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodríguez-Corrales JÁ, Josan JS. 2017. Resazurin Live Cell Assay: Setup and Fine-Tuning for Reliable Cytotoxicity Results. Methods Mol. Biol. Clifton NJ 1647:207–219. [DOI] [PubMed] [Google Scholar]
- 35.Farndale RW, Buttle DJ, Barrett AJ. 1986. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim. Biophys. Acta 883(2):173–177. [DOI] [PubMed] [Google Scholar]
- 36.Dourte LM, Pathmanathan L, Mienaltowski MJ, et al. 2013. Mechanical, compositional, and structural properties of the mouse patellar tendon with changes in biglycan gene expression. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 31(9):1430–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singer VL, Jones LJ, Yue ST, Haugland RP. 1997. Characterization of PicoGreen reagent and development of a fluorescence-based solution assay for double-stranded DNA quantitation. Anal. Biochem 249(2):228–238. [DOI] [PubMed] [Google Scholar]
- 38.Huebner KD, Shrive NG, Frank CB. 2014. Dexamethasone inhibits inflammation and cartilage damage in a new model of post-traumatic osteoarthritis. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 32(4):566–572. [DOI] [PubMed] [Google Scholar]
- 39.Roach BL, Kelmendi-Doko A, Balutis EC, et al. 2016. Dexamethasone Release from Within Engineered Cartilage as a Chondroprotective Strategy Against Interleukin-1α. Tissue Eng. Part A 22(7–8):621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dean BJF, Carr AJ. 2016. The Effects of Glucocorticoid on Tendon and Tendon Derived Cells. Adv. Exp. Med. Biol 920:239–246. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Keenan C, Wang JH-C. 2013. The effects of dexamethasone on human patellar tendon stem cells: implications for dexamethasone treatment of tendon injury. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 31(1):105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen W, Tang H, Zhou M, et al. 2015. Dexamethasone inhibits the differentiation of rat tendon stem cells into tenocytes by targeting the scleraxis gene. J. Steroid Biochem. Mol. Biol 152:16–24. [DOI] [PubMed] [Google Scholar]
- 43.Blomgran P, Hammerman M, Aspenberg P. 2017. Systemic corticosteroids improve tendon healing when given after the early inflammatory phase. Sci. Rep 7(1):12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong MWN, Lui WT, Fu SC, Lee KM. 2009. The effect of glucocorticoids on tendon cell viability in human tendon explants. Acta Orthop. 80(3):363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong MWN, Tang YYN, Lee SKM, Fu BSC. 2005. Glucocorticoids suppress proteoglycan production by human tenocytes. Acta Orthop. 76(6):927–931. [DOI] [PubMed] [Google Scholar]
- 46.Saito S, Katoh M, Masumoto M, et al. 1999. Dexamethasone inhibits collagen degradation induced by the combination of interleukin-1 and plasminogen in cartilage explant culture. Biol. Pharm. Bull 22(7):727–730. [DOI] [PubMed] [Google Scholar]
- 47.Garvican ER, Vaughan-Thomas A, Redmond C, et al. 2010. MMP-mediated collagen breakdown induced by activated protein C in equine cartilage is reduced by corticosteroids. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 28(3):370–378. [DOI] [PubMed] [Google Scholar]
- 48.Poulsen RC, Watts AC, Murphy RJ, et al. 2014. Glucocorticoids induce senescence in primary human tenocytes by inhibition of sirtuin 1 and activation of the p53/p21 pathway: in vivo and in vitro evidence. Ann. Rheum. Dis 73(7):1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xue E, Zhang Y, Song B, et al. 2016. Effect of autophagy induced by dexamethasone on senescence in chondrocytes. Mol. Med. Rep 14(4):3037–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hernandez-Segura A, Nehme J, Demaria M. 2018. Hallmarks of Cellular Senescence. Trends Cell Biol. 28(6):436–453. [DOI] [PubMed] [Google Scholar]
- 51.Wang JH-C, Thampatty BP, Lin J-S, Im H-J. 2007. Mechanoregulation of gene expression in fibroblasts. Gene 391(1–2):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lavagnino M, Wall ME, Little D, et al. 2015. Tendon mechanobiology: Current knowledge and future research opportunities. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 33(6):813–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Killian M, Lim C, Thomopoulos S, et al. 2013. The effect of unloading on gene expression of healthy and injured rotator cuffs. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 31(8):1240–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hannafin JA, Arnoczky SP, Hoonjan A, Torzilli PA. 1995. Effect of stress deprivation and cyclic tensile loading on the material and morphologic properties of canine flexor digitorum profundus tendon: an in vitro study. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 13(6):907–914. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.