

Abstract
In mouse models of cancer, the inhibition of a set of regulatory proteins improves checkpoint-blockade therapy by causing regulatory T cells to produce the cytokine interferon-γ.
Immune surveillance is the first line of the body’s defence against the uncontrolled proliferation of tumour cells, as leukocytes such as natural killer (NK) cells and cytotoxic CD8+ T lymphocytes (CTL) efficiently kill neoplastic cells. But tumour cells can explore immunosuppressive networks and evade anti-tumour immunity1. As an endogenous control mechanism over autoimmunity that can nevertheless also result in impaired anti-tumour immunity, forkhead-box P3 (FOXP3)+ CD25+ regulatory T (TREG) cells suppress the effector function of NK cells, CTLs and antigen-presenting cells (APCs) through multiple pathways2 (Fig. 1). In light of their immune suppressor activities2–4, and given that high numbers of TREG cells in the tumour microenvironment are associated with poor prognosis, TREG cells are an attractive target for anticancer therapy.
Fig. 1 |. Effect of TREG cells on immune cells.
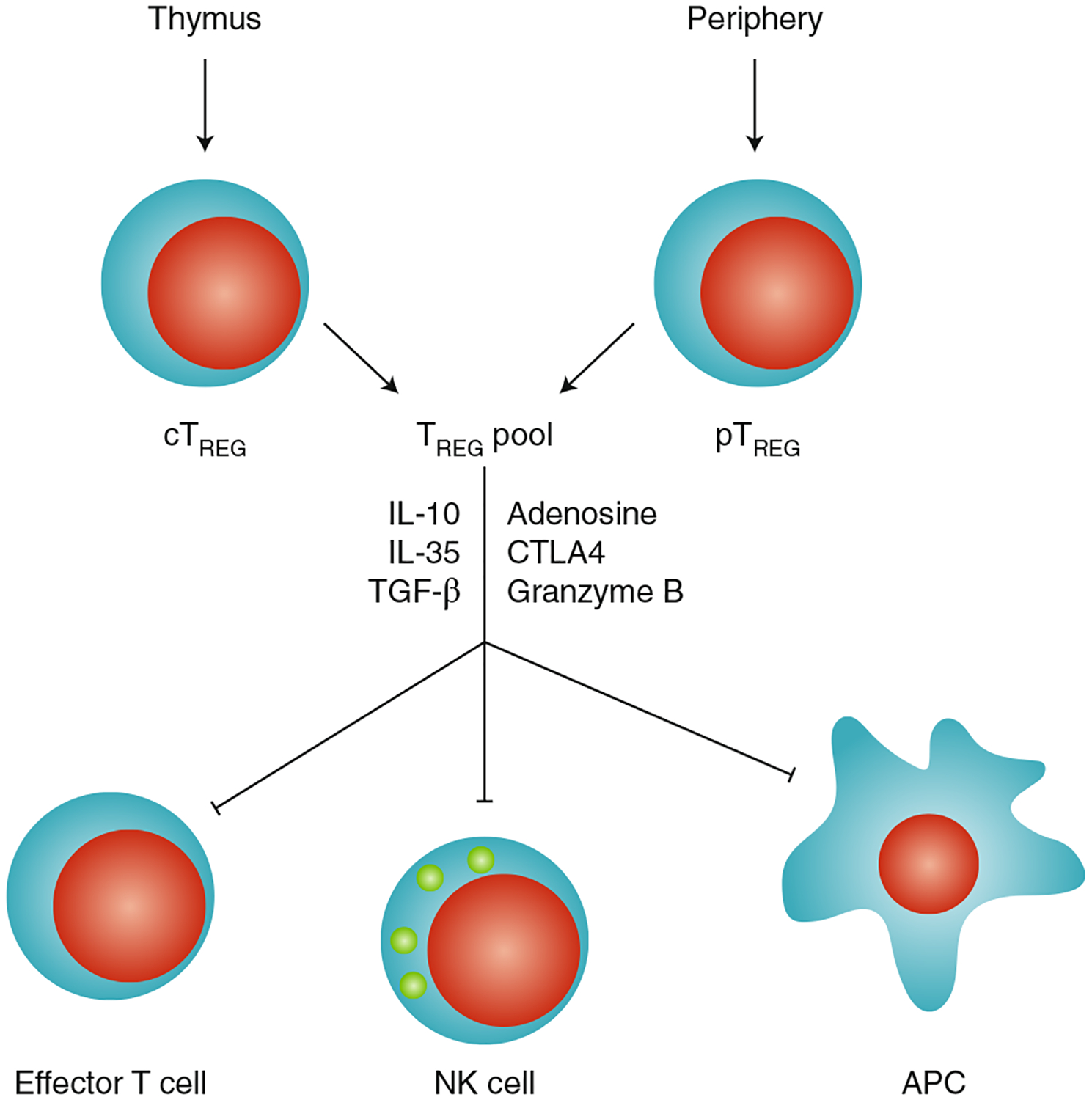
TREG cells are developed in the thymus (central TREG cells; cTREG) or generated and expanded outside the thymus (peripheral TREG cells; pTREG); can directly target antigen-presenting cells, NK cells and effector T cells; and mediate immunosuppression via distinct molecular mechanisms.
In humans, TREG-cell deficiency caused by mutations in the TREG-cell master transcription regulator FOXP3 results in IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome — a severe and systemic autoimmune disorder. The dramatic effects of systemic TREG-cell deficiency suggest that the complete removal of TREG cells is not a feasible approach for cancer immunotherapy. In a study published in Nature, Thorsten Mempel and colleagues now report that the disruption of the T-cell receptor (TCR) signalosome complex CARMA1–BCL10–MALT1 (CBM) in TREG cells causes a reversion in the cell’s phenotype, from immune suppression to immune activation, leading to the production of interferon (IFN)-γ in the tumour microenvironment and enhancing local anti-tumour activity5.
Mempel and colleagues identified a role for the CBM signalosome in TREG cells by crossing Foxp3YFP–Cre mice to Carma1flox/flox mice, which produced offspring with TREG-cell-specific Carma1 deficiency. In these mice, genetic deletion of Carma1 in TREG cells did not affect the expression of FOXP3 or other markers of TREG cells, but triggered the expression of the immune-effector transcription factors T-box transcription factor TBX21 (T-bet) and RAR-related orphan receptor-γt (ROR-γt), as well as the production of the pro-inflammatory cytokines tumour necrosis factor (TNF), interleukin (IL)-17 and IFN-γ. Importantly, the monoallelic deletion of Carma1 in TREG cells also resulted in tumour control while avoiding systemic autoimmunity in mice bearing either poorly immunogenic D4M.3A melanoma or immunogenic MC38 adenocarcinoma. In these models, IFN-γ production by TREG cells was critical for controlling tumour progression in a process restricted to the tumour microenvironment.
To explore the mechanism underlying the anti-tumour effect of Carma1–/– TREG cells, Mempel and colleagues explored the effects of the acute deletion of Carma1 in the tamoxifen-induced Foxp3Cre–ERT2 Carma1flox/flox model. In agreement with prior data, tumour growth was arrested in these animals only after tamoxifen administration. Notably, macrophages from Foxp3Cre–ERT2 Carma1flox/flox tumour-bearing animals expressed higher levels of major histocompatibility complex (MHC) class II when compared with Foxp3Cre–ERT2 Carma1+/+ control mice. Concurrently, tumour cells expressed higher levels of MHC-I, making them more sensitive to CTL-dependent killing. However, tumour cells also expressed higher levels of programmed death-ligand 1 (PD-L1, also known as B7-H1), which may have contained the anti-tumour immune response. This was found to be the case as, while Foxp3Cre–ERT2 Carma1+/+ animals responded moderately to PD-1 blockade, Foxp3Cre–ERT2Carma1flox/flox mice manifested significantly enhanced tumour regression in response to the checkpoint inhibitor.
The lack of pharmacological inhibitors for CARMA1 prevents the non-genetic interrogation of the function of this factor in TREG cells, but inhibitors exist for MALT1, an enzymatic component of the CBM complex. Administration of two MALT1 inhibitors (mepazine and MI-2) in the D4M.3A tumour model triggered a phenotype that approximates that of conditional deletion of Carma1 in TREG cells, including an increase in the frequency of T cells producing IFN-γ and TNF, and the elevated expression of MHC-I and PD-L1 on cancer cells. Furthermore, when D4M.3A melanoma-bearing male mice were treated with mepazine and anti-PD-1, the combination therapy yielded synergistic anti-tumour activity. Hence, the authors conclude that the inhibition of the CARMA1–BCL10–MALT1 signalosome induces an inflammatory phenotype in TREG cells and generates potent anti-tumour immunity (Fig. 2).
Fig. 2 |. Effect of the cBm signalosome on the phenotype of TREG cells.
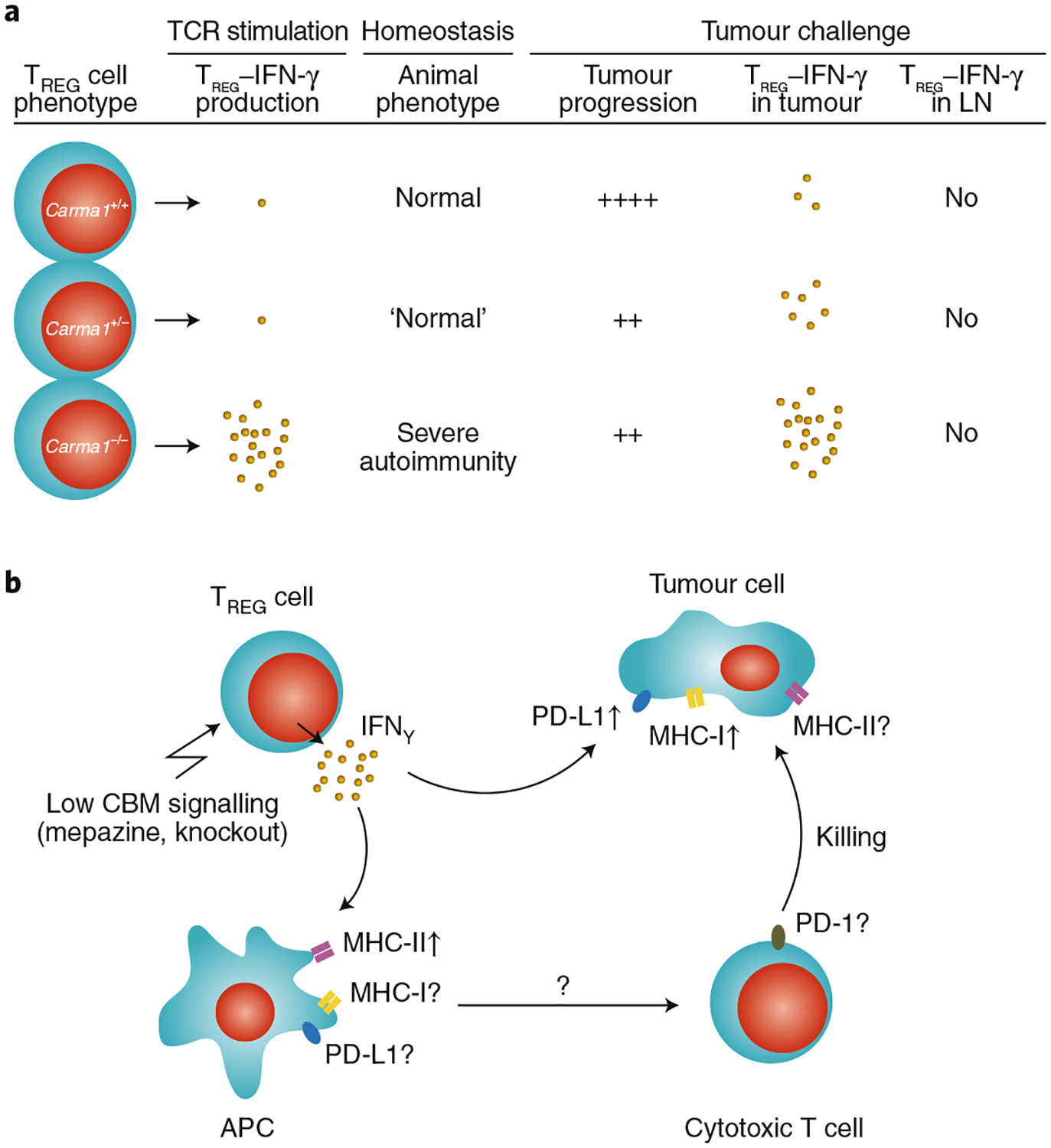
a, Expression levels of the CBM-complex scaffold protein CARMA1 are associated with different levels of IFN-γ production in TREG cells following in vitro stimulation and in vivo tumour challenge. LN, lymph node. b, The genetic or pharmacological loss of CBM signalling results in the increased production of IFN-γ in tumour-associated TREG cells. IFN-γ production by TREG cells may affect MHC-I, MHC-II and PD-L1 on tumour cells and on antigen-presenting cells. ↑, increased expression; ?, unknown effects.
Human TREG cells can express inflammatory and effector cytokines, including IL-8, IL-17 and IFN-γ in the tumour microenvironment and in inflammatory conditions6–8. Thus, that Mempel and colleagues observed IFN-γ+ TREG cells in the mouse tumour microenvironment is not surprising. However, that CARMA1 can dramatically alter the functional properties of FOXP3+ cells to enable IFN-γ expression in TREG cells is an important observation. Although the specific deletion of neuropilin-1 results in IFN-γ expression in TREG cells9, whether the CBM signalosome and neuropilin-1 are connected in regulating the TREG phenotype is unknown.
As TREG cells are regulated by the strength of the TCR signal10, the CBM signalosome may quantitatively and qualitatively alter TCR signalling in TREG cells. This potential mechanistic relationship would partially explain why TREG cells express IFN-γ in the tumour and not in lymph nodes. In addition, it is known that the CBM signalosome can activate several downstream pathways, including nuclear factor-κB (NF-κB), activator protein 1 and mammalian target of rapamycin (mTOR). Mempel and co-authors provided evidence that NF-κB has no role in their models. Given that the integrity of TREG cells is particularly regulated by the balance of mTORC1 and mTORC2 (ref.11), that the phenotypic plasticity of TREG cells can be regulated by the crosstalk of metabolic pathways, and that IFN-γ production is linked to the metabolic state of T cells12, it is sound to investigate the mechanistic association between CBM signalosome components, mTOR and IFN-γ expression in TREG cells. The authors show that CARMA1 deficiency induces the expression of T-bet and ROR-γt; it is therefore tempting to speculate whether CARMA1–CBM has a role in the interplay between the three key T-cell transcription factors FOXP3, T-bet and ROR-γt in TREG cells, and whether this interplay may determine the functional fate of TREG cells.
The observation of an increase in MHC-I expression in macrophages, and of PD-L1 expression on tumour cells under stimulation by IFN-γ+ TREG cells, offers a mechanism for the therapeutic efficacy of the phenotypically reverted TREG cells and would explain why PD-1 blockade concomitant with Carma1-deletion leads to the strong rejection of tumours. However, it is unclear whether IFN-γ from IFN-γ+ TREG cells also led to the induction of PD-L1 expression on macrophages or myeloid dendritic cells (mDCs). PD-L1+ antigen-presenting cells are the primary targets of PD-L1 and PD-1 blockade therapy13–15, and several studies have documented that PD-L1 is expressed on macrophages and mDCs in human tumour-draining lymph nodes and in the tumour microenvironments. Ultimately, multiple environmental, genetic and metabolic determinants may shape the outcome of PD-1 and PD-L1 blockade therapy in different experimental and clinical settings16. A clear understanding of the IFN-γ+ TREG cell’s interacting partners may help to develop new mechanistically informed therapeutic strategies for cancer patients.
References
- 1.Zou W Nat. Rev. Cancer 5, 263–274 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Zou W Nat. Rev. Immunol 6, 295–307 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Curiel TJ et al. Nat. Med 10, 942–949 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Zou L et al. Cancer Res. 64, 8451–8455 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Di Pilato M et al. Nature 570, 112–116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kryczek I et al. J. Immunol 186, 4388–4395 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Duhen T et al. Blood 119, 4430–4440 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kryczek I et al. Oncoimmunology 5, e1105430 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Overacre-Delgoffe AE et al. Cell 169, 1130–1141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakaguchi S Nat. Immunol 6, 345–352 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Zeng H et al. Nature 499, 485–490 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang CH et al. Cell 153, 1239–1251 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curiel TJ et al. Nat. Med 9, 562–567 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Lin H et al. J. Clin. Invest 128, 805–815 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang H et al. J. Clin. Invest 128, 580–588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou W, Wolchok JD & Chen L Sci. Transl. Med 8, 328rv324 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]