

Abstract
The Diels‐Alder reaction of cyclopentadiene with methyl acrylate catalyzed by AlCl3 has been theoretically investigated. M06‐2X level DFT calculations have shown that the formation of two C−C bonds is asynchronous in the cycloaddition both in the endo path and in the exo path, thus making a good contrast to the well‐known concept of [4+2] reactions based on the orbital symmetry arguments. It was found that the catalyst facilitates the cycloaddition and brings a higher endo selectivity in the highly asynchronous process, as compared with the reaction of the diene and the dienophile without the catalyst.
Keywords: cycloaddition reactions, Lewis acids, density functional calculations, orbital interactions
Asynchronous bonds: The reactivity and the stereoselectivity in the Lewis acid catalyzed Diels‐Alder reaction between cyclopentadiene and methyl acrylate are controlled by a combination of several interaction terms.
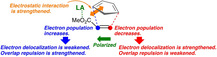
The Lewis acid catalyzed Diels‐Alder reaction is a powerful tool for synthetic organic chemistry.1 In the catalytic cycloaddition between dienes and dienophiles, Lewis acid catalysts have been shown to accelerate the reactions and make the reaction highly stereo‐ and/or regio‐selective.1, 2 Houk and co‐worker ascribed the reason why the Lewis acid facilitates the reaction with normal electron demand to the energy lowering of the LUMO in the dienophile part in the presence of Lewis acids.3 Electron delocalization from the diene part to the dienophile part is thus strengthened by the attachment of a Lewis acid to the dienophile. This idea has widely been accepted until now.4, 5
Our previous study of the Diels‐Alder reaction catalyzed by the Lewis acid activated oxazaborolidine, showed, however, that the weakening of the overlap repulsion between the occupied orbitals of diene and those of dienophile is another important outcome of attaching the catalyst.6 Here, we examine in detail the mechanism of activation of the endo‐cycloaddition by a Lewis acid catalyst, AlCl3, in the reaction between cyclopentadiene and methyl acrylate (Scheme 1), in which the endo/exo selectivity has been shown experimentally to be improved by adding the catalyst.7
Scheme 1.

The Diels‐Alder reaction between cyclopentadiene and methyl acrylate catalyzed by AlCl3.7
We investigated first the cycloaddition in the absence of Lewis acid by using M06‐2X level DFT calculations.8, 9 Six transition state structures were obtained at the M06‐2X/6‐311(d,p) level of theory as shown in Figure 1.10, 11 Among the three transition states leading to the endo addition, called here TSendo‐1, TSendo‐2 and TSendo‐3, TSendo‐1 was calculated to be the lowest in energy. Calculations at the other levels of theory also show the same trend, though the barrier height obtained changes to some extent depending on the level of calculations as seen in Table 1. The most preferred transition state giving the exo addition, TSexo‐1, is located only 0.4 kcal/mol above TSendo‐1. The energy difference is very small and, therefore, it is suggested that the stereoselectivity would not be high (cal. endo : exo=0.69 : 0.31 at 273 K),12 fairly in agreement with the experimental results.7
Figure 1.

Transition state structures for the reaction without AlCl3 at the M06‐2X/6‐311G(d,p) level of theory. Bond distances are in Å. Gibbs free energies at 273 K relative to TSendo‐1 are in kcal/mol.
Table 1.
Gibbs free energy (273 K) of the transition states for the reaction in the absence of AlCl3 relative to cyclopentadiene and methyl acrylate (kcal/mol).
|
|
TSendo‐1 |
TSendo‐2 |
TSendo‐3 |
TSexo‐1 |
TSexo‐2 |
TSexo‐3 |
endo : exo |
|---|---|---|---|---|---|---|---|
|
M06‐2X/6‐311G(d,p) |
22.9 |
30.6 |
24.3 |
23.3 |
30.8 |
25.0 |
0.69 : 0.31 |
|
M06‐2X/6‐311++G(3df,3pd)//M06‐2X/6‐311G(d,p) |
24.4 |
31.3 |
25.8 |
24.9 |
31.7 |
26.7 |
0.75 : 0.25 |
|
M06‐2X(IEF‐PCM)/6‐311++G(3df,3pd) //M06‐2X/6‐311G(d,p)[a] |
24.6 |
29.3 |
25.7 |
25.8 |
30.2 |
27.1 |
0.90 : 0.10 |
|
SCS‐MP2/6‐311G(d,p)//MP2/6‐311G(d,p) |
23.8 |
32.0 |
25.2 |
24.5 |
32.8 |
26.0 |
0.79 : 0.21 |
|
CCSD(T)/6‐311G(d,p)//MP2/6‐311G(d,p) |
24.7 |
32.6 |
26.1 |
25.3 |
33.4 |
26.9 |
0.77 : 0.23 |
|
SCS‐MP2/6‐311++G(3df,3pd)//MP2/6‐311G(d,p) |
21.8 |
28.9 |
23.2 |
22.7 |
29.8 |
24.3 |
0.83 : 0.17 |
|
RHF/6‐311G(d,p) |
51.7 |
61.0 |
53.2 |
52.1 |
61.4 |
54.0 |
0.71 : 0.29 |
[a] Dichloromethane is chosen as a solvent.
The AlCl3‐catalyzed reaction system gives also the six transition‐state structures shown in Figure 2.13 Among the three transition‐state structures giving the endo product, TS′endo‐1, TS′endo‐2, and TS′endo‐3, the first has the lowest energy. In contrast to the non‐catalyzed case, TS′exo‐1 and TS′exo‐3 are shown to be very similar in energy among the three exo transition‐state structures with an attached AlCl3. The difference in energy between TS′endo‐1 and TS′exo‐3 is 2.8 kcal/mol, which is considerably larger than that between TSendo‐1 and TSexo‐1. The attachment of AlCl3 is shown to lead to a larger energy difference between the endo and exo transition states and, therefore, to a higher stereoselectivity (calc. endo : exo=0.99 : 0.01 at 273 K) also in good agreement with the experimental observations.12 Calculations at the other levels of theory show the same trend except for the PCM calculations (Table 2). The three endo transition‐state structures are shown to be located more closely in energy and the three exo transition‐state structures are so in dichloromethane, as compared with the results in vacuo. The energy difference between the endo and exo transition states remains similar to those obtained without the effect of solvent.
Figure 2.

Transition state structures for the reaction with AlCl3 at the M06‐2X/6‐311G(d,p) level of theory. Bond distances are in Å. Gibbs free energies at 273 K relative to TS′endo‐1 are in kcal/mol.
Table 2.
Gibbs free energy (273 K) of the transition states for the reaction in the presence of AlCl3 relative to cyclopentadiene and AlCl3‐attached methyl acrylate (kcal/mol).
|
|
TS′endo‐1 |
TS′endo‐2 |
TS′endo‐3 |
TS′exo‐1 |
TS′exo‐2 |
TS′exo‐3 |
endo : exo |
|---|---|---|---|---|---|---|---|
|
M06‐2X/6‐311G(d,p) |
13.1 |
16.7 |
13.7 |
16.0 |
19.0 |
15.9 |
0.99 : 0.01 |
|
M06‐2X/6‐311++G(3df,3pd)//M06‐2X/6‐311G(d,p) |
14.8 |
18.6 |
15.5 |
17.2 |
20.7 |
17.8 |
0.99 : 0.01 |
|
M06‐2X(IEF‐PCM)/6‐311++G(3df,3pd) //M06‐2X/6‐311G(d,p)[a] |
16.1 |
15.8 |
15.9 |
19.1 |
18.0 |
18.5 |
0.99 : 0.01 |
|
SCS‐MP2/6‐311G(d,p)//MP2/6‐311G(d,p) |
15.8 |
19.6 |
16.2 |
18.8 |
22.0 |
17.5 |
0.97 : 0.03 |
|
CCSD(T)/6‐311G(d,p)//MP2/6‐311G(d,p) |
16.4 |
20.1 |
16.7 |
19.3 |
22.6 |
18.5 |
0.98 : 0.02 |
|
SCS−MP2/6‐311++G(3df,3pd)//MP2/6‐311G(d,p) |
12.5 |
16.8 |
13.3 |
16.0 |
19.4 |
15.3 |
0.99 : 0.01 |
|
RHF/6‐311G(d,p) |
41.0 |
43.1 |
41.4 |
43.2 |
46.1 |
42.6 |
0.94 : 0.06 |
[a] Dichloromethane is chosen as a solvent.
The relative Gibbs free energy diagram is shown in Figure 3. The energy of TSendo‐1 relative to the two reactant molecules in an isolated state which corresponds to the activation energy is calculated here to be 22.9 kcal/mol. On the other hand, the energy difference between the reactant molecules (cyclopentadiene+AlCl3‐attached methyl acrylate) and the transition state TS′endo‐1 is 13.1 kcal/mol. The activation energy for AlCl3‐catalyzed reaction is much lower than that for the non‐catalyzed reaction. The calculations demonstrate not only that the endo selectivity is enhanced but also that the reaction is accelerated by an attachment of AlCl3.
Figure 3.

Relative Gibbs free energy diagram (273 K) at the M06‐2X/6‐311G(d,p) level of theory (kcal/mol).
For the transition state TS′endo‐1, IRC calculations were performed.14 The calculations show clearly that the formation of the two C−C bonds is highly asynchronous in the presence of AlCl3, as illustrated in Figure 4(a). The C2−C6 bond, which is located farther from the methoxycarbonyl group, is formed much faster than the other bond, C3−C7. The C2−C6 bond is 2.01 angstrom at the transition state, but the C3−C7 bond reaches the same bond length at a much later stage of the reaction, s ∼4.0 amu1/2 bohr. The fragment charge profile based on NPA charges, presented in Figure 4(b), demonstrates that electronic charge is shifted from the diene fragment to the dienophile fragment at an early stage of the reaction involving the transition state, and the net electronic charges on the fragments begin to be reduced at the later stage, after passing the point, s ∼1.8 amu1/2 bohr. This signifies that the C2−C6 bond formation is brought about primarily by electron delocalization from the diene part to the dienophile part, while the C3−C7 bond formation is associated mainly with electron delocalization in an opposite direction. In this point, the Lewis‐acid catalyzed Diels‐Alder cycloaddition studied here makes a clear contrast to the well‐established concept of symmetry‐allowed [4+2] cycloadditions.15 The attached AlCl3 part is seen to retain almost the same amount of negative charge throughout the reaction coordinate in this case.
Figure 4.

(a) Change in bond lengths along IRC of TS′endo‐1. (b) Change in fragment charges at along IRC of TS′endo‐1.
Houk and co‐worker proposed that the attached Lewis acid lowers the energy of LUMO of dienophile and, hence, accelerates the Diels‐Alder reaction of normal electron demand.3 One should note now that the dienophile molecule is polarized by the attachment of the Lewis acid molecule, as to reduce the π‐type electron population on the terminal carbon, C6, and increase the π‐type electron population on the other carbon, C7, of the C=C bond. This change in electron population is reflected in the electron‐accepting orbital of the dienophile that shows a considerably larger amplitude on C6 and in the electron‐donating orbital that shows a larger amplitude on C7, in comparison with the dienophile without the catalyst (see, Figures S2 and S3 in Supporting Information).16 The unoccupied and occupied interaction orbitals are lowered in energy by 2.05 eV and 1.79 eV, respectively, by attaching AlCl3. Electron delocalization from the diene to the dienophile, dominantly onto C6, is thus strengthened, whereas that from the dienophile part to the diene part, dominantly from C7, is not enhanced. In addition, the increase in electron population on C7 leads to the strengthening of repulsion with C3 of the diene that arises from the overlap between the occupied orbitals of the diene and those of the dienophile. On the other hand, the overlap repulsion between C6 and C2 is lightened. As a result, asynchronous formation of new C−C bonds should become marked in the catalyzed case. This view of bond formation is fully supported by a partitioning of overlap population of the C2−C6 and C3−C7 bonds into the repulsive and attractive orbital interactions (see, Tables S2 and S3 in Supporting Information).
In the Diels‐Alder reactions, the endo transition state is placed generally under a stronger overlap repulsion between the occupied orbitals of the diene part and those of the dienophile part, but the repulsion is reduced by introducing electron‐withdrawing groups into the dienophile part, as we have already seen in the cycloaddition between cyclopentadiene and maleic anhydride.17 There the electrostatic attraction and delocalization‐polarization are shown to be strong enough to cover the overlap repulsion that have been suppressed considerably by the electron deficiency in the dienophile ring. The “normal electron demand” in the reaction and the high endo selectivity are connected to each other at this point.17, 18 Introduction of electron‐releasing groups into the diene part in place of introducing electron‐withdrawing groups into the dienophile part is not a way to achieve high endo selectivity, as the path will suffer from an intense overlap repulsion. Though the barrier height obtained in this study depends slightly on the level of DFT calculations, the difference in the activation Gibbs free energy between TSendo‐1 and TSexo‐1 remains in a narrow range, 0.4–1.2 kcal/mol (see Table 1), and that between TS′endo‐1 and TS′exo‐1 does so in the range of 2.2–3.5 kcal/mol (see Table 2), indicating that the endo transition state is preferred at all the levels of calculation. The RHF/6‐311G(d,p) level calculation also gives similar results, as it did in the calculations on the cycloaddition between cyclopentadiene and maleic anhydride previously studied.17
To see next the reason why the endo‐path is preferred in this highly asynchronous process, let us compare the reacting system with AlCl3 and the system without AlCl3 that are produced by removing the AlCl3 part from TS′endo‐1 and TS′exo‐1 , respectively, freezing the geometry of other atoms as they were in those structures. A simple partitioning analysis of the interaction energy at the RHF/6‐311G(d,p) level19 and an analysis of the M06‐2X/6‐311G(d,p) level DFT interaction energy by a scheme proposed by Su and Li20 indicate that the difference in energy between TS′endo‐1 and TS′exo‐1 , comes not from the orbital interaction terms but mainly from the electrostatic attraction.21 The sum of the repulsive and attractive orbital interaction terms, i. e., exchange and overlap repulsive interactions and electron delocalization between the diene and the dienophile with the associated polarization within the two parts, is very similar in magnitude in the endo‐ and exo‐paths (see, Tables S4 and S5, and Figure S4 in Supporting Information). The electrostatic attraction between the diene part and the dienophile part is stronger in the endo structure than in the exo structure, the attraction between the positive charge on C8 in the dienophile and the negative charges on C4 and C5 atoms in the diene part being added in the endo structure. Thus, the endo‐addition is preferred even in the absence of AlCl3. The difference in electrostatic attraction between the two transition states becomes more significant, because the positive charges on C8 gets larger in the dienophile polarized by attached AlCl3 (the electrostatic potential maps and atomic charges of the reactants are shown in Figures S8 and S9, respectively, in Supporting Information). The energy difference between TS′endo‐3 and TS′exo‐3 can also be interpreted in this manner (Tables S6 and S7, and Figure S5 in Supporting Information).22 In conclusion, electron delocalization strengthened in the presence of the Lewis acid lowers the barrier height of the cycloaddition to accelerate the reaction both in the endo‐ and exo‐paths, while the electrostatic attraction favors the endo‐path.
In summary, the reactivity and stereoselectivity in the Diels‐Alder reaction between cyclopentadiene and methyl acrylate catalyzed by a Lewis acid are controlled by a combination of several interaction terms. An AlCl3 molecule attached to the carbonyl oxygen atom of methyl acrylate lowers the energy of orbitals in acrylate and also polarizes the molecule.23 The unoccupied interaction orbital of the dienophile that plays the dominant role in electron delocalization from the diene is lowered in energy and shows a larger amplitude on of the two reaction sites, C6, which is placed farther from the carbonyl group. Thus, the electron‐accepting ability of the terminal carbon is enhanced. In contrast, the occupied interaction orbital tends to show a larger amplitude on the other reaction site, C7, but the orbital is also lowered considerably in energy by the presence of AlCl3. The electron‐donating ability of that carbon is weakened. The electron redistribution caused by the attached Lewis acid reduces overlap repulsion in the C2−C6 bond region and strengthens the repulsion in the C3−C7 bond region. As a consequence, the formation of new C−C bonds is highly asynchronous in the Lewis‐acid catalyzed Diels‐Alder reaction between cyclopentadiene and methyl acrylate, the formation of the C2−C6 bond preceding the formation of the C3−C7 bond. Lower barrier heights are provided both for the endo‐path and for the exo‐path than those in the reaction without AlCl3. Here again the electrostatic interaction is shown to play an important role in bringing the high endo selectivity to this cycloaddition, as we have seen previously in the Diels‐Alder reactions in which the formation of two C−C bonds take place in a synchronous manner.17, 24
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP17K05795.
K. Sakata, H. Fujimoto, ChemistryOpen 2020, 9, 662.
References
- 1. Nicolaou K. C., Snyder S. A., Montagnon T., Vassilikogiannakis G., Angew. Chem. Int. Ed. 2002, 41, 1668–1698; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1742–1773. [Google Scholar]
- 2. Du H., Ding K., Handbook of Cyclization Reactions; Ma, S. Ed.; Wiley-VCH: Weinheim ; 2009, p 1–57. [Google Scholar]
- 3. Houk K. N., Strozier R. W., J. Am. Chem. Soc. 1973, 95, 4094–4096. [Google Scholar]
- 4. Fleming I., Molecular Orbitals and Organic Chemical Reactions: Reference Edition; Wiley: West Sussex ; 2010. [Google Scholar]
- 5.
- 5a. Jørgensen K. A., Cycloaddition Reactions in Organic Synthesis; S. Kobayashi, K. A. Jørgensen, Eds.; Wiley-VCH: Weinheim: 2002; p 301–327; [Google Scholar]
- 5b. Ess D. H., Jones G. O., Houk K. N., Adv. Synth. Catal. 2006, 348, 2337–2361; [Google Scholar]
- 5c. Birney D. M., Houk K. N., J. Am. Chem. Soc. 1990, 112, 4127–4133; [Google Scholar]
- 5d. Yamabe S., Dai T., Minato T., J. Am. Chem. Soc. 1995, 117, 10994–10997; [Google Scholar]
- 5e. Dai W.-M., Lau C. W., Chung S. H., Wu Y.-D., J. Org. Chem. 1995, 60, 8128–8129; [Google Scholar]
- 5f. García J. I., Mayoral J. A., Salvatella L., J. Am. Chem. Soc. 1996, 118, 11680–11681; [Google Scholar]
- 5g. Ishihara K., Kondo S., Kurihara H., Yamamoto H., Ohashi S., Inagaki S., J. Org. Chem. 1997, 62, 3026–3027; [DOI] [PubMed] [Google Scholar]
- 5h. García J. I., Martínez-Merino V., Mayoral J. A., Salvatella L., J. Am. Chem. Soc. 1998, 120, 2415–2420; [Google Scholar]
- 5i. Singleton D. A., Merrigan S. R., Beno B. R., Houk K. N., Tetrahedron Lett. 1999, 40, 5817–5821; [Google Scholar]
- 5j. Yamabe S., Minato S. T., J. Org. Chem. 2000, 65, 1830–1841; [DOI] [PubMed] [Google Scholar]
- 5k. Avalos M., Babiano R., Bravo J. L., Cintas P., Jiménez J. L., Palacios J. C., Silva M. A., J. Org. Chem. 2000, 65, 6613–6619; [DOI] [PubMed] [Google Scholar]
- 5l. Alves C. N., Carneiro A. S., Andrés J., Domingo L. R., Tetrahedron 2006, 62, 5502–5509; [Google Scholar]
- 5m. Berski S., Andrés J., Silvi B., Domingo L. R., J. Phys. Chem. A 2006, 110, 13939–13947; [DOI] [PubMed] [Google Scholar]
- 5n. Sun H., Zhang D., Ma C., Liu C., Int. J. Quantum Chem. 2007, 107, 1875–1885; [Google Scholar]
- 5o. Xia Y., Yin D., Rong C., Xu Q., Yin D., Liu S., J. Phys. Chem. A 2008, 112, 9970–9977; [DOI] [PubMed] [Google Scholar]
- 5p. Hayden A. E., DeChancie J., George A. H., Dai M., Yu M., Danishefsky S. J., Houk K. N., J. Org. Chem. 2009, 74, 6770–6776. [DOI] [PubMed] [Google Scholar]
- 6. Sakata K., Fujimoto H., J. Org. Chem. 2013, 78, 3095–3103. [DOI] [PubMed] [Google Scholar]
- 7. Sauer J., Kredel J., Tetrahedron Lett. 1966, 7, 731–736. [Google Scholar]
- 8.
- 8a. Zhao Y., Truhlar D. G., Theor. Chem. Acc. 2008, 120, 215–241; [Google Scholar]
- 8b. Zhao Y., Truhlar D. G., Acc. Chem. Res. 2008, 41, 157–167. [DOI] [PubMed] [Google Scholar]
- 9.For the assessment of M06-2X functional for the Diels-Alder reaction systems, see: Pieniazek S. N., Clemente F. R., Houk K. N., Angew. Chem. Int. Ed. 2008, 47, 7746–7749; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 7860–7863. [Google Scholar]
- 10.Cartesian 6D-type Gaussian functions were used.
- 11.Another structure of the endo transition state (TSendo-4) was also found. However, the Gibbs free energy is higher than that of TSendo-1 by 15.5 kcal/mol. See, Figure S1 in Supporting Information.
- 12.Six transition states were taken into account for the calculation of the ratio.
- 13.The other transition state structures (TS′endo-4, TS′endo-5 and TS′exo-4) were also found. However, these free energies are much higher than that of TSendo-1 by 14.5, 12.8 and 16.0 kcal/mol, respectively. See, Figure S1 in Supporting Information.
- 14.
- 14a. Fukui K., Acc. Chem. Res. 1981, 14, 363–368; [Google Scholar]
- 14b. Hratchian H. P., Schlegel H. B., J. Chem. Phys. 2004, 120, 9918–9924. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Hoffmann R., Woodward R. B., J. Am. Chem. Soc. 1965, 87, 2046–2048; [Google Scholar]
- 15b. Woodward R. B., Hoffmann R., The Conservation of Orbital Symmetry; Verlag Chemie: Weinheim, Germany: 1970. [Google Scholar]
- 16.
- 16a. Fukui K., Koga N., Fujimoto H., J. Am. Chem. Soc. 1981, 103, 196–197; [Google Scholar]
- 16b. Fujimoto H., Acc. Chem. Soc. 1987, 20, 12519–12526. [Google Scholar]
- 17. Sakata K., Fujimoto H., Eur. J. Org. Chem. 2016, 4265–4278. [Google Scholar]
- 18. Fujimoto H., Inagaki S., Fukui K., J. Am. Chem. Soc. 1976, 98, 2670–2671. [Google Scholar]
- 19. Fukui K., Fujimoto H., Bull. Chem. Soc. Jpn. 1969, 41, 1989–1997. [Google Scholar]
- 20. Su P., Li H., J. Chem. Phys. 2009, 131, 014102. [DOI] [PubMed] [Google Scholar]
- 21.Analysis of the interaction energy at the B3LYP/6-311G(d,p)//M06-2X/6-311G(d,p) level shows the same tendency as that made at the M06-2X/6-311G(d,p) level. See Figures S6 and S7 in Supporting Information.
- 22.Much the same results were obtained also for the AlCl3-catalyzed reaction between 1,3-butadiene and methyl acrylate, indicating that the methylene moiety of the C1 atom in cyclopentadiene has little influence on the stereoselectivity. See, Figure S10 in Supporting Information.
- 23.Polarization effect by Lewis acids, see: Sakata K., Fujimoto H., J. Am. Chem. Soc. 2008, 130, 12519–12526. [DOI] [PubMed] [Google Scholar]
- 24. Imade M., Hirao H., Omoto K., Fujimoto H., J. Org. Chem. 1999, 64, 6697–6701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary