

Abstract
Current cutaneous T-cell lymphoma (CTCL) therapies are marked by an abbreviated response, subsequent drug resistance, and poor prognosis for patients with advanced disease. An understanding of molecular regulators involved in CTCL is needed to develop effective targeted therapies. One candidate regulator is p38γ, a mitogen-activated protein kinase crucial for malignant T-cell activity and growth. p38γ gene expression is selectively increased in CTCL patient samples and cell lines but not in healthy T cells. In addition, gene silencing of p38γ reduced CTCL cell viability, showing a key role in CTCL pathogenesis. Screening p38γ inhibitors is critical for understanding the mechanism of CTCL tumorigenesis and developing therapeutic applications. We prioritized a potent p38γ inhibitor (F7, also known as PIK75) through a high-throughput kinase inhibitor screen. At nanomolar concentrations, PIK75, a multiple kinase inhibitor, selectively killed CD4+ malignant CTCL cells but spared healthy CD4+ cells; induced significant reduction of tumor size in mouse xenografts; and effectively inhibited p38γ enzymatic activity and phosphorylation of its substrate, DLGH1, in CTCL cells and mouse xenografts. Here, we report that PIK75 has a potential clinical application to serve as a scaffold molecule for the development of a more selective p38γ inhibitor.
INTRODUCTION
Cutaneous T-cell lymphoma (CTCL) is a severe, disfiguring, and incurable malignancy with a poor prognosis for patients with advanced disease. Current therapies are associated with an abbreviated response and subsequent drug resistance (Hwang et al., 2008; Wilcox, 2016). Unlike many cancers, CTCL pathogenesis remains poorly understood. Until recently, no molecular drivers had been identified, prohibiting the development of driver-based targeted therapies. Thus, identifying critical pathways and molecular drivers of CTCL is essential to understanding progression of the disease and developing effective therapies that improve quality of life and outcome for CTCL patients.
We recently showed enrichment of transcripts involved in the T-cell receptor (TCR) and mitogen-activated protein kinase (MAPK) pathways in CTCL and identified the MAPK p38β isoform as a potential therapeutic target for CTCL (Bliss-Moreau et al., 2015). That study prompted us to characterize the potential of other isoforms of p38 to be therapeutic targets in CTCL. One candidate is p38γ, a 367-amino acid protein that is highly expressed in skeletal muscle, with no detectable expression in normal hematopoietic cells or tissues of the immune system, including lymph nodes and spleen (Li et al., 1996; Wang et al., 1997).
DLGH1, an important scaffolding protein that directs T-cell signaling through the nuclear factor of activated T-cells pathway rather than through the NF-κB pathway (Round et al., 2007; Sabio et al., 2005), is a substrate of p38γ. In HeLa cells, p38γ phosphorylates DLGH1 on serine 158, and the phosphorylation can release DLGH1 (SAP97) from its cofactor GKAP for further related function in HeLa cells (Sabio et al., 2005). Thus, DLGH1, which signals early in the TCR signaling pathway, may influence the TCR pathway through its phosphorylation by the unexpected presence of p38γ kinase in cancerous T cells, such as CTCL cells.
To date, there is only one p38γ inhibitor in clinical practice. Pirfenidone (the orphan drug Esbriet; Genentech, South San Francisco, CA), a drug approved by the US Food and Drug Administration for treatment of pulmonary fibrosis, is a p38γ inhibitor that blocks TGF-β synthesis (Moran, 2011). It inhibits p38γ expression in mice but requires a very high daily dosage (500 mg/kg per day in drinking water) (Yin et al., 2015), which raises questions about its clinical value against cancer. Therefore, more potent p38γ inhibitors may be of benefit for the therapy of this disease. In this study, we show a potential role for p38γ in malignant T-cell activity and growth, identify a potent small molecule inhibitor (F7, also known as PIK75) through high-throughput screening of a kinase inhibitor library, and describe the unique effects of F7/PIK75 on CTCL and p38γ along with other kinases.
RESULTS
p38γ is elevated in CTCL and is important for cell viability
Given that we previously described a role for p38β in CTCL (Bliss-Moreau et al., 2015), we evaluated the role of other p38 isoforms in CTCL. To examine expression of the p38 isoforms in CTCL, we first analyzed a publicly available RNA sequencing database [phs000725] for Sézary Syndrome (SS) (Wang et al., 2015) and microarray databases (GSE17601, n = 32 for SS; GSE12902, n = 22 for mycosis fungoides) (Iqbal et al., 2010). We found that mRNA expression of p38γ as well as p38β from both RNA sequencing (Figure 1a) and microarray analysis (see Supplementary Figure S1a online) was significantly increased and that of p38α was significantly reduced in both CTCL SS patients compared with healthy donors and primary CD4+ T cells (GSE19069, n = 8). It is known that although p38α, p38β, and p38δ are expressed in normal healthy T cells, p38γ is undetectable (Hale et al., 1999). We used quantitative real-time reverse transcriptaseePCR (qRT-PCR) to show that p38γ mRNA level, despite being lowest among other p38 isoforms (see Supplementary Figure S1b), was significantly elevated in CD4+ T cells from two SS patients compared with that of two healthy donors (two-sample t test, P value = 0.0049) (see Supplementary Figure S1c).
Figure 1. p38γ is elevated in CTCL and is important for viability.
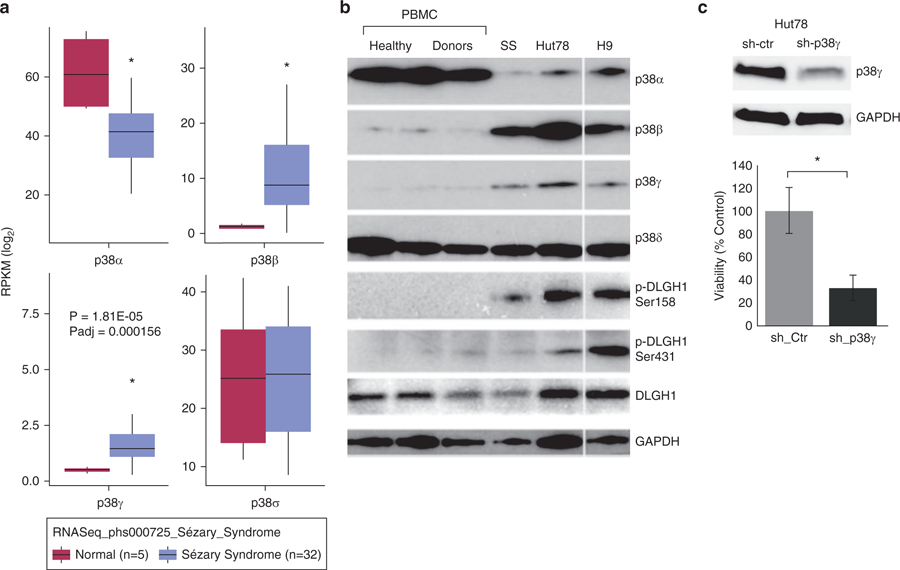
(a) RNA sequencing database phs000725 was downloaded from the dbGAP database with permission. Differential expression analysis of four p38 isoforms between CD4+ T cells of healthy donors (n = 5, pink) and that of patients with Sézary syndrome (n = 32, blue. *P < 0.005) using DESeq 2 package in R. Y-axis indicates log fold changes of expressed genes’ RPKM (reads per kilobase of transcript per million mapped reads). (b) Western blot was used to visualize protein expression of indicated p38 isoforms in peripheral blood mononuclear cells (PBMCs) of healthy donors (n = 3) and one patient with Sézary syndrome (SS), as well as whole cell lysates of Hut78 and H9 CTCL cell lines. White space indicates that the blotting of H9 sample are from different parts of the same gel with the same exposure. DLGH1 is a downstream target of p38γ kinase activity, indicated by phosphorylation at the 158 residue (p-DLGH1 Ser158); phosphorylation at the unrelated 431 residue (p-DLGH1 Ser431) is shown as a control. GAPDH is a control for protein loading. (c) Hut78 cells were transduced with lentiviral particles that harbor shRNA against p38γ or control shRNA. (Top) Western blot was used to visualize protein expression of p38γ. GAPDH is a control for protein loading. (Bottom) Cell viability was measured by trypan blue exclusion, and data are presented as a percentage of control-treated cells. Three replicates were performed for each sample. *P < 0.05. CTCL, cutaneous T-cell lymphoma; Ctr, control; sh, short hairpin RNA.
We used Western blot to evaluate protein expression of p38γ in a CTCL SS patient sample and CTCL cell lines (Hut78, H9, and HH). We showed elevated protein expression of p38γ in one SS patient peripheral blood mononuclear cell (PBMC) sample and two CTCL cell lines compared with PBMCs from three healthy donors, who did not express p38γ (Figure 1b). p38α protein levels were reduced, and those of p38δ remain unchanged. p38β protein expression (Figure 1b), as well as mRNA level (see Supplementary Figure S1a and b), were elevated in CTCL cells compared with healthy donor cells, which is consistent with our previous finding (Bliss-Moreau et al., 2015).
Given that p38γ is elevated in CTCL cells, we investigated the effects of p38γ expression on CTCL viability by silencing p38γ via small interfering RNA or short hairpin RNA (shRNA), using Hut78 cells as a model cell line for CTCL. We showed that both transient transfection with two validated p38γ small interfering RNAs (see Supplementary Figure S1d) and transduction with lentiviral shRNA against p38γ significantly reduced viability compared with the scrambled control (P < 0.05, Figure 1c). We used Western blot to verify the reduced protein expression of p38γ (Figure 1c), and that knockdown is specific to p38γ compared with other p38 isoforms (see Supplementary Figure S1e). We also confirmed that shRNA silencing of p38γ significantly reduced viability in another SS cell line, H9, with similar effects (see Supplementary Figure S1f).
Identification of a p38γ inhibitor with unique features in CTCL
Given that gene silencing of p38γ reduced viability of CTCL cells and few p38γ inhibitors are presently known, identifying novel, potent, and specific inhibitors of p38γ has great potential for clinical application in CTCL. We used high-throughput screening of a commercially available kinase inhibitor library (EMD Biosciences, San Diego, CA) and identified three candidates with activity against p38γ: A10 (also known as Ro3306), A11 (known as PHA-767491), and F7/PIK75 (Figure 2a).
Figure 2. Screening a kinase inhibitor library for p38γ inhibitors led to the selection of F7/PIK75.
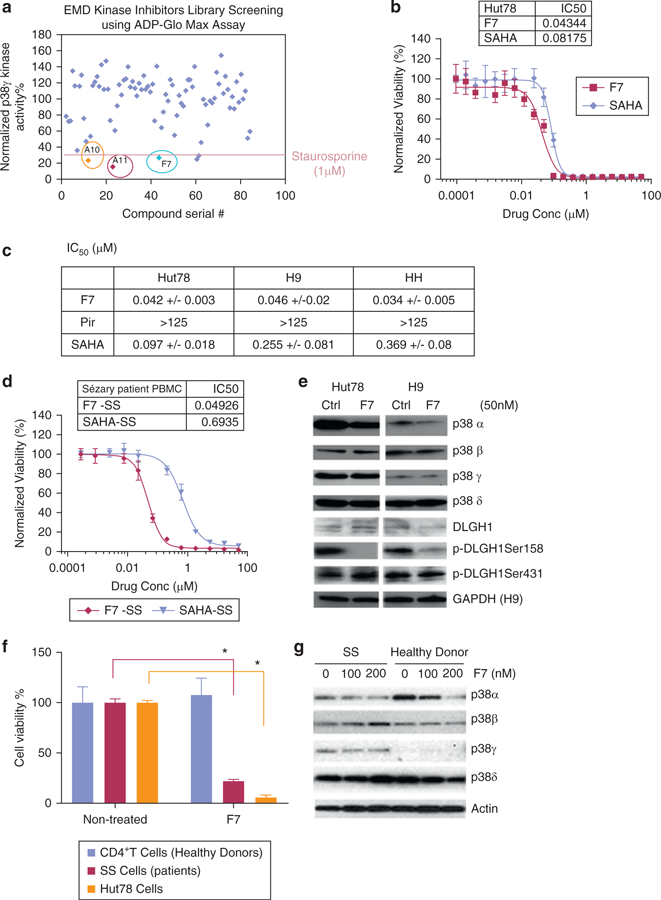
(a) A 260 kinase inhibitor library (EMD Biosciences, San Diego, CA) was screened for p38γ activity using ADP-Glo Max Assay (normalized to DMSO control). The three most potent candidates (A10, A11, and F7) are indicated. 1 μmol/L staurosporine was used as an internal positive control. (b) Data represented are normalized to DMSO control in Hut78 cells treated with varying concentrations of F7/PIK75 or SAHA (an FDA-approved drug for treatment of CTCL, used as a control). Inset table shows calculated IC50 values (μmol/L). (c) Determination of IC50 (μmol/L) in Hut78, H9, and HH cells treated with F7/PIK75, pirfenidone (Pir), or SAHA for 72 hours. *p < 0.05. (d) Cell viability was normalized to DMSO control in PBMCs isolated from SS patients and treated with varying concentrations of F7/PIK75 or SAHA for 72 hours. Inset table shows calculated IC50 values (μmol/L). *p < 0.05. (e) Western blot was used to visualize protein expression of indicated p38 isoforms, phosphorylated DLGH1 Ser158 and Ser431, and actin (loading control) in Hut78 and H9 CTCL cells treated with 50 nmol/L F7/PIK75, using indicated antibodies. (f) Cell viability was measured in CD4+ T cells from healthy donors (n = 2) and SS patient cells (n = 2) treated with 100 nmol/L of F7/PIK75 or DMSO control. Data presented as normalized to untreated controls. (g) Western blot for CD4+ T cells from a healthy donor or an SS patient treated with F7/PIK75 (100 nmol/L or 200 nmol/L) or DMSO control to indicate expression of p38 isoforms and actin as the loading control. All experiments were repeated in three independent experiments, and data represented are the average of triplicate experiments. CellTiterGlo Cell Viability Assay (Promega, Madison, WI) was used to measure viability in b–d. EMD, EMD Biosciences (San Diego, CA); FDA, US Food and Drug Administration; IC50, half maximal inhibitory concentration; M, mol/L; SS, Sézary syndrome.
To test whether the selected compounds caused death in cancer cell lines, we first profiled these three hits across the NCI60 cell line collection. In general, PIK75 was far more potent than A10 or A11 in many types of cancer cells, including solid tumor and hematopoietic cell lines (see Supplementary Table S2 online). Therefore, we centered our subsequent studies on PIK75. PIK75 also showed a dose-dependent reduction of proliferation in CTCL cell lines (Hut78, HH, and H9) and SS cells from patient PBMCs, with a similar half maximal inhibitory concentration (IC50) range (Figure 2b–d). We compared the potency of PIK75 and SAHA (vorinostat, as a hydroxymate histone deacetylase inhibitor), a drug approved by the US Food and Drug Administration for CTCL. PIK75 exceeded SAHA in the efficacy of killing CTCL cells (Figure 2b–d). PIK75 also exceeded pirfenidone greatly in both p38γ kinase activity and cell toxicity to the CTCL cells. The IC50 of pirfenidone against p38γ kinase activity is over 125 μmol/L and the IC50 of pirfenidone in Hut78 cells is over 125 μmol/L (see Supplementary Figure S2a–c online).
To determine whether F7/PIK75 was selective for CTCL over healthy cells, we tested its toxicity in SS patient cells (n = 2) and CTCL cell lines, as well as in normal CD4+ T cells from healthy donors. We showed that 100 nmol/L F7/PIK75 significantly reduced viability of patient SS and Hut78 cells but spared healthy CD4+ T cells (two-sample t test, P < 0.05) (Figure 2f). Cell death caused by F7/PIK75 was via apoptosis in both CTCL cell lines and the SS patient sample (see Supplementary Table S3 online). Furthermore, Western blot results of CD4+ cells from both healthy donors and SS patients indicated that p38γ remained undetectable in healthy donor cells but elevated in SS (Figure 2g). F7/PIK75 inhibited phosphorylation of DLGH1 at Ser158 (pDLGH1-Ser158, the specific site of p38γ phosphorylation) (Sabio et al., 2005), in SS cells (see Supplementary Figure S2c), and in Hut78 and H9 cells (Figure 2e). This suggests that F7/PIK75 inhibits the ability of the p38γ kinase to phosphorylate its substrates (e.g., DLGH1) in CTCL cells.
F7/PIK75 inhibits p38γ kinase activity in vitro and is adenosine triphosphate dose dependent
To show that F7/PIK75 specifically inhibits the kinase activity of p38γ among other p38 isoforms, we performed in vitro ADP-Glo kinase assays (Promega, Madison, WI). We observed a dose-dependent inhibition of p38γ and p38δ kinase activity, but not p38α and p38β, by F7/PIK75 (Figure 3a). We tested the F7/PIK75 compound from different sources (data not shown) and observed a consistent effect with the calculated IC50 values ranging from 4.1e29 nmol/L for p38γ and 32.1 nmol/Le119.98 nmol/L for p38δ, with p38γ being approximately 5-fold more sensitive than p38δ. We further confirmed these data using radiometric kinase assays. At 50 nmol/L and 200 nmol/L, F7/PIK75 showed a 22.5% and 59% inhibitory effect, respectively, on p38γ, compared with 5.4% and 34.9% on p38δ; neither concentration inhibited p38α or β (see Supplementary Table S4 online). Both assays suggest that the specific effects of F7/PIK75 on p38γ relative to p38δ are more pronounced at lower doses.
Figure 3. F7 targets p38γ kinase activity in vitro and in an ATP-dependent manner.
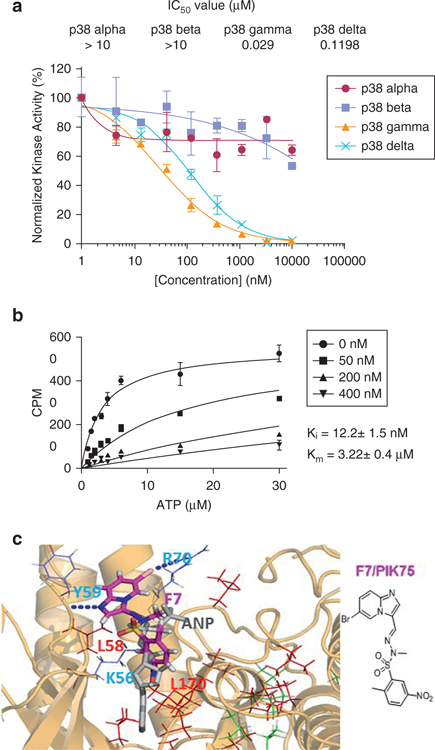
(a) ADP-Glo in vitro kinase assay was used to calculate IC50 for F7/PIK75 inhibition of kinase activity of the four p38 isoforms, normalized to DMSO control. Calculated IC50 values (μmol/L) are indicated. (b) Time-resolved fluorescence energy transfer was used to measure in vitro enzyme kinetics of inhibition of p38γ kinase at indicated concentrations of F7. CPM (counts per minute) corresponds to product formation level. The error bar of the measurements represents standard deviation of triplicate data. Solid lines represent data fitting to the competitive inhibition model. Calculated Ki and Km values are indicated. (c) Mapping of the CSPs induced by F7/PIK75 binding to p38γ on the docked structure of p38γ in complex with F7. F7/PIK75 forms three hydrogen bonds with K56/Y59/R70, which are displayed as blue dots. The ANP molecule x-ray structure is displayed as grey sticks for comparison. The residues with the largest line-broadening effects are indicated in red, and those with significant CSPs (> 0.05 ppm) are indicated in green with their sidechains shown in stick form. L58 and L170 are within a 3-Å distance of F7/PIK75. ANP, an analogue of ATP molecule with extra nitrogen bonds (an non-hydrolyzable ATP analogue); ATP, adenosine triphosphate; CSP, chemical shift perturbation; Ctrl, control; IC50, half maximal inhibitory concentration; Ki, enzyme constants for inhibitors; Km, enzyme constants for substrates; M, mol/L; SS, Sézary syndrome.
To confirm that F7/PIK75 binds to the adenosine triphosphate (ATP)-binding pocket of p38γ, we performed an in vitro kinase assay using increasing concentrations of ATP in the presence of p38γ and a synthetic peptide substrate. We performed time-resolved fluorescence energy transfer using a peptide substrate, ULight-4E-BP1 (PerkinElmer, Waltham, MA), derived from the p38γ phosphorylation site of 4E-BP1 (Pons et al., 2012). We determined the ATP Km (enzyme constants for substrates) and inhibitor Ki (enzyme constants for inhibitors) to be 3.2 ± 0.4 μmol/L and 12.21 ± 1.5 nmol/L, respectively (Figure 3b). The ATP Km for p38γ is nearly identical to that described in the product manual for the peptide substrate. The Ki value indicates strong binding and inhibition of p38γ by F7/PIK75. Together, the enzyme kinetics indicate that F7/PIK75 inhibits p38γ by a competitive mechanism, that is, competing with ATP binding to the enzyme.
We used NMR methods to identify the F7/PIK75 binding site on p38γ. F7/PIK75 induced extensive NMR chemical shift perturbations (CSPs, calculated as described in Methods) and line broadening in the 1H-13C HMQC spectrum (see Supplementary Figure S3 online). We used our in-house–developed All-Around Docking methodology (Nam et al., 2013) to model PIK75 binding to p38γ. The three-dimensional structural binding model predicted that PIK75 binds to the ATP-binding pocket of p38γ protein through three hydrogen bonds at K56, Y59, and R70 in the ATP pocket (displayed as blue dots in Figure 3c) based on the binding score calculation (Glide XP docking score of −8.0kcal/mol) (Glide; Schrödinger, New York, NY). The residues that showed line broadening effects of NMR data are indicated in red in the structure of p38γ (Figure 3c); residues that showed significant NMR CSPs are indicated in green. Because the most significant CSP occurs around the ATP-binding pocket, these results, combined with the enzyme kinetic studies, indicate that PIK75 binds to the ATP-binding pocket of p38γ.
F7/PIK75 targets p38γ kinase activity in vivo and reduces xenograft tumors
Because the effects of kinase inhibition in vitro can differ from kinase inhibition in vivo due to a number of factors, such as retention in the cytosol (or other compartment of the cells) because of phosphorylation of kinase substrates of p38, such as NFATs (Alam et al., 2014), we further investigated the effects of PIK75 in a CTCL xenograft model. We treated mice harboring Hut78 xenografts with PIK75 or vehicle control by intraperitoneal injection and harvested the tumors. We showed that PIK75 significantly inhibited tumor growth at 2 mg/kg (P = 0.015) and 10 mg/kg (P = 0.025) (Figure 4a). We then monitored p38γ activity using pDLGH1-Ser158. We showed that pDLGH1-Ser158 was significantly reduced in xenografts when mice were treated with PIK75 at 10 mg/kg; in contrast, untreated control mice xenografts expressed pDLGH1-Ser158 (Figure 4b).
Figure 4. Lead compound F7/PIK75 targets p38γ kinase activity in vivo and reduces tumor volume in a dose-dependent manner in Hut78 cell xenograft mice.
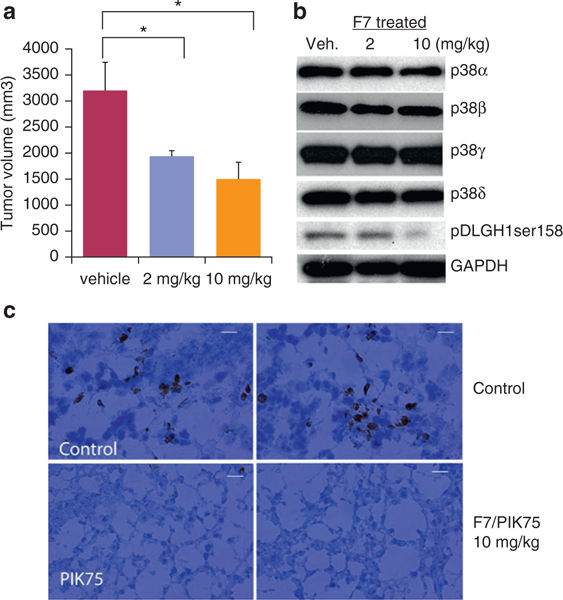
(a) Tumor volume was measured in tumors excised from Hut78 xenograft mice treated with vehicle control or 2 mg/kg or 10 mg/kg F7 for 8 days. n = 7 mice per treatment, three tumors were measured per mouse. *P <0.05. (b) Western blot was used to visualize protein expression of indicated p38 isoforms, phosphorylated DLGH1 Ser158, and GAPDH (loading control) in tumor sections from all Hut78 xenograft mice treated with vehicle control or 2 mg/kg or 10 mg/kg F7/PIK75. (c) Immunohistochemistry was performed on tumor sections from all Hut78 xenograft mice treated with vehicle control or 10 mg/kg F7/PIK75. Representative images for staining with pDLGH1-Ser158 polyclonal antibody (brown color). Scale bar = 20 μm. Veh, vehicle.
To confirm that PIK75 inhibited p38γ kinase activity in vivo, we also performed immunohistochemistry on tumor tissues from xenografted mice and showed that pDLGH1-Ser158 staining was dramatically reduced in PIK75-treated tumors compared with controls (Figure 4c). Taken together, our data indicate that PIK75 reduces cell growth and inhibits p38γ kinase activity both in vitro and in vivo.
PIK75 inhibits multiple kinases including p38γ and phosphatidylinositol 3 kinase p110a
Because PIK75 was originally identified as a phosphatidylinositol 3 kinase (PI3K) p110α inhibitor with an IC50 of 5.8 nmol/L (Zheng et al., 2011), we measured the IC50 of both PI3K p110α and p38γ; we showed IC50s of 4.1 and 30 nmol/L, respectively (see Supplementary Figure S4 online). To determine whether PI3K p110α targeting by F7/PIK75 contributes to cell death in CTCL, we first used lentiviral shRNA to silence PI3K p110α in Hut78 cells. To our surprise, unlike knockdown of p38γ (Figure 1c, and see Supplementary Table S1 and Figure S1e and f), cell viability was not affected by gene silencing of PI3K p110α isoform (Figure 5d). This suggests that in CTCL, F7/PIK75 causes cell death by targeting p38γ rather than targeting PI3K p110α. To further confirm this, we evaluated cell toxicity effects in CTCL cells with three other potent PI3K p110α specific inhibitors, A66, GDC0941, and BEZ235. GDC-0941 (pictilisib, which also targets mTOR) is a Pan-PI3K inhibitor (Wallin et al., 2012) and BEZ 235 (dactolisib, which also targets ATR)) is a dual inhibitor of PI3K and mTOR inhibitor, which blocks the dysfunctional activation of the PI3K pathway (Maira et al., 2008). A66 is a unique p110α-specific inhibitor that has over 100-fold selectivity over other class-I PI3K isoforms (IC50 of 11 nmol/L) (see Supplementary Figure S4). These inhibitors have low IC50s (Figure 5b) against PI3K p110α in cell-free assays but no inhibitory kinase activities against p38γ (Figure 5c). F7/PIK75 had the most potent inhibitory effect on Hut78 cells compared with the other three PI3K inhibitors (Figure 5a). GDC0941 and BEZ235 showed only modest reduction of cell viability. Western blot analysis indicated that A66 has dose-dependent inhibition of phosphorylation of ATK at ser473 in Hut78 cells (4-hour treatment) (Figure 5e) but has no influence on the viability of Hut78 cells up to approximately10 μmol/L in 72 hours (Figure 5a). Consistent with our shRNA results, PI3K p110α inhibition alone does not contribute to CTCL cell toxicity, whereas p38γ inhibition causes cell death in CTCL.
Figure 5. Lead compound F7/PIK75 targets activity of multiple kinases, including pI3K and p38γ.
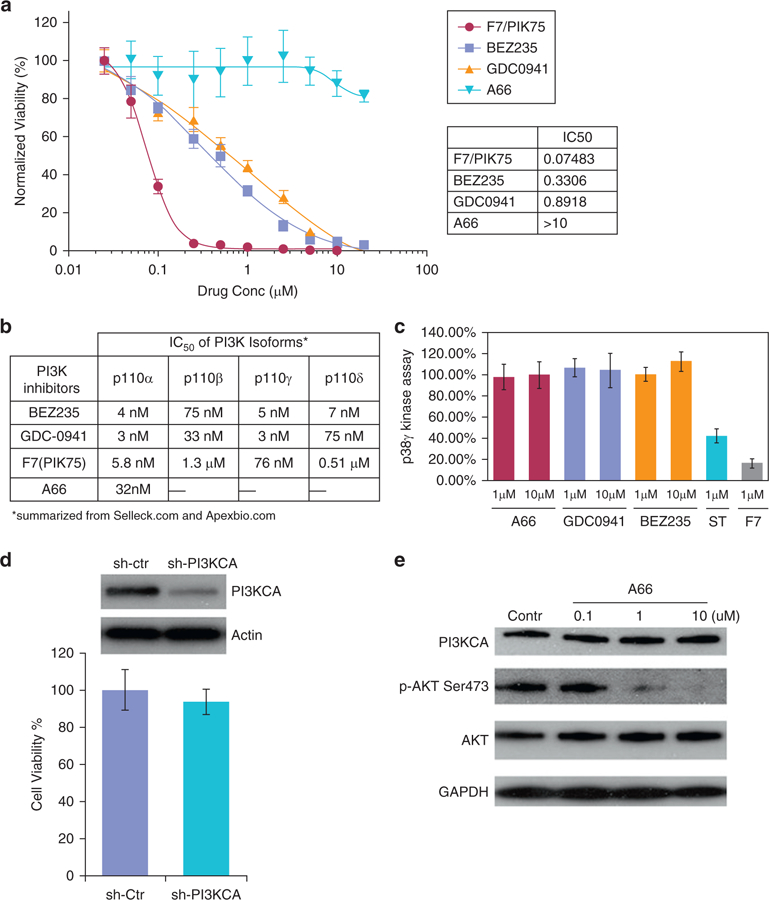
Cell viability was measured and normalized to DMSO controls in Hut78 cells treated with varying concentrations of three potent PI3K-specific inhibitors (BEZ235, GDC-0941, or A66) and F7/PIK75. Inset table shows calculated IC50 values. (a) Comparison of IC50 for inhibition of PI3K kinase activity by three PI3K-specific inhibitors (BEZ235, GDC-0941, or A66) and F7/PIK75. This table is summarized from publicly available databases (summarized from the companies of Selleck [Houston, TX] and Apexbio [Boston, MA]). (b) Cell-freeebased p38γ kinase assay using ADP Glo was performed with indicated concentrations (1 μmol/L and 10 μmol/L) of three PI3K-specific inhibitors (A66, GDC-0941, or BEZ235) and 1 μmol/L of F7/PIK75. Staurosporine (1 μmol/L), a pan-kinase inhibitor, was used as a positive control. (c) Hut78 cells were transduced with shRNA against PI3K 110α or control shRNA. (Top) Western blot was used to visualize protein expression of PI3K 110α. Actin is a control for protein loading. (Bottom) Trypan blue exclusion was used to measure cell viability as a percentage of control-treated cells. Three replicates were performed. *P < 0.05. (a) Western blot was used to visualize A66 effects on Hut78 cells, indicated by protein expression level of downstream targets of PI3Kp110α. GAPDH is a control for protein loading. Contr, control; Ctr, control; IC50 half maximal inhibitory concentration; M, mol/L; PI3K, phosphatidylinositol 3 kinase; sh, short hairpin; uM, μmol/L.
DISCUSSION
Several studies have identified p38γ inhibitors as promising therapeutic agents for many tumor types; such as colon (Yin et al., 2015), prostate (Browne et al., 2016), esophageal (Zheng et al., 2016), and breast cancers (Qi et al., 2015). In addition to activity against CTCL, the p38γ inhibitor we identified, F7/PIK75, also shows activity against cells derived from several types of cancers, including prostate, ovarian, and breast cancers (see Supplementary Table S2). In comparison, the known p38γ inhibitor pirfenidone (see Supplementary Figure S2a and b) had an IC50 for cell killing greater than 125 μmol/L on the CTCL cells and for p38γ kinase activity greater than 125 μmol/L, which is minimal compared with the effects of PIK75 on CTCL cells. Our data further showed many unique features of PIK75 that warrant further study of PIK75 as a potential anticancer treatment, particularly for CTCL.
Here, we report that F7/PIK75, a multiple kinase inhibitor, works in part through p38γ inhibition, either directly or indirectly, the mechanism of which may be through targeting the TCR signaling pathway in CTCL. It binds and inhibits p38γ at nanomolar concentrations (Figure 3a–c); more importantly, it selectively killed CTCL patient sample cells but spared healthy CD4+ T cells (Figure 2f and g). A likely explanation for this is that CTCL cancer cells are more vulnerable to p38γ inhibitors, because p38γ is elevated in CTCL but is not expressed in normal T cells.
PIK75 also targets other kinases (Lemke et al., 2014), including PI3K p110α. Our data show that it inhibits in vitro activity of p38γ at a nanomolar range (IC50 values of 29 nmol/L) (Figure 3a), which is the same magnitude as that of PI3K p110α (4.1 nmol/L) (see Supplementary Figure S4). A66 shows potent inhibitory effects on in vitro kinase activity of PI3K p110α (IC50 values of 11 nmol/L) (see Supplementary Figure S4), but does not inhibit in vitro activity of p38γ (Figure 5c). Surprisingly, silencing of PI3K p110α via shRNA or kinase inhibition via the specific inhibitor A66 does not affect cell viability in CTCL (Figure 5a), suggesting that p38γ plays a more critical role than PI3K p110α in CTCL and that cell death caused by PIK75 was due to targeting p38γ rather than PI3K p110α. However, the mechanism of specifically which inhibition of PI3Kp110α by A66 results in blocking phosphorylation of Akt at ser473 (Figure 5e) needs further investigation. The p110δ isoform of PI3K, not the p110α isoform, predominantly expressed in T cells, and that p110δ isoform plays more important roles in T-cell proliferation (Soond et al., 2010). However, p110δ is less sensitive to PIK75 (IC50 = 510 nmol/L) than p110α isoforms of PI3K (IC50 = 5.8 nmol/L) (Figure 5b). The PI3K pathway is known to be one of the most frequently mutated pathways in human cancer and is critical for driving cancer cell progression (Hanahan and Weinberg, 2011). A recent study (Lien et al., 2017) illustrated that a PI3K-dependent but AKT-independent pathway is just as important as the PI3K/AKT-dependent pathway in promoting carcinogenesis and progression. PI3K/AKT dependence provides a therapeutic target for cancers (Courtney et al., 2010), which is also true in CTCL (Querfeld et al., 2006). Our NanoString RNA analysis (NanoString Technologies, Seattle, WA) data followed by Ingenuity Pathway Analysis (Qiagen, Venlo, Netherlands) shows that the activation Z score of PI3K signaling (both AKT dependent and AKT independent) is −3.108, whereas that of AKT-dependent PI3K signaling is −0.63, indicating that the AKT-independent PI3K pathway is much more affected by F7/PIK75 (50 nmol/L) in CTCL cells than that of AKT-dependent PI3K pathway (see Supplementary Table S5 online).
F7/PIK75 targets other kinases in vitro as well, including GSK3 and PKCβ (Lemke et al., 2014), potential therapeutic targets we have previously studied in CTCL cells. The combined inhibition of GSK3 and PKCβ synergistically kills CTCL cells through the p38-TCR pathway (Bliss-Moreau et al., 2015), although the mechanism remains unclear. However, we observed reduction of both p38γ and p38β at the transcriptional level in this combined inhibition, which may contribute to the cell death. In addition, p38γ has many substrates, which may result in different signaling pathways in cancers, as our NanoString RNA analysis has shown (see Supplementary Table S5).
F7/PIK75 is known to have inhibitory activity against other kinases such as the CMGC kinase family, an evolutionarily conserved kinase group across all eukaryotes, including cyclin-dependent kinases, MAPKs, glycogen synthase kinases, and cyclin-dependent kinase–like kinases (Lemke et al., 2014). Therefore, we do not rule out possibility of other kinase involvement in cell killing in CTCL by F7/PIK75. However, we argue that its multitargeting nature does not diminish the importance of its activities on p38γ in CTCL. In fact, PI3K p110α is more sensitive to F7/PIK75 than that of p38γ in cell-free–based analysis (see Supplementary Figure S4), our shRNA data indicate that it is p38γ inhibition (Figure 1c), but not PI3K p110α inhibition (Figure 5a and d) that contributes to the Hut78 cell death in CTCL. Also, we have previously shown that inhibition in CTCL of PKCβ, another target of F7/PIK75, increases apoptosis in vitro (Querfeld et al., 2006), but it was not effective in in vivo patient preclinical and clinical studies (Querfeld et al., 2011). In contrast, F7/PIK75 exhibits a greater clinical potential for drug development in CTCL preclinical studies, as shown through its in vivo mouse studies reducing the size of Hut78 cells xenografts and targeting p38γ kinase activity.
In addition, in our animal studies we did not observe any toxic effects, especially notable given the much lower dosages required compared with pirfenidone, which requires 500 mg/kg dosages (Kozono et al., 2013; Qi et al., 2015). With a dosage as low as 10 mg/kg of F7/PIK75 every 2 days, we showed significantly inhibited tumor growth in xenograft mice within 8 days (Figure 4a) and reduced phosphorylation of the p38γ substrate DLGH1 (Figure 4c). This suggests that F7/PIK75 inhibition of p38γ kinase activity in CTCL plays a role in the reduction of tumor size in xenograft mice and marks F7/PIK75 as a potential lead compound for optimizing p38γ-specific inhibition in the future. Nevertheless, there are potential precautions that clinical application of a p38γ inhibitor could have cytotoxic effects on skeletal muscles, where p38γ is highly expressed (Ho et al., 2004). However, we argue that in other cell types, the mechanistic role of p38γ (e.g., nonkinase activities [Qi et al., 2004]) differs dramatically from its role that in CTCL in which a unique TCR signaling pathway is being targeted. Further studies are needed to address the F7/PIK75 synergistic effects on p38γ with other kinases or its indirect inhibition upstream of the MAPK pathway in CTCL.
MATERIALS AND METHODS
Compound, samples, and cell culture
PIKP110a inhibitors, PIK75, A66, GDC0941, and BEZ235 are from Selleck (Houston, TX). Isolation of PBMCs or CD4+ T cells from both patients and healthy donors and culture of CTCL cell lines (Hut78, HH, and H9) were performed as previously described (Bliss-Moreau et al., 2015). All patient tissue collection and research use adhered to protocols approved by the institutional review and privacy boards at City of Hope, in accordance with the Declaration of Helsinki. All participants signed written informed consent.
qRT-PCR
Total RNA was generated using Trizol reagent (Life Technologies, Carlsbad, CA), followed by double DNase treatment and column purification using Qiagen RNeasy Clean-up (Qiagen, Germantown, MD). The Invitrogen Superscript III First-Strand Synthesis System (Invitrogen, Waltham, MA) was used for cDNA synthesis and TaqMan 5700 Sequence Detection System (Applied Biosystems, Foster City, CA) was used for qRT-PCR. All PCR reactions were run in triplicate. The amplified transcripts were quantified using the comparative ΔCT method as described (Zhang et al., 2007). cDNA and primers for MAPK11, 12, 13, and 14 and GAPDH (see Supplementary Table S1) were added to SYBR Green PCR Master Mix (SYBR Green I Dye, AmpliTaq DNA polymerase, deoxynucleotides with deoxyuridine triphosphate, and optimal buffer components: Applied Biosystems) and subjected to PCR amplification (1 cycle at 95°C for 3 minutes, 35 cycles at 95°C for 15 seconds and 55°C for 30 seconds, and 1 cycle at 50°C for 2 minutes).
Viability assays using trypan blue exclusion and CellTiterGlo Cell Viability Assay
Cell viability was calculated by diluting cell suspensions 1:1 in 0.4% trypan blue solution (Sigma-Aldrich, St. Louis, MO) and counting the number of viable cells using a TC20 automated cell counter (Bio-Rad, Irvine, CA) that automatically excludes the number of nonviable cells stained with trypan blue per total cells. CellTiterGlo Cell Viability Assay (Promega) method was used as described previously. All data points are an average of triplicate experiment.
Small interfering RNA
For p38γ silencing, Hut78 cells were transfected using X-tremeGENE transfection reagent (Roche, Indianapolis, IN) and 100 μmol/L of specific human siRNAs against p38γ (GenScript, Piscataway, NJ): si_p38gA (Cat#SR304235A) sequences–5′ GGAAG CGUGUUACUUACAAAG-AGGT3′–; si_p38gB (Cat#SR304235B) sequences–5′ CGGAGAUGAUCACAGGCAAGAC-GCT3′–.
shRNA
MISSION shRNA Lentiviral Transduction Particles (company validated) expressing five shRNAs in pLKO.1-puro shRNA vector that targets four exons of the human p38γ gene (MAPK12) and scrambled transduction particles (pLKO.1-puro shRNA vector-only) were purchased from Sigma-Aldrich. Four shRNA lentiviral particles each contain unique 29mer target human PIK3CA-specific shRNA (catalog #TL310428V) and one Lenti shRNA scramble control particles (catalog #TR30021V) are from OriGene Technologies, Inc. (Rockville, MD). Hut78 and H9 cells growing exponentially (70%–80% confluent) were seeded into 6-well plates (2 × 105 cells/well) before transduction. Viral transduction efficiencies were improved by adding polybrene (hexadimethrine bromide, Sigma-Aldrich) to a final concentration of 8–10 μg/ml. Hut78 and H9 cells were transduced at a multiplicity of infection of 2. Hut78 and H9 cells were incubated overnight with the medium containing lentiviral particles and polybrene; then the medium containing lentiviral particles was removed from wells and replaced with fresh medium. Cells were collected on day 5 after transduction before RNA extraction and protein isolation.
In vitro kinase assay (ADP-Glo)
We identified p38γ inhibitors using in vitro kinase assay by screening a library of kinase inhibitors: the library consists of 244 compounds on three plates (EMD catalog #539744, #539745, and #539746) that are mostly ATP mimics. All compounds are cell permeable, reversible, and well-characterized. For the biochemical screening, kinase assays in vitro were performed using ADP-Glo kit (Promega) following the manufacturer’s instructions. All data points are the average of triplicate experiments unless stated otherwise, and all compounds were tested and show no inhibition of luciferase activities when using the ADP-Glo kit. Briefly, for in vitro assay experiments, human recombinant p38α, -β, -γ, or -δ proteins (active full-length) were from SignalChem (Richmond, BC, Canada), and followed the protocol of the company. The p38 kinase was pre-incubated with compound F7 in a dose-dependent manner for 10 minutes before synthetic peptide substrates (IPTTPITTTYFFFKKK) were added at a final concentration of 0.2 μg/μl, followed by addition of ATP. Then, ADP-Glo Reagent was incubated in the mixture at room temperature for 40 minutes, followed by incubation of Kinase Detection Reagent (Promega) for another 30 minutes. IC50 values were determined using CalcuSyn software (Biosoft, Cambridge, UK).
Radiometric in vitro kinase assay
Radiometric in vitro kinase assay was performed by SignalChem using F7 at two concentrations (50 nmol/L and 200 nmol/L) according to the company’s proprietary research methodologies.
Enzyme kinetics
The inhibition mechanism of the compound was measured using the time-resolved fluorescence energy transfer method (Ergin et al., 2016). Assays were conducted in a 384-well black, round-bottom plate in kinase reaction buffer (50 mmol/L HEPES, pH 7.5; 10 mmol/L MgCl2; 1 mmol/L EGTA, 100 μmol/L Na3VO4; 0.01% Tween-20; 0.5 mmol/L dithiothreitol). p38γ kinase (700 ng/ml) was mixed with ULightT-4E-BP1 peptide (50 nmopl/L) (PerkinElmer) and varying concentrations of ATP (1, 1.5, 2, 3, 4, 6, 15, and 30 μmol/L) and F7 (0, 50, 200, 400 nmol/L). Time course data were collected by stopping the kinase reaction at different times by adding detection buffer containing Europium-anti-phospho-4E-BP1 antibody (4 nmol/L, PerkinElmer). Fluorescence signals were measured at 665 nm with a 50-ms delay after excitation at 320 nm using a CLARIOstar microplate reader (BMG Labtech, Ortenberg, Germany). The signal ratio at 665/620 nmol/L was used for data analysis. The inhibition mechanism and kinetic rate constants were analyzed using GraphPad Prism 7 software (GraphPad, La Jolla, CA).
NMR studies
2D; Ileδ1-[13CH3]; Leu, Val-[13CH3, 12CD3]elabeled p38γ sample was purified and prepared for NMR studies. The sample contained 20 mmol/L protein in buffer (20 μmol/L sodium phosphate, pH 7.3; 100 mmol/L NaCl; 0.03% NaN3; and 10% D2O). The compound was added to a final concentration of 200 μmol/L. The same volume of DMSO was used as a control. 1H-13C HMQC spectra were collected with 2048 × 128 complex points at 25°C for 5 hours on a Bruker Ascend 700 spectrometer equipped with a cryoprobe. The spectra were processed and analyzed with the software NMRPipe (Delaglio et al., 1995) and Sparky (UCSF, San Francisco, CA), respectively. NMR CSPs were calculated as
where ΔδH and ΔδC are the chemical shift differences between the free and bound states in the proton and carbon dimensions, respectively. For p38γ resonance assignments, NMR experiments were acquired using 15N–; 13C–; 2D–; Ileδ1-[13CH3] –; and Leu, Val-[13CH3, 12CD3]elabeled p38γ sample. Triple-resonance spectra, including HNCA, HNCOCA, HNCACB, HNCOCACB, HNCO, and HNCACO, were used for the backbone assignments, as we described previously (Wang et al., 2015). Methyl groups of Ile, Val, and Leu were assigned using carbon-13 nuclear overhauser effect spectroscopy and methyl-backbone correlation experiments (Tugarinov and Kay, 2003). The additional assignments of backbone amides and methyls were transferred from the deposited assignments (Biological Magnetic Resonance Bank entry: 26732 and 26733) (Aoto et al., 2016).
Modeling of F7/PIK75 binding to p38γ protein by using All-Around Docking methodology
We implemented our in-house All-Around Docking methodology to predict the best binding site and binding pose of F7 on p38γ protein. Based on the Glide software (Schrödinger) for docking, All-Around Docking allows a small molecule to search the whole surface of a target protein for the best docking site with the lowest docking score. The structure of the p38γ protein in complex with the ANP molecule phosphoaminophosphonic acid-adenylate ester was used as the docking target (RCSB protein databank ID: 1cm8), as shown in Figure 3c.
IC50 value determination of cytotoxicity of F7/PIK75 against human NCI-60 cell lines and CTCL cells
To determine the cytotoxicity of F7/PIK75 using our in-house NCI-60 cancer cell line panel assay at City of Hope, MTS assays (Promega) were performed, and cell viability was determined as described previously (Yang et al., 2014). For determination in CTCL cells, CellTiterGlo Cell Viability Assay (Promega) was used to measure viability of Hut78, HH, and H9 cells, and SS patient samples. Absorbance was monitored at 490 nm using an automated BMG PHERAstar plate reader (BMG Labtech). IC50 values were determined using CalcuSyn software (Biosoft). All experiments are repeated in three independent experiments, and data represented are the average of triplicate experiments.
CTCL xenograft tumor model
All animals were housed and handled in accordance with the guidelines of City of Hope Institutional Animal Care and Use Committee. The experiments described here were specifically approved by Institutional Animal Care and Use Committee protocols #07049 and #17118. Female and male 6-week-old NSG mice (NOD-scid IL2Rγ null) were purchased from Jackson Laboratory (Sacramento, CA). Hut78 cells were used for developing CTCL xenograft tumors. Briefly, 5 million cells in 100 μl (50% Matrigel/50% phosphate buffered saline; Sigma-Aldrich) were injected subcutaneously into the right flank of each mouse, and tumor development was monitored every other day. Once the tumors are were palpable and reached 100 mm3 in volume, treatment was commenced. Male and female mice, aged 8–12 weeks and with mass of 20–25 g, were divided into 3 groups (seven mice each group): control group 1 received saline containing 2% DMSO, group 2 was treated with 2 mg/kg F7/PIK75, and group 3 was treated with 10 mg/kg F7/PIK75. Mice were treated every 2 days via intraperitoneal injection. Tumors were measured twice per week using calipers. Animals were sedated and killed once tumors reached 30 mm in diameter or if any animal lost more than 20% body weight or exhibited any severe pain/distress signs that matched premature death criteria.
Western blot
Western blots were performed as described previously (Bliss-Moreau et al., 2015). Rabbit primary antibodies (Cell Signaling Technology [CST], Danvers, MA) were used at the following dilutions: anti-p38α, -p38β, -p38γ, and -p38δ (1:1,000); anti-β-tubulin (1:2,000), anti-p38 GAPDH 1:1,000; anti-β-actin (8H10D10) mouse mAb (1:2,000, CST); anti-DLGH1 total protein; and anti-pDLGH1 at serine 158 and 431 (total SAP97, S285B SAP97 phospho-Ser158 and 431, affinity purified sheep polyclonal antibody, University of Dundee, Scotland, 1:1;000). horseradish peroxidase-conjugated goat anti-rabbit antibody #7074 and anti-mouse IgG horseradish peroxidase-linked antibody #7076 (CST, 1:2,000) were used as secondary antibodies. For detection of p38γ protein level in SS patient samples, we used a monoclonal antibody that close to N-terminus of human p38γ MAPK (SAPK3) from Abcam (ab205926).
Immunohistochemistry
Xenograft tumors were prepared in paraffin, and representative paraffin blocks were chosen for immunohistochemical analysis, performed by City of Hope Pathology Core (Solid Tumor) as described previously (Chu et al., 2011), using primary anti-pDLGH1 phospho-Ser158 polyclonal antibody (S285B SAP97, 1:500) and secondary anti-sheepehorseradish peroxidase antibodies (1:2,000).
NanoString nCounter gene expression quantification and validation
100 ng of RNA isolated from Hut78 cells, as suggested by the NanoString protocol, was used in the experiment. All samples were validated. Data were analyzed using nSolver 3.0 digital analyzer software and R program (NanoString Technologies).
Statistical analysis
All experimental data are shown as mean ± standard error of the mean unless indicated otherwise. The statistical significance of differences, that is, in cell viability assays and mRNA expression of target genes, were assessed by Student t test (SPSS; IBM, Armonk, NY) or one-way analysis of variance (GraphPad PRISM, version 3.0, GraphPad). Differences were considered significant at P less than 0.05. Sign test, a nonparametric test, was used for inhibitory IC50 of NCI 60 cells by p38 γ candidates, significant at P less than 0.005.
Supplementary Material
ACKNOWLEDGMENTS
We thank City of Hope Pathology Core (Solid Tumor) for sectioning and for pDLGH1 staining of xenograft tumors. Research reported in this publication included work performed in the Bioinformatics, High Throughput Screening, Synthetic and Biopolymer Chemistry, Pathology (Solid Tumor), and Molecular Pathology Cores supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also thank Lu Chen, City of Hope Department of Information Sciences, for statistical consulting and Sarah T. Wilkinson for manuscript editing.
Abbreviations:
- ATP
adenosine triphosphate
- CSP
chemical shift perturbation
- CTCL
cutaneous T-cell lymphoma
- IC50
half maximal inhibitory concentration
- MAPK
mitogen-activated protein kinase
- NMR
nuclear magnetic resonance
- PBMC
peripheral blood mononuclear cell
- PI3K
phosphatidylinositol 3 kinase
- shRNA
short hairpin RNA
- SS
Sézary syndrome
- TCR
T-cell receptor
Footnotes
CONFLICT OF INTEREST
Yuan Chen owns equity in SUMO Biosciences, Inc. The remaining authors state no conflict of interest.
SUPPLEMENTARY MATERIALS
Supplementary material is linked to the online version of the paper at www.jidonline.org, and at https://doi.org/10.1016/j.jid.2018.04.030.
REFERENCES
- Alam MS, Gaida MM, Ogawa Y, Kolios AG, Lasitschka F, Ashwell JD. Counter-regulation of T cell effector function by differentially activated p38. J Exp Med 2014;211:1257–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto PC, Martin BT, Wright PE. NMR characterization of information flow and allosteric communities in the MAP kinase p38gamma. Sci Rep 2016;6: 28655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss-Moreau M, Coarfa C, Gunaratne PH, Guitart J, Krett NL, Rosen ST. Identification of p38β as a therapeutic target for the treatment of Sézary syndrome. J Invest Dermatol 2015;135:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne AJ, Gobel A, Thiele S, Hofbauer LC, Rauner M, Rachner TD. p38 MAPK regulates the Wnt inhibitor Dickkopf-1 in osteotropic prostate cancer cells. Cell Death Dis 2016;7:e2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu PG, Chung L, Weiss LM, Lau SK. Determining the site of origin of mucinous adenocarcinoma: an immunohistochemical study of 175 cases. Am J Surg Pathol 2011;35:1830–6. [DOI] [PubMed] [Google Scholar]
- Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol 2010;28:1075–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 1995;6:277–93. [DOI] [PubMed] [Google Scholar]
- Ergin E, Dogan A, Parmaksiz M, Elcin AE, Elcin YM. Time-resolved fluorescence resonance energy transfer [TR-FRET] assays for biochemical processes. Curr Pharm Biotechnol 2016;17:1222–30. [DOI] [PubMed] [Google Scholar]
- Hale KK, Trollinger D, Rihanek M, Manthey CL. Differential expression and activation of p38 mitogen-activated protein kinase alpha, beta, gamma, and delta in inflammatory cell lineages. J Immunol 1999;162:4246–52. [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- Ho RC, Alcazar O, Fujii N, Hirshman MF, Goodyear LJ. p38gamma MAPK regulation of glucose transporter expression and glucose uptake in L6 myotubes and mouse skeletal muscle. Am J Physiol Regul Integr Comp Physiol 2004;286:R342–9. [DOI] [PubMed] [Google Scholar]
- Hwang ST, Janik JE, Jaffe ES, Wilson WH. Mycosis fungoides and Sezary syndrome. Lancet 2008;371(9616):945–57. [DOI] [PubMed] [Google Scholar]
- Iqbal J, Weisenburger DD, Greiner TC, Vose JM, McKeithan T, Kucuk C, et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood 2010;115:1026–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozono S, Ohuchida K, Eguchi D, Ikenaga N, Fujiwara K, Cui L, et al. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res 2013;73:2345–56. [DOI] [PubMed] [Google Scholar]
- Lemke J, von Karstedt S, Abd El Hay M, Conti A, Arce F, Montinaro A, et al. Selective CDK9 inhibition overcomes TRAIL resistance by concomitant suppression of cFlip and Mcl-1. Cell Death Differ 2014;21:491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jiang Y, Ulevitch RJ, Han J. The primary structure of p38 gamma: a new member of p38 group of MAP kinases. Biochem Biophys Res Commun 1996;228:334–40. [DOI] [PubMed] [Google Scholar]
- Lien EC, Dibble CC, Toker A. PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol 2017;45:62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 2008;7:1851–63. [DOI] [PubMed] [Google Scholar]
- Moran N p38 kinase inhibitor approved for idiopathic pulmonary fibrosis. Nat Biotechnol 2011;29:301. [DOI] [PubMed] [Google Scholar]
- Nam S, Wen W, Schroeder A, Herrmann A, Yu H, Cheng X, et al. Dual inhibition of Janus and Src family kinases by novel indirubin derivative blocks constitutively-activated Stat3 signaling associated with apoptosis of human pancreatic cancer cells. Mol Oncol 2013;7:369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons B, Armengol G, Livingstone M, Lopez L, Coch L, Sonenberg N, et al. Association between LRRK2 and 4E-BP1 protein levels in normal and malignant cells. Oncol Rep 2012;27:225–31. [DOI] [PubMed] [Google Scholar]
- Qi X, Tang J, Pramanik R, Schultz RM, Shirasawa S, Sasazuki T, et al. p38 MAPK activation selectively induces cell death in K-ras-mutated human colon cancer cells through regulation of vitamin D receptor. J Biol Chem 2004;279:22138–44. [DOI] [PubMed] [Google Scholar]
- Qi X, Yin N, Ma S, Lepp A, Tang J, Jing W, et al. p38gamma MAPK is a therapeutic target for triple-negative breast cancer by stimulation of cancer stem-like cell expansion. Stem Cells 2015;33:2738–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querfeld C, Kuzel TM, Kim YH. Multicenter phase II trial of enzastaurin in patients with relapsed or refractory advanced cutaneous T-cell lymphoma. Leuk Lymphoma 2011;52:1474–80. [DOI] [PubMed] [Google Scholar]
- Querfeld C, Rizvi MA, Kuzel TM. The selective protein kinase C beta inhibitor enzastaurin induces apoptosis in cutaneous T-cell lymphoma cell lines through the AKT pathway. J Invest Dermatol 2006;126:1641–7. [DOI] [PubMed] [Google Scholar]
- Round JL, Humphries LA, Tomassian T, Mittelstadt P, Zhang M, Miceli MC. Scaffold protein Dlgh1 coordinates alternative p38 kinase activation, directing T cell receptor signals toward NFAT but not NF-kappaB transcription factors. Nat Immunol 2007;8:154–61. [DOI] [PubMed] [Google Scholar]
- Sabio G, Arthur JSC, Kuma Y, Peggie M, Carr J, Murray-Tait V, et al. p38γ regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP. EMBO J 2005;24:1134–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soond DR, Bjorgo E, Moltu K, Dale VQ, Patton DT, Torgersen KM, et al. PI3K p110delta regulates T-cell cytokine production during primary and secondary immune responses in mice and humans. Blood 2010;115: 2203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugarinov V, Kay LE. Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. J Am Chem Soc 2003;125:13868–78. [DOI] [PubMed] [Google Scholar]
- Wallin JJ, Guan J, Prior WW, Lee LB, Berry L, Belmont LD, et al. GDC-0941, a novel class I selective PI3K inhibitor, enhances the efficacy of docetaxel in human breast cancer models by increasing cell death in vitro and in vivo. Clin Cancer Res 2012;18:3901–11. [DOI] [PubMed] [Google Scholar]
- Wang L, Ni X, Covington KR, Yang BY, Shiu J, Zhang X, et al. Genomic profiling of Sezary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat Genet 2015;47:1426–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XS, Diener K, Manthey CL, Wang S, Rosenzweig B, Bray J, et al. Molecular cloning and characterization of a novel p38 mitogen-activated protein kinase. J Biol Chem 1997;272:23668–74. [DOI] [PubMed] [Google Scholar]
- Wilcox RA. Cutaneous T-cell lymphoma: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol 2016;91:151–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Nam S, Brown CE, Zhao R, Starr R, Horne DA, et al. A novel berbamine derivative inhibits cell viability and induces apoptosis in cancer stem-like cells of human glioblastoma, via up-regulation of miRNA-4284 and JNK/AP-1 signaling. PLoS One 2014;9(4):e94443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin N, Qi X, Tsai S, Lu Y, Basir Z, Oshima K, et al. p38gamma MAPK is required for inflammation-associated colon tumorigenesis. Oncogene 2015;35:1039–48. [DOI] [PubMed] [Google Scholar]
- Zhang L, Handel MV, Schartner JM, Hagar A, Allen G, Curet M, et al. Regulation of IL-10 expression by upstream stimulating factor (USF-1) in glioma-associated microglia. J Neuroimmunol 2007;184:188–97. [DOI] [PubMed] [Google Scholar]
- Zheng S, Yang C, Liu T, Liu Q, Dai F, Sheyhidin I, et al. Clinicopathological significance of p38beta, p38gamma, and p38delta and its biological roles in esophageal squamous cell carcinoma. Tumour Biol 2016;37:7255–66. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Amran SI, Thompson PE, Jennings IG. Isoform-selective inhibition of phosphoinositide 3-kinase: identification of a new region of nonconserved amino acids critical for p110alpha inhibition. Mol Pharmacol 2011;80: 657–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.