

Abstract
During the last decades, much attention has been focused on SNEDDS approach to resolve concerns of BCS II class drugs with accentuation on upgrading the solubility and bioavailability. The present hypothesis confirms the theory that SNEDDS can reduce the impact of food on Candesartan solubilization, thereby offering the potential for improved oral delivery without co-administration with meals. The present studies describe quality-by-design-based development and characterization of Candesartan loaded SNEDDS for improving its pharmacodynamic potential. D-optimal mixture design was used for systematic optimization of SNEDDS, which showed globule size of 13.91 nm, more rapid drug release rate of >90% in 30 min and 16 s for self-emulsification. The optimized formulations were extensively evaluated, where an in vitro drug release study indicated up to 1.99- and 1.10-fold enhancement in dissolution rate from SNEDDS over pure drug and marketed tablet. In vivo pharmacodynamic investigation also showed superior antihypertensive potential of SNEDDS in normalizing serum lipid levels as compared to pure drug and marketed tablet that was executed on male Wistar rats. Overall, this paper reports successful systematic development of candesartan-loaded SNEDDS with distinctly improved biopharmaceutical performance. This research work interpreted a major role of SNEDDS for enhancing the rate of dissolution and bioavailability of poorly water soluble drugs.
Keywords: Candesartan, D-optimal mixture design, food-effect, in vitro lipolysis, SNEDDS, pharmacodynamic study
1. Introduction
Candesartan (kan” de sar’ tan) is a BCS II class drug that is widely used alone or in combination with other agents for therapy of hypertension and heart failure. It inhibits the renin-angiotensin system by blocking the angiotensin II type 1 receptor, which prevents the vasoconstriction and volume expansion induced by circulating angiotensin II, resulting in its antihypertensive potential. It is commercially available in 4, 8, 16 and 32 mg tablets generically (Candesar/Candosa/blopress/Camperten), under the trade name Atacand. It may be brought into play to treat hypertension, isolated systolic hypertension, left ventricular hypertrophy and diabetic nephropathy. It may also be used as a second-line drug for the treatment of heart failure, systolic dysfunction, myocardial infarction and coronary artery disease. Its typical dose is 16–32 mg “quaque die” in adults which is used for the long term (Zhao & Wang, 2018).
It has the most effective antihypertensive pharmacological response. Its poor aqueous solubility results in its slow rate of dissolution and its less oral bioavailability (15%). Thus, improving its dissolution can result in improved oral bioavailability (Alshora et al., 2018).
Regardless of numerous novel inventions for delivering active pharmacotherapeutic compounds, drug administration through oral route is most desired among patients of all age groups. The acceptability of this versatile and natural oral route is attributed to ease of administration, cost-effectiveness, and improved compliance by patients (Pal et al., 2013).
SNEDDS is a novel approach in drug delivery and solves deficiency related to the delivery of BCS II class medicaments (Thomas et al., 2013; Verma et al., 2017). These are described as clear systems that consist of oils, surfactants, co-surfactant, which result in ultrafine oil/water emulsion with mean globule size distribution <100 nm upon emulsification in the gastric milieu (Gahlawat et al., 2019). They help in possessing higher solubilization capacity, leading to the addition of medicament inside the oil phase (Khalifa et al., 2019; Tong et al., 2019; Kuncahyo et al., 2019). The excipients contained in the SNEDDS tend to facilitate bioavailability of the drugs not merely by improving drug solubility and permeability but by circumventing the metabolism by liver microsomes and inhibiting P-gp efflux, along with the ability to facilitate lymphatic drug absorption. Several literature studies on various self-emulsifying formulations have reported potential improvement in the bioavailability of various drugs (Kalantari et al., 2017; Alhasani et al., 2019; Patki et al., 2019; Alskär et al., 2018).
Based on these considerations, the main aim of this research is use of quality-by-design (QbD) approach for the systematic drug product development that helps in attaining consistent quality and robust performance.
QbD approach provides product and process understanding for continuous improvement. Among diverse elements of QbD, the experimental designs are considered as a pivotal tool, which provides maximal information using minimal experimentation (Heshmati et al., 2013).
2. Materials and methods
2.1. Materials
Candesartan pure drug was a generous gift from Sun Pharmaceuticals Laboratories, Gurugram, Haryana. Capmul PG-8 and Kolliphor EL were kindly supplied by IMCD India Private Limited, Delhi. Transcutol P (Gattefosse) and Pancreatin (Loba Chemie). The marketed tablet (Candesar 16 mg batch no. 9040580) was dispensed from a community pharmacy. Other materials and reagents used in this report were of analytically research grade.
2.2. Methods
2.2.1. Investigations of candesartan solubility in excipients
To determine and plot the possible emulsifying regions, it is necessary to elucidate the solubility of Candesartan in different oil or surfactant components (Gué et al., 2016).
The saturation solubility study of Candesartan in various vehicles was investigated. An excessive quantity of Candesartan was incorporated into each ingredient (2 g) in screw-capped glass vials. Vortex mixer (Genius, India) was used to assist the proper blending of Candesartan and vehicles (Gamal et al., 2017).
For shaking of the mixture, thermostatically controlled shaker (Calton) was used at 100 rpm for 72 h at 25 ± 0.5 °C. After removal of samples, centrifugation was done at 5000 rpm for 30 min. The supernatant was collected from the solution and a 0.45 μm membrane filter (Millipore) was used for filtration. The concentration of Candesartan was deliberately using a ultra-violet spectrophotometer (UV 1700, Shimadzu, Japan) at 254 nm. The experiment was repeated in triplicates (ElShagea et al., 2019). Self-emulsification capacity of surfactant and oil was investigated for choosing their best combination. 10 ml of each surfactant solution (10% w/w aqueous solution) was titrated with each oil (Borhade et al., 2008). Volume of oil when it converted emulsions clarity into turbid, was noted and combination was selected, which offered the highest quantity of oil emulsified (Bharti et al., 2018; Verma et al., 2018).
1:1 (Smix) was formulated with each co-surfactant for selection of co-surfactant and various formulations were developed with chosen oil and Smix. 500 mg of each formulation was blended with 500 ml of distilled water and resultant’s emulsion clarity was noted down (Lee et al., 2018).
2.2.2. Construction of pseudo-ternary phase diagram
It was plotted (in the presence of medicament) with surfactant, co-surfactant and oil, and every one of them speaks to a side of the triangle. Ternary blends with shifting organizations of these three ingredients were readied, bringing about an aggregate sum of 1 g. Smix were blended in five proportions, to be specific; 1:1, 1:2, 2:1, 3:1 and 1:3. Oil and Smix proportion were blended completely in nine diverse weight proportions from 1:9 to 9:1 in various glass vials with the goal that most extreme proportions were formulated for the examining to outline the limits of phase accuracy created in this diagram (Panigrahi et al., 2019). Its outlines were created utilizing the water dilution method. The development of the nanoemulsion was outwardly seen as transparent/clear and effectively flowable/dispersible with low consistency o/w nanoemulsion and set apart on it. The measure of every part (oil, Smix) now was recorded and introduced in it (Johnson et al., 2009; FDA, 2012). It was built utilizing Chemix software (Chemix Version 4.50) (Kim et al., 2018).
2.2.3. Design of experiment (DOE) to optimize SNEDDS
Recently, many statistical experimental designs have been utilized for more expertly improved plans utilizing fewer investigations, and to gauge the relative significance among other factors. Among various statistical optimization tools, D-optimal mixture design is one of the most mainstream surface approaches for optimizing SNEDDS since; it limits the difference related to the assessment of coefficients in a model and delivers the ideal subset by taking into account the criteria for boosting data grid determinants. In addition, this design considers the total system of SNEDDS as 100%, while other designs do not consider (Son et al., 2018; Mura et al., 2005).
The components were X1 as oil percentage (Capmul PG-8), X2 as surfactant percentage (Kolliphor EL), and X3 as a co-surfactant percentage (Transcutol P) to formulate SNEDDS with least globule size. The design of the experiment helped us both analyze and record the response (Y) as outcomes, namely globule size (Y1), %CDR (Y2) and self-emulsification time (Y3). Design Expert ® Software, Trial Version, was used to harmonize the regression equations further to calculate the recorded responses (Hosny et al., 2019).
2.2.3.1. Determination of globule size
Zetasizer ZS nano series; Malvern Instruments, Malvern, UK based on Photo correlation scattering was used for the assessment of globule size of the nano-emulsion after 100 folds dilution of SNEDDS formulation with distilled water (Eleftheriadis et al., 2019).
2.2.3.2. Dissolution testing
For this test, 900 ml of 0.35% Polysorbate 20 in 0.05 M Phosphate buffer media of pH 6.5 ± 0.05 at 37 °C ± 0.5 °C was utilized as dissolution media in USP II apparatus (Distek, USA) at 50 rpm (Pal et al., 2016). The capsule containing SNEDDS equivalent to 16 mg of Candesartan was incorporated into the buffer media after initiating rotation of the paddle. Aliquots (5 ml) were withdrawn after 30 min and analyzed by UV spectroscopy at λmax 254 nm (Patel et al., 2019). The experiment was repeated in triplicates.
2.2.3.3. Emulsification time
By the reported method, a self-emulsification study was carried out on each of the mixtures. Briefly, 1 ml of optimized SNEDDS was added into 500 ml of Millipore water and agitated at approximately 100 rpm with a magnetic stirrer. Emulsion formation and dispersibility time was noted (Rangaraj et al., 2019).
2.2.4. Evaluation parameters
Based on optimization results, optimized formulation was chosen to carry out characterization and further investigations such as transmittance test, cloud point, globule size and zeta potential.
2.2.4.1. % transmittance test
While preparing SNEDDS formulation for the oral route, there are chances of precipitation of the medicament following dilution in lumen of the gut and for that % transmittance is measured. 1 g of SNEDDS was diluted with 100 ml Millipore water and measurement was done at λmax 254 nm using UV spectrophotometer 1700, Shimadzu, Japan and performed in triplicates using water as blank (Zhang et al., 2019).
2.2.4.2. Robustness to dilution
Robustness was investigated following 100 times dilution of optimized formulation with various mediums including 0.1 N HCl, Acetate buffer (pH 4.5) and phosphate buffer (pH 6.8). After storing these samples for 24 h, they were checked for phase partitioning or precipitation of medicament (Ahsan & Verma, 2017).
2.2.4.3. Viscosity measurement
Hyrdromotion viscometer (Brookfield Engineering, USA) was used for measuring the viscosity of the optimized SNEDDS formulation. This test confirms whether the nano-emulsion is o/w or w/o type. If nano-emulsion has a high viscosity, then it indicates that it is w/o type and vice-versa (Abhijit et al., 2007; Wu et al., 2015).
2.2.4.4. Cloud point (TCloud) determination
Measurement was done, following 100 times dilution of 1 g optimized formulation with double distilled water and kept in a water bath for gradual increment in temperature of formulation (5 °C increments) (Shoshtari et al., 2010; Agrawal et al., 2015).
2.2.4.5. Determination of drug content
For this, drug content was extracted after its 10 times dilution with methanol (v/v) and centrifugation was performed for 30 min at 10,000 rpm. Then, supernatant was diluted with methanol (2.5 folds) which was analyzed for drug content through UV spectrophotometer 1700, Shimadzu, Japan at 254 nm and performed in triplicates (Baloch et al., 2019).
2.2.4.6. Measurement of globule size, polydispersity index (PDI) and zeta potential
The optimized formulation was diluted freshly at a ratio of 1:100 w/v and blended for 1 min before analysis of globule size, PDI and zeta potential measured by Zetasizer ZS nano series; Malvern Instruments, Malvern, UK. This was performed in triplicates and depicted as mean ± standard deviation (Bang et al., 2019; Alwadei et al., 2019; Enin, 2015).
2.2.4.7. Multi-media dissolution testing
The SNEDDS formulations (equivalent to 16 mg of Candesartan, size “00” capsules) were dropped in dissolution medium of pH 1.2, 4.5 and 6.8 at 37˚C ± 0.5 °C in USP apparatus II (paddle) (Distek, USA). Aliquots (5 ml) were withdrawn at predetermined time points, an equal volume of fresh buffer media was incorporated after each sampling and 0.45-μm Millipore membrane filter was used for filtration. Drug release was measured using UV spectrophotometrically at 254 nm after appropriate dilution with media against equivalent proportions of excipients as blank in triplicates (Jakab et al., 2018).
2.2.4.8. Comparative study of in vitro dissolution testing of optimized formulation with pure medicament and marketed formulation
In vitro dissolution testing was performed with pure Candesartan, optimized SNEDDS and marketed tablet (Candesar 16 mg batch no. 9040580) in the “USP type-II dissolution apparatus (Distek, USA) as per dissolution conditions specified by FDA guidelines”. Each formulation was kept in 0.35% Polysorbate 20 in 0.05 M Phosphate buffer media of pH 6.5 ± 0.05 at 37 °C ± 0.5 °C at 50 rpm. After 5, 10, 15, 30 and 60 min, 5 ml of aliquots were analyzed using UV spectrophotometer at 242 nm. After every sampling, fresh buffer was utilized as replacement media.
2.2.4.9. Investigation of food effect by dynamic in vitro lipolysis
The literature reported that SNEDDS avoids the food effect in terms of drug discharge. For proving this theory, the dissolution of SNEDDS formulation was conducted in modified Fa/FeSSIF V-2 media to mimic in vivo milieu.
For best clinical pertinence, it is essential to lead in vitro analysis of medicament to imitate in vivo environment as intently as could reasonably be expected. This investigation is valuable for two specific rationales. Firstly, quantification of rate and degree of lipolysis by pH-stat titration, which can set up how the formulation can be influenced by equilibrium solubility and dispersion qualities of SNEDDS. Also, after the response is ended, the post lipolysis item can be examined to foresee how much content of the medicament is in solubilized or precipitated form. This model can dependably foresee the capacity of such formulations to upgrade oral assimilation of medicaments that have poor aqueous solubility.
In the present investigation, the dynamic in vitro lipolysis investigation was a rendition of the strategy recently depicted by Mohsin (2012). Each 520 mg of optimized formulation was dispersed into 36 ml of FaSSIF V-2 and FeSSIF V-2 whose composition is shown in Table 1 (Mosgaard et al., 2015; Xiao et al., 2016; Sassene et al., 2014). Concentrations of Ca++, bile, phospholipid (PL), and sodium chloride (NaCl) were preferred to imitate typical concentrations occurring in FaSSIF V-2/FeSSIF V-2. During the early phase of dispersion, 6.5/5.8 ± 0.05 of pH was adjusted with NaOH or HCl.
Table 1.
Composition of biorelevant media used during in vitro lipolysis.
| Chemical | FaSSIF V-2 | FeSSIF V-2 |
|---|---|---|
| Sodium taurocholate (mM) | 3 | 10 |
| Glyceryl monooleate (mM) | – | 2 |
| Sodium oleate (mM) | – | 5 |
| Lecithin (mM) | 0.2 | 0.8 |
| Maleic acid (mM) | 19.12 | 55.02 |
| NaCl (mM) | 68.62 | 125.5 |
| NaOH (mM) | 34.8 | 81.65 |
| pH | 6.5 | 5.8 |
FaSSIF V2: fasted state simulated intestinal fluid V-2; FeSSIF V-2: fed state simulated intestinal fluid V2.
The stirring process was utilized for the emulsification of SNEDDS formulations on a magnetic stirrer with a hot plate at 37 °C, earlier to the incorporation of enzyme. 4 ml of pancreatic extract [formulated by suspending pancreatin powder (1 g) in digestion buffer (5 ml) and vortex blending for 15 min. Ultracentrifugation was performed and supernatant of pH 6.5/5.8 containing 800 TBU/ml of pancreatic lipase/co-lipase] addition initiates lipolysis which was continuous for next 30 min with a pH-stat titration unit (Metrohm, Switzerland), which was maintained a constant pH of 6.8/5.8. During lipolysis, production of fatty acids (FAs) results in an elevation in pH of biorelevant media, 0.2 M NaOH solution was utilized for maintaining pH. The progress of drug release in digestion buffers was monitored directly by UV analysis at 254 nm (Williams et al., 2012; Alshamsan et al., 2018). By following this protocol, the present strategy was seen as robust and estimated values were reproducible.
2.2.4.10. Stability studies
30 capsules containing optimized Candesartan SNEDDS (each capsule contains Candesartan SNEDDS equivalent to Candesartan 16 mg) were packed in 60 cc HDPE Bottle and were placed in stability chamber (Thermolab, India) at 40 ± 2 °C/75 ± 5% RH for 6 months after sealing bottles. After 1 M/3M/6M, samples were removed and evaluated in terms of description, drug release and disintegration time of formulation (Izham et al., 2019).
2.2.4.11. Pharmacodynamic studies
Candesartan has a dose-dependent pharmacodynamic effect and that’s why comparative in vivo study was investigated with the marketed tablet dosage form.
The ethical permission for the pharmacodynamic study of Candesartan SNEDDS formulation in rats was granted by the “Institutional Animal Ethical Committee (IAEC), Maharshi Dayanand University, India (Reg. no. 1767/RE/S/14/CPCSEA, vide reference no. 153-165 dated 14/12/2018). Male Wistar rats having weight 150–200 g were purchased from Lala Lajpat Rai University of Veterinary and Animal Sciences, Hisar. Animals were maintained as per the guidelines of CPCSEA, India”. All animals were kept in plastic cages; six animals per cage were provided accommodation with 12 h of light/dark cycle, at 25 ± 2˚C, with pelleted food, tapwater and libitum were fed.
All animals were adapted to research facility environment for 1 week prior to experimentation and fasted for 12 h before the experiment, they were made available with libitum access to water. This investigation in rats was carried out according to the method as depicted in previous literature (Kumar & Nanda, 2018) with a few modifications. The animals were separated into five groups (total 30 rats; each group having 6 rats), i.e., “control treatment group (CTG), placebo treatment group (PTG), reference treatment group (RTG), test (TTG) and marketed treatment group (MTG)”.
The effect of Candesartan loaded SNEDDS (TTG) on lipid profile was determined by comparison with Candesartan drug (RTG) and SNEDDS without Candesartan (PTG). Marketed, test, reference and placebo formulation was diluted with 2.0% acacia solution. Each treatment group received 18% NaCl solution as a dose of 10 ml/kg/day bodyweight daily for 4 weeks (Mao et al., 2015). TTGs, RTGs, MTGs, and PTGs additionally receive test formulation, reference formulation, marketed and placebo formulation, respectively, for 4 weeks. The administered oral dose of the test product and reference product was equivalent to 0.3 mg/kg/day of Candesartan (Gleiter et al., 2006).
Alteration in MSBP was measured (0 days and after 28 days) with noninvasive blood pressure (NIBP) (AD Instruments, Australia) by using the tail-cuff method for each treatment group. One-way analysis of variance (ANOVA) with the Dunnet test was implemented to evaluate the differences in the mean of different groups using the Graph Pad version 5.0 statistical analysis software. Data are shown in mean ± standard deviation. The statistical significance level was acceptable at p < 0.05.
3. Results and discussion
3.1. Solubility study
For the determination of stability of the formulation, solubility of medicament in ingredients plays a significant function because many formulations undergo precipitation before experiencing in situ solubilization. High drug solubilization is very significant for increasing the efficiency of drug loading into carriers with concomitant improvement in oral bioavailability (Parmar et al., 2011). In addition, for the development of an effective Candesartan SNEDDS, its prescribed amount should be miscible in its selected excipients with the least amount of the mixture (Qi et al., 2011). The results of Candesartan solubility in various ingredients are shown in Figure 1.
Figure 1.

Solubility of Candesartan in various oils.
The self-emulsification feature depends on the selection of a suitable combination of ingredients. This study showed that Kolliphore EL with the highest quantity of Capmul PG-8 had been emulsified as shown in Table 2. That’s why; Kolliphore EL and Capmul PG-8 combination was selected.
Table 2.
Emulsification of oils with different surfactant.
| Surfactant (10% solution) | Oils | Volume of oil emulsified (mL) |
|---|---|---|
| Kolliphor EL | Capmul PG-8 | 0.70 |
| Kolliphor RH 40 | Capmul PG-8 | 0.50 |
| Kolliphor EL | Capmul MCM EP | 0.40 |
| Kolliphor RH 40 | Capmul MCM EP | 0.60 |
Transcutol P is selected as a co-surfactant because it showed greater nanoemulsion region as compare to PEG 400 as shown in Table 3.
Table 3.
Identification of nanoemulsion region (transparent) based on visual observation with different co-surfactants.
| %Oil | %Smix | Nanoemulsion
region |
|
|---|---|---|---|
| Smix Kolliphore EL : Transcutol P (1:1) |
Smix Kolliphor EL:PEG 400 (1:1) |
||
| 10 | 90 | Transparent | Transparent |
| 20 | 80 | Transparent | Transparent |
| 30 | 70 | Transparent | Transparent |
| 40 | 60 | Transparent | Turbid |
| 50 | 50 | Transparent | Turbid |
| 60 | 40 | Turbid | Turbid |
| 70 | 30 | Turbid | Turbid |
| 80 | 20 | Turbid | Turbid |
| 90 | 10 | Turbid | Turbid |
Turbid: nonnanoemulsion region.
Transparent: nanoemulsion region.
The surfactant creates a layer around oil droplets and diminishes surface tension between aqueous and oil phase. Additional, elevation of the concentration of surfactant results in enhancement of the spontaneity of self-emulsification. Elevation in co-surfactant concentration diminishes the area for the formation of emulsion, but it has minimal impact on dropping interfacial tension (Nepal et al., 2010). A higher value of HLB is necessary for creating o/w type emulsion. Co-surfactant is used in the Candesartan preparation mainly to minimize the surfactant ratio in the formulation (Zhao et al., 2010). Transcutol P was incorporated in the formulation to increase the solubilization of the model lipophilic drug compounds.
3.2. Pseudo-ternary phase diagram
It was plotted in the presence of Candesartan to recognize the self-nanoemulsifying region and for the selection of an appropriate concentration of ingredients for the development of SNEDDS. It plays a significant function to study phase behavior of formed nanoemulsions (Balakumar et al., 2013). It was constructed by using water dilution method with different amount of oil (5-90%), Smix (1:1, 1:2, 2:1, 3:1 and 1:3) and transparency for the formation of nanoemulsion as shown in Table 4. Resulted data was used for the construction of a ternary phase diagram where each vertex represents 100% of that specific ingredient. In Figure 2, the shaded area presented transparent and low viscosity nanoemulsion area in it.
Table 4.
Result of water dilution method of Oil + Smix with Candesartan.
| S. no. | % Oil | % Surfactant | % Co-surfactant | Observation |
|---|---|---|---|---|
| Smix ratio 1:1 | ||||
| 1. | 5 | 55 | 55 | Transparent |
| 2. | 10 | 45 | 45 | Transparent |
| 3. | 20 | 40 | 40 | Transparent |
| 4. | 30 | 35 | 35 | Transparent/bluish |
| 5. | 40 | 30 | 30 | Transparent/bluish |
| 6. | 50 | 25 | 25 | Turbid |
| 7. | 60 | 20 | 20 | Turbid |
| 8. | 70 | 15 | 15 | Turbid |
| 9. | 80 | 10 | 10 | Turbid |
| 10. | 90 | 5 | 5 | Turbid |
| Smix ratio 2:1 | ||||
| 1. | 5 | 63.34 | 31.66 | Turbid |
| 2. | 10 | 60 | 30 | Transparent |
| 3. | 20 | 53.30 | 26.70 | Transparent |
| 4. | 30 | 46.70 | 23.30 | Transparent/bluish |
| 5. | 40 | 40 | 20 | Transparent/bluish |
| 6. | 50 | 33.30 | 16.70 | Turbid |
| 7. | 60 | 26.70 | 13.30 | Turbid |
| 8. | 70 | 20 | 10 | Turbid |
| 9. | 80 | 13.30 | 6.70 | Turbid |
| 10. | 90 | 6.70 | 3.30 | Turbid |
| Smix ratio 1:2 | ||||
| 1. | 5 | 31.66 | 63.34 | Transparent |
| 2. | 10 | 30 | 60 | Transparent |
| 3. | 20 | 26.70 | 53.30 | Transparent |
| 4. | 30 | 23.30 | 46.70 | Transparent/bluish |
| 5. | 40 | 20 | 40 | Transparent/bluish |
| 6. | 50 | 16.70 | 33.30 | Turbid |
| 7. | 60 | 13.30 | 26.70 | Turbid |
| 8. | 70 | 10 | 20 | Turbid |
| 9. | 80 | 6.70 | 13.30 | Turbid |
| 10. | 90 | 3.30 | 6.70 | Turbid |
| Smix ratio 3:1 | ||||
| 1. | 5 | 71.25 | 23.75 | Turbid |
| 2. | 10 | 67.5 | 22.5 | Turbid |
| 3. | 20 | 60 | 20 | Transparent/bluish |
| 4. | 30 | 52.5 | 17.5 | Turbid |
| 5. | 40 | 45 | 15 | Turbid |
| 6. | 50 | 37.5 | 12.5 | Turbid |
| 7. | 60 | 30 | 10 | Turbid |
| 8. | 70 | 22.5 | 7.5 | Turbid |
| 9. | 80 | 15 | 5 | Turbid |
| 10. | 90 | 7.5 | 2.5 | Turbid |
| Smix ratio 1:3 | ||||
| 1. | 5 | 23.75 | 71.25 | Turbid |
| 2. | 10 | 22.5 | 67.5 | Transparent |
| 3. | 20 | 20 | 60 | Transparent/bluish |
| 4. | 30 | 17.5 | 52.5 | Turbid |
| 5. | 40 | 15 | 45 | Turbid |
| 6. | 50 | 12.5 | 37.5 | Turbid |
| 7. | 60 | 10 | 30 | Turbid |
| 8. | 70 | 7.5 | 22.5 | Turbid |
| 9. | 80 | 5 | 15 | Turbid |
| 10. | 90 | 2.5 | 7.5 | Turbid |
Figure 2.

Pseudo ternary phase diagram.
3.3. Mixture design tool in the optimization and statistical analysis
For optimization Candesartan-loaded SNEDDS composition, a mixture design was used using Design Expert® software Trial Version. As shown in Table 5, fourteen experimental runs were found according to this design with two center points. Y1 ranged from 11.39 to 119.8 nm, Y2 from 86 to 98.5% and Y3 ranged from 15 to 41 s. The effect of different proportions of components on globule size, drug release and self-emulsification could be explained by the following equations:
Table 5.
Composition of various SMEDDS formulation suggested by Design Expert® in the study and response.
| Formulation code |
Excipients
ratio |
Y1 (nm) | Y2 (%) | Y3 (s) | ||
|---|---|---|---|---|---|---|
| X1 (%) | X2 (%) | X3 (%) | ||||
| F1 | 0.10 | 0.40 | 0.50 | 13.68 | 93.2 | 26 |
| F2 | 0.16 | 0.25 | 0.59 | 60.94 | 95 | 41 |
| F3 | 0.17 | 0.40 | 0.43 | 52.23 | 91 | 25 |
| F4 | 0.05 | 0.48 | 0.47 | 11.39 | 90 | 15 |
| F5 | 0.10 | 0.25 | 0.65 | 22.69 | 97 | 39 |
| F6 | 0.05 | 0.38 | 0.57 | 15.89 | 94.5 | 17 |
| F7 | 0.25 | 0.5 | 0.25 | 87.39 | 86 | 24 |
| F8 | 0.18 | 0.30 | 0.52 | 40.79 | 94.9 | 32 |
| F9 | 0.23 | 0.25 | 0.52 | 119.8 | 91.7 | 36 |
| F10 | 0.25 | 0.35 | 0.40 | 111.1 | 91 | 28 |
| F11 | 0.05 | 0.32 | 0.63 | 13.91 | 98.5 | 18 |
| F12 | 0.21 | 0.44 | 0.35 | 105 | 87.7 | 21 |
| F13 | 0.10 | 0.40 | 0.50 | 13.6 | 93 | 26 |
| F14 | 0.14 | 0.5 | 0.36 | 14.45 | 88 | 20 |
Independent variable: X1 as oil percentage (Capmul MCM EP), X2 as surfactant percentage (Tween 20), and X3 as a co-surfactant percentage (Transcutol P). Dependent variables: globule size (Y1), %CDR (Y2) and self-emulsification time (Y3).
The equation of the fitted model for
Globular size:
| (1) |
%CDR:
| (2) |
Self-emulsification time:
| (3) |
Where
X1 = Conc. of Capmul PG-8 (Oil)
X2 = Conc. of Kolliphor EL (Surfactant)
X3 = Conc. of Transcutol P (Co-surfactant)
2-D contour plots and 3-D response plots are depicted in Figures 3 and 4 which explains the effects of X1, X2 and X3 on variables Y1, Y2 and Y3 responses. It was observed that increment in the concentration of oil results into increment in globule size and decline in drug discharge rate and self-emulsification time also increases. But, an increase in concentration of surfactant resulted in decrease of globule size, increment in drug release rate and decrease in self-emulsification time. While Figure 5 shows the actual versus predicted graph for responses that summarized that actual and predicted responses are approximately very close. Within the triangle image, the area other than gray indicates minimum globule size area, maximum %CDR and minimum self-emulsification time.
Figure 3.

2D counter plot for (a) globule size, (b) % CDR and (c) self-emulsification time.
Figure 4.

3D response plot for (a) globule size, (b) % CDR and (c) self-emulsification time.
Figure 5.

Prediction profiler (a) globule size, (b) % CDR and (c) self-emulsification time.
Both 2-D, 3-D contours and Equations (1, 2, 3) indicated high ratios of oil that had significantly decreased the globule size, while surfactant and co-surfactant increased it up to a limit in the formulation. The same occurs in response Y3 and Y2 up to a limit, and then it starts to increase as shown by the prediction profiler in Figure 6. Equations 1, 2, and 3 of regression helped formulate the optimized formulation. The results of ANOVA are depicted in Table 6.
Figure 6.

Actual versus predicted graph for response: (a) globule size, (b) % CDR and (c) self-emulsification time.
Table 6.
Result of ANOVA.
| Result of
ANOVA | ||||||
|---|---|---|---|---|---|---|
| Response | Sum of squares | df | Mean square | F value | p Value | Model |
| Y1 | 21674.05 | 9 | 2408.23 | 62.90 | 0.0006 | Significant |
| Y2 | 147.68 | 9 | 16.41 | 767.34 | <0.0001 | Significant |
| Y3 | 864.26 | 9 | 96.03 | 648.47 | <0.0001 | Significant |
The combined application of RSM and the desirability approach results into a more powerful method for finding an optimal balance between the responses. This combination has resulted in a new method called “Desirability Optimization Methodology or DOM” (Derringer, 1994). Desirability index is used for factor optimization in multi-response system that is based on the transformation of all the obtained responses from different scales into a scale-free value (Amdoun et al., 2018). The values of desirability functions lie between 0 and 1. The value 1 corresponds to the optimal performance for the investigating factors, while the value 0 is attributed when the factors result an undesirable response. The desirability index of the formulation was 1 which confirmed that the investigating factors resulted in the optimal performance of the formulation as shown in Figure 7 (Jeong & Kim, 2009).
Figure 7.

Desirability index for optimization of formulation.
The optimized formulation of Candesrtan loaded SNEDDS consist of 5% Capmul PG-8, 32% Kolliphor EL and 63% Transcutol P with globule size of 13.91 nm, 98.5% drug release within 30 min and 18 s self-emulsification time with desirability index value of 1.
3.4. Evaluation parameters
3.4.1. % Transmittance
It was determined to evaluate the stability of the optimized nanoemulsion of SNEDDS. It also gave a proposal about the features of formulation such as size and uniformity of the globules. It was found to be 99.98 ± 0.5% which confirmed its clarity after dispersion into buffer media. Also, it confirmed that there are no chances of drug precipitation and optimized formulation had good solubilization capacity after dispersion.
3.4.2. Robustness to dilution
The generation of uniform nano-emulsion from SNEDDS is very significant in various mediums as medicaments may precipitate out in vivo which may have an impact on the assimilation of medicaments. Optimized formulation was exposed to various media after 100 times dilution to mimic the in vivo conditions. Even after 24 h, the optimized formulation did not show any signs precipitation, haziness or separation of phase which made certain the stability of formulation. These outcomes ensured the prospect of a uniform profile of drug discharge during in vivo conditions.
3.4.3. Viscosity
Viscosity of optimized formulation was found to be 168 ± 5 cps which was measured by Brookfield hydromotion viscometer in triplicates. This confirmed that this formulation can be easily transferred to any container or a capsule shell for its storage.
3.4.4. Cloud point (TCloud) determination
It helps in examining the impact of temperature on the phase behavior of formulation which is one of the serious issues related to nanoemulsions, particularly when using nonionic surfactants. “It is the temperature above which the formulation transparency turns into cloudiness. An ideal formulation should remain as a single-phase clear system at its storage temperature and the temperature of its proposed use.” At high temperature, phase separation can arise because of the decline solubility of the surfactant in aqueous. It can decline both drug solubilization and formulation stability that’s why the cloud point should be over 37 °C of the formulation (Verma et al., 2017). The cloud point for the optimized SNEDDS formulation was much higher (70 °C) which shows that this formulation is stable at physiological temperature.
3.4.5. Drug content
Drug content was measured by VU spectrophotometrically in optimized SNEDDS formulation which was found to be 100.05 ± 1.2% which confirms the accuracy of dose in the formulation.
3.4.6. Globule size, PDI and zeta potential
Globule size is of the mainly significant qualities of nanoemulsion for stability assessment and a basic advance in the pathway of improving assimilation of medicament. Its smaller size results in greater interfacial surface area for assimilation of medicament and enhanced bioavailability. Hence, its smaller size may govern the effective discharge of medicament (Eltobshi et al., 2018). The globule size of optimized formulations specifies that droplets of emulsion are in nanometric range (13.91 nm) with a PDI value less than 0.5 which indicates uniformity in the globule size distribution and zeta potential value of –0.32 mV as shown in Figure 8.
Figure 8.

(a) Globule size and PDI. (b) Zeta potential of optimized formulation.
The stability of colloidal dispersions depends on the value of zeta potential which one is its significance. For smaller globules, high zeta potential will confirm electrically stability because increment in surface charge opposes the aggregation of particles. When the potential is high, repulsion exceeds attraction and the dispersion will not be deflocculated or break. In the present study, the zeta potential of optimized formulations was negatively charged due to the presence of nonionic surfactants that create a -vely charged interface at neutral pH (Choi et al., 2014, Shakeel et al., 2013).
3.4.7. Multi-media dissolution testing
The in vitro dissolution profile of optimized formulation was investigated in various dissolution media whose results are shown in Figure 9 and Table 7 (Zhang et al., 2010).
Figure 9.

Multi-media dissolution testing of Candesartan loaded SNEDDS (n = 3).
Table 7.
Multi-media dissolution testing of Candesartan loaded SNEDDS (n = 3).
| Time (min) | 0.1 N HCl | 4.5 acetate buffer | 6.5 phosphate buffer |
|---|---|---|---|
| 0 | 0 | 0 | 0 |
| 5 | 55 ± 2.3 | 62 ± 1.2 | 70 ± 1.1 |
| 10 | 79 ± 1.4 | 80 ± 1.7 | 86 ± 0.5 |
| 15 | 92 ± 2.1 | 95 ± 1.9 | 99 ± 1.2 |
| 30 | 97 ± 1.3 | 97.5 ± 0.7 | 99.8 ± 1.8 |
| 45 | 97.2 ± 0.9 | 98 ± 2.5 | 99.8 ± 2.3 |
| 60 | 97.3 ± 1.0 | 98 ± 2.8 | 99.9 ± 2.5 |
Data are presented as the mean ± SD.
It was concluded that drug discharge reached over 80% in 15 min in all media. Though, a mild decline or fluctuation in drug release was found at pH 1.2 and 4.5. Overall, the optimized formulation resulted in extremely improved drug release in multi-media dissolution testing.
3.4.8. Comparative dissolution testing of optimized formulation with marketed tablet and pure drug
This study was conducted with optimized formulation, marketed tablet and pure drug. It was summarized that the rate of drug discharge for the optimized formulation is more than the marketed tablet and pure drug from the results as summarized in Figure 10 and Table 8.
Figure 10.
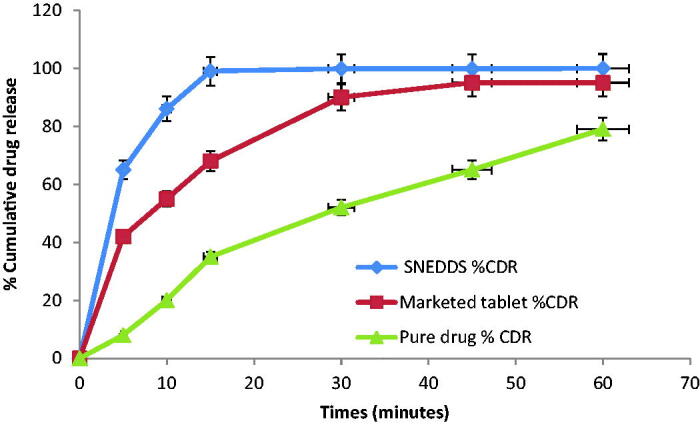
Comparative dissolution study of Candesartan loaded SNEDDS, marketed tablet and pure drug (n = 3).
Table 8.
Comparative dissolution study of Candesartan loaded SNEDDS, marketed tablet and pure drug (n = 3).
| Time (min) | SNEDDS (%) | Marketed tablet (%) | Pure drug (%) |
|---|---|---|---|
| 0 | 0 | 0 | 0 |
| 5 | 70 ± 1.1 | 42 ± 2.1 | 8 ± 2.4 |
| 10 | 86 ± 0.5 | 55 ± 1.9 | 20 ± 1.5 |
| 15 | 99 ± 1.2 | 68 ± 2.5 | 35 ± 1.7 |
| 30 | 99.8 ± 1.8 | 90 ± 2.7 | 50 ± 1.8 |
| 45 | 99.8 ± 2.3 | 95 ± 2.9 | 51 ± 2.7 |
| 60 | 99.9 ± 2.5 | 95 ± 1.0 | 52 ± 2.0 |
Data are presented as the mean ± SD.
This analysis showed up to 1.99 and 1.10-folds improvement in dissolution rate from optimized SNEDDS over pure drug and marketed tablet. These outcomes resemble with data which is obtained by several other researchers. This enhanced dissolution was likely ascribed to the accompanying basis. First, the crystalline structure of API alters into an amorphous state in which one is thermodynamically stable and offers solid-to-liquid phase transition effortlessly in SNEDDS. It is well established that this conversion of form enhances its rate of dissolution owing to elevated disorder and high energy form of the amorphous state (Liu et al., 2010, Verma & Kaushik, 2019, Kassem et al., 2016). Another reason is due to the existence of drugs as solubilized molecules inside nanoemulsion globules and nanosized suspended drug particles forming SNEDDS. All the samples showed faster drug release than unprocessed raw Candesartan because this aerophilization had relatively fewer effects on the dissolution than the creation of a high energy amorphous phase, decline in particle size and decline of surface tension of the dissolution medium.
3.4.9. Assessment of food effect with dynamic in vitro lipolysis
One of the most mind-boggling and inadequately comprehended parts of SNEDDS is that they interact with GI content which has a direct impact on their performance. Digestion of dietary TG in the small intestine (SI) is generally extremely quick and various nonionic esters act as substrates of pancreatic lipase or other esterases. This process may aid the dispersion of the medicament in the existence of BS/PLs from SNEDDS and advances its retention. Therefore, lipid digestion examination can be vital because they forecast the chance of precipitation of the formulation and medicament in the intestinal lumen.
Alterations in solubilization capacity that arises throughout this process were of great significance to evaluate food effects through in vitro lipolysis (Alshamsan et al., 2018). During this investigation, it was vital to examine if there was any chance of precipitation of medicament or loss of medicament arising within 30 min. The results of the fasting state confirmed that Candesartan was present in solubilized form in the optimized formulation which leads to approximately 97.2 ± 0.7% drug discharge. While similar outcomes were obtained under fed state where the drug discharge was estimated to be 96.33 ± 0.9% which suggested that optimized formulation was able to keep Candesartan in solubilized form which is crucial for assimilation of drug. So, SNEDDS avoids the food effect in terms of drug discharge which has been reported in the literature was found to be a true hypothesis in the case of SNEDDS formulation. This suggests that SNEDDS overcome the influence of food on drug discharge. Thus, SNEDDS would enhance patient compliance, specifically in patients who are not able to take their medicines with food.
During lipolysis, the continuous digestion of the SNEDDS and generation of digestion products leads to a decline in the solubilization capacity and precipitation of Candesartan. Since, the lipid to drug ratio is higher for the SNEDDS that results into its higher the solubilizing capacity.
3.4.10. Stability study
The optimized formulation was physically stable in terms of description/drug release/disintegration time. Stability data for Candesartan SNEDDS formulation has been given in Table 9. From stability data, it was observed that there are no significant differences in physicochemical parameters of Candesartan SNEDDS formulation from initial to 6 M accelerated stability condition (40 °C/75%RH) and hence, it was concluded that Candesartan SNEDDS formulation is stable.
Table 9.
Stability data of Candesartan SNEDDS formulation (n = 6).
| Test parameter | Initial | 40 °C/75% RH/1M | 40 °C/75% RH/3M | 40 °C/75% RH/6M |
|---|---|---|---|---|
| Description | Whitish colored capsules containing clear liquid | Whitish colored capsules containing clear liquid | Whitish colored capsules containing clear liquid | Whitish colored capsules containing clear liquid |
| % CDR | 99.9 ± 2.5 | 98.8 ± 2.8 | 98.0 ± 2.7 | 97.5 ± 2.0 |
| Disintegration time | 5 ± 0.5 min | 5 ± 0.5 min | 6 ± 1 min | 6 ± 1.0 min |
%CDR: percentage of cumulative drug release. Data are presented as the mean ± SD.
3.4.11. Pharmacodynamic study
Pharmacodynamic study was carried out with optimized Candesartan loaded SNEDDS formulation (F11). Pharmacodynamics study results for MSBP (Mean systolic blood pressure) for each group have been given in Table 10 that were collected by NIBP (AD Instruments, Australia). The one-way ANOVA with Dunnet analysis showed a significant difference in the percentages of parameters between the positive CTG, PTG, MTG, RTG and Test treatment group (p < 0.05).
Table 10.
MSBP profile in experimental animals with mean ± std. deviation (n = 6).
| Parameter | 0 Day | Control | Placebo | SNEDDS | Marketed Tablet | Pure drug |
|---|---|---|---|---|---|---|
| MSBP | 119.7 ± 3.39 | 119.5 ± 3.62 | 164.7 ± 3.78*** | 126.8 ± 2.14** | 133.5 ± 2.43*** | 143.8 ± 2.79*** |
MSBP: mean systolic blood pressure. Data are presented as the mean ± SD.
***p < 0.001, **p < 0.01, and *p < 0.05 (*as compared to control).
p < 0.001 (highly significant), p < 0.01 (significant), and p < 0.05 (less significant).
Following the administration of a high-fat diet for 28 days, all the animal groups revealed a considerable rise in the systolic blood pressure levels signifying the hypertension. Treatment with TTG showed remarkable alteration in the levels of systolic blood pressure as illustrated in Figure 11. All the treatment formulations revealed the initiation of their pharmacodynamic effects in varying the systolic blood pressure levels with a statistically significant difference observed among the total duration of treatment period 28th day (p <0.05).
Figure 11.
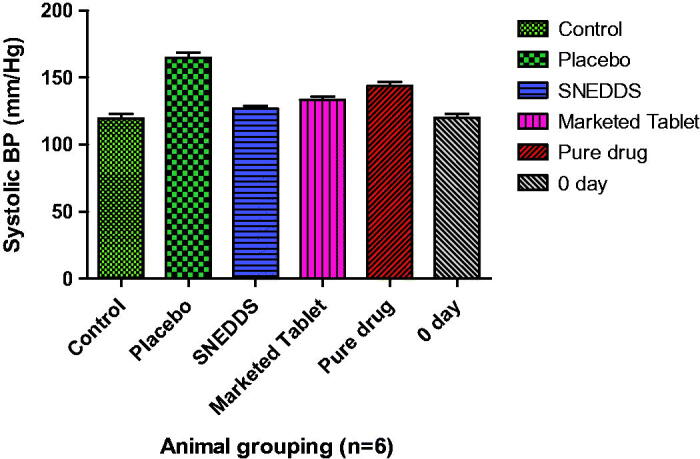
Comparative systolic blood pressure of each treatment group (n = 6).
It was observed that TTG decline the serum CH level more significantly as compared to PTG (p < 0.001), MTG (p <0.01) and RTG (p <0.05) in comparison to control group.
These data suggested that the drug was more efficient when administered as SNEDDS. These findings proved that SNEDDS can better maintain the potential of Candesartan at an equivalent dose to that of the standard drug solution and marketed tablet. Test formulation has an appreciable effect on the systolic blood pressure profiles of experimental animals in comparison to reference and marketed formulation. Thus, test formulation confirmed extensively better in vivo performance than reference formulation in terms of pharmacodynamic parameters.
4. Conclusion
The novel approach was developed for SNEDDS by selecting the optimum concentration of ingredients using a systematic ‘‘DoE’’ methodology of D-optimal mixture design. It has been reported that SNEDDS formulation had a quicker dissolution rate w.r.t. pure drug and marketed tablets which could be attributed to nano globule size and negative value of zeta potential for SNEDDS, which in turn provide greater surface area for the discharge of medicament. The optimized SNEDDS had minimal globule size with the highest rate of drug release. There was no significant difference in the level of Candesartan solubilization under fed and fasting conditions which depicted that SNEDDS can eradicate the influence of food on drug solubilization in vitro. The present investigation has also established that SNEDDS principle is also effective in rats; a significantly improved pharmacodynamic was found when dosed in a SNEDDS compared to a pure drug and marketed tablet with the same dose of drug. Hence, this approach established a considerable improvement in the oral bioavailability of highly lipophilic drugs through the use of SNEDDS. The present study entails the potential effectiveness of SNEDDS with improved release profile and avoidance of food effects and improved pharmacodynamic of medicament w.r.t. pure drug and marketed tablet. The results obtained from a strong rationale for further preclinical studies indicates the potential of SNEDDS as an alternative to oral delivery of Candesartan with enhanced bioavailability and patient compliance and minimal side effects.
Funding Statement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Acknowledgments
The authors thank Sun Pharmaceuticals Pvt. Ltd. Gurugram, Central Investigation Laboratories, Maharshi Dayanand University, Rohtak and Guru Jambheshwar Science and Technology University, Hissar for their co-operation during this project. A word of special thanks goes to AD Instruments, South Asia, India (Pvt. Ltd.), New Delhi and Cuckos Pharmaceutical Pvt. Ltd., Bahadurgarh for providing help during this research project.
Ethical approval and consent to participate
Reg. no. 1767/RE/S/14/CPCSEA, vide reference no. 153-165 dated 14/12/2018.
Human and animal rights
Animals were used for this research investigation under Reg. no. 1767/RE/S/14/CPCSEA, vide reference no. 153-165 dated 14/12/2018.
Availability of data and material
The author authenticates that the data supporting the results and findings of this study are existing within the article.
Consent for publication
Not applicable.
Disclosure statement
There is no conflict of interest financial or otherwise.
References
- Abhijit A, Nagarsenker MS. (2007). Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int. J. Pharm. 329:166–72. [DOI] [PubMed] [Google Scholar]
- Agrawal AG, Kumar A, Gide PS. (2015). Formulation of solid self-nanoemulsifying drug delivery systems using N-methyl pyrrolidone as cosolvent. Drug Dev. Ind. Pharm 41:594–604. [DOI] [PubMed] [Google Scholar]
- Ahsan MN, Verma PR. (2017). Solidified self nano-emulsifying drug delivery system of rosuvastatin calcium to treat diet-induced hyperlipidemia in rat: In vitro and in vivo evaluations. Ther. Deliv 8:125–36. [DOI] [PubMed] [Google Scholar]
- Alhasani KF, Kazi M, Abbas M, et al. (2019). Self-nanoemulsifying ramipril tablets: a novel delivery system for the enhancement of drug dissolution and stability. Int. J. Nanomed Volume14:5435–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alshamsan A, Kazi M, Badran MM, Alanazi FK. (2018). Role of alternative lipid excipients in the design of self-nanoemulsifying formulations for fenofibrate: characterization, in vitro dispersion, digestion and ex vivo gut permeation studies. Front. Pharmacol 9:1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alshora DH, Ibrahim MA, Elzayat E, et al. (2018). Rosuvastatin calcium nanoparticles: improving bioavailability by formulation and stabilization codesign. PLoS One 13:e0200218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alskär LC, Keemink J, Johannesson J, et al. (2018). Impact of drug physicochemical properties on lipolysis-triggered drug supersaturation and precipitation from lipid-based formulations. Mol. Pharmaceutics 1, 15:4733–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alwadei M, Kazi M, Alanazi FK. (2019). Novel oral dosage regimen based on self-nanoemulsifying drug delivery systems for codelivery of phytochemicals – Curcumin and thymoquinone. Saudi Pharm. J 27:866–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amdoun R, Khelifi L, Khelifi-Slaoui M, et al. (2018). The desirability optimization methodology; a tool to predict two antagonist responses in biotechnological systems: Case case of biomass growth and Hyoscyamine content in elicited Datura starmonium hairy roots. Iranian J. Biotech 16:e1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakumar K, Raghavan CV, Selvan NT, et al. (2013). Self-nanoemulsifying drug delivery system (SNEDDS) of rosuvastatin calcium: Design, formulation, bioavailability and pharmacokinetic evaluation. Colloids Surf. B: Biointerfaces 112:337–43. [DOI] [PubMed] [Google Scholar]
- Baloch J, Sohail MF, Sarwar HS, et al. (2019). Self-nanoemulsifying drug delivery system (SNEDDS) for improved oral bioavailability of chlorpromazine: In vitro and in vivo evaluation. Medicina (Kaunas) 55:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang SP, Yeon CY, Adhikari N, et al. (2019). Cyclosporine A eyedrops with self-nanoemulsifying drug delivery systems have improved physicochemical properties and efficacy against dry eye disease in a murine dry eye model. PLoS One 14:e0224805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharti D, Pandey P, Verma R, Kaushik D. (2018). Development and characterization of rosuvastatin loaded self-emulsifying drug delivery system. Appl. Clinical Res. Clinical Trials Reg. Affairs 5:1–8. [Google Scholar]
- Borhade V, Nair H, Hegde D. (2008). Design and evaluation of self-microemulsifying drug delivery system (SMEDDS) of tacrolimus. AAPS PharmSciTech 9:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KO, Aditya NP, Ko S. (2014). Effect of aqueous pH and electrolyte concentration on structure, stability and flow behavior of non-ionic surfactant based solid lipid nanoparticles. Food Chem 147:239–44. [DOI] [PubMed] [Google Scholar]
- Derringer GC. (1994). A balancing act: optimizing a product’s. properties. Quality Prog 21:51–8. [Google Scholar]
- Eleftheriadis GK, Mantelou P, Karavasili C, et al. (2019). Development and characterization of a self-nanoemulsifying drug delivery system comprised of rice bran oil for poorly soluble drugs. AAPS PharmSciTech 20:78. [DOI] [PubMed] [Google Scholar]
- ElShagea HN, ElKasabgy NA, Fahmy RH, Basalious EB. (2019). Freeze-dried self nanoemulsifying self-nanosuspension (SNESNS): a new approach for the preparation of a highly drug-loaded dosage form. AAPS PharmSciTech 20:258. [DOI] [PubMed] [Google Scholar]
- Eltobshi AA, Mohamed EA, Abdelghani GM, Nouh AT. (2018). Self-nanoemulsifying drug-delivery systems for potentiated anti-inflammatory activity of diacerein. Int.J. Nanomed Volume13:6585–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enin HA. (2015). Self-nanoemulsifying drug-delivery system for improved oral bioavailability of rosuvastatin using natural oil antihyperlipdemic. Drug Dev. Ind. Pharm 41:1047–56. [DOI] [PubMed] [Google Scholar]
- FDA. 2012. Quality by design for ANDAs: an example for immediate-release dosage forms. pp. 1–107. Available from: https://www.fda.gov/media/83664/download.
- Gahlawat N, Verma R, Kaushik D. (2019). Recent developments in self-microemulsifying drug delivery system: An overview. Asian J. Pharm 13: 59–72. [Google Scholar]
- Gamal W, Fahmy RH, Mohamed MI. (2017). Development of novel amisulpride-loaded liquid self-nanoemulsifying drug delivery systems via dual tackling of its solubility and intestinal permeability. Drug Dev. Ind. Pharm 43:1530–48. [DOI] [PubMed] [Google Scholar]
- Gleiter CH, Jägle C, Gresser U, Mörike K. (2006). Candesartan. Cardiovascular Drug Reviews, Neva Press, Branford, Connecticut 22:263–84. [DOI] [PubMed] [Google Scholar]
- Gué E, Since M, Ropars S, et al. (2016). Evaluation of the versatile character of a nanoemulsion formulation. Int. J. Pharm 498:49–65. [DOI] [PubMed] [Google Scholar]
- Heshmati N, Cheng X, Eisenbrand G, Fricker G. (2013). Enhancement of oral bioavailability of E804 by self-nanoemulsifying drug delivery system (SNEDDS) in rats. J. Pharm. Sci 102:3792–9. [DOI] [PubMed] [Google Scholar]
- Hosny KM, Aldawsari HM, Bahmdan RH, et al. (2019). Preparation, optimization, and evaluation of hyaluronic acid-based hydrogel loaded with miconazole self-nanoemulsion for the treatment of oral thrush. AAPS PharmSciTech 20:1–12. 10.1016/j.jconrel.2010.04.029. [DOI] [PubMed] [Google Scholar]
- Izham MHM, Hussin Y, Aziz MNM, et al. (2019). Preparation and characterization of self nano-emulsifying drug delivery system loaded with citral and its antiproliferative effect on colorectal cells in vitro. Nanomaterials 9:E1028. 1028: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakab G, Fülöp V, Bozó T, et al. (2018). Optimization of quality attributes and atomic force microscopy imaging of reconstituted nanodroplets in baicalin loaded self-nanoemulsifying formulations. Pharmaceutics 10:275. 10040 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong IJ, Kim KJ. (2009). An interactive desirability function method to multiresponse optimization. Eur. J. Oper. Res 195:412–26. [Google Scholar]
- Johnson B, Nornoo AO, Zheng H, et al. (2009). Oral microemulsions of paclitaxel: in situ and pharmacokinetic studies. Eur. J. Pharm. Biopharm 71:310–7. [DOI] [PubMed] [Google Scholar]
- Kalantari A, Kósa D, Nemes D, et al. (2017). Self-nanoemulsifying drug delivery systems containing Plantago lanceolata-An assessment of their antioxidant and anti-inflammatory effects. Molecules 22:1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassem AA, Mohsen AM, Ahmed RS, Essam TM. (2016). Self-nanoemulsifying drug delivery system (SNEDDS) with enhanced solubilization of nystatin for treatment of oral candidiasis: Design, optimization, in vitro and in vivo evaluation. J. Mol. Liquids 218:219–32. [Google Scholar]
- Khalifa MKA, Salem HA, Shawky SM, et al. (2019). Enhancement of zaleplon oral bioavailability using optimized self-nano emulsifying drug delivery systems and its effect on sleep quality among a sample of psychiatric patients. Drug Deliv 26:1243–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim RM, Jang DJ, Kim YC, et al. (2018). Flurbiprofen-loaded solid SNEDDS preconcentrate for the enhanced solubility, in vitro dissolution and bioavailability in rats. Pharmaceutics 10:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Nanda A. (2018). Design and optimization of simvastatin self-microemulsifying drug delivery system for enhanced therapeutic potential. Asian J. Pharm 12: S159–S165. [Google Scholar]
- Kuncahyo I, Choiri S, Fudholi A, et al. (2019). Assessment of fractional factorial design for the selection and screening of appropriate components of a self-nanoemulsifying drug delivery system formulation. Adv. Pharm. Bull 9:609–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Kim HY, Cho YH, et al. (2018). Development and evaluation of raloxifene-hydrochloride-loaded supersaturatable SMEDDS containing an acidifier. Pharmaceutics 10:78–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sun C, Hao Y, et al. (2010). Mechanism of dissolution enhancement and bioavailability of poorly water soluble celecoxib by preparing stable amorphous nanoparticles. J Pharm Pharm Sci 13:589–606. [DOI] [PubMed] [Google Scholar]
- Mao LM, Qi XW, Hao JH, et al. (2015). In vitro, ex vivo and in vivo anti-hypertensive activity of Chrysophyllum cainito L. extract. Int. J. Clinical Exper. Med 8:17912–21. [PMC free article] [PubMed] [Google Scholar]
- Mohsin K. (2012). Design of lipid-based formulations for oral administration of poorly water-soluble drug fenofibrate: Effects effects of digestion. AAPS PharmSciTech 13:637–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosgaard MD, Sassene P, Mu H, et al. (2015). Development of a high-throughput in vitro lipolysis model for rapid screening lipid-based drug delivery systems. Eur. J. Pharm. Biopharm 94:493–500. [DOI] [PubMed] [Google Scholar]
- Mura P, Furlanetto S, Cirri M, et al. (2005). Optimization of glibenclamide tablet composition through the combined use of differential scanning calorimetry and D-optimal mixture experimental design. J. Pharm. Biomed. Anal 37:65–71. [DOI] [PubMed] [Google Scholar]
- Nepal PR, Han HK, Choi HK. (2010). Preparation and in vitro–in vivo evaluation of Witepsol H35 based self-nanoemulsifying drug delivery systems (SNEDDS) of coenzyme Q (10. ). Eur. J. Pharm. Sci 39:224–32. [DOI] [PubMed] [Google Scholar]
- Pal S, Nagy S, Bozo T, et al. (2013). Technological and biopharmaceutical optimization of nystatin release from a multiparticulate based bioadhesive drug delivery system. Eur. J. Pharm. Sci 49:258–64. [DOI] [PubMed] [Google Scholar]
- Pal TP, Saha D, Maity S. (2016). Bioequivalence modulation with modified starch in orodispersible tablets in comparison to marketed conventional tablets of rosuvastatin calcium. Eur. J. Pharm. Med. Res 2016:3(4), 236–249. [Google Scholar]
- Panigrahi KC, Patra N, Rao MEB. (2019). Quality by design enabled development of oral self-nanoemulsifying drug delivery system of a novel calcimimetic cinacalcet HCl using a porous carrier: In vitro and in vivo characterisation. AAPS PharmSciTech 20:216. [DOI] [PubMed] [Google Scholar]
- Parmar N, Singla N, Amin S, Kohli K. (2011). Study of cosurfactant effect on nanoemulsifying area and development of lercanidipine loaded (SNEDDS) self-nanoemulsifying drug delivery system. Colloids Surf. B: Biointerfaces 86:327–38.21550214 [Google Scholar]
- Patel MH, Mundada VP, Sawant KK. (2019). Novel drug delivery approach via self-microemulsifying drug delivery system for enhancing oral bioavailability of asenapine maleate: Optimization, characterization, cell uptake, and in vivo pharmacokinetic studies. AAPS PharmSciTech 20:1–8. [DOI] [PubMed] [Google Scholar]
- Patki M, Giusto K, Gorasiya S, et al. (2019). 17-α hydroxyprogesterone nanoemulsifying preconcentrate-loaded vaginal tablet: A a novel non-invasive approach for the prevention of preterm birth. Pharmaceutics 11:335. 11070335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Wang L, Zhu J, et al. (2011). Self-double-emulsifying drug delivery system (SDEDDS): A new way for oral delivery of drugs with high solubility and low permeability. Int. J. Pharm 409:245–51. [DOI] [PubMed] [Google Scholar]
- Rangaraj N, Shah S, Maruthi AJ, et al. (2019). Quality by design approach for the development of self-emulsifying systems for oral delivery of febuxostat: pharmacokinetic and pharmacodynamic evaluation. AAPS PharmSciTech 20:267. [DOI] [PubMed] [Google Scholar]
- Sassene P, Kleberg K, Williams HD, et al. (2014). Toward establishment of standardized in vitro tests for LbDDSs, Part 6: Effect of varying pancreatin and calcium levels. AAPS J 16:1344–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakeel F, Haq N, El-Badry M, et al. (2013). Ultra fine super self-nanoemulsifying drug delivery system (SNEDDS) enhanced solubility and dissolution of indomethacin. J. Mol. Liq 180:89–94. [Google Scholar]
- Shoshtari S, Wen J, Alany RG. (2010). Formulation and physicochemical characterization of imwitor 308 based self microemulsifying drug delivery systems. Chem. Pharm. Bull 58:1332–8. [DOI] [PubMed] [Google Scholar]
- Son HY, Chae BR, Choi JY, et al. (2018). Optimization of selfmicroemulsifying drug delivery system for phospholipid complex of telmisartan using D-optimal mixture design. PLoS One 13:e0208339–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas N, Holm R, Garmer M, et al. (2013). Supersaturated self-nanoemulsifying drug delivery systems (Super-SNEDDS) enhance the bioavailability of the poorly water-soluble drug simvastatin in dogs. AAPS J 15:219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Zhang Q, Shi Z, Wang J. (2019). Mechanisms of oral absorption improvement for insoluble drugs by the combination of phospholipid complex and SNEDDS. Drug Deliv 26:1155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Kaushik D. (2019). Development, optimization, characterization and impact of in vitro lipolysis on drug release of telmisartan loaded SMEDDS. Drug Deliv. Letters 9:330–40. [Google Scholar]
- Verma R, Mittal V, Kaushik D. (2017). Self-microemulsifying drug delivery system: A vital approach for bioavailability enhancement. Int. J. ChemTech Res 10:515–28. [Google Scholar]
- Verma R, Mittal V, Kaushik D. (2018). Quality based design approach for improving oral bioavailability of valsartan loaded SMEDDS and study of impact of lipolysis on the drug diffusion. Drug Deliv. Letters 8:130–9. [Google Scholar]
- Williams HD, Anby MU, Sassene P, et al. (2012). Toward the establishment of standardized in vitro tests for lipid-based formulations, Part 2. The effect of bile salt concentration and drug loading on the performance of type I, II, IIIA, IIIB, and IV formulations during in vitro digestion. Mol. Pharm 101:3286–300. [DOI] [PubMed] [Google Scholar]
- Wu L, Qiao Y, Wang L, et al. (2015). A self-microemulsifying drug delivery system (SMEDDS) for a novel medicative compound against depression: a preparation and bioavailability study in rats. AAPS PharmSciTech 16:1051–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Yi T, Liu Y, Zhou Z. (2016). The in vitro lipolysis of lipid-based drug delivery systems: A newly identified relationship between drug release and liquid crystalline phase. Bio. Med. Res. Int 2016:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wen X, Dai Y, Xia Y. (2019). Mechanistic studies on the absorption enhancement of a self-nanoemulsifying drug delivery system loaded with norisoboldine-phospholipid complex. Int. J. Nanomed Volume14:7095–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhi Z, Jiang T, et al. (2010). Spherical mesoporous silica nanoparticles for loading and release of the poorly water-soluble drug telmisartan. J. Contr. Releas 145:257–63. [DOI] [PubMed] [Google Scholar]
- Zhao X, Wang X. (2018). Candesartan targeting of angiotensin II type 1 receptor demonstrates benefits for hypertension in pregnancy via the NF‑κB signaling pathwayNF-κB signaling pathway. Mol. Med. Rep 18:705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wang C, Chow AH, et al. (2010). Self-nanoemulsifying drug delivery system (SNEDDS) for oral delivery of zedoary essential oil: Formulation formulation and bioavailability studies. Int. J. Pharm 383:170–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The author authenticates that the data supporting the results and findings of this study are existing within the article.