

Abstract
Because of the emergence of drug-resistant tumor cells, successful treatments of human malignancies have been difficult to achieve in the clinic. In spite of various approaches to overcome multi drug resistance, it has remained challenging and elusive. It is, therefore, necessary to define and understand the mechanisms of drug-induced tumor cell killing for the future development of anticancer agents and for rationally designed combination chemotherapies. The clinically active antitumor drugs, topotecan, doxorubicin, etoposide, and procarbazine are currently used for the treatment of human tumors. Therefore, a great deal research has been carried to understand mechanisms of actions of these agents both in the laboratory and in the clinic. These drugs are also extensively metabolized in tumor cells to various reactive species and generate oxygen free radical species (ROS) that initiate lipid peroxidation and induce DNA damage. However, the roles of ROS in the mechanism of cytotoxicity remain unappreciated in the clinic. In addition to ROS, various reactive nitrogen species (RNS) are also formed in tumor cells and in vivo. However, the importance of RNS in cancer treatment is not clear and has remained poorly defined. This review discusses the current understanding of the formation and the significance of ROS and RNS in the mechanisms of various clinically active anticancer drugs.
INTRODUCTION
There is a significant interest in the formation and consequences of free radicals in biological systems [1–4]. Free radicals molecules contain unpaired electrons and are produced during normal cell metabolism [5, 6]. In vivo, inflammatory cells and xanthine/xanthine oxidase system have been identified as sources for the generation of free radicals. Free radicals are also formed during metabolism of various anticancer drugs and xenobiotics in vivo and in tumor cells by cytochrome P450 and peroxidases. The bio-activations of these compounds result in the formation of either a carbon- or nitrogen-centered primary radical. Because these radicals have unpaired electrons, they are not stable and react rapidly with a wide variety of cellular macromolecules, including protein and DNA. Furthermore, in the presence of oxygen, these free radical intermediates react with O2 and generate various oxygen reactive species (superoxide anion radical, hydrogen peroxide and reactive hydroxyl radical, commonly known as ROS). The reactive •OH is formed following metal ion-catalysis of hydrogen peroxide (scheme 1) which has been shown to be pH dependent and this catalysis is efficient between pH 3 and 8.
SCHEME 1:
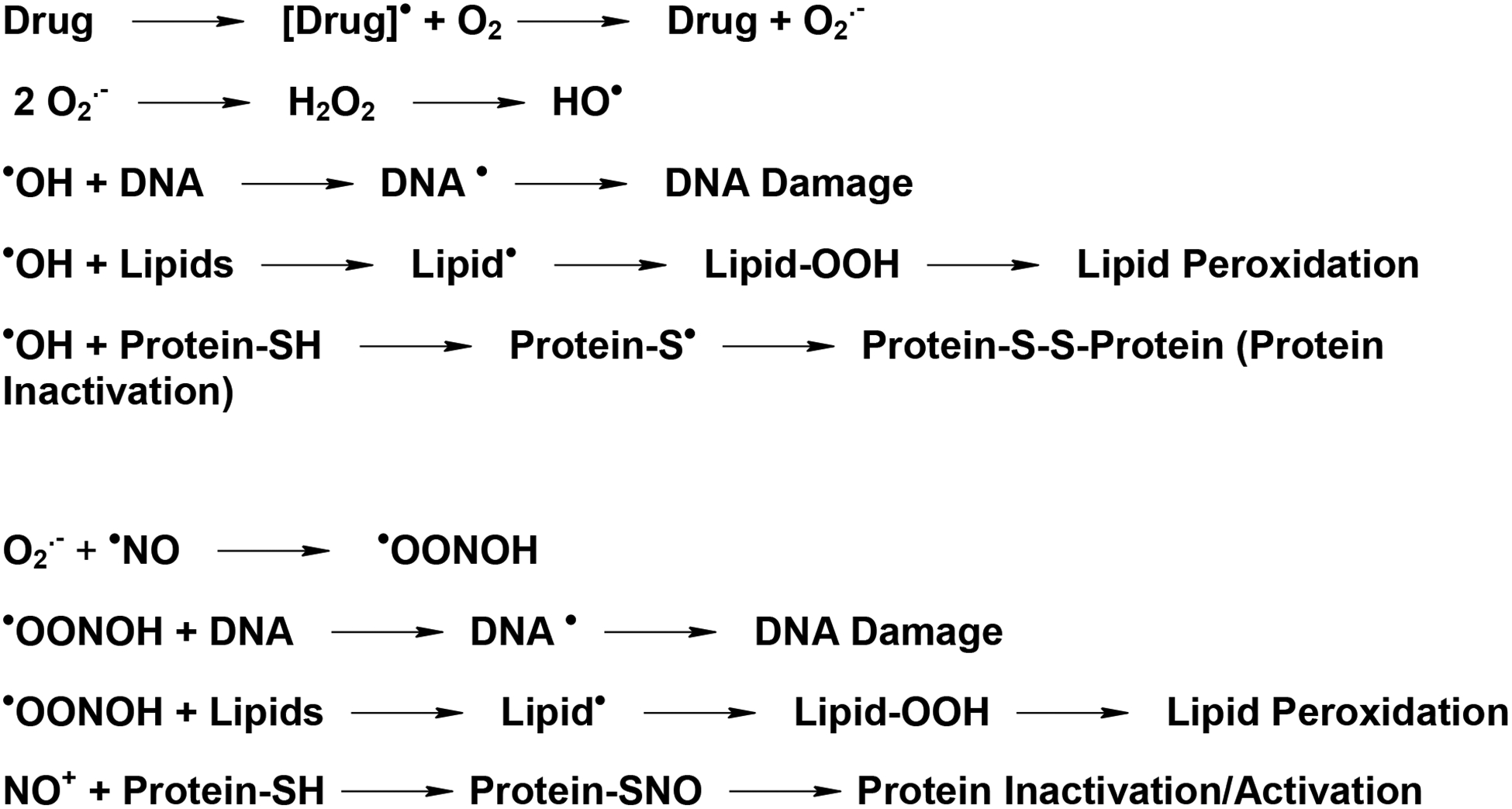
Activation and the formation of free radical intermediates from anticancer drugs and subsequent damage to cellular macromolecules induced by ROS and RNS.
In addition to ROS, reactive nitrogen species (RNS) derived from nitric oxide (•NO) e.g., NO+, N2O3, and −OONO, are also formed in cells. Nitric oxide is a short-lived free radical molecule which easily diffuses in cells and is synthesized by nitric oxide synthase (NOS) from L-arginine. Nitric oxide is an important cellular messenger and has been reported to plays a significant role in vasodilatation, apoptosis, and the innate immune response [7]. As a signaling molecule, •NO has been shown to interact with the heme moiety of soluble guanyl cyclase, resulting in the activation and production of second messenger cyclic GMP [7]. Furthermore, additional actions of •NO also result from the reaction of RNS with protein -SH groups (S-nitrosylation) and introduction of nitroso groups to form S-nitrosothiols (-SNO) (Scheme-1). It has been shown that the nitrosation of proteins is involved in protein stabilization or inactivation as well as in cell signaling [8–10].
ROS and RNS are continuously generated during normal cell functions in vivo; these reactive intermediates are removed by extensive cellular protective mechanisms (e.g., reduced glutathione, ascorbate, SOD, catalase and selenium-dependent glutathione peroxidases) and thus, they do not pose significant risks to human health. However, in the absence of proper removable of ROS/RNS, these reactive species have been shown to cause damages to cellular proteins, lipids (lipid peroxidation) and DNA (formation of 8-oxo-deoxyguanosine, other oxidized DNA molecules), and inducing oxidative or nitrosative stress. These events lead to cellular toxicity, tumor formation or cell death (Scheme-1).
A number of anticancer drugs, e.g., topotecan, doxorubicin, etoposide and procarbazine are currently used for the treatment of a wide variety of malignancies in the clinic [11–16]. Topotecan (TPT), doxorubicin (DOX) and etoposide (VP-16) belong to a class of drugs known as topoisomerase poisons that induce the formation of highly cytotoxic double-strand DNA breaks for their antitumor activities [17]. While the main mechanism(s) of cell death by these agents is due to the formation of DNA double-strand breaks mediated by topoisomerase I (TPT) and II (DOX, VP-16), several other mechanisms are now known, e.g., enzymatic activation to reactive species that also induce cellular damage and cell death. Procarbazine, a hydrazine derivative, has been shown to undergo extensive metabolism to form various reactive species that cause cellular damage and tumor cell death. Thus, bioactivation of anticancer drugs and the generation of reactive species (ROS and RNS) appears to be a common mechanism for actions of these drugs. This review examines activation, formation, and roles of ROS and RNS in the mechanisms of action (s) of certain anticancer drugs that may induce cell damage and ultimately lead to cell death.
DOXORUBICIN
Doxorubicin is extensively used for the treatment of both hematological and solid human tumors in the clinic [13, 18]. It contains both an anthraquinone chromophore (Figure 1) and a quinone-hydroquinone structure. While various cellular enzymes, e.g., NADPH cytochrome P450 reductase, xanthine oxidase, DT-diophorase and nitric oxide synthase are known to reduce the quinone-hydroquinone moiety of doxorubicin [19, 20], the cytochrome P450 reductase/NADPH system is considered to be the main reductive activation pathway for doxorubicin in tumor cells (Figure 1) [21–23].
Figure 1.
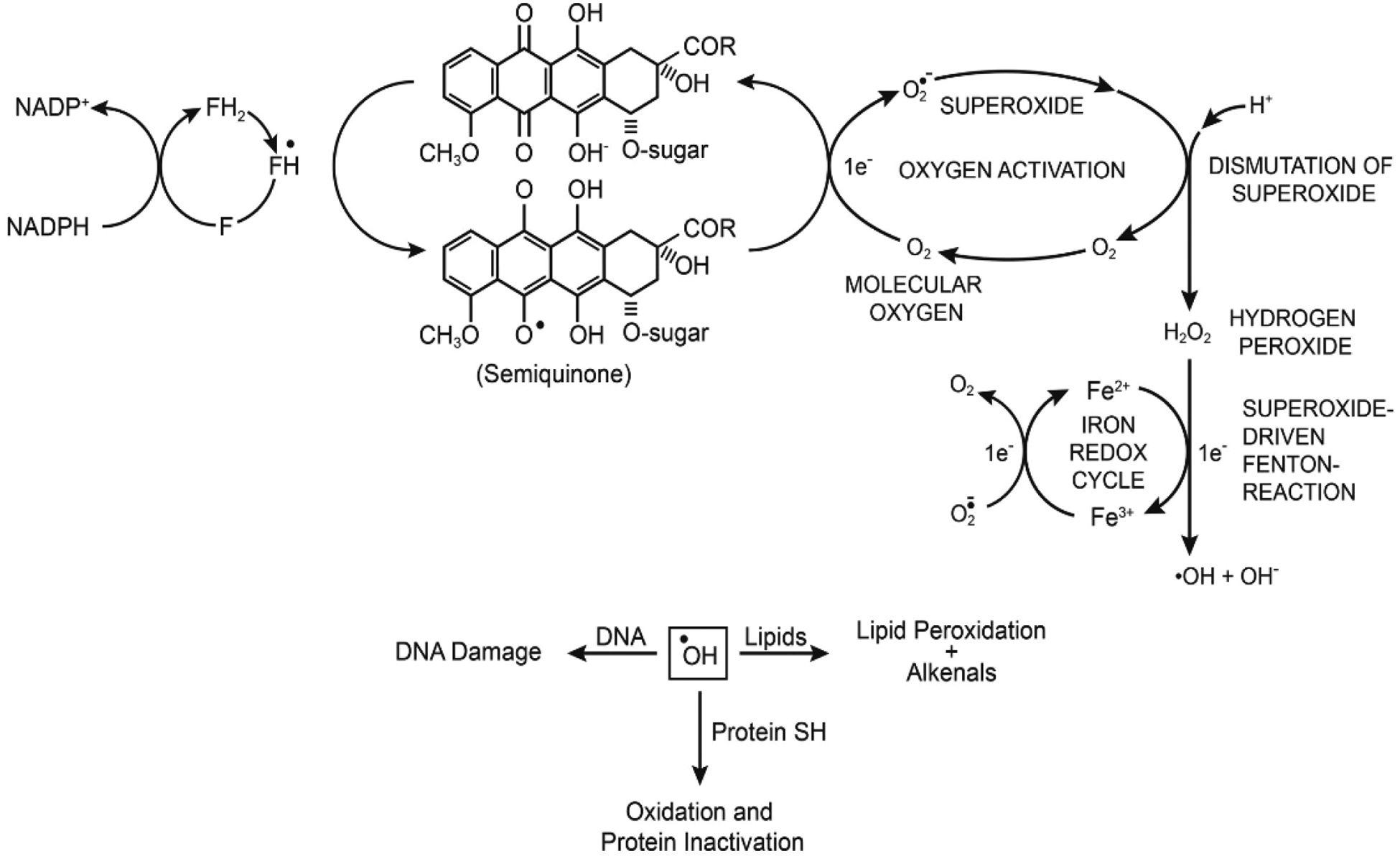
Reductive activation of doxorubicin to semiquinone radical, formation of ROS, and induction of DNA damage, lipid peroxidation and protein oxidation.
Under anaerobic conditions, the doxorubicin semiquinone radical has been detected in biological samples by EPR [23, 24]. However, it rapidly undergoes redox-cycling in the presence of O2 to generate superoxide anion, hydrogen peroxide, and hydroxyl radical, and regenerates the parent drug [20–22, 25]. ●OH has been shown to induce DNA damage and initiates peroxidation of cellular lipids which forms other toxic metabolites that bind to DNA and proteins [26, 27]. The role of ROS in doxorubicin cytotoxicity has remained a matter of disagreement in spite of significant amounts of research supporting the formation of ROS in doxorubicin-induced tumor cell death [26–29]. This debate stems from the fact that doxorubicin is effective at nanomolar concentrations, while the EPR-based detection of free radicals requires micromolar concentrations of the drug. In addition, since the formation of ●OH from H2O2 is metal ion dependent, very little free Fe3+ is present in tumor cells for ●OH formation.
These disagreements are easily resolved as the detection of ROS in cells requires significantly higher concentrations of doxorubicin because of the limited sensitivity of EPR for the detection of free radical intermediates. Moreover, ROS formed in tumor cells and tissues are rapidly destroyed due to the presence of high amounts of reduced glutathione and other sulfhydryl compounds in cells. Furthermore, detoxifying enzymes (SOD, catalase and glutathione peroxidases) are also present in tumor cells that remove superoxide, hydrogen peroxide and hydroperoxides, respectively, and further reducing detectable levels of ROS. The formation of H2O2 in tumor cells and tissues from nanomolar concentrations of doxorubicin has been confirmed by florescence detection methods [30]. It should also be noted that depletion of glutathione by BSO in most tumor cells results in significantly higher amounts of ROS generation and increase doxorubicin cytotoxicity, suggesting ROS are formed and participate in tumor cell death by doxorubicin [31, 32].
Because doxorubicin requires bioactivation to form ROS, it is also possible that certain tumor cells cannot activate doxorubicin to the semiquinone radical for the formation of ROS as was found with doxorubicin-resistant MCF-7 tumor cells [33, 34]. Furthermore, significantly smaller amounts of doxorubicin-dependent ROS are formed and detected due to lesser amounts of doxorubicin present in tumor cells due to increased activities of both ABS transporter proteins and detoxifying enzymes (SOD, catalase, glutathione peroxidase and glutathione transferase) in resistant tumor cells [33–36]. Overexpression of MnSOD has been shown to inhibit the growth of tumor cells [37]. When combined with doxorubicin, MnSOD significantly increases tumor cell death by doxorubicin which is further increased by BCNU, an inhibitor of glutathione reductase [38–40]. Of interest is the finding that doxorubicin-sensitive human breast MCF-7 tumor cells are more sensitive to H2O2 than the resistant MCF-7 tumor cells [32]. These observations strongly suggest that ROS are formed and that H2O2 is the key intermediate for tumor cell killing by doxorubicin.
Although higher amounts of copper and iron are present in human tumors, the role of iron in doxorubicin cytotoxicity is complex [41, 42]. Because doxorubicin-Fe complexes do not cross cell membrane, nor they are actively transported in tumor cells, it is believed these complexes do not catalyze or participate in the reduction of H2O2 inside tumor cells and form ●OH to induce cell death. It is possible, however; that Fe-doxorubicin complexes are formed within tumor cells as Fe ions are released from dying tumor cells. Furthermore, under anaerobic conditions, the semiquinone radical of doxorubicin has been reported to release iron from ferritin [43]. These observations would then suggest that doxorubicin-Fe complexes can be formed in tumors to generate ●OH and, ultimately cause cell death. We have found that RNS inhibits both catalytic and cleavage activities of topo II. However, the cytotoxicity of doxorubicin was not significantly modulated in several human tumor cell lines, indicating a non-topo II-dependent mechanism for doxorubicin cytotoxicity, likely a ROS-dependent cell death in these tumor cells [44].
ETOPOSIDE (VP-16,213)
Etoposide (VP-16, Figure-2) is active against a wide variety of tumors, including lymphoma and testicular tumors [14]. It is a topo poison and induces the formation of topo II-mediated double-stranded DNA breaks in tumor cells, causing tumor cell death [45–47]. VP-16 is metabolized by cytochrome P450, horseradish peroxidase, and tyrosinase to a VP-16 phenoxy radical, o-quinone-VP-16 (VP-16-Q, and o-dihydroxy VP-16 (DHVP) Figure 2) [48–52]. The presence of the 4’-OH in VP-16 has been found to be essential for the formation of VP-16•, its metabolites, and for the antitumor activity of VP-16.
Figure 2:
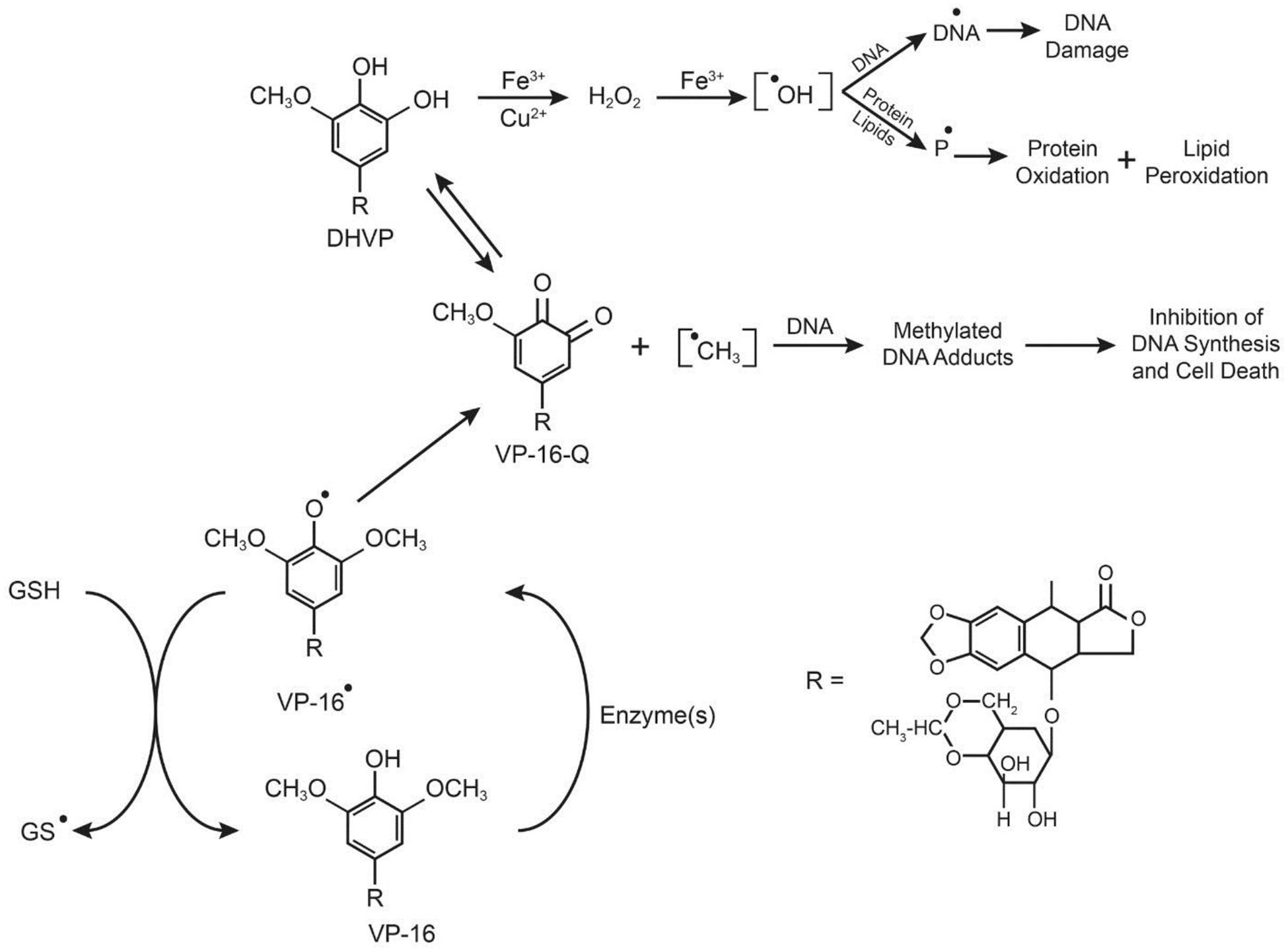
Enzymatic activation of VP-16 and formation of VP-16 radical, o-quinone, and dihydroxy-VP-16.
Metal chelation of DHVP with either copper or iron ions induces the formation of •OH from H2O2, resulting in significant damage to DNA [26, 53]. The DHVP metabolite is also autoxidized to produce H2O2 and •OH, and in the presence of metal ions the rate of •OH formation is significantly increased from H2O2 [48]. Treatment of tumor cells or mice in vivo with VP-16 results in the formation of GSSG from the oxidation of GSH by VP-16• [54]. This observation suggests that: (a) oxidative stress is induced in tumor cells from the depletion of GSH by VP-16, which may lead to damage to cellular lipids (lipid peroxidation) or to enzymes necessary for cell survival, and (b) products of lipid peroxidation (e.g., aldehydes) may bind to DNA, inhibiting DNA synthesis and cell death. Thus, the synergistic interactions observed in the clinic between VP-16 and ionizing radiation or photosensitizers may result from this oxidative stress induced by glutathione depletion by VP-16 or its metabolites [55, 56].
Topotecan
Topotecan (TPT, Figure-3), a water soluble derivative of camptothecin, is an important anticancer agent for the treatment of various human malignancies in the clinic [12, 57]. It is a topo I poison, and it stabilizes transient complexes formed between topo I and DNA, leading to the formation of double-strand DNA breaks in tumor cells, and cell death. Induction of oxidative stress [58–60] and inhibition of hypoxia-inducible factors by TPT have also been suggested to play a role in tumor cells death [61, 62]. Treatment of MCF-7 tumor cells with TPT leads to decreases in glutathione levels with increases in lipid peroxidation. Furthermore, higher levels of antioxidant enzymes, superoxide dismutase, and glutathione peroxidase, have also been observed following treatment of MCF-7 cells with TPT, indicating increased formation of ROS and oxidative stress [58–60]. It is interesting to note that ROS generated by arsenic trioxide have been suggested to increase the formation of DNA-topo I complexes [61], while H2O2 cytotoxicity has been reported to be mediated, in part, by topo I [63]. These observations, taken together, clearly indicate that ROS are formed following TPT treatment and contribute to topo I-mediated DNA damage and cytotoxicity.
Figure-3:
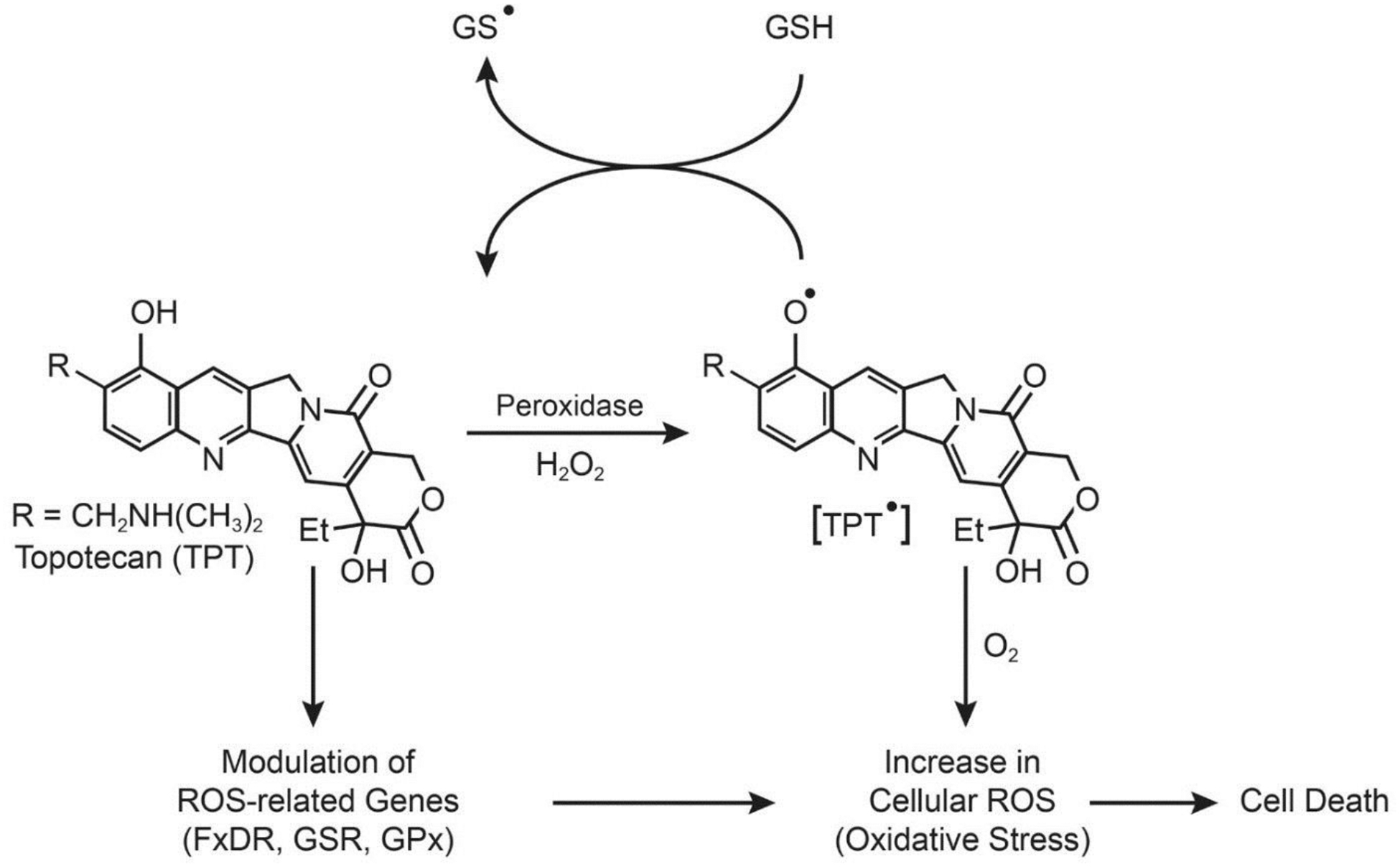
Formation of topotecan radical, oxidation of glutathione and modulation of ROS-sensing genes in tumor cells.
We have recently reported that TPT is oxidized by H2O2 and various peroxidases to a TPT radical (TPT•) that reacts with both glutathione and cysteine to form GS•and Cys• radicals, respectively, and regenerates TPT (Figure-3) [64]. We have found that unlike doxorubicin, the TPT• can be generated in the presence of DNA (i.e., bound/intercalated TPT) and react with GSH. We have also shown that ascorbic acid is highly synergistic with TPT in MCF-7 breast cancer cells. Ascorbic acid is known to generate H2O2 which is taken up by tumor cells, leading to the formation of •OH in the presence of metal ions [65–67]. Our recent studies based on gene expression profiling following TPT treatment in MCF-7 cells have shown that key ROS-related genes (glutathione reductase, glutathione peroxidase, ferredoxin reductase, methionine sulfoxide reductase,) are differentially regulated by TPT, suggesting that oxidative stress is indeed induced by ROS, and plays an important role in TPT cytotoxicity (manuscript in submission). A ROS-based mechanism of TPT cytotoxicity is summarized in Figure-3.
PROCARBAZINE
Procarbazine, a hydrazine derivative and a pro-drug, requires activation for its antitumor activities. Procarbazine is used in the treatment of Hodgkin’s lymphoma, malignant melanoma, and brain tumors in children. It has been reported to be metabolized by cytochrome P450 and monoamine oxidase to its azo derivative and, subsequently, to the azoxy derivative [68–70]. It has been shown that from the azoxy derivative of procarbazine, methyl carbonium ion (CH3+) is formed which then reacts with DNA and proteins, inhibiting DNA and protein synthesis, and causing tumor cell death [71]. Free radical intermediates have also been detected during microsomal P450 and peroxidative metabolism of procarbazine (Figure-4) [72]. The identity of these species has been confirmed by spin-trapping techniques [72]. It has been shown that a nitrogen-centered radical formed by one-electron oxidation of procarbazine is the obligatory intermediate for the formation of reactive CH3• and PhCH2• (Figure-4). The highly reactive CH3• and PhCH2• can then bind irreversibly with DNA and proteins, inhibiting both DNA and protein synthesis, and causing cells death (Figure-4).
Figure 4:
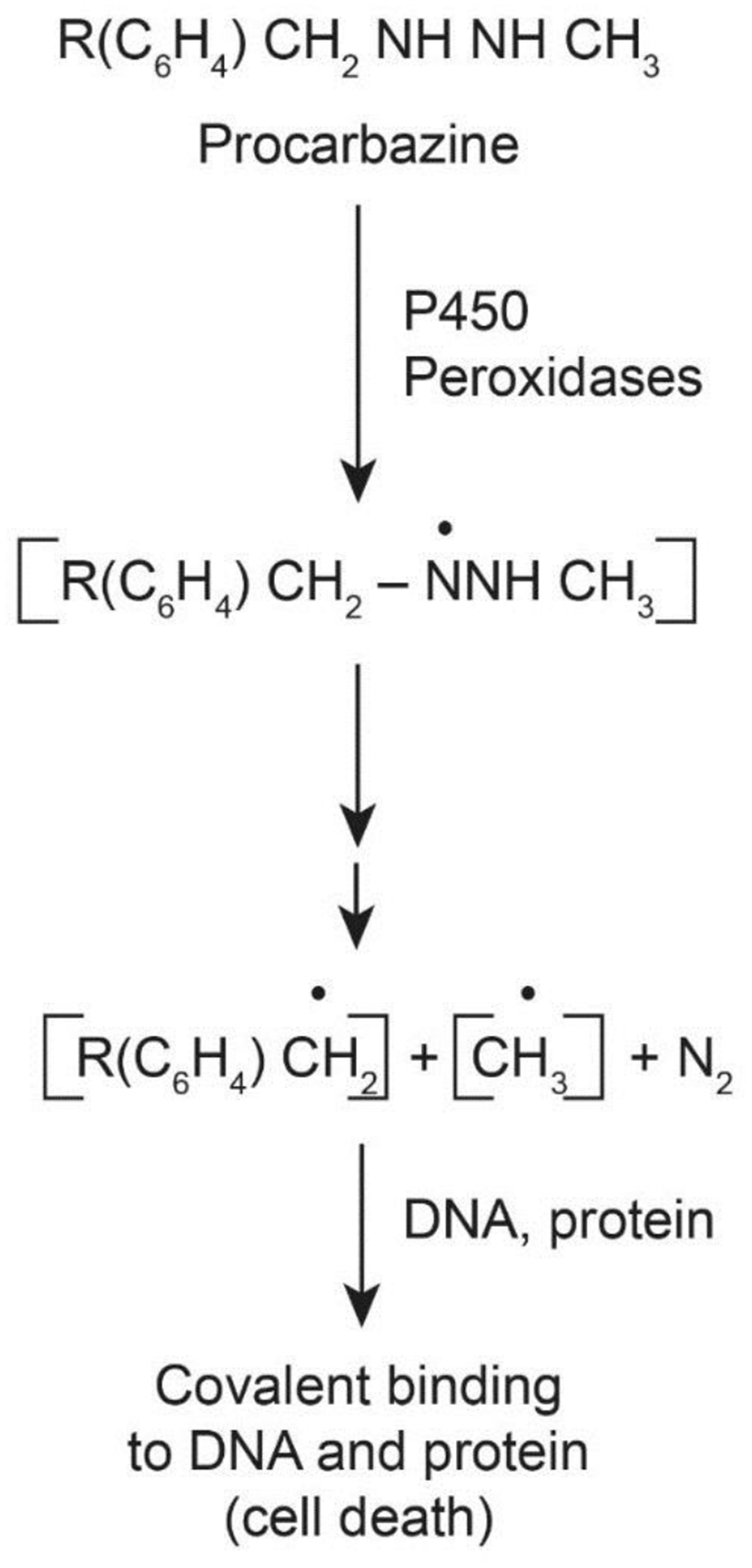
Formation of reactive free radical intermediates from procarbazine following metabolic activation
Reactive Nitrogen Species
Nitric oxide was discovered as an endothelial relaxing factor in late 1970. Nitric oxide is continuously generated in vivo from arginine by nitric oxide synthase in nanomolar quantities. However, during infection (and following the induction of iNOS), •NO concentration in cells is significantly increased. •NO rapidly reacts with O2 and forms various reactive metabolites (RNS) that induce cellular damage and cell death. •NO/RNS have been shown to inhibit the growth of human melanoma A375 tumor cells in vitro [73]. Since then a considerable amount of research has been carried out to bring NO-donors to the clinic for the treatment of various diseases, including cancers. Because •NO/RNS are cytotoxic to tumor cells various NO-donors have been developed that can generate high concentrations of •NO/RNS in tissue and tumor cells. NO-donors have been reported to be synergistic with cis-platin in CHO cells [74] and to enhance cytotoxicity of various other anticancer drugs both in vitro and in vivo [75–81]. DETA-NO and nitroglycerin (GTN) enhance doxorubicin cytotoxicity and reverse hypoxia-induced resistance to doxorubicin [82]. However, NO-donors with a short half-life are not effective modulators of doxorubicin cytotoxicity against several human tumor cells [44, 83]. Several excellent reviews are available highlighting the importance of NO-donors as anticancer agents [79, 84–87].
Various tumor specific NO-donors have been prepared, e.g., esterase-sensitive diazeniumdiolates-based [88] and O2-aryl diazeniumdiolates-based NO-donors [89] based on the idea that NO/RNS delivered specifically to tumors would be more cytotoxic. These No-donors release •NO/RNS in vivo following reaction with either tumor esterases or GSH/GST systems, respectively [89–91]. JS-K, as a single agent, is active against many human tumors both in vitro and in vivo [89, 92, 93].
It is interesting to note that •NO/RNS are also known to induce cis-platin resistance in several human tumor cell lines [94, 95]. It has been reported that this NO-induced resistance is caused by the stabilization of bcl2 protein, resulting in the inhibition of apoptosis [94]. RNS has been reported to induce VP-16 resistance by directly reacting with VP-16 and forming noncytotoxic metabolites of VP-16 [83]. RNS have shown to nitrosylates topo I in breast MCF-7 tumor and colon tumor HT-29 cells, inducing significant resistance to camptothecin only in MCF-7 cells [44, 96]. This development of resistance to camptothecin was found to result from wtp53-dependent upregulation and stabilization of bcl2 protein in MCF-7 cells by •NO /RNS [96]. We have shown that RNS nitrosylate also topo II, leading to an inhibition of its functional activities and inducing resistance to various topo II poisons in MCF-7 breast tumor cells [44]. We have recently shown that RNS inhibit the ATPase activity of topo II, resulting in decreases in DNA damage and resistance to several topo II poisons [64]. Increased •NO formation has been reported to induce interferon (IFN-ϒ) and lead to altered cell migration and development of resistance to taxol in MDA-231 breast cancer cells. IFN-ϒ induces NOS2, •NO and IL-6 formation in MDA-231 cells [97] as it is linked to more aggressive, clinically resistant tumors [98, 99].
One of the most promising effects of •NO/RNS is that they can also reverse multi-drug resistance (MDR). MDR cells overexpress ATP-dependent ABC transporters, e.g., p-170-glycoprotein (P-gp), breast cancer resistance protein (BCRP) and multi-drug resistance proteins (MRP’s). These efflux proteins remove intracellular drugs in an energy-dependent manner. The reversal of doxorubicin resistance by •NO/RNS has been described in various ABC transporter-overexpressing cell lines (HT-29-dx and K562-dx); this effect is observed when •NO/RNS increase drug accumulation resulting from nitration of tyrosines in the MRP3 transporter protein [100, 101]. •NO/RNS have also been reported to increase the accumulation of daunorubicin in leukemia K562 cells which constitutively express anti-apoptotic bcl2 and survivin proteins and are resistant to daunorubicin [102]. K562 tumor cells overexpress both MRP and lung-resistance proteins which are involved in removing and redistributing drugs away from the nucleus [103, 104], thus reducing effective drug concentrations in the nucleus.
The ATPase activity of P-gp is also inhibited by ●NO/RNS, resulting in significantly increased accumulation of drugs in a P-gp-overexpressing NCI/ADR-RES cell line. This increase in drug accumulation significantly reversed both adriamycin and taxol resistance in NCI/ADR-RES cells [105]. We found that ●NO/RNS treatment of MDR cells also enhanced •OH formation from adriamycin, resulting from the increased drug accumulation [105]. Recently we have used JS-K, a tumor-specific NO-donor, to study the reversal of drug resistance in both P-gp- and BCRP-overexpressing human tumor cells [106]. JS-K was found to be extremely effective in reversing adriamycin resistance in NCI/ADR-RES, however, it was also highly resistant to BCRP-overexpressing MCF-7/MX tumor cells. In that study, we found that ●NO/RNS inhibits the ATPase activity of BCRP, resulting in significant increases in the accumulation of Hoechst 33342 dye, and topotecan, leading to reversal of topotecan and mitoxantrone resistance to MCF-7/MX cells. A mechanism-based modification of cysteines in the ATP binding site by RNS is shown in Figure-5. It is believed that the modification of cysteines to NO-Cysteines in both P-gp and BCRP in ATP binding sites by RNS leads to a decrease in ATP binding and an increase in drug accumulation and cytotoxicity (Figure-5).
Figure 5:

Under normal conditions drugs (D) are exported out of the cells by P-gp or BCRP following ATP binding (A). In the presence of RNS, binding of ATP is significantly decreased due to modifications of cysteines (S-NO) in the ATP binding site, resulting in enhanced drug accumulation and cytotoxicity (B).
CONCLUSIONS
It is clear that doxorubicin is reductively activated to its semiquinone radical in tumor cells. The semiquinone radical generates various other reactive species that alkylate DNA and proteins, leading to in a plethora of unwanted cellular stresses and ultimately causing tumor cell death. The mechanism of doxorubicin cytotoxicity, however, is dependent upon both the cell type and the presence of O2. In the absence of O2, especially under hypoxic conditions, formation of the covalent binding species is favored. In contrast, under aerobic conditions; the formation of covalently binding species is significantly reduced due to reactions of the semiquinone radicals with O2, and in which case ROS-mediated tumor cell killing dominates. ROS have been detected in tumor cells and their presence are known to damage cellular macromolecules, leading to tumor cell death. Because doxorubicin is active at low concentrations in vivo, it has been suggested that the topo II is more important in doxorubicin cytotoxicity and ROS-dependent mechanisms do not contribute significantly. This is more so in highly hypoxic cells where there is very little O2, as ROS formation requires O2. It should be mentioned, however, that under highly hypoxic conditions and when tumor cells are not dividing, the concentration of topo II is also very low as topo II is cell-cycle dependent, and at low topo II protein levels, doxorubicin is inactive. Thus, tumor cell killing by doxorubicin is depend upon both cell type and O2 concentrations, and in tumor with rich O2 environments, ROS will play an important role in doxorubicin cytotoxicity.
Free radical intermediates are also formed in tumor cells from VP-16 following its metabolism, which may be important in the mechanism of tumor cell killing in vivo. We found that the inhibition of tyrosinase activity decreases both VP-16 activation and its cytotoxicity in tumor cells, indicating a free radical-based mechanism for VP-16 cytotoxicity. It is interesting to note that CH3• is released during the oxidation of VP-16 by peroxidases to form of VP-16-quinone through the intermediacy of VP-16•. •CH3 has been shown to alkylate DNA and proteins, inhibiting DNA synthesis, leading to tumor cell death. However, to date studies related to the formation of •CH3 from VP-16 has not been carried out and thus, its role in VP-16 cytotoxicity is not known. Oxidative metabolism of VP-16 to VP-16• and to its reactive metabolites (DHVP and o-VP-Q) in tumor cells may be important in tumor cell killing as metabolites are also topo II-active. Although the formation of ROS has been reported from DHVP in the presence of metal ions, no significant data are currently available in the clinic for the formation of ROS or their role in cytotoxicity.
Regarding topotecan, our recent studies show that TPT radical is formed from TPT following its one-electron oxidation in tumor cells, suggesting induction of oxidative stress by ROS formed by TPT. Furthermore, our gene expression study strongly indicates that various ROS-sensing genes are differentially regulated by TPT in MCF-7 cells. These observations suggest that ROS are formed and that they play a significant role in tumor cell death by TPT. Our observations also suggest that the formation of ROS by TPT is involved in the mechanism of synergistic interactions between topotecan and ionizing radiation in the clinic.
It is now clear that RNS are cytotoxic to tumor cells and that NO-donors enhance the antitumor activities of various clinically active antitumor agents against a variety of human tumors. Although various NO-donors have found good success in the clinic for the treatment of heart-related complications [107], NO-donors are currently not utilized for the treatment of cancers, and only a limited number of trials have been carried out [108, 109]. There are a number of reasons for this, as NO-donors are toxic to the host and are not tumor-specific. Furthermore, while •NO/RNS are cytotoxic to some tumors, they also cause resistance to certain anticancer drugs. Tumor-specific NO-donors that are target specific and require intracellular activation to release •NO/RNS, e.g., JS-K, offer more promise for future development.
In the clinic, one must overcome multi-drug resistance as it is an important determinant for a successful therapy. At present, the use of NO-donors in combinations with other active drugs is extremely promising directions for the treatment of resistance tumors. It appears to this authors that it is important to develop newer cancer therapies with tumor site-specific NO-donors that release •NO/RNS following intracellular activation. Tumor-specific intracellularly activated NO-donors may inhibit ATPase activities of resistant cells and thus would be suitable for targeting MDR and cancer stem cells for the reversal of drug resistance in the clinic. It is also possible that selective target-specific NO-donors could be delivered to tumors in vivo by newer techniques including lipid encapsulation or nanotechnologies for better overall tumor responses with standard chemotherapeutic agents. Recently, Sun et al. [110] have utilized NO conjugated with anti-CD24 antibody against hepatic carcinoma and found that this NO-donor is highly selective against tumors.
Acknowledgements:
We thank Drs. Maria Kadiiska, Erik Tokar, and Ann Motten for their critical evaluation of the manuscript.
Funding: This research was supported [in part] by the intramural research program of the National Institute of Environmental Health Sciences, NIH (Grant E505013922). Statements contained herein do not necessarily represent the statements, opinions, or conclusions of NIEHS, NIH, or the US Government.
Footnotes
Conflict of Interest: The author declares no actual or potential conflicts of interest.
REFERENCES
- [1].Halliwell B, The chemistry of free radicals, Toxicol Ind Health 9(1–2) (1993) 1–21. [DOI] [PubMed] [Google Scholar]
- [2].Davies MJ, The oxidative environment and protein damage, Biochimica et biophysica acta 1703(2) (2005) 93–109. [DOI] [PubMed] [Google Scholar]
- [3].Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J, Free radicals and antioxidants in normal physiological functions and human disease, The international journal of biochemistry & cell biology 39(1) (2007) 44–84. [DOI] [PubMed] [Google Scholar]
- [4].Phaniendra A, Jestadi DB, Periyasamy L, Free radicals: properties, sources, targets, and their implication in various diseases, Indian journal of clinical biochemistry : IJCB 30(1) (2015) 11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Loschen G, Flohe L, Chance B, Respiratory chain linked H(2)O(2) production in pigeon heart mitochondria, FEBS letters 18(2) (1971) 261–264. [DOI] [PubMed] [Google Scholar]
- [6].Nohl H, Hegner D, Do mitochondria produce oxygen radicals in vivo?, European journal of biochemistry 82(2) (1978) 563–7. [DOI] [PubMed] [Google Scholar]
- [7].Murad F, Nitric oxide signaling: would you believe that a simple free radical could be a second messenger, autacoid, paracrine substance, neurotransmitter, and hormone?, Recent progress in hormone research 53 (1998) 43–59; discussion 59–60. [PubMed] [Google Scholar]
- [8].Gaston B, Nitric oxide and thiol groups, Biochimica et biophysica acta 1411(2–3) (1999) 323–33. [DOI] [PubMed] [Google Scholar]
- [9].Aranda E, Lopez-Pedrera C, De La Haba-Rodriguez JR, Rodriguez-Ariza A, Nitric oxide and cancer: the emerging role of S-nitrosylation, Current molecular medicine 12(1) (2012) 50–67. [DOI] [PubMed] [Google Scholar]
- [10].Stamler JS, Lamas S, Fang FC, Nitrosylation. the prototypic redox-based signaling mechanism, Cell 106(6) (2001) 675–83. [DOI] [PubMed] [Google Scholar]
- [11].Horita N, Yamamoto M, Sato T, Tsukahara T, Nagakura H, Tashiro K, Shibata Y, Watanabe H, Nagai K, Inoue M, Nakashima K, Ushio R, Shinkai M, Kudo M, Kaneko T, Topotecan for Relapsed Small-cell Lung Cancer: Systematic Review and Meta-Analysis of 1347 Patients, Scientific reports 5 (2015) 15437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Muderspach LI, Blessing JA, Levenback C, Moore JL Jr., A Phase II study of topotecan in patients with squamous cell carcinoma of the cervix: a gynecologic oncology group study, Gynecologic oncology 81(2) (2001) 213–5. [DOI] [PubMed] [Google Scholar]
- [13].Weiss RB, The anthracyclines: will we ever find a better doxorubicin?, Seminars in oncology 19(6) (1992) 670–86. [PubMed] [Google Scholar]
- [14].O’Dwyer PJ, Leyland-Jones B, Alonso MT, Marsoni S, Wittes RE, Etoposide (VP-16–213). Current status of an active anticancer drug, The New England journal of medicine 312(11) (1985) 692–700. [DOI] [PubMed] [Google Scholar]
- [15].Devita VT Jr., Serpick AA, Carbone PP, Combination chemotherapy in the treatment of advanced Hodgkin’s disease, Annals of internal medicine 73(6) (1970) 881–95. [DOI] [PubMed] [Google Scholar]
- [16].Spivack SD, Drugs 5 years later: procarbazine, Annals of internal medicine 81(6) (1974) 795–800. [DOI] [PubMed] [Google Scholar]
- [17].Nitiss JL, DNA topoisomerase II and its growing repertoire of biological functions, Nature reviews. Cancer 9(5) (2009) 327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wiernik PH, Advances in treatment for Hodgkin’s disease, The Mount Sinai journal of medicine, New York 59(5) (1992) 405–7. [PubMed] [Google Scholar]
- [19].Bachur NR, Gordon SL, Gee MV, A general mechanism for microsomal activation of quinone anticancer agents to free radicals, Cancer research 38(6) (1978) 1745–50. [PubMed] [Google Scholar]
- [20].Bachur NR, Gordon SL, Gee MV, Kon H, NADPH cytochrome P-450 reductase activation of quinone anticancer agents to free radicals, Proceedings of the National Academy of Sciences of the United States of America 76(2) (1979) 954–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sato S, Iwaizumi M, Handa K, Tamura Y, Electron spin resonance study on the mode of generation of free radicals of daunomycin, adriamycin, and carboquone in NAD(P)H-microsome system, Gan 68(5) (1977) 603–8. [PubMed] [Google Scholar]
- [22].Kalyanaraman B, Morehouse KM, Mason RP, An electron paramagnetic resonance study of the interactions between the adriamycin semiquinone, hydrogen peroxide, iron-chelators, and radical scavengers, Archives of biochemistry and biophysics 286(1) (1991) 164–70. [DOI] [PubMed] [Google Scholar]
- [23].Schreiber J, Mottley C, Sinha BK, Kalyanaraman B, Mason RP, One-electron reduction of daunomycin, daunomycinone, and 7-deoxydaunomycine by the xanthine xanthine-oxidase system - Detection of semiquinone free-radicals by electron-spin-resonance, J. Am. Chem. Soc 109(2) (1987) 348–351. [Google Scholar]
- [24].Kalyanaraman B, Perez-Reyes E, Mason RP, Spin-trapping and direct electron spin resonance investigations of the redox metabolism of quinone anticancer drugs, Biochimica et biophysica acta 630(1) (1980) 119–30. [DOI] [PubMed] [Google Scholar]
- [25].Doroshow JH, Role of hydrogen peroxide and hydroxyl radical formation in the killing of Ehrlich tumor cells by anticancer quinones, Proceedings of the National Academy of Sciences of the United States of America 83(12) (1986) 4514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Myers CE, Mimnaugh EG, Yeh GC, Sinha BK, Biochemical Mechanisms of Tumor Cell Kill by the Anthracyclines, in: Lown JW (Ed.), Anthracycline and Anthracenedione-based Anticancer Agents, Elsevier, New York, 1988, pp. 527–569. [Google Scholar]
- [27].Sinha BK, Mimnaugh EG, Free radicals and anticancer drug resistance: oxygen free radicals in the mechanisms of drug cytotoxicity and resistance by certain tumors, Free radical biology & medicine 8(6) (1990) 567–81. [DOI] [PubMed] [Google Scholar]
- [28].Gewirtz DA, A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin, Biochemical pharmacology 57(7) (1999) 727–41. [DOI] [PubMed] [Google Scholar]
- [29].Keizer HG, Pinedo HM, Schuurhuis GJ, Joenje H, Doxorubicin (adriamycin): a critical review of free radical-dependent mechanisms of cytotoxicity, Pharmacology & therapeutics 47(2) (1990) 219–31. [DOI] [PubMed] [Google Scholar]
- [30].Ubezio P, Civoli F, Flow cytometric detection of hydrogen peroxide production induced by doxorubicin in cancer cells, Free radical biology & medicine 16(4) (1994) 509–16. [DOI] [PubMed] [Google Scholar]
- [31].Kramer RA, Zakher J, Kim G, Role of the glutathione redox cycle in acquired and de novo multidrug resistance, Science 241(4866) (1988) 694–7. [DOI] [PubMed] [Google Scholar]
- [32].Dusre L, Mimnaugh EG, Myers CE, Sinha BK, Potentiation of doxorubicin cytotoxicity by buthionine sulfoximine in multidrug-resistant human breast tumor cells, Cancer research 49(3) (1989) 511–5. [PubMed] [Google Scholar]
- [33].Batist G, Tulpule A, Sinha BK, Katki AG, Myers CE, Cowan KH, Overexpression of a novel anionic glutathione transferase in multidrug-resistant human breast cancer cells, The Journal of biological chemistry 261(33) (1986) 15544–9. [PubMed] [Google Scholar]
- [34].Mimnaugh EG, Fairchild CR, Fruehauf JP, Sinha BK, Biochemical and pharmacological characterization of MCF-7 drug-sensitive and AdrR multidrug-resistant human breast tumor xenografts in athymic nude mice, Biochemical pharmacology 42(2) (1991) 391–402. [DOI] [PubMed] [Google Scholar]
- [35].Benchekroun MN, Sinha BK, Robert J, Doxorubicin-induced oxygen free radical formation in sensitive and doxorubicin-resistant variants of rat glioblastoma cell lines [corrected and republished erratum originally printed in FEBS Lett 1993 May 17;322(3):295–8], FEBS letters 326(1–3) (1993) 302–5. [DOI] [PubMed] [Google Scholar]
- [36].Cowan KH, Batist G, Tulpule A, Sinha BK, Myers CE, Similar biochemical changes associated with multidrug resistance in human breast cancer cells and carcinogen-induced resistance to xenobiotics in rats, Proceedings of the National Academy of Sciences of the United States of America 83(24) (1986) 9328–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Oberley LW, Mechanism of the tumor suppressive effect of MnSOD overexpression, Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 59(4) (2005) 143–8. [DOI] [PubMed] [Google Scholar]
- [38].Weydert CJ, Waugh TA, Ritchie JM, Iyer KS, Smith JL, Li L, Spitz DR, Oberley LW, Overexpression of manganese or copper-zinc superoxide dismutase inhibits breast cancer growth, Free radical biology & medicine 41(2) (2006) 226–37. [DOI] [PubMed] [Google Scholar]
- [39].Sun W, Kalen AL, Smith BJ, Cullen JJ, Oberley LW, Enhancing the antitumor activity of adriamycin and ionizing radiation, Cancer research 69(10) (2009) 4294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sun WG, Weydert CJ, Zhang Y, Yu L, Liu J, Spitz DR, Cullen JJ, Oberley LW, Superoxide Enhances the Antitumor Combination of AdMnSOD Plus BCNU in Breast Cancer, Cancers 2(1) (2010) 68–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cohen Y, Epelbaum R, Haim N, McShan D, Zinder O, The value of serum copper levels in non-Hodgkin’s lymphoma, Cancer 53(2) (1984) 296–300. [DOI] [PubMed] [Google Scholar]
- [42].Carpentieri U, Myers J, Thorpe L, Daeschner CW 3rd, Haggard ME, Copper, zinc, and iron in normal and leukemic lymphocytes from children, Cancer research 46(2) (1986) 981–4. [PubMed] [Google Scholar]
- [43].Monteiro HP, Vile GF, Winterbourn CC, Release of iron from ferritin by semiquinone, anthracycline, bipyridyl, and nitroaromatic radicals, Free radical biology & medicine 6(6) (1989) 587–91. [DOI] [PubMed] [Google Scholar]
- [44].Kumar A, Ehrenshaft M, Tokar EJ, Mason RP, Sinha BK, Nitric oxide inhibits topoisomerase II activity and induces resistance to topoisomerase II-poisons in human tumor cells, Biochimica et biophysica acta 1860(7) (2016) 1519–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang AH, Gao YG, Liaw YC, Li YK, Formaldehyde cross-links daunorubicin and DNA efficiently: HPLC and X-ray diffraction studies, Biochemistry 30(16) (1991) 3812–5. [DOI] [PubMed] [Google Scholar]
- [46].Froelich-Ammon SJ, Osheroff N, Topoisomerase poisons: harnessing the dark side of enzyme mechanism, The Journal of biological chemistry 270(37) (1995) 21429–32. [DOI] [PubMed] [Google Scholar]
- [47].Sinha BK, Topoisomerase inhibitors. A review of their therapeutic potential in cancer, Drugs 49(1) (1995) 11–9. [DOI] [PubMed] [Google Scholar]
- [48].Kalyanaraman B, Nemec J, Sinha BK, Characterization of free radicals produced during oxidation of etoposide (VP-16) and its catechol and quinone derivatives. An ESR Study, Biochemistry 28(11) (1989) 4839–46. [DOI] [PubMed] [Google Scholar]
- [49].Haim N, Roman J, Nemec J, Sinha BK, Peroxidative free radical formation and O-demethylation of etoposide(VP-16) and teniposide(VM-26), Biochemical and biophysical research communications 135(1) (1986) 215–20. [DOI] [PubMed] [Google Scholar]
- [50].Haim N, Nemec J, Roman J, Sinha BK, In vitro metabolism of etoposide (VP-16–213) by liver microsomes and irreversible binding of reactive intermediates to microsomal proteins, Biochemical pharmacology 36(4) (1987) 527–36. [DOI] [PubMed] [Google Scholar]
- [51].Usui N, Sinha BK, Tyrosinase-induced free radical formation from VP-16,213: relationship to cytotoxicity, Free radical research communications 10(4–5) (1990) 287–93. [DOI] [PubMed] [Google Scholar]
- [52].van Maanen JM, de Vries J, Pappie D, van den Akker E, Lafleur VM, Retel J, van der Greef J, Pinedo HM, Cytochrome P-450-mediated O-demethylation: a route in the metabolic activation of etoposide (VP-16–213), Cancer research 47(17) (1987) 4658–62. [PubMed] [Google Scholar]
- [53].Sinha BK, Antholine WM, Kalyanaraman B, Eliot HM, Copper ion-dependent oxy-radical mediated DNA damage from dihydroxy derivative of etoposide, Biochim Biophys Acta 1096(1) (1990) 81–3. [DOI] [PubMed] [Google Scholar]
- [54].Katki AG, Kalyanaraman B, Sinha BK, Interactions of the antitumor drug, etoposide, with reduced thiols in vitro and in vivo, Chemico-biological interactions 62(3) (1987) 237–47. [DOI] [PubMed] [Google Scholar]
- [55].Haddock MG, Ames MM, Bonner JA, Assessing the interaction of irradiation with etoposide or idarubicin, Mayo Clinic proceedings 70(11) (1995) 1053–60. [DOI] [PubMed] [Google Scholar]
- [56].Gantchev TG, Brasseur N, van Lier JE, Combination toxicity of etoposide (VP-16) and photosensitisation with a water-soluble aluminium phthalocyanine in K562 human leukaemic cells, British journal of cancer 74(10) (1996) 1570–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Robati M, Holtz D, Dunton CJ, A review of topotecan in combination chemotherapy for advanced cervical cancer, Therapeutics and clinical risk management 4(1) (2008) 213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Akbas SH, Timur M, Ozben T, The effect of quercetin on topotecan cytotoxicity in MCF-7 and MDA-MB 231 human breast cancer cells, The Journal of surgical research 125(1) (2005) 49–55. [DOI] [PubMed] [Google Scholar]
- [59].Kisa U, Caglayan O, Kacmaz M, The effects of topotecan on lipid peroxidation and antioxidant enzyme levels in rabbit liver tissue, Redox report : communications in free radical research 10(2) (2005) 79–82. [DOI] [PubMed] [Google Scholar]
- [60].Timur M, Akbas SH, Ozben T, The effect of Topotecan on oxidative stress in MCF-7 human breast cancer cell line, Acta biochimica Polonica 52(4) (2005) 897–902. [PubMed] [Google Scholar]
- [61].Rapisarda A, Uranchimeg B, Sordet O, Pommier Y, Shoemaker RH, Melillo G, Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implications, Cancer research 64(4) (2004) 1475–82. [DOI] [PubMed] [Google Scholar]
- [62].Puppo M, Battaglia F, Ottaviano C, Delfino S, Ribatti D, Varesio L, Bosco MC, Topotecan inhibits vascular endothelial growth factor production and angiogenic activity induced by hypoxia in human neuroblastoma by targeting hypoxia-inducible factor-1alpha and -2alpha, Molecular cancer therapeutics 7(7) (2008) 1974–84. [DOI] [PubMed] [Google Scholar]
- [63].Daroui P, Desai SD, Li TK, Liu AA, Liu LF, Hydrogen peroxide induces topoisomerase I-mediated DNA damage and cell death, The Journal of biological chemistry 279(15) (2004) 14587–94. [DOI] [PubMed] [Google Scholar]
- [64].Sinha BK, Kumar A, Mason RP, Nitric oxide inhibits ATPase activity and induces resistance to topoisomerase II-poisons in human MCF-7 breast tumor cells, Biochemistry and biophysics reports 10 (2017) 252–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Clement MV, Ramalingam J, Long LH, Halliwell B, The in vitro cytotoxicity of ascorbate depends on the culture medium used to perform the assay and involves hydrogen peroxide, Antioxidants & redox signaling 3(1) (2001) 157–63. [DOI] [PubMed] [Google Scholar]
- [66].Basudhar D, Glynn SA, Greer M, Somasundaram V, No JH, Scheiblin DA, Garrido P, Heinz WF, Ryan AE, Weiss JM, Cheng RYS, Ridnour LA, Lockett SJ, McVicar DW, Ambs S, Wink DA, Coexpression of NOS2 and COX2 accelerates tumor growth and reduces survival in estrogen receptor-negative breast cancer, Proceedings of the National Academy of Sciences of the United States of America 114(49) (2017) 13030–13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bonavida B, Garban H, Nitric oxide-mediated sensitization of resistant tumor cells to apoptosis by chemo-immunotherapeutics, Redox biology 6 (2015) 486–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sinha BK, Mason RP, Biotransformation of Hydrazine Dervatives in the Mechanism of Toxicity, Journal of drug metabolism & toxicology 5(3) (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Dunn DL, Lubet RA, Prough RA, Oxidative metabolism of N-isopropyl-alpha-(2-methylhydrazino)-p-toluamide hydrochloride (procarbazine) by rat liver microsomes, Cancer research 39(11) (1979) 4555–63. [PubMed] [Google Scholar]
- [70].Prough RA, Brown MI, Dannan GA, Guengerich FP, Major isozymes of rat liver microsomal cytochrome P-450 involved in the N-oxidation of N-isopropyl-alpha-(2-methylazo)-p-toluamide, the azo derivative of procarbazine, Cancer research 44(2) (1984) 543–8. [PubMed] [Google Scholar]
- [71].Erikson JM, Tweedie DJ, Ducore JM, Prough RA, Cytotoxicity and DNA damage caused by the azoxy metabolites of procarbazine in L1210 tumor cells, Cancer research 49(1) (1989) 127–33. [PubMed] [Google Scholar]
- [72].Sinha BK, Metabolic activation of procarbazine. Evidence for carbon-centered free-radical intermediates, Biochemical pharmacology 33(17) (1984) 2777–81. [DOI] [PubMed] [Google Scholar]
- [73].Maragos CM, Wang JM, Hrabie JA, Oppenheim JJ, Keefer LK, Nitric oxide/nucleophile complexes inhibit the in vitro proliferation of A375 melanoma cells via nitric oxide release, Cancer research 53(3) (1993) 564–8. [PubMed] [Google Scholar]
- [74].Wink DA, Cook JA, Christodoulou D, Krishna MC, Pacelli R, Kim S, DeGraff W, Gamson J, Vodovotz Y, Russo A, Mitchell JB, Nitric oxide and some nitric oxide donor compounds enhance the cytotoxicity of cisplatin, Nitric oxide : biology and chemistry / official journal of the Nitric Oxide Society 1(1) (1997) 88–94. [DOI] [PubMed] [Google Scholar]
- [75].Bratasz A, Selvendiran K, Wasowicz T, Bobko A, Khramtsov VV, Ignarro LJ, Kuppusamy P, NCX-4040, a nitric oxide-releasing aspirin, sensitizes drug-resistant human ovarian xenograft tumors to cisplatin by depletion of cellular thiols, Journal of translational medicine 6 (2008) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Hirst D, Robson T, Nitric oxide in cancer therapeutics: interaction with cytotoxic chemotherapy, Current pharmaceutical design 16(4) (2010) 411–20. [DOI] [PubMed] [Google Scholar]
- [77].Huerta S, Chilka S, Bonavida B, Nitric oxide donors: novel cancer therapeutics (review), International journal of oncology 33(5) (2008) 909–27. [PubMed] [Google Scholar]
- [78].Huerta S, Baay-Guzman G, Gonzalez-Bonilla CR, Livingston EH, Huerta-Yepez S, Bonavida B, In vitro and in vivo sensitization of SW620 metastatic colon cancer cells to CDDP-induced apoptosis by the nitric oxide donor DETANONOate: Involvement of AIF, Nitric oxide : biology and chemistry / official journal of the Nitric Oxide Society 20(3) (2009) 182–94. [DOI] [PubMed] [Google Scholar]
- [79].Bonavida B, Baritaki S, Huerta-Yepez S, Vega MI, Chatterjee D, Yeung K, Novel therapeutic applications of nitric oxide donors in cancer: roles in chemo- and immunosensitization to apoptosis and inhibition of metastases, Nitric oxide : biology and chemistry / official journal of the Nitric Oxide Society 19(2) (2008) 152–7. [DOI] [PubMed] [Google Scholar]
- [80].Adams DJ, Levesque MC, Weinberg JB, Smith KL, Flowers JL, Moore J, Colvin OM, Silber R, Nitric oxide enhancement of fludarabine cytotoxicity for B-CLL lymphocytes, Leukemia 15(12) (2001) 1852–9. [DOI] [PubMed] [Google Scholar]
- [81].Matthews NE, Adams MA, Maxwell LR, Gofton TE, Graham CH, Nitric oxide-mediated regulation of chemosensitivity in cancer cells, Journal of the National Cancer Institute 93(24) (2001) 1879–85. [DOI] [PubMed] [Google Scholar]
- [82].Frederiksen LJ, Siemens DR, Heaton JP, Maxwell LR, Adams MA, Graham CH, Hypoxia induced resistance to doxorubicin in prostate cancer cells is inhibited by low concentrations of glyceryl trinitrate, The Journal of urology 170(3) (2003) 1003–7. [DOI] [PubMed] [Google Scholar]
- [83].Sinha BK, Bhattacharjee S, Chatterjee S, Jiang J, Motten AG, Kumar A, Espey MG, Mason RP, Role of nitric oxide in the chemistry and anticancer activity of etoposide (VP-16,213), Chemical research in toxicology 26(3) (2013) 379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Huang Z, Fu J, Zhang Y, Nitric Oxide Donor-Based Cancer Therapy: Advances and Prospects, Journal of medicinal chemistry 60(18) (2017) 7617–7635. [DOI] [PubMed] [Google Scholar]
- [85].Sinha BK, Nitric oxide: Friend or foe in cancer chemotherapy and drug reistance: a perspective, Journal of cancer science & therapy 8(10) (2016) 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Muscara MN, Wallace JL, Nitric Oxide. V. therapeutic potential of nitric oxide donors and inhibitors, The American journal of physiology 276(6 Pt 1) (1999) G1313–6. [DOI] [PubMed] [Google Scholar]
- [87].Reynolds MM, Witzeling SD, Damodaran VB, Medeiros TN, Knodle RD, Edwards MA, Lookian PP, Brown MA, Applications for nitric oxide in halting proliferation of tumor cells, Biochemical and biophysical research communications 431(4) (2013) 647–51. [DOI] [PubMed] [Google Scholar]
- [88].Saavedra JE, Shami PJ, Wang LY, Davies KM, Booth MN, Citro ML, Keefer LK, Esterase-sensitive nitric oxide donors of the diazeniumdiolate family: in vitro antileukemic activity, Journal of medicinal chemistry 43(2) (2000) 261–9. [DOI] [PubMed] [Google Scholar]
- [89].Shami PJ, Saavedra JE, Wang LY, Bonifant CL, Diwan BA, Singh SV, Gu Y, Fox SD, Buzard GS, Citro ML, Waterhouse DJ, Davies KM, Ji X, Keefer LK, JS-K, a glutathione/glutathione S-transferase-activated nitric oxide donor of the diazeniumdiolate class with potent antineoplastic activity, Molecular cancer therapeutics 2(4) (2003) 409–17. [PubMed] [Google Scholar]
- [90].Shami PJ, Maciag AE, Eddington JK, Udupi V, Kosak KM, Saavedra JE, Keefer LK, JS-K, an arylating nitric oxide (NO) donor, has synergistic anti-leukemic activity with cytarabine (ARA-C), Leukemia research 33(11) (2009) 1525–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Weyerbrock A, Baumer B, Papazoglou A, Growth inhibition and chemosensitization of exogenous nitric oxide released from NONOates in glioma cells in vitro, Journal of neurosurgery 110(1) (2009) 128–36. [DOI] [PubMed] [Google Scholar]
- [92].Maciag AE, Nandurdikar RS, Hong SY, Chakrapani H, Diwan B, Morris NL, Shami PJ, Shiao YH, Anderson LM, Keefer LK, Saavedra JE, Activation of the c-Jun N-terminal kinase/activating transcription factor 3 (ATF3) pathway characterizes effective arylated diazeniumdiolate-based nitric oxide-releasing anticancer prodrugs, Journal of medicinal chemistry 54(22) (2011) 7751–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kiziltepe T, Hideshima T, Ishitsuka K, Ocio EM, Raje N, Catley L, Li CQ, Trudel LJ, Yasui H, Vallet S, Kutok JL, Chauhan D, Mitsiades CS, Saavedra JE, Wogan GN, Keefer LK, Shami PJ, Anderson KC, JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells, Blood 110(2) (2007) 709–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Chanvorachote P, Nimmannit U, Stehlik C, Wang L, Jiang BH, Ongpipatanakul B, Rojanasakul Y, Nitric oxide regulates cell sensitivity to cisplatin-induced apoptosis through S-nitrosylation and inhibition of Bcl-2 ubiquitination, Cancer research 66(12) (2006) 6353–60. [DOI] [PubMed] [Google Scholar]
- [95].Godoy LC, Anderson CT, Chowdhury R, Trudel LJ, Wogan GN, Endogenously produced nitric oxide mitigates sensitivity of melanoma cells to cisplatin, Proceedings of the National Academy of Sciences of the United States of America 109(50) (2012) 20373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Sharma NK, Kumar A, Kumari A, Tokar EJ, Waalkes MP, Bortner CD, Williams J, Ehrenshaft M, Mason RP, Sinha BK, Nitric Oxide Down-Regulates Topoisomerase I and Induces Camptothecin Resistance in Human Breast MCF-7 Tumor Cells, PloS one 10(11) (2015) e0141897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Glynn SA, Boersma BJ, Dorsey TH, Yi M, Yfantis HG, Ridnour LA, Martin DN, Switzer CH, Hudson RS, Wink DA, Lee DH, Stephens RM, Ambs S, Increased NOS2 predicts poor survival in estrogen receptor-negative breast cancer patients, The Journal of clinical investigation 120(11) (2010) 3843–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DS, Baker SW, Khodarev N, Su AW, Shaikh AY, Roach P, Kreike B, Roizman B, Bergh J, Pawitan Y, van de Vijver MJ, Minn AJ, An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer, Proceedings of the National Academy of Sciences of the United States of America 105(47) (2008) 18490–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Chapat C, Kolytcheff C, Le Romancer M, Auboeuf D, De La Grange P, Chettab K, Sentis S, Corbo L, hCAF1/CNOT7 regulates interferon signalling by targeting STAT1, The EMBO journal 32(5) (2013) 688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Riganti C, Miraglia E, Viarisio D, Costamagna C, Pescarmona G, Ghigo D, Bosia A, Nitric oxide reverts the resistance to doxorubicin in human colon cancer cells by inhibiting the drug efflux, Cancer research 65(2) (2005) 516–25. [PubMed] [Google Scholar]
- [101].De Boo S, Kopecka J, Brusa D, Gazzano E, Matera L, Ghigo D, Bosia A, Riganti C, iNOS activity is necessary for the cytotoxic and immunogenic effects of doxorubicin in human colon cancer cells, Molecular cancer 8 (2009) 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Curta JC, de Moraes AC, Licinio MA, Costa A, Santos-Silva MC, Effect of nitric oxide on the daunorubicin efflux mechanism in K562 cells, Cell biology international 36(6) (2012) 529–35. [DOI] [PubMed] [Google Scholar]
- [103].Dalton WS, Scheper RJ, Lung resistance-related protein: determining its role in multidrug resistance, Journal of the National Cancer Institute 91(19) (1999) 1604–5. [DOI] [PubMed] [Google Scholar]
- [104].Kitazono M, Okumura H, Ikeda R, Sumizawa T, Furukawa T, Nagayama S, Seto K, Aikou T, Akiyama S, Reversal of LRP-associated drug resistance in colon carcinoma SW-620 cells, International journal of cancer 91(1) (2001) 126–31. [DOI] [PubMed] [Google Scholar]
- [105].Sinha BK, Bortner CD, Mason RP, Cannon RE, Nitric oxide reverses drug resistance by inhibiting ATPase activity of p-glycoprotein in human multi-drug resistant cancer cells, Biochimica et biophysica acta. General subjects 1862(12) (2018) 2806–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Sinha BK, Perera L, Cannon RE, Reversal of drug resistance by JS-K and nitric oxide in ABCB1- and ABCG2-expressing multi-drug resistant human tumor cells, Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 120 (2019) 109468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Miller MR, Megson IL, Recent developments in nitric oxide donor drugs, British journal of pharmacology 151(3) (2007) 305–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Yasuda H, Yamaya M, Nakayama K, Sasaki T, Ebihara S, Kanda A, Asada M, Inoue D, Suzuki T, Okazaki T, Takahashi H, Yoshida M, Kaneta T, Ishizawa K, Yamanda S, Tomita N, Yamasaki M, Kikuchi A, Kubo H, Sasaki H, Randomized phase II trial comparing nitroglycerin plus vinorelbine and cisplatin with vinorelbine and cisplatin alone in previously untreated stage IIIB/IV non-small-cell lung cancer, Journal of clinical oncology : official journal of the American Society of Clinical Oncology 24(4) (2006) 688–94. [DOI] [PubMed] [Google Scholar]
- [109].Siemens DR, Heaton JP, Adams MA, Kawakami J, Graham CH, Phase II study of nitric oxide donor for men with increasing prostate-specific antigen level after surgery or radiotherapy for prostate cancer, Urology 74(4) (2009) 878–83. [DOI] [PubMed] [Google Scholar]
- [110].Sun F, Wang Y, Luo X, Ma Z, Xu Y, Zhang X, Lv T, Zhang Y, Wang M, Huang Z, Zhang J, Anti-CD24 Antibody-Nitric Oxide Conjugate Selectively and Potently Suppresses Hepatic Carcinoma, Cancer research 79(13) (2019) 3395–3405. [DOI] [PubMed] [Google Scholar]