

Abstract
Background:
The cell culture technique has become a routine and a popular method for its wide applications in the field of cell biology and biotechnology and in medical research. Isolation of primary cells over the cancer cells is an essential component of cell culture technology as they are the reliable source to understand normal physiological, morphological and molecular process of human cells. As fibroblasts are the prominent cells of the connective tissue of oral mucosa, many disease entities and histogenesis are linked to fibroblasts. Culture of oral fibroblast cells helps the oral biologists and researchers to study the morphological and molecular process in the oral diseases.
Aim:
The aim of our experiment is to isolate and culture the human buccal mucosal fibroblast cells from healthy individuals using a combination of explant–enzymatic method and characterization of the cells by short tandem repeat (STR) profiling.
Materials and Methods:
The tissue samples were collected from healthy individuals undergoing routine impacted third molar extraction. A combination of explant–enzymatic technique was used for the isolation from the tissue samples. The cells were further subcultured, maintained and stored as per the standard protocols. Thus, to confirm the oral fibroblasts of human origin and its uniqueness, they were characterized using STR profiling.
Results and Conclusion:
Using the combination technique, we were successful in isolating the cells at a faster rate by detachment of cells on day 3 and confluency on day 10. The morphological assessment and STR profiling further confirmed that the isolated cell lines resemble human fibroblast cells.
Keywords: Cell lines primary culture, enzymatic technique, explant culture, human buccal mucosal fibroblast, short tandem repeats
INTRODUCTION
In recent years, the tissue culture technology has a great influence in human society. Cell culture technique has become popular and a widely used method in various fields like in understanding cell biology, medical research, in the field of pharmacology to test efficacy and toxicity of new drugs, manufacture of vaccines and also in assisted reproductive technology. Isolation of the primary cell lines and their primary cultures allows researchers to study the morphological, cellular and functional behavior of the cells.[1]
Developed and established cell lines have disadvantage because there are chances to carry several copies of mutations. This could be due to cross contamination or inappropriate exchange of cell lines from laboratories against ethical guidelines. Although primary cell line has finite life span, it is always preferred to study the process of cell cycle, apoptosis and cell repair in a controlled condition to understand the original characteristics and functions of the cells for further clinical translation. Thus, it is the need of the hour to establish standardized protocol for primary culture in the field of cell biology.[2]
There are many established standard protocols for the primary culture of systemic normal and neoplastic cells. Availability of standard protocols for the isolation of normal primary cell lines of human cells such as keratinocytes, skin fibroblasts, epithelial cells and embryonic cells has been well documented. Surprisingly, there is very sparse literature regarding the primary culture of human buccal mucosal fibroblasts.[3]
Fibroblasts are the important cells in the connective tissue of the oral mucosa. They play an important role in the synthesis of structural proteins and extracellular matrix, help in the differentiation of the adjacent epithelium by production of epithelial growth factors and regulate the inflammatory process by secreting the chemokines and cytokines. Hence, to further study the mechanisms of cell functioning and the morphology, a need for the primary culture of human oral fibroblast has been a concern. Primary cultures of fibroblasts allow researchers to obtain cells that can be grown in controlled in vitro conditions by retaining their original characteristics and functions.[4]
Establishing a primary cell lines of oral fibroblast can help the oral biologists in understanding the proper structural characteristics, growth rate and morphological alteration to check the toxicity level with carcinogens and also to assess the response of specific novel treatment.[5]
One of the best examples of alteration of fibroblast leading to irreversible disease is oral submucous fibrosis. There are several in vitro studies on fibroblasts to assess the effect of arecoline toxicity. A study done by Jheng et al. in 1999 found that there were morphological alterations when treated with different concentrations of arecoline. 6 A similar study was carried out by Abhishek et al., where they concluded that different concentrations of arecoline showed a decrease in the cell count of the gingival fibroblasts. Hence, developing accurate primary cell line of oral mucosal fibroblasts can further help in validating and performing several in vitro studies to link with novel therapeutic targets.[7,8]
Over the past decade, two tissue culture techniques widely used are enzymatic and direct explant techniques described by Bernice in 1994 and Kedjarune et al. in 2001. Daniels et al. in 1996 described a detailed protocol for enzymatic technique where they used an enzyme for the isolation of the cells from tissue sample using trypsin or diaspase enzyme. Whereas in the direct explant technique, the tissue samples are processed without using enzyme which involves fewer steps compared with the enzymatic technique.[9,10] Moreover, it has been observed that there are advantages and disadvantages mentioned about both the techniques. To overcome these, we decided to develop primary cell lines by a combination of both the techniques.[11,12]
Hence, the aim of our study was to establish a detailed protocol in developing a standard primary cell line of human buccal mucosal fibroblasts (HBMFs) by a combination of explant–enzymatic technique, study their morphological and growth characteristics and further characterize the cell lines by DNA/short tandem repeat (STR) profiling.[13]
MATERIALS AND METHODS
Ethical consideration
Ethical clearance for the study was obtained from the Ethics Committee of KLE Academy of Higher Education and Research (KLEU/Ethic/2016-2017/D-227). Informed consent was also obtained from all the patients involved in our study. Confidentiality of the documentation of the detailed case history, clinical details and personnel information of the patients was maintained and has not been revealed even in this paper.
Tissue sample collection
Tissue samples for primary HBMFs were obtained from 10 healthy human subjects (age ranging from 18 to 55 years), undergoing routine extraction of impacted third molar at the Department of Oral and Maxillofacial Surgery, KLE V K Institute of Dental Sciences, Belagavi. The tissues were carried from the oral surgery department to the cell culture laboratory in an Eppendorf tube containing the cell culture media [Figure 1a] which also acted as the transport media (Dulbecco's Modified Eagle's Medium, Gibco [pH 7.2]) and were stored at 4°C before processing.
Figure 1.

Photomicrograph of images (a-d) depicting steps involved in the tissue processing like collection of sample in sterilized Eppendorf tube containing dish containing the culture media (a), process of mincing of tissue sample in the laminar air flow chamber with proper arrangement of sterilized instruments (b), tissue samples are minced or processed in a glass Petri dish using a BP blade No. 22 into small pieces of 1 mm × 1 mm in size (c and d)
The cell culture technique
The tissue processing was carried out in a biosafety cabinet and all the sterilization protocols were maintained before the experiments as mentioned in Table 1. The tissue specimens obtained were first washed and disinfected in phosphate-buffered saline (pH 7.3–7.4) solution for 2–3 min and then washed in culture media. Tissues were then minced into 1 mm × 1 mm pieces with the help of sterile BP blade No. 22 in a sterilized Petri dish containing the culture media (DMEM) [Figure 1b-e]. Trypsin (0.25%) containing 0.02% ethylenediaminetetraacetic acid (EDTA) (HiMedia) was added for the separation of the cells, which further helps in the passaging of the cells. The minced tissue was then centrifuged at 4000 rpm for 3 min (Eppendorf 5418R) resulting in the formation of pellet to which the fresh media was added and the sediment was seeded in a 6-well culture plate. The culture plate with the minced tissue was flooded with culture media containing DMEM media pH 7.2 supplemented with fetal calf serum (FCS). To prevent the growth of microorganisms, 100 U/ml penicillin, 100 μg/ml streptomycin and 1% amphotericin B (Gibco-BRL, New York, USA) were added to culture media. The culture plate was then incubated at 37°C in a humidified chamber of 95% air and 5% CO2 incubator New Brunswick - Eppendorf company, German (Bangalore distributers).
Table 1.
Sterilization protocol to prevent contamination
| Procedure | Measures taken to prevent contamination |
|---|---|
| Sterilization and maintenance of culture laboratory | Before any procedure, the entire cell culture laboratory was properly cleaned with disinfectants The walls, floors, outer surface of culture hood, refrigerators, CO2 incubators and other equipment were thoroughly cleaned with disinfectant After cleaning the laboratory, UV light for the entire laboratory was kept on for 1 h Fumigation of laboratory was done at the every 10 days |
| Sterilization of the laminar air flow hood | UV light was switched on 15-30 min before and after the experiment The laminar chamber was cleaned with 70% ethanol as a disinfectant prior and after the experiment The chemicals and instruments were constantly wiped with 70% ethanol before placing them in the laminar air flow chamber |
| Sterilization of CO2 chamber | Fresh distilled water was regularly replaced every 15 days with a pinch of cupric sulfate dissolved in it The inner surface of incubator was cleaned with 70% ethanol every alternate day to prevent contamination of the flasks and culture plates All the flasks and culture plates were thoroughly wiped with 70% ethanol before placing them in incubator |
| Sterilization of accessory instruments | Instruments such as BP blade holder, kidney trays, tissue holding forceps and the Petri plates were sterilized using autoclave before the tissue processing at a temperature of 121°C for 30 min at 15 psi pressure |
| Procurement and transfer of the tissue sample to culture laboratory | The tissue samples were procured from patients undergoing impaction of third molars under the guidance of oral surgeon with aseptic precautions taken while surgery The samples were collected and transferred in the sterilized vials containing DMEM media to the culture laboratories |
| Tissue processing | Sterilization of the laminar air flow chamber as mentioned above The optimum operating area should be maintained The instruments before the tissue processing should be neatly organized Use of laboratory coats, gloves, masks and head caps was used to prevent contamination The tissue is first washed with PBS 3-4 times to remove the blood and any other contamination Mincing of the tissue sample was done using preautoclaved and sterilized instruments Cleansing of the operator hands was done with 70% ethanol often during the tissue processing helps avoid contamination from external factors |
| Subculturing | Once the primary cell lines are established, the cultures were maintained with complete DMEM media containing 1%-2% of antibiotics to avoid contamination The tissue culture plates were observed frequently for any contamination, if any contamination was found the plates were discarded immediately |
| Maintaining and preservation of cell lines | Sterilized Eppendorf tubes were used for the preservation of cell lines The cell lines to be preserved were transferred to Eppendorf tubes containing 90% FBS and 10% DMSO under sterilized aseptic condition The Eppendorf tubes with the cell lines and preservation media were thoroughly sealed with paraffin wax with cell name and coding |
UV: Ultraviolet, DMEM: Dulbecco’s Modified Eagle’s Medium, PBS: Phosphate-buffered saline, FBS: Fetal bovine serum, DMSO: Dimethyl sulfoxide, BP: Bard-Parker Company
Monitoring of primary cell cultures and contamination
Daily observation was done to check for any contamination and explant dislodgment and the overall radial migration of primary cells from the explants. Constant monitoring for any microbial or fungal contamination was done under an inverted microscope. Replacement of old medium by new complete media was also done based on the color change of media. If microbial contamination was detected in any dish or flask, the entire content was immediately discarded. In our experiment, no contamination was found with six tissue samples; however, four samples were detected with contamination. On day 3 of culture, the migration of primary cells was seen, and a monolayer of cells was observed. The primary cell lines were moved from the culture plate to a culture flask. Tissue fragments were covered with culture medium containing fetal calf serum and antibiotics, and the process of culture was continued until the primary outgrowing cells reached around 70%–80% confluency in the culture flasks.
Establishing secondary cultures/passaging
During the subculturing or passaging, the cells morphologically spindle in shape were identified as fibroblasts. On reaching confluency, the cells surrounding the explant tissue further expanded. The medium was poured out of the culture flasks and the cell surfaces washed gently with phosphate-buffered saline (PBS) three times. The cells were removed from the cultures by trypsinization with 0.25% trypsin and 0.05% EDTA. After 2–3 min, the cells attained round shape and began to detach from the plastic surface of the culture flask. The cell suspension along with media were then transferred to a 1.5-ml Eppendorf tube and subjected for centrifugation. Fresh complete media was added for culture flasks to expand cell numbers in a new T25 culture flask. Cultures were grown at 37°C in a humidified atmosphere containing 5% CO2. The culture medium was changed once in every 48 h. The 2nd–4th passages of cultures of these cells were frozen for further STR profiling, as the cells were in lag phase of growth.
Cryopreservation of fibroblasts
Cryopreservation of the buccal mucosal fibroblasts was performed according to the standard protocol of cryopreservation of the cell lines using dimethyl sulfoxide (DMSO), which permits long-term storage of cells in liquid nitrogen. Once the cells reached the confluency, the cells of the 4th–5th passage are selected and routine trypsinization of cells is done with 0.25% of trypsin. The cells were subjected for centrifugation at 4000 rpm for 3 min. The cell pellet was resuspended in freezing medium consisting of 90% fetal bovine serum (FBS) and 10% DMSO. Cell suspensions were aliquoted into cryogenic storage vials and frozen subsequently at 2°C–4°C for 1 h, −20°C for 1 h and −80°C in a deep freezer overnight.
Characterization of the primary cell lines of human buccal mucosal fibroblast
Morphological characterization of the primary cell lines
Once the primary cell lines were established, the cells were observed under an inverted microscope on day 1–day 8 and the morphological changes were noted. The morphological variation of the cells was recorded as F1 which are spindle-shaped cells, F2 which are epithelioid-shaped cells and F3 as stellate-shaped cells.[3]
Characterization of cell lines by short tandem repeat profiling
The cells were characterized for STR/DNA profiling from DNA Forensics Laboratory, New Delhi. The cells from the confluent flask with passage 3rd or 4th were selected for the DNA profiling. The cell pellet was prepared according to the standard protocol provided by the laboratory and transferred to the laboratory. Before sending the cell lines to the laboratory, the cells were named and coded for the convenience and future use as KLED-BF18. The cell pellet was then properly sealed and transferred to the DNA Forensics Laboratory for further processing. Once the primary cell lines were established, the cells were observed under an inverted microscope on day 1–day 8 and the morphological changes were noted. The morphological variation of the cells was recorded as F1 which are spindle-shaped cells, F2 which are epithelioid-shaped cells and F3 as stellate-shaped cells.[3]
RESULTS AND OBSERVATION
The results showed that six out of ten primary cultures of oral fibroblast cells done by a combination of explant–enzymatic technique were successful. In our experiment, we were able to isolate the fibroblast cells within 3–5 days and 70%–80% of confluency reached around 8–10 days of culture. The remaining tissue samples or the cells were discarded due to contamination.
First day: Primary culture
The culture showed mixture of cells which include round/spherical clumps of cells, red blood cell (RBC) and white blood cell (WBC). The primary culture of the tissue sample also showed some areas of tissue degeneration with fibrin and round cells [Figure 2a]. On observation, the culture plates showed the absence of contamination. The culture was then kept for incubation for 24 h overnight.
Figure 2.
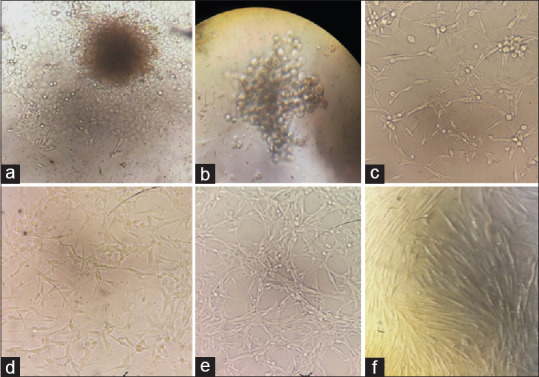
Photomicrograph of images showing the isolation and primary outgrowth of the cells from the explant tissue. Cells migrating and outgrowing from the explant tissue on day 2 (a). Round clump of cells seen indicating the outgrowth of the explant tissue and multiplication of cell lines on day 3 (b). Small spindle-shaped fibroblast cells (F1) with mixture of spherical dividing cells (c). Cultures showing majority of F1-shaped fibroblasts with few F2- and F3-shaped cells (d). The fibroblasts reach the confluency around day 10 with mostly F1-shaped fibroblast cells (e and f)
Second day: Primary culture
The culture plates showed the same round cells with a slight decrease in the number of RBC and WBC. The spherical cells were seen detaching and an outgrowth of the primary tissue was observed [Figure 2b]. Primary cultures were maintained with the addition of fresh media, FBS and antibiotics to avoid and control the contamination.
Third–fifth day
The cultures showed a series of changes. The fibroblasts were attached to the base of the culture wells and small spindle-shaped cells (F1 type) were observed on the periphery of the culture plates [Figure 2c]. A mixture of cells were observed showing from round to spherical cells; small extension of the cells was observed, resembling that spindleshaped cells (F1) were noted.[3] There was absence of contamination observation of the culture.
Sixth–tenth day: Secondary culture/passaging
The cultures showed majority of long spindle-shaped cells (F1) extending and covering all the surfaces of the wells. The cell population also showed stellate-shaped cells (F3) with fibroblastic extensions, and small round dividing primary cells were noted [Figure 2d]. The buccal fibroblast cell lines reached around 60%–70% confluency on day 8. On day 10, the cell lines were subcultured and passaging was done and labeled as T1 and T2 flask. The cells were then maintained and incubated with constant monitoring for any contamination.
Eleventh–fourteenth day: Secondary culture
On observation under an inverted microscope, subculture T1 flask showed a monolayer of long spindle-shaped cells (F1 type), and T2 flask showed the cells with confluency of 70% with some areas showing small dividing spherical cells [Figure 2e and f]. The subcultured flasks were then incubated with appropriate media, FCS and antibiotics for the further growth and storage of the cell lines.
Growth curve assay
The growth curve assay was carried to assess the cell growth pattern and proliferation of the fibroblast cell lines. The results of the growth curve assay are mentioned in Table 2, and the graph was plotted [Figure 3]. Our experiment started by seeding around 7000 cells per well in a 12-well culture plate; the cells were allowed to grow and observed using an inverted microscope under ×40 magnification and counted daily for 5 days. The cell count was done every day using trypan blue count assay and an average was recorded at the end of the week. The cells in the last well were recorded as 182,000 cells per well. The experiment shows an increase in the number of cells and the growth pattern of cell lines from day 0 to day 5.
Table 2.
Growth curve assay
| Number of days | Number of cells/well |
|---|---|
| Day 0 | 7000 |
| Day 1 | 22,000 |
| Day 2 | 156,000 |
| Day 3 | 171,000 |
| Day 4 | 172,000 |
| Day 5 | 182,000 |
Figure 3.
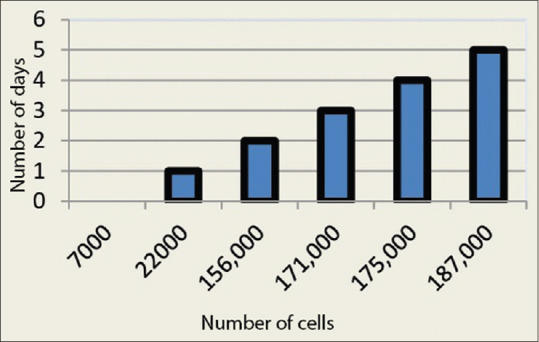
Linear bar graph describing the growth characteristics of primary cell lines of human buccal mucosal fibroblast where the number of cells dividing (X) was plotted against the number of days (Y)
Results of characterization of cell lines
Morphological assessment of primary cell lines of human buccal mucosal fibroblast
The cells were monitored for the morphological changes from day 1 to 14. There were no cell changes observed on day 1 and 2 as they retained round to spherical shape. On day 3–5, the cells showed cellular extensions, resembling spindle-shaped morphology of fibroblast cells. The cultures showed the cells resembling mature spindle-shaped fibroblast (F1) [Figure 4a] on day 7–10. The epithelioid (F2)-shaped cells were observed at around day 10 with mixture of very less number of stellate (F3)-shaped cells [Figure 4b].
Figure 4.
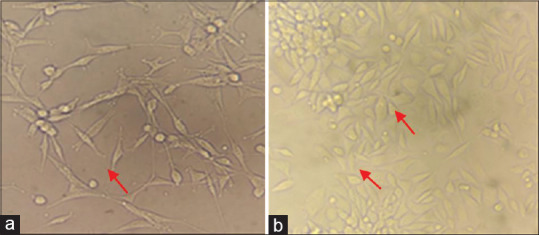
Morphological changes of fibroblasts noted during culture. Predominant spindle-shaped fibroblasts (F1) (a). Mixture of cells with few number of both stellate-shaped (F3) and epithelioid-shaped fibroblasts (F2) (b)
Results of short tandem repeat/DNA profiling for cell line authentication report
In our study, we observed that the STR profiles of DNA isolated from the cell pellet of primary cell lines of HBMF had some unique features as compared to other standard cell lines considered. Table 3 describes the loci of the DNA of our sample of the cell line KLED-BF18 comparing it with standard fibroblast cell lines. Table 4 shows the locus point that are matching with the standard fibroblast cell lines such as CRL 7201, CRL-7226, CRL7553 and CRL 7554, whereas the cells are showing less resemblance with loci of cells of epithelial origin such as JCRB0178.0, JCRB0178.1 and JCRB0234.0. Thus, the overall results of the STR profiling of our sample of the cell lines state that the established cell line is of human origin, with unique features not resembling to any previous human genes and showing the characteristics of cell of fibroblastic origin.
Table 3.
STR profile report table
| Test result for submitted sample | ||
|---|---|---|
| Loci | Query profile (KLED-BF18) | |
| TH01 | 6 | 12 |
| D5S818 | 11 | 13 |
| D13S317 | 9 | 11 |
| D7S820 | 10 | 12 |
| D16S539 | 10 | 12 |
| CSF1PO | 12 | |
| VWA | 14 | 16 |
| TPOX | 9 | 11 |
| Amelogein | X | X |
Table 4.
Comparative table of STR profile data
| EV | CELL NUMBER | CELL NAME | D5S818 | D13S317 | D7S820 | LOCUS NAMES | TH01 | AM | TPOX | CSF1PO | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| D16S539 | VWA | ||||||||||
| Query (Your Cell) | 11,13 | 9,11 | 10,12 | 10,12 | 14,16 | 6,8 | X, X | 9,11 | 12,12 | ||
| 0.67 (24/36) | 675 | LS | 12,13 | 9,11 | 8,10 | 10,11 | 14,16 | 6,7 | X, X | 8,8 | 12,12 |
| 0.67 (24/36) | CRL-7201 | HS235.SK | 11,12 | 11,13 | 10,13 | 10,12 | 14,16 | 6,9 | X, X | 9,11 | 11,14 |
| 0.67 (24/36) | CRL-7226 | HS280.T | 11,13 | 9,11 | 11,12 | 10,11 | 16,17 | 7,9.3 | X, X | 8,11 | 10,12 |
| 0.67 (24/36) | CRL-7553 | HS821.SK | 12,13 | 9,11 | 10,12 | 10,12 | 14,15 | 7,9.3 | X, X | 8,11 | 12,13 |
| 0.67 (24/36) | CRL-7554 | HS821.T | 12,13 | 9,11 | 10,12 | 10,12 | 14,15 | 6,9.3 | X, X | 8,11 | 12,13 |
| 0.67 (24/36) | JCRB0178.0 | KP-3 | 13,13 | 9,11 | 10,12 | 9,13 | 17,18 | 6,9.3 | X, X | 9,11 | 12,12 |
| 0.67 (24/36) | JCRB0178.1 | KP-3L | 13,13 | 9,11 | 10,12 | 9,13 | 17,18 | 6,9.3 | X, X | 9,11 | 12,12 |
| 0.67 (24/36) | JCRB0234.0 | TYK-NU | 12,13 | 10,11 | 10,10 | 9,10 | 14,16 | 9,9 | X, X | 9,11 | 12,12 |
DISCUSSION
Animal cell is a fundamental unit of life and form an incredible tool in biological research. It acts as a model system for understanding physiological processes and screening of toxic or therapeutic compounds for use in medical treatments. The cells play a major role in the production of functional enzyme, growth factor and vaccines. Cell culturing is a process of maintaining cells of multicellular organisms outside their original body under precise conditions. Over the last three decades, there have been advances in the field of tissue culture and tissue engineering technology. The isolation of primary cells from whole tissues for biological studies is becoming widely recognized in studying and understanding the mechanism of cell cycle, apoptosis and DNA repair. To isolate and establish a cell line with standardized protocols has now become an essential tool in biomedical research and tissue culture technology.[1,2]
Primary cells have the capacity to undergo replicative senescence or aneuploidization which makes their isolation and infinite growth difficult. Therefore, new cultures need to be established regularly with proper monitoring and maintenance. The concerns of Primary cell cultivation, the need for a high quantity of primary buccal fibroblasts in obtaining the greatest number of clonogenic cells, cell performance, and the best culture lifespan cells has been in demand since decades has lead to development of standard protocols for primary cultures of cell lines. Literature and standardized protocols are available for the isolation of rodent embryonic fibroblasts, gingival fibroblasts and oral keratinocytes, but there is minimum literature available on the isolation and culturing of HBMFs.[3]
For obtaining the primary culture of cells, Bolls and Lee suggested two basic techniques involved and they are enzymatic and explant method. Billingham and Reynolds proposed a technique for the separation of epithelial cells using an enzyme (trypsin), thus called the enzymatic method. The tissue dissociation or enzymatic method involves proteolytic enzymes to obtain a single cell suspension or subsequent subcultures. Trypsin which is a proteolytic enzyme breaks down the protein by inactivating the adhesion molecules and integrins to detach the cells from the plate and float.[7,8]
Trypsin along with EDTA is used in cell culture where EDTA acts as a chelating agent that weakens the cell matrix interaction. Trypsin is the most widely used enzyme for dissociation of cells from tissues, adhesive cells from the flasks and for rapid passaging of large cell suspension. However, in a study done by Stems et al. and Plaskach et al. in 1993, their results indicated that trypsinization or enzymatic method can cause the modification in the adhesive properties of the cells, dysregulation of the cell functions, loss of cell activity, alteration of the cell membrane permeability and disruption of the cell resulting in the cell deformity. A study done by Mexican et al. concluded that exposure to high concentration of trypsin can disrupt the cell morphology and their adhesive properties, which is a common problem in subculture.[9,10] P K Nanda et al. conducted a study to assess the enzymatic disintegration of the tissue samples for establishing the primary culture; different concentrations of trypsin (0.12% and 0.25%) with regard to time were used. The effect of different concentrations of trypsin showed that the tissue samples partly dissociated with 0.12% and completely dissociated with 0.25% of trypsin. Further, the cells showed morphological alteration and failure of attachment of cells to flasks in the subsequent subculture.[11,12]
According to literature, a minimum concentration of 0.25%–0.5% of trypsin is considered safe for tissue culture studies. In our experiment, trypsin of minimum concentration of 0.25% with EDTA was used for isolation and subculture of the cell lines. Its concentrations did not alter the biological behavior of the cells, and allowed the removal of other contaminating cells, such as epithelial cells, RBCs and other inflammatory cells. We would like to propose that the combination of explant–enzymatic technique showed that 0.25% of trypsin was appropriate for the detachment and developing a primary cell line faster and accurate.[14]
In 1910, Carrel and Burrows described a method for the extraction of epithelial cells called direct explant method where the cells are isolated directly from the tissue samples without using dissociating enzymes such as trypsin. However, they concluded that the cells were not able to dissociate easily from the explant tissue and it took long duration for the first cells to come out from the parent explant tissue. The main disadvantage of the explant technique is that the cells remain in clusters, which becomes difficult for the disassociation of the tissue and fails in the development of monolayer. The explant technique also results in the appearance of undesired cells.
In a study done by Orazizadeh et al. in 2015, the keratinocytes were able to get detached from the explant tissue within 24 h whereas the fibroblasts were isolated after 4 days of culture.[12,14]
Similarly, another study done by Punita et al. in year 2018 found that the success rate of outgrowth of fibroblast cell from skin biopsy was 4–12 days of processing of the tissue samples. Comparing with these studies, we were successful in isolating the buccal mucosal fibroblast cell lines using a combination of explant–enzymatic method, which helped the cells to detach and migrate from the main explant tissue sample on day 3 of our experiment. The combination method of tissue culture in our study showed that cells were easily separated from the main tissue and were able to grow at faster rate without any morphological alteration.[14]
The major concern in our study was the prevention and maintenance of primary cultures from contamination. The tissue samples obtained from the oral cavity are always associated with bacterial contamination, which hinders the rate of growth of primary cell lines. O W Merten described about bacterial and viral contamination and incorporated some measure to prevent the contamination like washing the tissue samples before the processing with PBS, using sterilized and autoclaved instruments and checking the raw materials for any contamination. They were successful in establishing the primary cell lines with absence of bacterial and viral growth in the cell lines.
In our experiment, measures were taken to prevent any bacterial or fungal contamination right from the procurement of the tissue samples to maintaining and subculturing of the cell lines. Based on our experiments, we would like to propose the list of measure to be taken in developing a primary cell line of BMF cell line [Table 1]. Of ten tissue samples, we were successful in isolating the fibroblast cell line from four tissue samples. The failure of other samples was due to contamination while tissue processing or from the source of sample.
On observation of the morphology of fibroblasts, we observed that F1 type of fibroblasts was seen on 3rd–5th day, whereas a combination of F1 and F2 type of fibroblasts was observed on 6–10th day. However, subsequently, we found predominantly spindle-shaped fibroblasts in all our cultures. Hence, appearance of F3-shaped cells during culture would suggest terminal stage of fibroblasts, synthesizing high amounts of collagen. Surprisingly, we found less number of F2- and F3-shaped fibroblasts during our culture, as these fibroblasts are predominantly seen with the effect of exogenous addition of arecoline.[15] The above findings further prove that our developed fibroblast cell lines are pure primary cell lines developed with no toxic effect during culture.
The cell lines were further subjected for authentication and characterization through DNA or STR profiling from DNA Forensics Laboratory, New Delhi, according to the standard protocol provided by the laboratory. STR profiling or DNA profiling is the most commonly and routinely used method for identification and authentication of human cell lines, which generates a specific molecular and genomic identity code of cell lines and stem cells. This can establish an identity to the specific individual cell lines and helps in further research and reproducibility of the cells for any clinical intervention. STR profiling is cost-effective and can be considered as a standard tool for cell line authentication. As the cell lines from the surgical specimens are difficult to isolate and generate subcultures, we thought to characterize and authenticate our primary cell lines using STR or DNA profiling. Hence, in our study, by STR analysis, we confirmed that cell lines are of human origin and resemble the fibroblast cells with unique loci of tandem repeats.[13]
Pure cultures of authenticated primary HBMF cell lines with subsequent subcultures were incubated, maintained and stored for the future application in the research and clinical aspects of dentistry.
CONCLUSION
The experiment was carried out to standardize the protocols for isolating and establishing the primary cell lines of HBMFs in large numbers from healthy individuals by a combination of explant–enzymatic technique. Obtaining primary cell lines of HBMF from the buccal tissues was very challenging as they were susceptible to source of contaminations. The above-mentioned standardized protocol helps in authentication and establishment of primary cell lines of HBMFs with morphological and genetic profiling.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We would like to acknowledge and thank Dr. Prabhakar Kore, Basic Science Research Centre, KLE Academy of Higher Education and Research, Belgaum, for providing the facilities for successful carrying out our experiment in the tissue culture laboratory.
REFERENCES
- 1.Southgate J, Williams HK, Ludwik K. Primary culture of human oral epithelial cells. J Lab Invest. 1987;2:211. [PubMed] [Google Scholar]
- 2.Daniels JT, Kearney JN, Ingham E. Human keratinocyte isolation and cell culture: A survey of current practices in the UK. Burns. 1996;22:35–9. doi: 10.1016/0305-4179(95)00085-2. [DOI] [PubMed] [Google Scholar]
- 3.Vaidya A, Gorbunova V. Establishing primary adult fibroblast cultures from rodents. J Vis Exp. 2010;44 doi: 10.3791/2033. pii: 2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wanichpakorn S, Kedjarune U. Primary cell culture from human oral tissue: Gingival keratinocytes, gingival fibroblasts and periodontal ligament fibroblasts Songklanakarin. J Sci Technol. 2010;32:327–31. [Google Scholar]
- 5.de Waal J, Olivier A, van Wyk CW, Maritz JS. The fibroblast population in oral submucous fibrosis. J Oral Pathol Med. 1997;26:69–74. doi: 10.1111/j.1600-0714.1997.tb00024.x. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee A, Kamath VV, Kotrashetti V, Bhatt K. Fibroblastic phenotype in oral submucous fibrosis – A cell culture analysis. Saudi J Pathol Microbiol. 2017;2:36–47. [Google Scholar]
- 7.Siengdee P, Klinhom S, Thitaram C, Nganvongpanit K. Isolation and culture of primary adult skin fibroblasts from the Asian elephant (Elephas maximus) PeerJ. 2018;6:e4302. doi: 10.7717/peerj.4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bayar GR, Aydıntuǧ YS, Günhan O, Oztürk K, Gülses A. Ex vivo produced oral mucosa equivalent by using the direct explant cell culture technique. Balkan Med J. 2012;29:295–300. doi: 10.5152/balkanmedj.2012.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bayar GR, Aydintug YS, Gulses AS, Elci P. A pilot study of the primary culture of the oral mucosa by the direct explant technique. J Oral health Dent Manage. 2011;2:20. [Google Scholar]
- 10.Srirama G, Bigliardi PL, Bigliardi-Qi M. Fibroblast heterogeneity and its implications for engineering organotypic skin models in vitro. Eur J Cell Biol. 2015;94:303–21. doi: 10.1016/j.ejcb.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Orazizadeh M, Hashemitabar M, Bahramzadeh S, Dehbashi FN, Saremy S. Comparison of the enzymatic and explant methods for the culture of keratinocytes isolated from human foreskin. Biomed Rep. 2015;3:304–8. doi: 10.3892/br.2015.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keira SM, Ferreira LM, Gragnani A, Duarte IS, Santos IAN. Experimental model for fibroblast culture. J Brazilian Soc Develop Res Surg. 2004;19 [Google Scholar]
- 13.Markovic O, Markovic N. Cell cross-contamination in cell cultures: The silent and neglected danger. In Vitro Cell Dev Biol Anim. 1998;34:1–8. doi: 10.1007/s11626-998-0040-y. [DOI] [PubMed] [Google Scholar]
- 14.Sriram G, Bigliardi PL, Bigliardi-Qi M. Fibroblast heterogeneity and its implications for engineering organotypic skin models in vitro. Eur J Cell Biol. 2015;94:483–512. doi: 10.1016/j.ejcb.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Reid Y, Storts D, Riss T, Minor L. Authentication of human cell lines by STR DNA profiling analysis. In: Sittampalam GS, Coussens NP, Nelson H, Arkin M, Auld D, Austin C, et al., editors. Bethesda (MD) Assay Guidance Manual; 2004. [Google Scholar]