

Abstract
Benzalkonium chlorides (BACs) are widely used as disinfectants in cleaning products, medical products, and the food processing industry. Despite a wide range of reported toxicities, limited studies have been conducted on the metabolism of these compounds in animal models and none in human-derived cells or tissues. In this work, we report on the metabolism of BACs in human liver microsomes (HLM) and by recombinant human hepatic cytochrome P450 (CYP) enzymes. BAC metabolism in HLM was NADPH-dependent and displayed apparent half-lives that increased with BAC alkyl chain length (C10 < C12 < C14 < C16), suggesting enhanced metabolic stability of the more lipophilic, longer chain BACs. Metabolites of d7-benzyl labeled BAC substrates retained all deuteriums and there was no evidence of N-dealkylation. MS/MS fragmentation of BAC metabolites confirmed oxidation occurs on the alkyl chain region. Major metabolites of C10-BAC were identified as ω-hydroxy-, (ω−1)-hydroxy-, (ω, ω−1)-diol-, (ω−1)-ketone-, and ω-carboxylic acid-C10-BAC by LC-MS comparison with synthetic standards. In a screen of hepatic CYP isoforms, recombinant CYP2D6, CYP4F2, and CYP4F12 consumed substantial quantities of BAC substrates and produced the major microsomal metabolites. The use of potent pan-CYP4 inhibitor HET0016, the specific CYP2D6 inhibitor quinidine, or both confirmed major contributions of CYP4- and CYP2D6-mediated metabolism in the microsomal disappearance of BACs. Kinetic characterization of C10-BAC metabolite formation in HLM demonstrated robust Michaelis-Menten kinetic parameters for ω-hydroxylation (Vmax = 380 pmol/min/mg, Km = 0.69 μM) and (ω−1)-hydroxylation (Vmax = 126 pmol/min/mg, Km = 0.13 μM) reactions. This work illustrates important roles for CYP4-mediated ω-hydroxylation and CYP2D6/CYP4-mediated (ω−1)-hydroxylation during the hepatic elimination of BACs, an environmental contaminant of emerging concern. Furthermore, we demonstrate that CYP-mediated oxidation of C10-BAC mitigates the potent inhibition of cholesterol biosynthesis exhibited by this short-chain BAC.
Keywords: Benzalkonium chlorides, cytochrome P450, in vitro hepatic metabolism, human liver microsomes, cholesterol biosynthesis inhibition
Graphical Abstract
Introduction
Benzalkonium chlorides (BACs; also known as alkyl dimethyl benzyl ammonium chloride) (Figure 1A) are common quaternary ammonium compounds (QACs) that have been widely used as antimicrobials for 70 years. BACs are relatively stable and persistent in the environment1–3 due to the very slow rates of decomposition of QACs by bacteria found in sewage and soil.4, 5 BACs are prevalent in cleaning products (such as Lysol and Clorox solutions, wipes, and hand sanitizers), medical products (such as eye and nasal drops), and in the food processing industry, suggesting humans may be systemically exposed to BACs through direct dermal/eye contact, inhalation, and ingestion.6–9 Furthermore, high levels of BACs have been found in grapefruit seed extracts10, food additives11, fruits, and in particular, processed food such as milk and other dairy products.12–14
Figure 1. Chemical structures and microsomal stability of BACs.
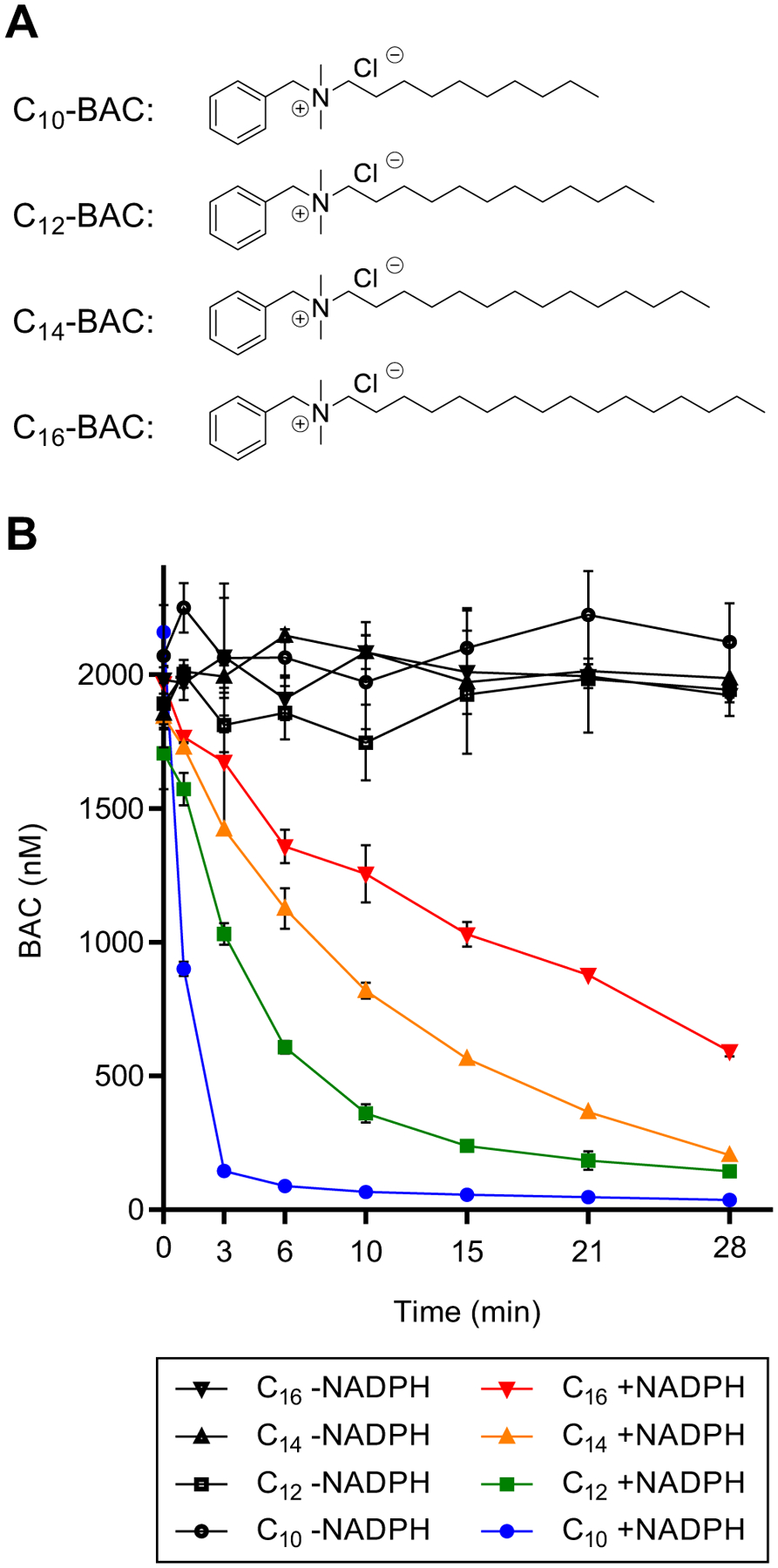
(A) Chemical structures of BACs with 10, 12, 14, and 16 carbon alkyl chain lengths. (B) Time-courses of NADPH-dependent depletion of BACs (2 μM) incubated separately in HLM (1 mg/mL) out to 28 minutes. Data points represent the mean ± SD (n = 3). When not visible, error bars are contained within the data point.
While BACs used to be generally recognized as safe (GRAS), this determination is under question by the FDA, which recently called for additional safety data on their usage in healthcare and consumer antiseptic products.15, 16 In fact, a wide range of cytotoxicity of BACs has been reported in various biological systems. BACs halted the development of over 50% of clam eggs and completely killed larvae at only 0.2 ppm (equivalent to 500 nM).17 BACs are also highly toxic to other aquatic life, including daphnids (LC50 from 0.1 to 1.0 ppm) and fish (LC50 from 0.5 to 5.0 ppm).2, 18 Furthermore, BACs are toxic to peripheral neurons, including enteric and ganglion neuronal cells, at μM concentrations.19, 20 A study by Hrubec, Hunt, and coworkers demonstrated that chronic consumption of BAC-containing food (60 or 120 mg/kg/day) led to significantly decreased fertility and fecundity in mice, as well as increased dam mortality.21 More recently, the same group reported that maternal exposure to a BAC-containing diet to the dams led to large increases in the incidence of neural tube defects in the embryos in a concentration dependent manner and at doses as low as 7.5 mg/kg/day.22 The same study showed that control mice did not produce any embryos with neural tube defects in an absolutely QAC-free room, whereas even ambient QAC exposure (in a facility where QACs were used as disinfectants) led to neural tube defects.22 We recently found that short-chain BACs potently inhibit the 3β-hydroxysterol-Δ7-reductase (DHCR7) in cholesterol biosynthesis at nM concentrations.23 This inhibition leads to the same biochemical defects as those observed in the cholesterol biosynthesis disorder, Smith-Lemli-Opitz syndrome, i.e., greatly elevated levels of a highly oxidizable cholesterol precursor, 7-dehydrocholesterol, and decreased levels of cholesterol.24–26 We further found all BACs disrupt lipid homeostasis in a neuronal cell line and in neonatal mouse brains exposed in utero, despite only nM levels of BACs being detected in neonatal tissues and maternal blood following 120 mg/kg/day dosing.27, 28 Thus, even low level tissue exposure to BACs could be detrimental to reproduction, development, and normal lipid metabolism.
While there exists a large and growing body of literature concerning the toxicology of BACs, information on the metabolism of BACs in mammalian species is still lacking. Single-dose intravenous injection of BACs to rats (7 mg/kg) led to a wide distribution of these compounds in various tissues (levels in μg/g of tissue shown in the parentheses), with the highest level observed in the kidney (50.5), followed by lung and spleen (15.4 each), serum (1.2), liver (0.9), and brain (0.2) after 30 min of administration.29 When BACs were administered orally (250 mg/kg), the levels of BAC reached their highest concentrations for the majority of the tissues after 24 hours (2 hours for liver), with the level in kidney (5.25) > lung (2.75) > liver (0.72) > blood (0.34) (not determined in brain).30, 31 Therefore, BACs are orally bioavailable and distribute broadly throughout tissues in vivo. However, the mechanisms of metabolism and disposition of BACs in humans have not been studied and this represents a barrier to our understanding of the systemic toxicology of BACs.
As the liver is the major organ that performs metabolic clearance of lipophilic xenobiotics, studying human hepatic metabolism of BACs is a logical first step to understand the relationship between BAC persistence and any potential toxicity to humans. Here, we investigate the metabolism of BACs in human liver microsomes and recombinant cytochrome P450 (CYP) isoforms and report: (1) the in vitro metabolic stability of BACs; (2) identification of major metabolites and the metabolic pathways; and (3) elucidation of the enzymes responsible for human hepatic clearance of BACs. Finally, we provide evidence that the two major metabolites of a short-chain BAC display greatly diminished potency in inhibiting cholesterol biosynthesis in comparison with the parent BAC, suggesting that BAC metabolism serves as a detoxification mechanism in terms of cholesterol biosynthesis inhibition. This work lays the foundation for testing our overarching hypothesis that metabolic clearance of BACs protects human health by limiting toxicity and facilitating excretion.
Materials and Methods
Materials.
Optima LC-MS grade acetonitrile, water, formic acid, and ammonium formate used in liquid chromatography were purchased from Fisher Scientific (Santa Clara, CA). BACs of various alkyl chain lengths (C10-, C12-, C14-, and C16-BAC), the DHCR7 inhibitor AY9944, and all synthetic reagents and starting materials were from Sigma-Aldrich (St. Louis, MO). Deuterated NMR solvents were from Cambridge Isotopes Laboratories, Inc. (Andover, MA). Human liver tissues were obtained from the School of Pharmacy Human Liver Bank at the University of Washington (Seattle, WA). Human recombinant CYP enzymes individually-expressed in Escherichia coli (EasyCYP Bactosomes) co-expressing human cytochrome P450 reductase and, where indicated as +b5, supplemented with purified human cytochrome b5 (CYPs 1A2, 2A6+b5, 2A13, 2B6+b5, 2C8+b5, 2C9+b5, 2C19+b5, 2D6, 2E1+b5, 2J2, 3A4+b5, 3A5+b5, 4A11+b5, 4F2+b5, and 4F3B+b5) and membrane protein from E. coli with empty expression plasmid (EasyCYP Control Bactosomes) were from XenoTech, LLC (Lenexa, KS). Human recombinant CYP4F12 containing human cytochrome P450 reductase and cytochrome b5 in insect microsomes isolated from baculovirus-transfected insect cells (CYP4F12 Supersomes) and negative control insect cell microsomes (Control Supersomes) were from Corning (Corning, NY). NADPH co-factor was from Oriental Yeast (Tokyo, Japan).
Synthesis.
Preparation of internal standards.
Deuterated (d7-benzyl) BACs (d7-C10-BAC, d7-C12-BAC, d7-C14-BAC, and d7-C16-BAC) were synthesized as described previously.23 C6-BAC was synthesized by an analogous procedure through nucleophilic coupling of N,N-dimethylbenzylamine and 1-chlorohexane in ethanol.32
Preparation of ω-hydroxy-C10-BAC.
To a stirred solution of N,N-dimethylbenzylamine (0.320 g, 2.37 mmol) in ethanol (50 mL), was added 10-chloro-1-decanol (0.481 g, 2.50 mmol). The resulting solution was heated to reflux and stirred for 24 hours. Ethanol was evaporated under reduced pressure and the solid obtained was recrystallized from hot acetone affording 0.505 g (65%) of white solid product. 1H-NMR (CDCl3, 500 MHz): 1.29 and 1.36 (br s, 12H), 1.56 (m, 2H), 1.81 (m, 2H), 3.30 (s, 6H), 3.53 (m, 2H), 3.64 (q, 2H, J = 6.1 Hz), 5.03 (s, 2H), 7.47 (m, 3H), 7.64 (d, 2H, J = 7.0 Hz); MS [M-Cl+] (C19H34NO): observed, 292.2639; theoretical: 292.2640.
Preparation of ω-alkene-C10-BAC.
First, 9-decen-1-ol was converted to 10-chloro-1-decene using 2-chloro-1,3-dimethylimidazolinium chloride as the chlorinating reagent according to the method of Isobe and Ishikawa33 and the product was isolated by silica gel chromatography (hexanes:ethyl acetate, 1:8).
The above 10-chloro-1-decene (0.190 g, 1.09 mmol) was added to a stirred solution of N,N-dimethylbenzylamine (0.142 g, 1.05 mmol) in ethanol (50 mL). The resulting solution was heated to reflux and stirred for 24 hours. Ethanol was evaporated under reduced pressure and the solid obtained was recrystallized from hot acetone affording 0.219 g (62%) of product. 1H-NMR (CDCl3, 500 MHz): 1.29 and 1.36 (br s, 10H), 1.80 (m, 2H), 2.03 (q, 2H, 7.0 Hz), 3.30 (s, 6H), 3.50 (m, 2H), 4.97 (m, 2H), 5.03 (s, 2H), 5.80 (m, 1H), 7.47 (m, 3H), 7.64 (d, 2H, J = 7.0 Hz); MS [M-Cl+] (C19H32N): observed, 274.2541; theoretical: 274.2535.
Preparation of (ω−1)-hydroxy-C10-BAC.
A solution of ω-alkene-C10-BAC (10 mg, 32 μmol) in aqueous 1M HCl (3 mL) was heated to 80˚C for 40 hours to effect hydration of the alkene by Markovnikov addition. The reaction mixture was then diluted with water:methanol (1:1) mixture (3 mL) and extracted 3 times with chloroform (5 mL). The combined organic extracts were evaporated to dryness. Crude product mixture was re-dissolved in chloroform and loaded onto a silica gel column. The product eluted with increasing strength of methanol in chloroform (increased in stages up to 20% methanol). The product-containing fractions were combined and evaporated to yield 4.5 mg (43%) of product as a clear oil. 1H-NMR (CDCl3, 500 MHz): 1.19 (d, 3H, 6.1 Hz), 1.31 and 1.39 (br s, 12H), 1.82 (m, 2H), 3.29 (s, 6H), 3.53 (m, 2H), 3.80 (br s, 1H), 5.00 (s, 2H), 7.48 (m, 3H), 7.64 (d, 2H, J = 7.0 Hz); MS [M-Cl+] (C19H34NO): observed, 292.2645; theoretical: 292.2640.
Preparation of (ω, ω−1)-dihydroxy-C10-BAC.
The epoxidation of ω-alkene-C10-BAC (10 mg, 32 μmol) was accomplished by the procedure of Bach et al.34 and this was followed by ring-opening of the epoxide in 1M NaOH heated to 80˚C for 2 hours to produce the vicinal diol. The aqueous product mixture was purified on a solid-phase C18 extraction cartridge by loading and washing with water and eluting with methanol to afford 2.1 mg (19%) of white solid product. 1H-NMR (CDCl3, 500 MHz): 1.35 and 1.41 (br s, 14H), 1.84 (m, 2H), 3.23 (s, 6H), 3.45 (m, 1H), 3.62 (m, 3H), 3.73 (m, 1H), 4.89 (s, 2H), 7.48 (m, 3H), 7.61 (d, 2H, J = 7.1 Hz); MS [M-Cl+] (C19H34NO2): observed, 308.2584; theoretical: 308.2590.
Preparation of (ω−1)-ketone-C10-BAC.
Dess-Martin periodinane (31 mg, 73 μmol) was added to a stirred solution of (ω−1)-hydroxy-C10-BAC (10 mg, 30 μmol) in chloroform (4 mL). After 24 hours of stirring at room temperature, the crude product mixture was loaded onto a silica gel column., rinsed with chloroform, and the product was eluted off silica with chloroform:methanol (8:2) mixture to yield 4.7 mg (48%) of white solid. 1H-NMR (CDCl3, 500 MHz): 1.27 and 1.35 (br s, 8H), 1.54 (m, 2H), 1.80 (m, 2H), 2.13 (s, 3H), 2.41 (t, 2H, J = 7.4 Hz), 3.27 (s, 6H), 3.50 (m, 2H), 4.97 (s, 2H), 7.46 (m, 3H), 7.62 (d, 2H, J = 7.0 Hz); MS [M-Cl+] (C19H32NO): observed, 290.2484; theoretical: 290.2484.
Preparation of ω-carboxylic acid-C10-BAC.
Dess-Martin periodinane (0.185 g, 0.436 mmol) was added to a stirred solution of ω-hydroxy-C10-BAC (0.095 g, 0.290 mmol) in chloroform (15 mL). After 1 hour of stirring at room temperature, saturated aqueous sodium bicarbonate (10 μL) was added to the reaction and the resulting mixture was stirred overnight. The reaction was quenched with saturated aqueous sodium bicarbonate (1 mL), aqueous 2M sodium thiosulfate (1 mL), and aqueous 1M NaOH (1 mL), stirred for 30 minutes, and then extracted 3 times with water (5 mL). The basic aqueous extract was acidified with 1M HCl and extracted 3 times with chloroform:methanol (9:1) mixture (5 mL). The extract was evaporated and crude product was re-dissolved in chloroform and loaded onto a silica gel column. After extensive rinsing with chloroform, the product was eluted off silica with chloroform:methanol (8:2) mixture to yield 70 mg (70%) of white solid. 1H-NMR (CDCl3, 500 MHz): 1.34 and 1.38 (br s, 10H), 1.66 (m, 2H), 1.84 (m, 2H), 2.38 (t, 2H, J = 6.7 Hz), 3.21 (s, 6H), 3.64 (m, 2H), 4.88 (s, 2H), 7.48 (m, 3H), 7.61 (d, 2H, J = 7.0 Hz); 1H-NMR (d6-DMSO, 500 MHz): 1.27 (br s, 10H), 1.49 (m, 2H), 1.78 (m, 2H), 2.18 (t, 2H, 7.2 Hz), 2.94 (s, 6H), 3.23 (m, 2H), 4.51 (s, 2H), 7.53 (br s, 5H), 12.00 (br s, carboxylic acid); MS [M-Cl+] (C19H32NO2): observed, 306.2431; theoretical: 306.2433.
Preparation of Human Liver Microsomes.
Freshly-thawed human liver tissues (pool of 4 donors: 2 male, 2 female) were homogenized in ice-cold buffer A (50 mM potassium phosphate, pH 7.4, 0.25 M sucrose). The homogenate was centrifuged at 12,000 g for 30 minutes at 4°C and the supernatant was collected. Centrifugation of the supernatant at 108,000 g for 70 minutes at 4°C yielded microsomal pellets. Pellets were rinsed and resuspended in buffer B (50 mM potassium phosphate, pH 7.4, 0.15 M KCl). Centrifugation of this suspension at 108,000 g for 70 minutes at 4°C yielded washed microsomal pellets that were rinsed and resuspended in buffer A and stored at −80°C until use. Microsomal protein and P450 concentrations were determined by BCA and spectral carbon-monoxide binding35 assays, respectively. The final microsomal preparation had a protein concentration of 15 mg/mL and specific P450 content of 315 pmol/mg protein.
Incubation Procedures.
Incubations with human liver microsomes or recombinant P450 enzymes were performed with a minimum of 3 replicates at 37°C and pH 7.4 in 100 mM potassium phosphate buffer (KPi) in polypropylene tubes or polypropylene 96-well PCR plates. Substrates and inhibitors were delivered from concentrated DMSO stocks. The percentage of DMSO (v/v) in the incubations was ≤ 0.5%. The concentration of microsomal protein (mg/mL), incubation times (minutes), and substrate concentrations (μM) used in incubations is specified in the Results sections and in the figure captions associated with each experiment. Recombinant CYP enzyme incubations contained 50 nM P450 and a protein concentration of 0.5 mg/mL. Before starting enzymatic reactions, incubations were pre-warmed at 37°C for 3 minutes in a shaking water bath and then initiated with NADPH co-factor dissolved in KPi or KPi as vehicle (-NADPH controls). Incubation conditions for capturing initial rates of product formation were optimized as follows: ≤ 3 minutes incubation length, product formation within the linear range, and low microsomal protein concentration (0.04 mg/mL) to minimize depletion of the initial substrate concentration to ≤ 20%. Incubations were terminated by addition of 1 volume equivalent of ice-cold acetonitrile spiked with internal standards. Quenched incubations were centrifuged (3,500 g, 15 minutes, 4°C) and an aliquot of supernatant was diluted into water:methanol (1:1) mixture for analysis by LC-MS.
Cell Culture, Treatment, and Sterol Analysis.
Mouse Neuro2a neuroblastoma cells were purchased from American Type Culture Collection (Rockville, MD) and maintained in DMEM supplemented with L-glutamine, 10% fetal bovine serum (FBS; Thermo Scientific HyClone, Logan, Utah), and penicillin/streptomycin at 37°C and 5% CO2. The cell treatment procedure and sterol analysis were carried out according to our published methods.23, 36 Briefly, cells were plated in 100 mm plates at cell density of 5 × 105 cells per plate and were allow to grow overnight before treatment. For treatment of the adhered cells, serum-containing media was replaced with DMEM high glucose media without serum, but with the addition of N2-supplement, L-glutamine and penicillin/streptomycin. The serum-free media contained one of the following small molecule treatments: AY9944 (positive control for Dhcr7 inhibition), C10-BAC, ω-OH-C10-BAC, or (ω−1)-C10-BAC (each at a concentration of 100 nM), or 0.1% DMSO as the vehicle control. After 72 hours of treatment duration, cells were harvested and protein concentrations were determined by BCA Protein assay kit prior to spiking internal standards, d7-cholesterol and 13C3-lanosterol (0.5 μg and 0.1 μg per 100 μg of protein respectively). Lipids were then extracted using the Folch method and analyzed by HLPC-MS/MS, according to our previously described method.36, 37
LC-MS Methods.
Targeted analysis of BACs and their metabolites by triple-quadrupole mass spectrometry.
Analyte separation was performed at a flow rate of 0.400 mL/min on a Thermo Hypersil GOLD C18 column (100×2.1 mm, 1.9-μm particle size) at ambient temperature. Mobile phase delivery and sample injection were managed by an Acquity UPLC system and autosampler (Waters Corporation, Milford, MA). A linear solvent gradient consists of solvent A (0.1% formic acid, 2 mM ammonium formate in water) and solvent B (acetonitrile) was used: 0 min, 20% B; 4.7 min, 38% B; 5.7 min, 90% B; 8.2 min, 90% B; 8.5–10.2 min, 20%. The eluted analytes were detected on an API-4000 Q-trap triple quadrupole mass spectrometer (Sciex, Foster City, CA) operated in ESI(+) mode with source temperature of 400°C and ion spray voltage set at 3500 V. Additional source parameters were set as curtain gas: 35 psi, collision activated dissociation: “medium”, gas 1 (nebulizer gas): 45 psi, gas 2 (heater gas): 50 psi, declustering potential: 50 V, entrance potential: 10 V, collision energy 31 V, and cell exit potential: 10 V. Analyte retention times (tR) and the precursor to product ion transitions monitored were: (ω,ω−1)-dihydroxy-C10-BAC [tR= 2.31 min; m/z 308.2→216.2]; (ω−1)-hydroxy-C10-BAC [tR= 4.12 min; m/z 292.2→200.2]; ω-hydroxy-C10-BAC [tR= 4.38 min; m/z 292.2→200.2]; C6-BAC [tR= 4.40 min; m/z 220.2→128.2]; ω-carboxylic acid-C10-BAC [tR= 4.41 min; m/z 306.2→214.2]; (ω−1)-ketone-C10-BAC [tR= 4.47 min; m/z 290.2→198.2]; d7-C10-BAC [tR= 7.00 min; m/z 283.2→184.2]; C10-BAC [tR= 7.04 min; m/z 276.2→184.2]; d7-C12-BAC [tR= 7.30 min; m/z 311.2→212.2]; C12-BAC [tR= 7.34 min; m/z 304.2→212.2]; d7-C14-BAC [tR= 7.69 min; m/z 339.2→240.2]; C14-BAC [tR= 7.73 min; m/z 332.2→240.2]; d7-C16-BAC [tR= 8.34 min; m/z 367.2→268.2]; C16-BAC [tR= 8.38 min; m/z 360.2→268.2]. Analyte peak areas were normalized to the appropriate internal standard peak area where C6-BAC served as internal standard for C10-BAC metabolites and d7-labeled BACs served as internal standards for each respective BAC parent compound.
Time-of-Flight High-Resolution Mass Spectrometry.
The LC conditions were the same as described above but with a different %B versus time schedule in the linear mobile phase gradient: 0 min, 10% B; 17 min, 85% B; 18 −20 min, 10% B. The eluted analytes were detected on a Synapt G2-Si ion mobility Q-TOF mass spectrometer (Waters Corporation, Milford, MA) operated in ESI-(+) mode with source temperature of 120°C and capillary voltage set at 0.8 kV. Additional source parameters were set as desolvation temperature: 350°C, cone gas flow: 15 L/hr, nebulizer gas flow: 6.5 bar, desolvation gas flow: 600 L/hr, cone voltage: 30 V, and source offset: 80 V. Ions of m/z value in the 50 – 1000 range were acquired with the mass analyzer set to “Resolution” mode. The mass detector was calibrated using sodium formate cluster ions by infusing a sodium formate solution. During data acquisition, leucine-enkephalin lock mass (MH+ 556.2771 m/z) was sampled at regular intervals and applied as a mass correction reference.
Data Analysis
Data are presented as the mean ± standard deviation (SD) and include a minimum of 3 replicates (n = 3) as specified in figure captions. Statistical significance was judged by two-tailed, unequal variances t-test: p ≤0.05 (*); p ≤0.01 (**); p ≤0.001 (***). Plotting of the data, statistical analyses, and nonlinear regression analyses were completed in GraphPad Prism (GraphPad Software, La Jolla, CA).
Results
Metabolic Stability of BACs in HLM.
We first examined the metabolic stability of individual BACs with varying alkyl chain lengths (C10-, C12-, C14-, and C16-BAC) (Figure 1A) in HLM (1 mg/mL) and found that all BACs were consumed rapidly (each BAC was incubated separately at 2 μM) (Figure 1B). Metabolism in HLM was NADPH-dependent as BACs were not consumed in the absence of NADPH. The rate of NADPH-dependent consumption followed the order of C10 > C12 > C14 > C16, with apparent half-lives of approximately 1, 3, 6, and 15 minutes. BACs of longer alkyl chain length exhibited greater metabolic stability in HLM than did BACs of shorter alkyl chain length.
Metabolism of BACs by Human Recombinant CYP Enzymes.
A panel of recombinantly expressed hepatic cytochrome P450 (CYP) isoforms was screened for NADPH-dependent consumption of C10-BAC and C16-BAC (2 μM initial concentration) to bracket the shortest and longest BAC alkyl chain lengths examined in this study. Neither BAC was consumed in the control preparations (bactosomes or supersomes without CYP enzyme) in the absence or presence of NADPH. For C10-BAC, CYPs 2D6, 3A4, and 4F12 consumed statistically-significant quantities of substrate relative to the control preparations (Figure 2A). Over the 30-minute incubation period with 50 nM of individual P450, CYPs 2D6 and 4F12 consumed essentially all of C10-BAC while CYP3A4 consumed a modest 15% of this substrate. For C16-BAC, CYPs 2A6, 2D6, 4F2, and 4F12 consumed statistically-significant quantities of substrate relative to the control preparation (Figure 2B). CYP2A6 consumed a modest 5% of C16-BAC whereas CYPs 2D6, 4F2, and 4F12 consumed 77%, 45%, and 88%, respectively.
Figure 2. BAC consumption by human recombinant CYP isoforms.
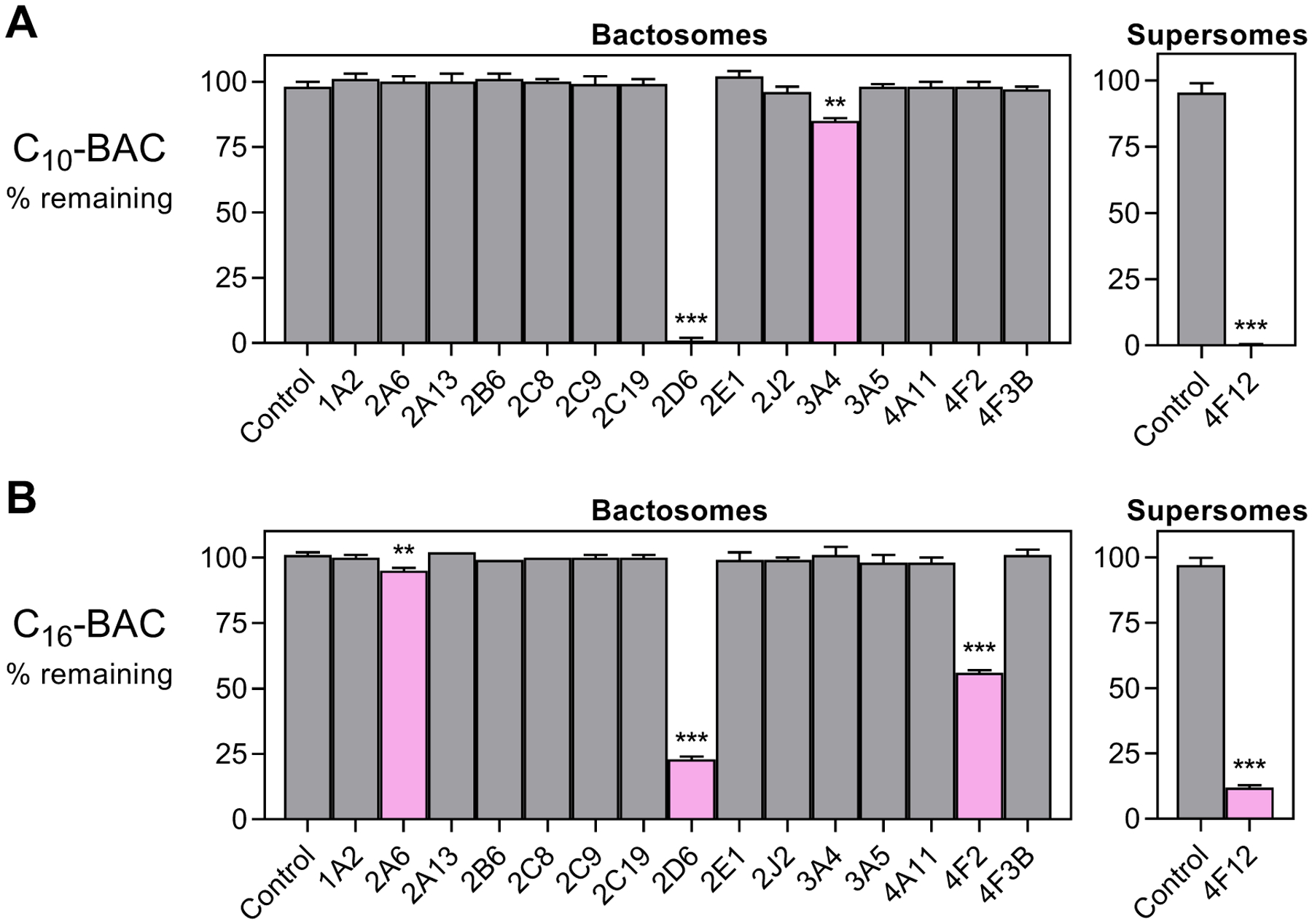
Recombinant CYP isoforms were screened for NADPH-dependent consumption of (A) C10-BAC (2 μM) and (B) C16-BAC (2 μM) in 30 minutes incubations with each individually-expressed CYP enzyme (50 nM P450). Refer to Materials and Methods for details on the commercially-available “Bactosomes” and “Supersomes” recombinant enzyme expression systems. Quantities represent the mean ± SD (n = 3). Bars colored magenta indicate statistically-significantly consumption of BAC substrate relative to the Control (without CYP) preparation: p ≤0.05 (*); p ≤0.01 (**); p ≤0.001 (***).
Structural Elucidation of BAC Metabolites.
Metabolite profiles generated in NADPH-fortified HLM were comprised of mono-, di-, and tri-hydroxylated BAC metabolites (+1O, +2O, and +3O) and further oxidized and desaturated BAC metabolites (+1O, −2H and +2O, −2H). The most abundant species were the primary +1O metabolites of BACs. For C10, C12, C14, and C16-BACs, these included a major +1O metabolite and a minor +1O metabolite of each BAC (Figure 3A). Comparison of the unlabeled +1O BAC metabolites with the corresponding metabolites of d7-BAC analogs (all deuterium atoms on the benzyl group; see Herron et al.23 for synthesis) confirmed complete retention of deuterium atoms across all observed metabolites (Figure 3B), not only for the major +1O metabolites, but for all detected microsomal metabolites of BACs as well (data not shown). In addition, MS/MS fragmentation of the parent BACs (Figure S1) and of non-labeled BAC metabolites (Figure S2–S7) gave rise to an unmodified benzyl cation fragment (91 m/z). Generally, all BAC metabolite MS/MS fragmentation can be summarized as consisting of a spectrum of [M]+, [M - benzyl]+, and [benzyl]+ ions. Furthermore, none of the detected metabolite m/z values suggested N-dealkylation. Therefore, the aromatic benzyl group and the carbon atoms positioned alpha to the quaternary nitrogen atom were not targeted during oxidative metabolism of BACs in HLM. Consequently, all of the observed oxidation and desaturation reactions were directed along the alkyl chain region of BACs.
Figure 3. Hydroxylated BAC metabolites produced in HLM.
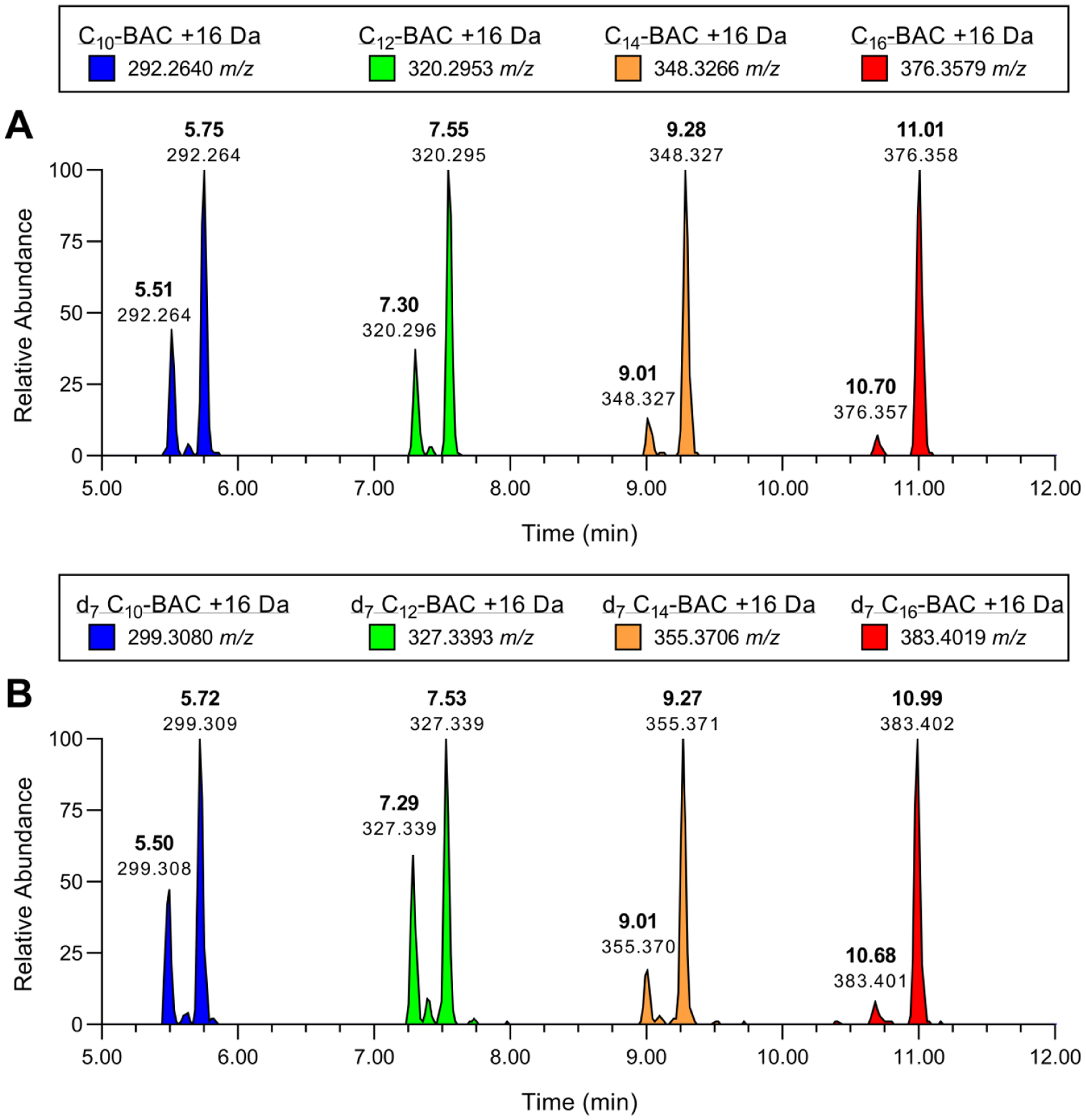
LC-MS extracted ion chromatograms displaying +1O metabolites of (A) d0-BACs and (B) d7-BACs produced by individually incubating each substrate (2 μM) in HLM (0.5 mg/mL) for 30 minutes with NADPH. Extracted theoretical m/z values (±0.005 m/z) of BAC +1O metabolites are indicated in each figure legend. Metabolite peaks are labeled with retention times and observed m/z values.
For C10-BAC, comparison with authentic metabolite standards confirmed the identities of several microsomal metabolites (Figure S7–S12) (see Materials and Methods and the last section of Results for descriptions of syntheses). Namely, these C10-BAC metabolites were a pair of ω- and (ω−1)-hydroxylated primary metabolites (Figure 4A), (ω, ω−1)-dihydroxy and (ω−1)-ketone secondary metabolites (Figures 4B and 4C), and the ω-carboxylic acid tertiary metabolite (Figure 4D). A number of very low abundance metabolites were also observed, indicating that carbon atoms at further internal positions were also oxidized (Figure S9–S12). While this hinted, qualitatively, at the great diversity of metabolites being generated in HLM, the identities of these metabolites were not pursued due to their lack of quantitative importance. Thus, as evidenced for C10-BAC, the ω- and (ω−1)-carbon atoms of the BAC alkyl chain undergo initial hydroxylation reactions and further sequential metabolism.
Figure 4. Identification of C10-BAC microsomal metabolites.
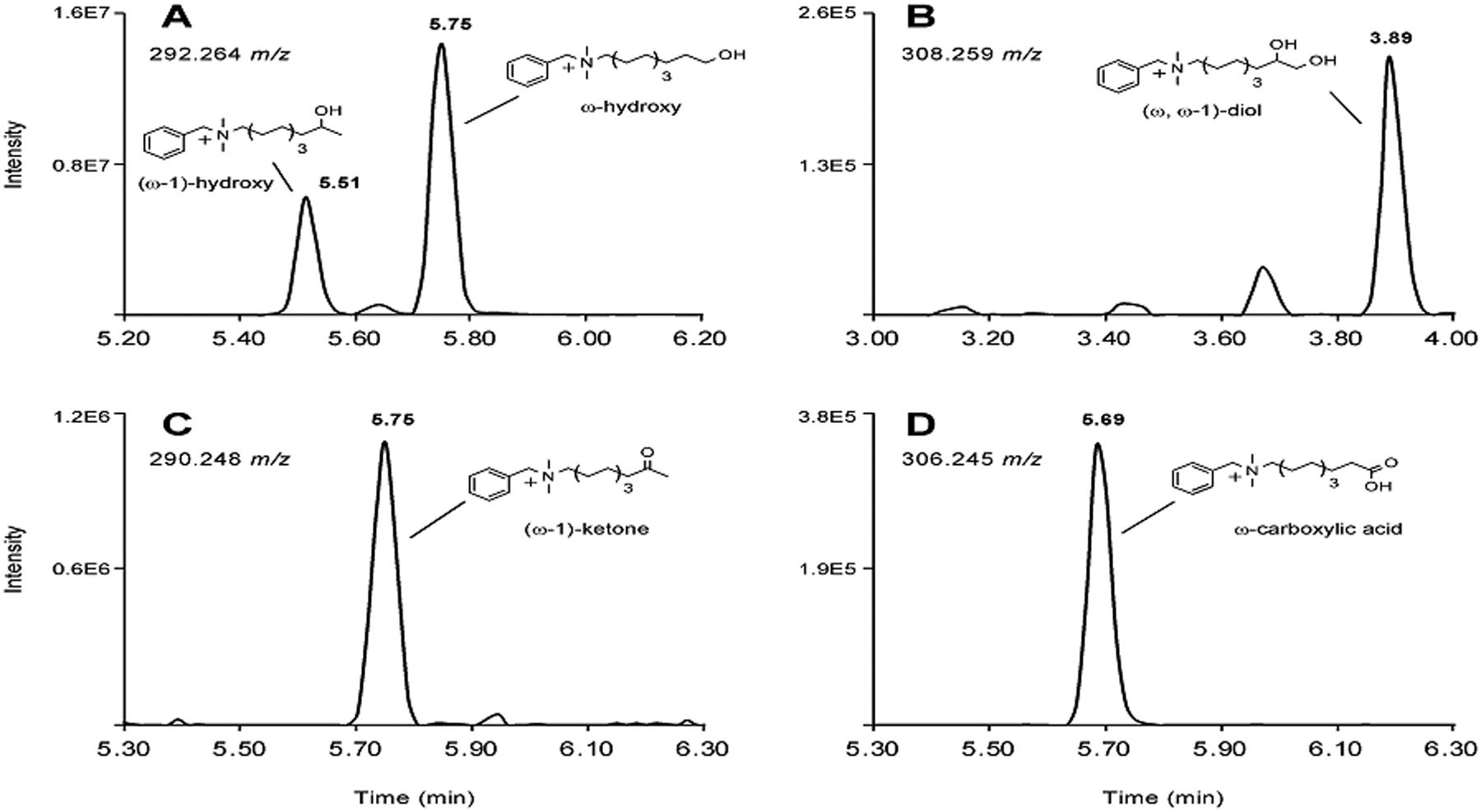
LC-MS extracted ion chromatograms displaying (A) +1O, (B) +2O, (C) +1O, −2H, and (D) +2O, −2H metabolites of C10-BAC produced by metabolism of C10-BAC (2 μM) in NADPH-supplemented HLM (0.5 mg/mL) after 30 minutes. Extracted theoretical m/z values (±0.005 m/z) of the specified metabolites are noted in each chromatogram. The identities of several metabolites were confirmed by comparison with synthetic standards prepared in-house and these analyte peaks are labeled with retention times and metabolite structures.
C12-, C14-, C16-BACs underwent similar CYP-mediated oxidation of the alkyl chain region (Figures S9 – S12) suggesting that these longer chain BACs have also likely been metabolized at the ω and (ω−1)-positions to hydroxy, dihydroxy, ketone, and carboxylic acid metabolites.
In recombinant CYP enzyme incubations, we found that CYPs 2D6 and 4F12 primarily produced (ω−1)-oxidized and further internally oxidized metabolites of C10-BAC with a notable absence of the major ω-hydroxy C10-BAC metabolite (Figures S13 and S14). Similarly, for C16-BAC, CYPs 2D6 and 4F12 appeared to form the minor, internally oxidized metabolites (Figures S15 and S16). Recombinant CYP4F2, on the other hand, produced the major +1O and +2O, −2H microsomal metabolites of C16-BAC (Figure S17) which, given the precedent set by C10-BAC and well-established role of CYP4F2 as an ω–hydroxylase, are presumably the ω-hydroxy and ω-carboxylic acid C16-BAC metabolites.
Inhibitory Effects of Quinidine and HET0016 on BAC Metabolism in HLM.
The screen of recombinant CYP isoforms discussed above established a role for CYP2D6 and CYP4F isoforms in BAC metabolism. In addition, ω-hydroxylation (a characteristic CYP4 reaction) of C10-BAC in HLM was confirmed with an authentic metabolite standard (described above), strongly implying the involvement of CYP4 enzymes. To measure the relative importance of CYP2D6- and CYP4-mediated microsomal metabolism of BACs, potent and selective inhibitors of these isoforms were applied to a BAC consumption assay in HLM (Figure 5). Each BAC substrate (2 μM) was incubated individually in HLM to establish the extent of NADPH-dependent consumption in the absence of any inhibitor. The inhibitors HET0016 (1 μM) and quinidine (1 μM) were then employed to specifically inhibit CYP4s and CYP2D6, respectively, either alone or in combination. For all four BAC alkyl chain lengths, HET0016 was the more effective inhibitor at blocking metabolism (Figure 5A). Quinidine exhibited a more modest inhibition of metabolism, which was not consistently statistically-significant for all BACs. However, relative to HET0016 alone, the combination of HET0016 and quinidine was particularly effective at blocking BAC metabolism with 93% ± 1%, 95% ± 3%, 98% ± 5%, and 95% ± 2% (mean ± SD, n = 3) of C10, C12, C14, and C16 substrate remaining compared with 55% ± 1%, 63 ± 3%, 60% ± 1%, and 67% ± 1% remaining, respectively, in the absence of any inhibitor. Thus, combined inhibition of CYP4 enzymes and CYP2D6 essentially blocks BAC metabolism in HLM, indicating that these isoforms serve key roles in determining BAC metabolic stability.
Figure 5. Inhibitory effects of quinidine and HET0016 on BAC consumption in HLM and recombinant CYPs.
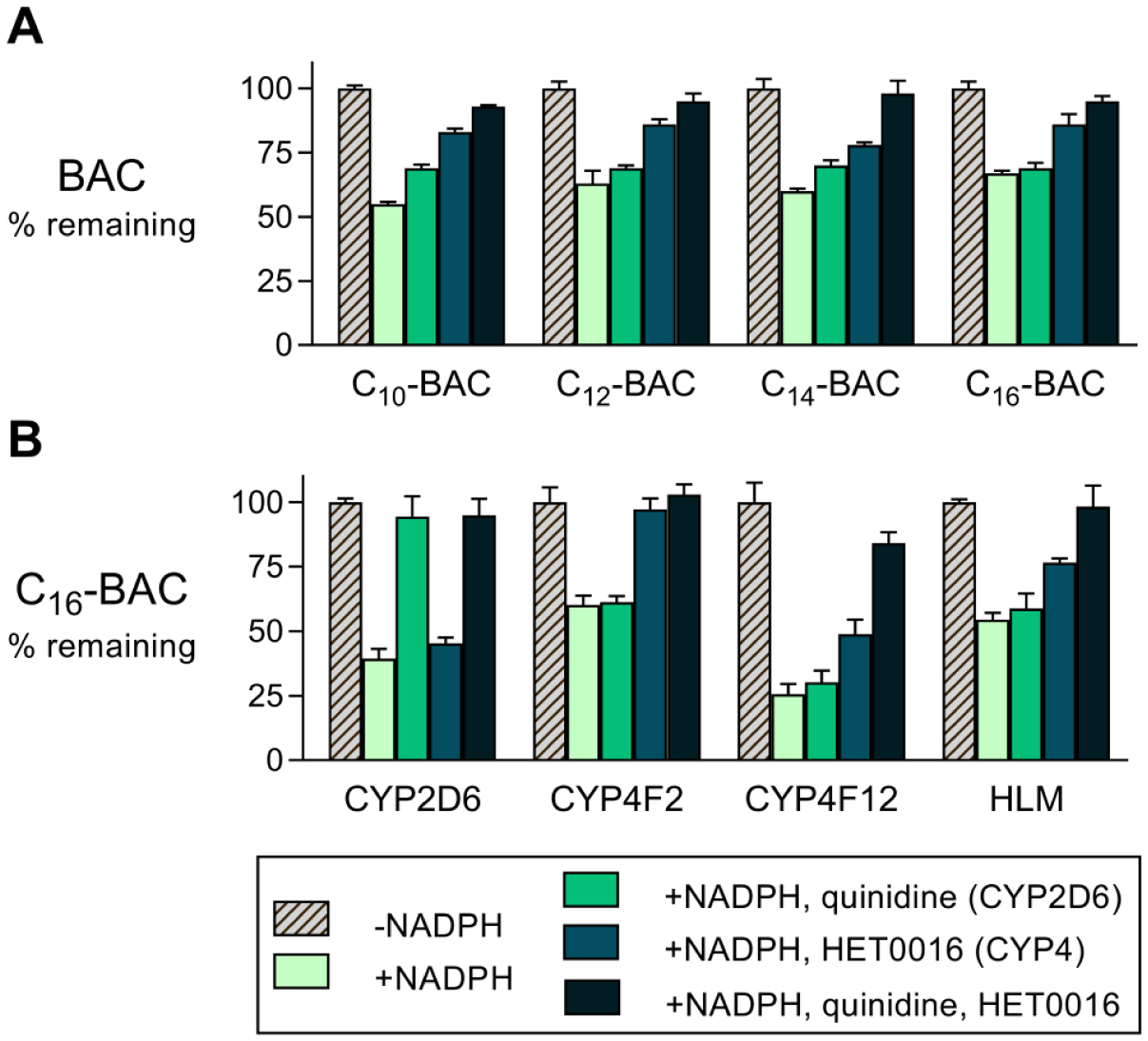
(A) Inhibition of BAC (2 μM) consumption in NADPH-supplemented HLM (0.5 mg/mL) by quinidine (1 μM; CYP2D6 inhibitor) and HET0016 (1 μM; CYP4 inhibitor) applied individually or in combination. Incubation times were 2, 5, 10, and 20 minutes, respectively, for C10-, C12-, C14-, and C16-BAC substrates. (B) Inhibition of C16-BAC (2 μM) consumption in recombinant CYP incubations (2D6, 4F2, and 4F12; 50 nM P450 enzyme) and in HLM (0.5 mg/mL) by the inhibitors quinidine (1 μM) and HET0016 (1 μM) in 20 minute incubations. Note, in panels A and B, the rightmost groups are identical experiments performed in HLM on different days to assess reproducibility of the inhibitor effects upon C16-BAC consumption. Each quantity represents the mean ± SD (n = 3).
The selectivity of the employed inhibitors for individual CYP isoforms will determine how the above results should be interpreted. Quinidine is a well-established and selective inhibitor of CYP2D6.38–40 HET0016 is a potent inhibitor of CYPs 4A11, 4F2, and 4F3B, but a less potent inhibitor of 4F12.41 Therefore, one may suspect that neither inhibitor effectively inhibited CYP4F12 activity in HLM experiments. To test this assumption and to assess the selectivity of the chosen inhibitors, C16-BAC was incubated with recombinant CYPs 2D6, 4F2, and 4F12 under identical conditions as was performed in HLM: 20 minute incubations with 2 μM substrate in the presence or absence of 1 μM inhibitor (Figure 5B). C16-BAC was the proper substrate to carry out this test as it is a substrate for all three CYP isoforms (Figure 2B). For reference, and to examine reproducibility, the identical experiment in HLM was reproduced on the same day and included in this dataset. This analysis confirms that quinidine, but not HET0016, was an effective inhibitor of recombinant CYP2D6 while HET0016, but not quinidine, was an effective inhibitor of recombinant CYP4F2 (Figure 5B). Both inhibitors offered essentially complete inhibition of their respective target enzymes. For recombinant CYP4F12, quinidine did not inhibit C16-BAC consumption while HET0016 displayed moderate inhibition of C16-BAC consumption. However, interestingly, values of C16-BAC percent remaining were 26% ± 4% (no inhibitor), 49% ± 6% (HET0016 alone), and 84% ± 4% (HET0016 + quinidine) (mean ± SD, n = 3). Thus, the combination of quinidine and HET0016 (1 μM each) was surprisingly effective at blocking C16-BAC consumption by recombinant CYP4F12, suggesting that this combination may have indeed been inhibitory toward CYP4F12 activity in our HLM experiments. On the same day that these inhibitors were evaluated against the recombinant CYP isoforms, we further established that the effects of each inhibitor on C16-BAC consumption in HLM were reasonably reproducible (Figure 5B) as can be seen by comparison of the rightmost groups in panels A and B of Figure 5 (identical experiments evaluating inhibitory effects on C16-BAC consumption in HLM, but performed on different days).
We next examined the susceptibility of C10-BAC metabolite formation in HLM to inhibition by quinidine and HET0016. First, time-courses of C10-BAC depletion in HLM (0.2 mg/mL) in the absence of any inhibitor beginning at 680 nM substrate were carried out to assess the mass balance contribution of ω- and (ω−1)-hydroxy metabolite formation and of further oxidized C10-BAC metabolites (Figure 6). As expected, both depletion of C10-BAC and metabolite formation were entirely NADPH-dependent. At the 4-minute time point, half of the initial C10-BAC concentration had been depleted to 341 ± 7 nM (Figure 6A). Of the approximate 340 nM decrease in substrate, a majority of that quantity was accounted for by the ω-hydroxy metabolite at 54% (185 out of 340 nM) and the (ω−1)-hydroxy metabolite at 22% (76 out of 340 nM). In sum, these two major metabolites accounted for 77% (261 out of 340 nM) of C10-BAC depletion that had occurred by the 4-minute time point. It was also apparent by 4 minutes that further oxidized sequential metabolites of ω- and (ω−1)-hydroxy C10-BAC, such as (ω, ω−1)-dihydroxy, ω-carboxylic acid, (ω−1)-hydroxy metabolites had begun to form, thereby depleting the hydroxylated species during their formation as indicated by the plateau in the two hydroxy products from the 8-minute to 16-minute time points (Figures 6B and 6C). By 16 minutes, C10-BAC depletion was essentially complete (Figure 6A). The mass balance of metabolite formed relative to substrate consumed was calculated as: 45% ω-hydroxy, 19% (ω−1)-hydroxy, 15% (ω−1)-ketone, <1% (ω, ω−1)-dihydroxy, and <1% ω-carboxylic acid. In total, approximately 80% of the mass balance could be accounted for with these five metabolites.
Figure 6. Inhibitory effects of quinidine and HET0016 on C10-BAC metabolite formation in HLM.
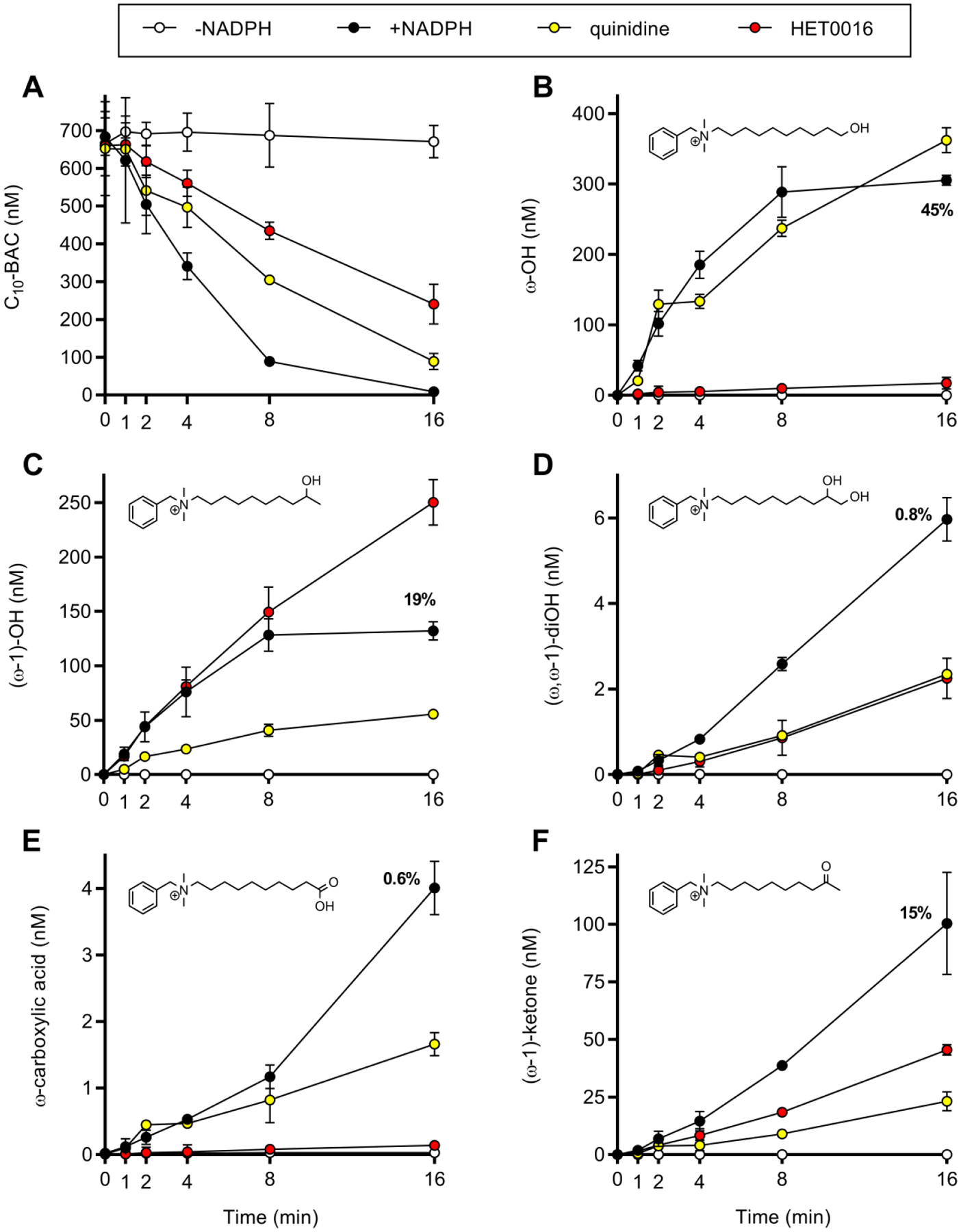
Time-courses of (A) C10-BAC (680 nM) depletion and (B) ω-hydroxy, (C) (ω−1)-hydroxy, (D) (ω, ω−1)-dihydroxy, (E) ω-carboxylic acid, and (F) (ω−1)-ketone metabolite formation in HLM (0.2 mg/mL) were measured in the absence (open circles) and presence (closed circles) of NADPH out to 16 minutes. The 16-minute data points for the +NADPH condition are annotated with the percentage amount of metabolite formed relative to the amount of C10-BAC consumed at 16 minutes. Time-courses were also performed in the presence of quinidine (1 μM; CYP2D6 inhibitor; yellow circles) and HET0016 (1 μM; CYP4 inhibitor; red circles). Data points represent the mean ± SD (n = 3). When not visible, error bars are contained within the data point.
To examine the roles of CYP4 and CYP2D6 in producing the above C10-BAC metabolites, the inhibitors HET0016 (1 μM) and quinidine (1 μM) were applied to incubations run in parallel. The resulting time courses in the presence of either inhibitor are overlaid in Figure 6. HET0016 inhibited ω-hydroxy metabolite formation, but not (ω−1)-hydroxy metabolite formation, while the opposite was true for quinidine (Figures 6B and 6C). This evidence suggests that one or more CYP4 isoforms catalyze C10-BAC ω-hydroxylation while CYP2D6 contributes to (ω−1)-hydroxylation. Further, in these incubations, it appears that HET0016 did not inhibit CYP2D6 and quinidine did not inhibit CYP4 ω-hydroxylase activity. The inhibitory effects on sequential metabolites were more nuanced. As HET0016 accomplished nearly complete inhibition of ω-hydroxylation, the nearly complete inhibition of ω-carboxylic acid formation by HET0016 in Figure 6E is logical. However, understanding why quinidine caused partial suppression of ω-carboxylic acid formation would require further study of sequential metabolism. It is also unclear why substantial (ω, ω−1)-dihydroxy formation still occurred in the presence of HET0016 (Figure 6D) despite nearly complete inhibition of ω-hydroxylation by HET0016 (Figure 6B). It can further be seen that HET0016 promoted (ω−1)-hydroxylation (Figure 6C), while simultaneously suppressing (ω−1)-ketone formation (Figure 6F). Therefore, while the effects of CYP4 and CYP2D6 inhibition appear straightforward for ω- and (ω−1)-hydroxylation itself, the effects of the inhibitors on sequential metabolic reactions appears to involve a more complex interplay of metabolite interactions with these CYP isoforms. Overall, while the effects of these inhibitors were mixed, HET0016 was more effective at inhibiting oxidations at the ω-position and quinidine was more effective at inhibiting oxidations at the (ω−1)-position.
Kinetic Characterization of C10-BAC Hydroxylation in HLM.
Hydroxylation of BACs in HLM was a major route of metabolic clearance with ω- and (ω−1)-hydroxylation generating the two major primary metabolites of C10-BAC (Figures 6B and 6C). Measuring the initial rates of these reactions at concentrations ranging from 0.1 to 128 μM C10-BAC yielded Michaelis-Menten plots that had clearly reached saturation at low μM concentrations (Figure 7). Very low microsomal protein concentration (0.04 mg/mL) was utilized in this experiment to minimize depletion of the substrate and experimental conditions were optimized to capture initial rates of product formation (see Materials and Methods). Kinetic parameters for C10-BAC ω-hydroxylation were characterized by a Vmax of 380 pmol/min/mg and Km value of 0.69 μM. The (ω−1)-hydroxylation process was characterized by a Vmax of 126 pmol/min/mg and Km value of 0.13 μM. Thus, microsomal intrinsic clearance values (Vmax/Km) were 551 μL/min/mg for ω-hydroxylation and 969 μL/min/mg for (ω−1)-hydroxylation.
Figure 7. C10-BAC ω- and (ω−1)-hydroxylation rates in HLM.
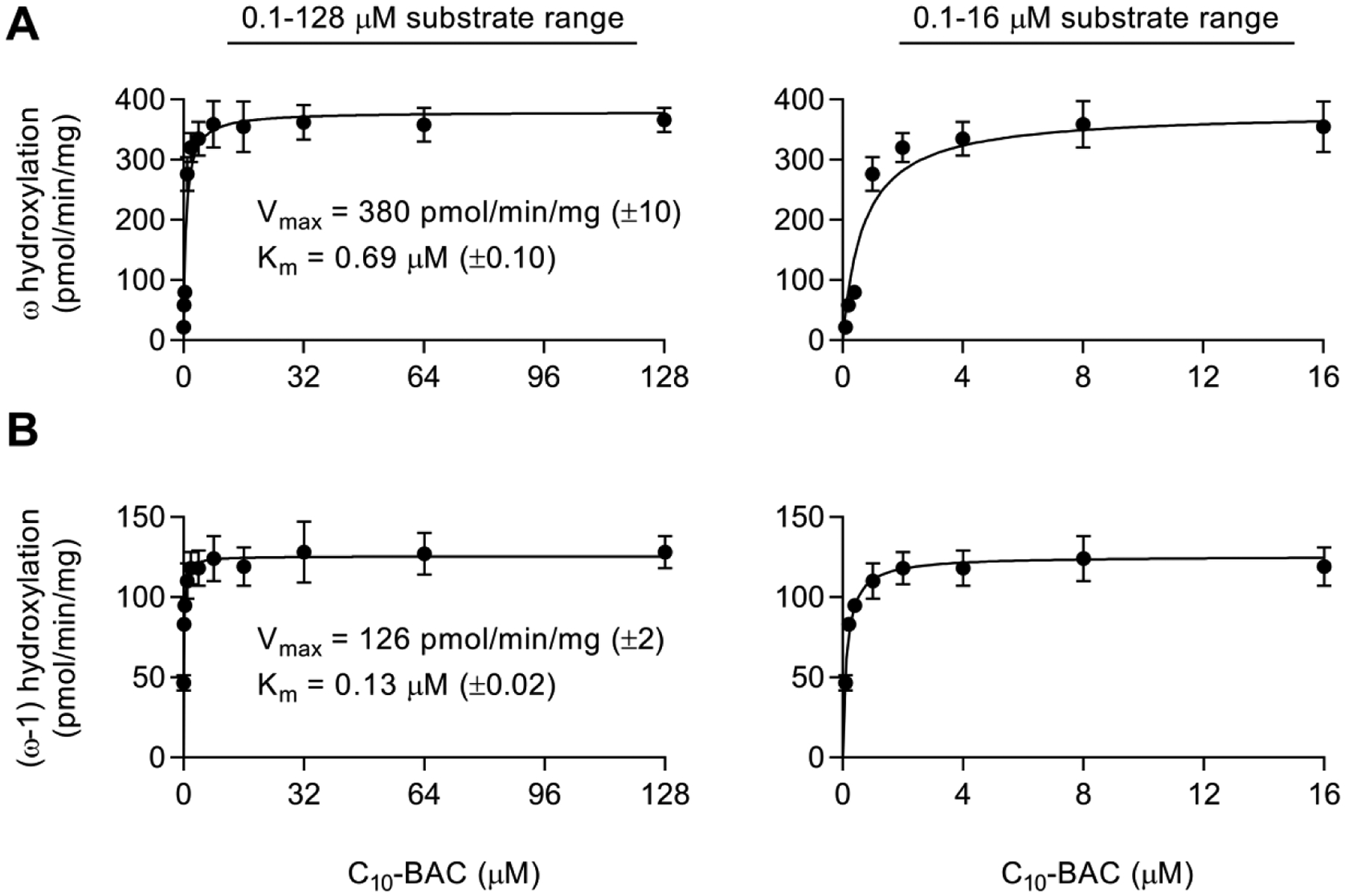
Initial rates of C10-BAC (A) ω-hydroxylation and (B) (ω−1)-hydroxylation in NADPH-supplemented HLM (0.04 mg/mL) determined at substrate concentrations of 0.1, 0.2, 0.4, 1, 2, 4, 8, 16, 32, 64, and 128 μM. In panels A and B, the left-side graphs display rates obtained from the full substrate concentration range and the right-side graphs display the rates of only the lower substrate concentrations (0.1–16 μM). Michaelis-Menten kinetic parameters (Vmax, Km) were estimated by nonlinear regression. Data points represent the mean ± SD (n = 3). When not visible, error bars are contained within the data point.
Biological Consequence of C10-BAC Hydroxylation on Inhibition of Sterol Biosynthesis
We previously demonstrated23 that short-chain BACs (C10 and C12) inhibit DHCR7 in the cholesterol biosynthesis pathway, similar to the small molecule AY9944 (a teratogen). To investigate whether BAC metabolites also inhibit cholesterol biosynthesis, we exposed Neuro2a cells to AY9944 (as positive control), C10-BAC, and the ω- and (ω−1)-hydroxylated metabolites of C10-BAC (each at 100 nM) and assessed the resulting changes in cellular sterol levels (Figure 8). After 72 hour exposure, cells exposed to AY9944 and C10-BAC displayed significant cellular accumulation of 7-dehydrocholesterol (7-DHC), 7-dehydrodesmosterol (7-DHD), 8-dehydrocholesterol, and zymosterol, and significant decreased level of cellular desmosterol and cholesterol (products of 7-DHD and 7-DHC, respectively, via Dhcr7). In contrast, these concerning sterol changes were greatly diminished in cells treated with the ω- and (ω−1)-hydroxy metabolites. The results demonstrate that although the two major metabolites of C10-BAC do retain some capacity to inhibit cholesterol biosynthesis, their potency in inhibiting Dhcr7 is minimum in comparison with the parent molecule, C10-BAC. For example, cellular accumulation of 7-DHC caused by the metabolites was over 2 orders of magnitude less than the accumulation caused by C10-BAC. As such, CYP-mediated BAC oxidation appears to play an overall detoxifying role with respect to the inhibition of cholesterol biosynthesis elicited by short-chain BACs. Additional comprehensive work will be pursued in order to assess the effect of CYP metabolism on the systemic toxicity of BACs.
Figure 8. Cellular sterol levels following exposure to AY9944, C10-BAC, and ω- and (ω−1)-hydroxylated C10-BAC metabolites.
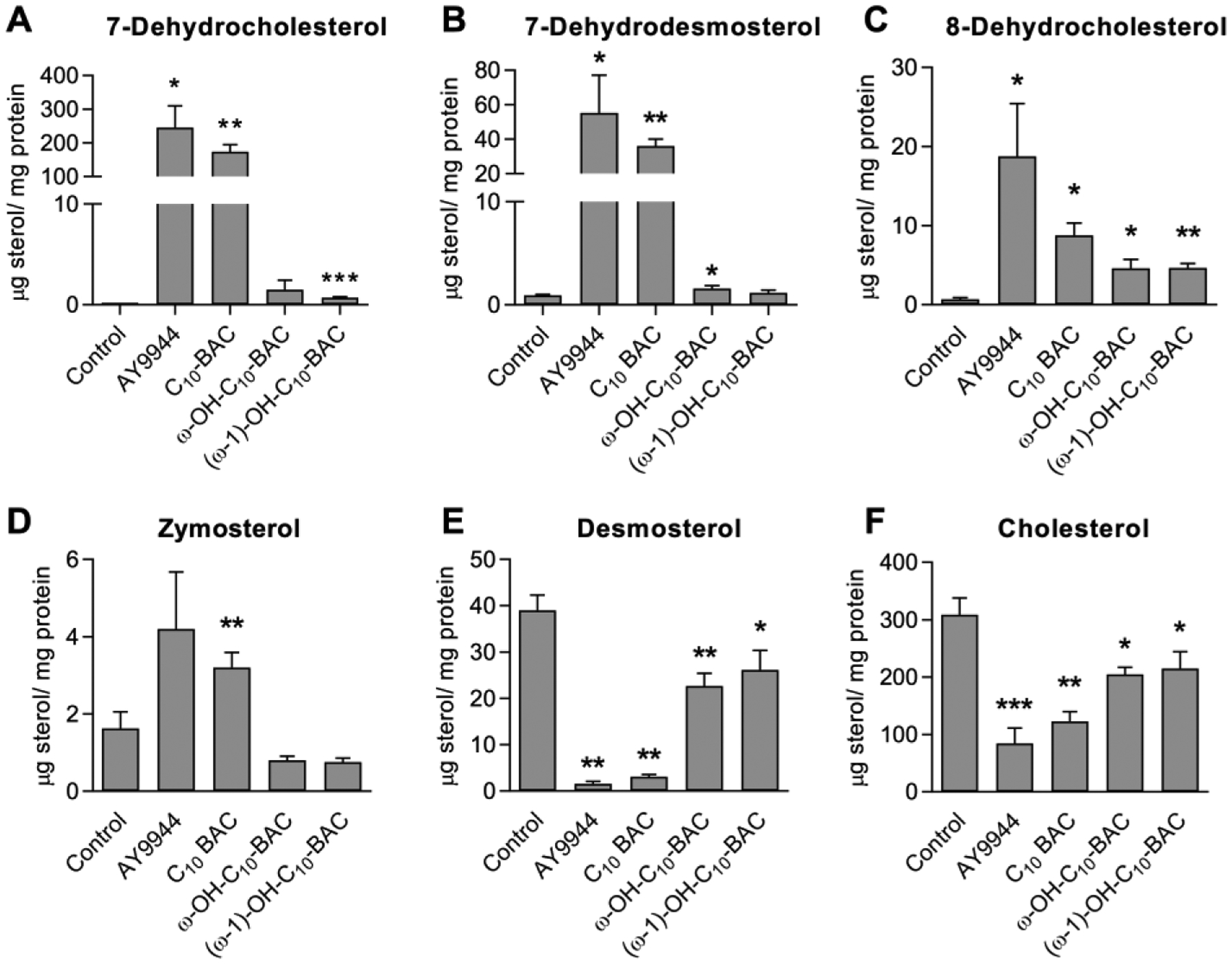
Neuro2a cells were exposed to 100 nM test compound over 72 hours in serum-free media: Control (0.1% DMSO), AY9944 (positive control; DHCR7 inhibitor), C10-BAC, ω-hydroxy C10-BAC, and (ω−1)-hydroxy C10-BAC. Levels of the indicated sterols in lipid extracts from treated cells were measured by LC-MS and normalized to protein content. Data represent the mean ± SD (n = 3). Statistical-significance is relative to the Control group: p ≤0.05 (*); p ≤0.01 (**); p ≤0.001 (***).
Synthesis of C10-BAC Metabolite Standards.
The ω-hydroxy derivative of C10-BAC was synthesized as described under Materials and Methods and the chemical structure was confirmed by 1H-NMR (Figure S18). The ω-alkene derivative of C10-BAC was synthesized, confirmed by 1H-NMR (Figure S19), and used as the starting material to synthesize additional C10-BAC metabolites. Heating the ω-alkene in aqueous hydrochloric acid generated (ω−1)-hydroxy-C10-BAC as the major hydration product as predicted by Markovnikov’s rule and as confirmed by 1H-NMR (Figure S20). A minor hydration product was also formed and assumed to be (ω−2)-hydroxy-C10-BAC formed through either migration of the double-bond or rearrangement of the carbocation intermediate. Epoxidation of the ω-alkene with hydrogen peroxide under basic conditions34 gave the terminal epoxide, which was then ring-opened in heated aqueous sodium hydroxide to give (ω, ω−1)-dihydroxy-C10-BAC (Figure S21). Dess-Martin oxidation of (ω−1)-hydroxy-C10-BAC produced (ω−1)-ketone-C10-BAC (Figure S22). Dess-Martin oxidation of ω-hydroxy-C10-BAC proceeded past the ω-aldehyde intermediate to yield ω-carboxylic acid-C10-BAC, which was confirmed by 1H-NMR in two solvents (Figures S23 and S24). Accurate mass m/z values of all the above synthetic standards were consistent with their respective molecular formulas as detailed under Materials and Methods. Further, the LC-MS retention times and accurate mass m/z values of each synthesized standard matched a metabolite of C10-BAC generated from incubation with HLM and NADPH.
Discussion
BACs are used as cationic surfactants, excipients, and antimicrobial preservatives in a variety of pharmaceutical, medical, and personal care products. These products cover a range of administration routes including ocular, nasal, inhaled, oromucosal, and epicutaneous. The oral bioavailability of BACs has been demonstrated in a human case study involving accidental ingestion of a BAC cleaning solution wherein ~0.5 μM BAC was detectable in patient serum.29 Despite a long history of use of these compounds and a high likelihood for systemic exposure in the general population, no prior in vivo or in vitro studies on the human metabolism of BACs exist in the literature.
The major goal of this study was to elucidate the mechanisms of BAC hepatic metabolism using human liver microsomes and recombinant hepatic CYP isoforms. The chemical structures of BACs contain an even-numbered carbon chain of varying length (C8 to C18) connected to a positively charged quaternary ammonium group bearing an additional benzyl group and two methyl groups. Here we studied the most commonly used BACs with alkyl chain lengths from C10 to C16 (Figure 1A). BAC structures preclude direct conjugation or oxidation of the nitrogen heteroatom, but BACs could feasibly be susceptible to N-dealkylation and C-H oxidation reactions catalyzed by CYP enzymes.
In this study, we provide the first evidence that BACs are extensively metabolized by CYP enzymes in HLM (Figure 1B) and that BAC metabolic stability in HLM increases with alkyl chain length. This result could indicate that longer chain BACs are relatively poorer substrates for the relevant CYP isoforms or may simply reflect that increased lipophilicity increases non-specific binding to lipid membranes of microsomes. Using recombinant CYP enzymes (Figure 2) and specific inhibitors (Figures 5 and 6), we identified CYP4 isoforms and CYP2D6 to be the major contributors to BAC depletion in HLM. Further, we utilized deuterium-labeled substrates and MS/MS fragmentation to confirm that all CYP-mediated oxidation targeted the alkyl chain region of BACs; we saw no evidence of N-dealkylation. In the case of C10-BAC, we confirmed that CYP-mediated oxidation led to ω- and (ω−1)-hydroxy C10-BAC as the major products (Figure 4). Based on the relative polarity of the ω- and (ω−1)-hydroxy products and the similar patterns between the products of C10-BAC and other BACs, we tentatively assign the earlier +1O peak being the (ω−1)-hydroxy metabolite and the latter major peak being the ω-hydroxy metabolite for other BACs.
To our knowledge, this is the first ever report to describe CYP4-catalyzed metabolism of quaternary ammonium substrates. The CYP4 family of isoforms typically recognizes the carboxylate anion in their substrate repertoire while BACs bear positively charged head groups, making BACs unusual CYP4 substrates in this regard. Formation of ω-hydroxy C10-BAC is similar to other CYP4-catalyzed reactions, such as ω-oxidation of long chain fatty acids42 and elongated lipids like vitamin K1.43
On the other hand, a number of previously reported CYP2D6 substrates contain a basic nitrogen atom, which is likely positively charged under physiological conditions.44 Furthermore, a quaternary nitrogen is also present in the CYP2D6 fluorescent probe substrate AMMC.45 Interestingly, CYP2D6 was also found to catalyze (ω−1)-hydroxylation of the N-octyl group of pactimibe,44 same as the reaction of C10-BAC reported here. For pactimibe, the (ω−1)-hydroxy metabolite has been confirmed with an authentic standard and ligand docking simulations supported positioning of the (ω−1)-carbon atom 5.0 Å from the heme iron of CYP2D6.44, 46
Following initial oxidation of BACs in HLM, sequential metabolism of the primary hydroxylated metabolites to further oxidized secondary and tertiary metabolites was evident. We observed the sequential oxidation of ω-hydroxy C10-BAC to ω-carboxylic acid C10-BAC, sequential oxidation of (ω−1)-hydroxy C10-BAC to (ω−1)-ketone C10-BAC, and a convergence of the sequential oxidations of ω- or (ω−1)-hydroxy C10-BAC leading to the (ω, ω−1)-dihydroxy metabolite. The progression of sequential metabolism was apparent in our metabolite formation time-course data as indicated by a plateau in the levels of ω-and (ω−1)-hydroxy C10-BAC primary metabolites and a sharp increase in the sequential oxidation products from 8- to 16-minutes (Figure 6). Interestingly, while treatment with pan-CYP4 inhibitor HET0016 completely suppressed the formation of ω-hydroxy C10-BAC (Figure 6B), it actually increased the amount of (ω−1)-hydroxy C10-BAC formed by 16 minutes (Figure 6C). This could be due to an increase in the fraction of substrate metabolized by CYP2D6 in the presence of HET0016 or, alternatively, due to an attenuation of further sequential metabolism of the (ω−1)-hydroxy metabolite to the vicinal diol, (ω−1)-ketone, or other sequential oxidation products that consume this intermediary metabolite. The CYP2D6-specific inhibitor quinidine, on the other hand, was primarily inhibitory toward products requiring oxidation at the (ω−1)-position. However, interestingly, quinidine was also capable of inhibiting ω-carboxylate formation (Figure 6E) despite showing no inhibition of ω-hydroxy C10-BAC formation (Figure 6B). These results suggest that sequential oxidation of BAC metabolites is complex and involves multiple CYP isoforms.
In recombinant CYP2D6 and CYP4F12 incubations with C10-BAC, we primarily saw formation of (ω−1)-oxidized products. Although some (ω,ω−1)-dihydroxy and ω-carboxylate did form in these incubations, the ω-hydroxy metabolite was absent from the metabolite profiles with 2D6 and 4F12. This result suggests that CYPs 2D6 and 4F12 prefer internal oxidation of the BAC substrate. We did not identify an ω-hydroxylase CYP enzyme of C10-BAC in this study from the available recombinant CYPs (Figure 2). However, when C16-BAC was incubated with recombinant CYP4F2 we observed formation of the major microsomal +1O and +2O, −2H metabolites. We suggest that these metabolites are likely ω-hydroxy and ω-carboxylate C16-BAC, respectively, given that CYP4F2 is an ω-hydroxylase and HET0016 is an effective inhibitor of C16-BAC consumption in HLM (Figure 5).
The individual members of the CYP4 family responsible for ω-hydroxylation of BACs remain to be completely delineated. The other hepatic CYP4 isoforms that were not present in our panel include CYPs 4F11 and 4V2, and CYP4V2 is not expressed to a meaningful extent in human liver. CYP4F11 contains the critical ester linkage between an I-helix glutamate residue and the prosthetic heme group, which is the CYP active site structural feature seen in CYP4 isoforms that preferentially oxidize the ω-carbon position of elongated lipid substrates.47–50 Therefore, CYP4F11 is known to ω-hydroxylate lipid substrates while CYP4 isoforms without this ester linkage, such as CYP4F12, tend to oxidize at internal ω−1 and ω−2 carbon positions.42, 51 Our examination of ω- and (ω−1)-hydroxy C10-BAC metabolite formation in the presence of the CYP4 and CYP2D6 selective inhibitors HET0016 and quinidine, respectively, revealed that ω-hydroxylation is a CYP4-mediated process (Figure 6B) and (ω−1)-hydroxylation is largely CYP2D6-mediated with the remaining uninhibited portion likely accounted for by CYP4F12 (Figure 6C). CYP4F11 is currently the most likely candidate CYP4 enzyme to account for C10-BAC ω-hydroxylation. Furthermore, CYP4F11 is recognized to metabolize medium chain fatty acids (C10 – C16) indicating that C10-BAC and other BAC substrates, are within the range of CYP4F11 chain length tolerance.
Currently, we do not fully understand the role of metabolism in BAC toxicity or whether metabolism can protect humans from either chronic or acute BAC exposure. Nonetheless, the substantial contributions of CYP2D6 and CYP4 enzymes to BAC metabolism suggest that certain human populations may be more susceptible to BAC exposure due to polymorphisms of the genes encoding these enzymes. For CYP2D6, 10–17% of human populations are intermediate metabolizers while 5 – 10% are poor metabolizers.52 For CYP4F2, a common CYP4F2*3 variant (~27% allele frequency among Caucasians) was found to increase warfarin dose requirement as it decreases ω-hydroxylation of vitamin K.43, 53–55 Furthermore, variants in CYP4F11 and CYP4F12 were also found to reduce enzyme activities and lead to altered clinical outcomes.55–57 Thus, individuals with decreased expression or function of CYP2D6 and CYP4F enzymes could have an impaired ability to metabolically clear BACs and, by extension, these individuals may be more susceptible to potential detrimental effects exerted by BACs. While the effect of CYP-metabolism on the systemic toxicities of BACs requires further study, here we have demonstrated that metabolism of C10-BAC mitigates the potent inhibition of cholesterol biosynthesis exhibited by short-chain BACs. This was confirmed by exposing cells to parent C10-BAC and the ω- and (ω−1)-hydroxy metabolites, and then quantifying the impact of these exposures upon cellular sterol levels (Figure 8). The results indicate that converting C10-BAC to oxidized metabolites ameliorate the potent inhibition of Dhcr7 by this compound.
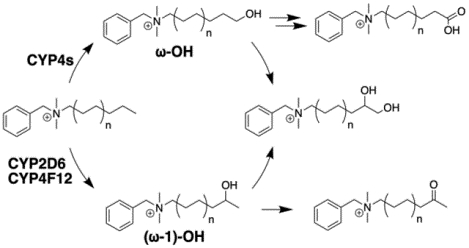
In conclusion, we propose that BAC metabolism by hepatic CYP enzymes can be primarily summarized as ω-hydroxylation by certain CYP4 family members, including CYP4F2, and (ω−1)-hydroxylation (or further internal oxidation) by CYPs 2D6 and 4F12, which is followed by sequential metabolism of the initial oxidation products (Figure 9). Polymorphisms affecting CYP2D6 and CYP4 function could be an important consideration when examining inter-individual variability in the susceptibility of distinct populations to BAC exposure, but further study is required to determine if these polymorphisms have a meaningful impact. Additionally, factors outside the scope of this study that may affect the overall in vivo persistence and toxicity of BACs include plasma protein binding, tissue binding, active transport, and Phase II conjugation reactions, and we intend to characterize these processes in future reports. As a first step, here we demonstrated Phase I oxidative metabolism of BACs by hepatic CYP enzymes and identified the major metabolic pathways. Identification of these BAC-metabolizing enzymes also shed light on potential local metabolism in the kidney as human kidney expresses all forms of the CYP4Fs42, 58 that can metabolize BACs. We further demonstrated that metabolism ameliorates potent inhibition of cholesterol biosynthesis by short-chain C10-BAC, thus highlighting the potential for metabolism to play a protective and detoxifying role.
Figure 9. Proposed summary of BAC metabolism by hepatic CYP enzymes.
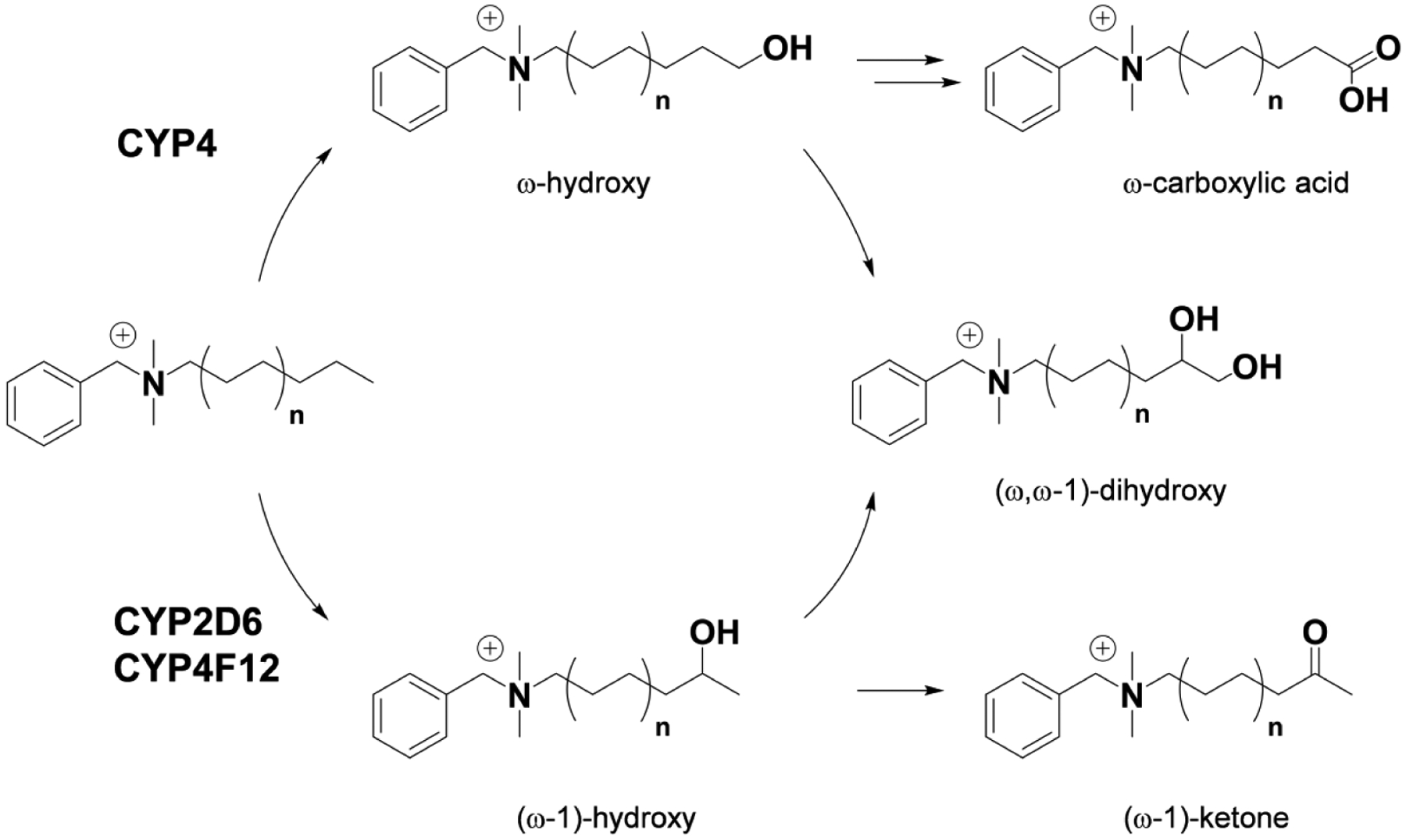
Metabolite structures were confirmed for C10-BAC (n=3) microsomal metabolism with authentic standards. Similar metabolism of longer alkyl chain BAC substrates (n=4, 5, and 6) and the assignment of the responsible CYP isoforms is supported by recombinant enzyme experiments and inhibitor studies described herein.
Supplementary Material
Acknowledgement
The authors wish to thank the University of Washington School of Pharmacy Mass Spectrometry Center for instrument assistance and Professor Allan Rettie for helpful discussion regarding CYP4 enzymes.
Funding Sources
This work was supported by a pilot grant from the University of Washington Center for Exposures, Diseases, Genomics, and Environment [National Institutes of Health P30ES007033], a National institutes of Health grant (R01HD092659) (to L.X.), startup funding from the Department of Medicinal Chemistry at the University of Washington (to L.X.), and the Environmental Pathology and Toxicology Training Grant [National Institute of Environmental Health Sciences T32-ES007032] (to R.P.S., J.M.H., and J.L.D.).
Abbreviations
- BAC(s)
benzalkonium chloride(s)
- CYP(s)
cytochrome(s) P450
- DHCR7
3β-hydroxysterol-Δ7-reductase
- DMSO
dimethyl sulfoxide
- HLM
human liver microsomes
- KPi
potassium phosphate buffer
- Km
Michaelis-Menten constant
- LC-MS
liquid chromatography-mass spectrometry
- QAC(s)
quaternary ammonium compound(s)
- SNP(s)
single nucleotide polymorphism(s)
- Vmax
maximum product formation rate
- Vmax/Km
intrinsic clearance
Footnotes
Supporting Information
LC-MS chromatograms, mass spectra, and 1H-NMR spectra of BAC metabolites
References
- (1).Neu TR (1996) Significance of bacterial surface-active compounds in interaction of bacteria with interfaces. Microbiological reviews 60, 151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kummerer K, Eitel A, Braun U, Hubner P, Daschner F, Mascart G, Milandri M, Reinthaler F, and Verhoef J (1997) Analysis of benzalkonium chloride in the effluent from European hospitals by solid-phase extraction and high-performance liquid chromatography with post-column ion-pairing and fluorescence detection. J Chromatogr A 774, 281–286. [DOI] [PubMed] [Google Scholar]
- (3).Tezel U, Pierson JA, and Pavlostathis SG (2006) Fate and effect of quaternary ammonium compounds on a mixed methanogenic culture. Water research 40, 3660–3668. [DOI] [PubMed] [Google Scholar]
- (4).Dean-Raymond D, and Alexander M (1977) Bacterial metabolism of quaternary ammonium compounds. Applied and environmental microbiology 33, 1037–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Games LM, King JE, and Larson RJ (1982) Fate and Distribution of a Quaternary Ammonium Surfactant, Octadecyltrimethylammonium Chloride (Otac), in Wastewater-Treatment. Environ Sci Technol 16, 483–488. [Google Scholar]
- (6).Holah JT, Taylor JH, Dawson DJ, and Hall KE (2002) Biocide use in the food industry and the disinfectant resistance of persistent strains of Listeria monocytogenes and Escherichia coli. Symp Ser Soc Appl Microbiol, 111S–120S. [PubMed] [Google Scholar]
- (7).Ratani SS, Siletzky RM, Dutta V, Yildirim S, Osborne JA, Lin W, Hitchins AD, Ward TJ, and Kathariou S (2012) Heavy metal and disinfectant resistance of Listeria monocytogenes from foods and food processing plants. Applied and environmental microbiology 78, 6938–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gilbert P, and Moore LE (2005) Cationic antiseptics: diversity of action under a common epithet. J Appl Microbiol 99, 703–715. [DOI] [PubMed] [Google Scholar]
- (9).McDonnell G, and Russell AD (1999) Antiseptics and disinfectants: activity, action, and resistance. Clin Microbiol Rev 12, 147–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Takeoka GR, Dao LT, Wong RY, and Harden LA (2005) Identification of benzalkonium chloride in commercial grapefruit seed extracts. J Agric Food Chem 53, 7630–7636. [DOI] [PubMed] [Google Scholar]
- (11).Kröckel L, Jira W, and Wild D (2003) Identification of benzalkonium chloride in food additives and its inefficacy against bacteria in minced meat and raw sausage batters. Eur Food Res Technol 216, 402–406. [Google Scholar]
- (12).BfR of Germany. (2012) Health assessment of benzalkonium chloride residues in food. BfR opinion No 032/2012, http://www.bfr.bund.de/cm/349/health-assessment-of-benzalkonium-chloride-residues-in-food.pdf. Last accessed Oct 13, 2019.
- (13).BfR of Germany. (2012) Health assessment of didecyldimethylammonium chloride (DDAC) residues in food. BfR opinion No 027/2012, http://www.bfr.bund.de/cm/349/health-assessment-of-didecyldimethylammonium-chloride-ddac-residues-in-food.pdf. Last accessed Oct 13, 2019.
- (14).Slimani K, Feret A, Pirotais Y, Maris P, Abjean JP, and Hurtaud-Pessel D (2017) Liquid chromatography-tandem mass spectrometry multiresidue method for the analysis of quaternary ammonium compounds in cheese and milk products: Development and validation using the total error approach. J Chromatogr A 1517, 86–96. [DOI] [PubMed] [Google Scholar]
- (15).US FDA. (2015) Safety and Effectiveness of Health Care Antiseptics; Topical Antimicrobial Drug Products for Over-the-Counter Human Use; Proposed Amendment of the Tentative Final Monograph; Reopening of Administrative Record. Federal Register 21 CFR Part 310, 80, 25165–25205. [Google Scholar]
- (16).US FDA. (2016) Safety and Effectiveness of Consumer Antiseptics; Topical Antimicrobial Drug Products for Over-the-Counter Human Use; Proposed Amendment of the Tentative Final Monograph; Reopening of Administrative Record Federal Register 21 CFR Part 310, 81, 42911–42937. [PubMed] [Google Scholar]
- (17).Davis HC, and Hidu H (1969) Effects of pesticides on embryonic development of clams and oysters and on survival and growth of the larvae. Fish. Bull 67, 393–404. [Google Scholar]
- (18).Nalecz-Jawecki G, Grabinska-Sota E, and Narkiewicz P (2003) The toxicity of cationic surfactants in four bioassays. Ecotoxicology and environmental safety 54, 87–91. [DOI] [PubMed] [Google Scholar]
- (19).Herman JR, and Bass P (1989) Enteric neuronal ablation: structure-activity relationship in a series of alkyldimethylbenzylammonium chlorides. Fundam Appl Toxicol 13, 576–584. [DOI] [PubMed] [Google Scholar]
- (20).Sarkar J, Chaudhary S, Namavari A, Ozturk O, Chang JH, Yco L, Sonawane S, Khanolkar V, Hallak J, and Jain S (2012) Corneal neurotoxicity due to topical benzalkonium chloride. Invest Ophthalmol Vis Sci 53, 1792–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Melin VE, Potineni H, Hunt P, Griswold J, Siems B, Werre SR, and Hrubec TC (2014) Exposure to common quaternary ammonium disinfectants decreases fertility in mice. Reproductive toxicology 50, 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hrubec TC, Melin VE, Shea CS, Ferguson EE, Garofola C, Repine CM, Chapman TW, Patel HR, Razvi RM, Sugrue JE, Potineni H, Magnin-Bissel G, and Hunt PA (2017) Ambient and Dosed Exposure to Quaternary Ammonium Disinfectants Causes Neural Tube Defects in Rodents. Birth Defects Res 109, 1166–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Herron J, Reese R, Tallman KA, Narayanaswamy R, Porter NA, and Xu L (2016) Identification of Environmental Quaternary Ammonium Compounds as Direct Inhibitors of Cholesterol Biosynthesis. Toxicol. Sci 151, 261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Xu L, Korade Z, Rosado DA, Liu W, Lamberson CR, and Porter NA (2011) An oxysterol biomarker for 7-dehydrocholesterol oxidation in cell/mouse models for Smith-Lemli-Opitz syndrome. J. Lipid Res 52, 1222–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Xu L, Korade Z, Rosado DA Jr., Mirnics K, and Porter NA (2013) Metabolism of oxysterols derived from nonenzymatic oxidation of 7-dehydrocholesterol in cells. J Lipid Res 54, 1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Xu L, and Porter NA (2015) Free radical oxidation of cholesterol and its precursors: Implications in cholesterol biosynthesis disorders. Free radical research 49, 835–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hines K, Herron J, and Xu L (2017) Assessment of Altered Lipid Homeostasis by HILIC-Ion Mobility-Mass Spectrometry-Based Lipidomics. J Lipid Res 58, 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Herron JM, Hines KM, Tomita H, Seguin RP, Cui JY, and Xu L (2019) Multi-omics investigation reveals benzalkonium chloride disinfectants alter sterol and lipid homeostasis in the mouse neonatal brain. Toxicol Sci, In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Xue Y, Hieda Y, Kimura K, Nishiyama T, and Adachi T (2002) Sensitive determination of benzalkonium chloride in blood and tissues using high-performance liquid chromatography with solid-phase extraction. Legal medicine (Tokyo, Japan) 4, 232–238. [DOI] [PubMed] [Google Scholar]
- (30).Xue Y, Hieda Y, Saito Y, Nomura T, Fujihara J, Takayama K, Kimura K, and Takeshita H (2004) Distribution and disposition of benzalkonium chloride following various routes of administration in rats. Toxicol Lett 148, 113–123. [DOI] [PubMed] [Google Scholar]
- (31).Xue Y, Hieda Y, Kimura K, Takayama K, Fujihara J, and Tsujino Y (2004) Kinetic characteristics and toxic effects of benzalkonium chloride following intravascular and oral administration in rats. J Chromatogr B Analyt Technol Biomed Life Sci 811, 53–58. [DOI] [PubMed] [Google Scholar]
- (32).Kuca K, Marek J, Stodulka P, Musilek K, Hanusova P, Hrabinova M, and Jun D (2007) Preparation of benzalkonium salts differing in the length of a side alkyl chain. Molecules (Basel, Switzerland) 12, 2341–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Isobe T, and Ishikawa T (1999) 2-Chloro-1,3-dimethylimidazolinium Chloride. 3. Utility for Chlorination, Oxidation, Reduction, and Rearrangement Reactions. The Journal of Organic Chemistry 64, 5832–5835. [Google Scholar]
- (34).Bach RD, Knight JW, Fowler KW, and Buchi G (1981) Epoxidation of Olefins by Hydrogen Peroxide-Acetonitrile: cis -Cyclooctene Oxide. Organic Syntheses 60, 63. [Google Scholar]
- (35).Omura T, and Sato R (1964) THE CARBON MONOXIDE-BINDING PIGMENT OF LIVER MICROSOMES. I. EVIDENCE FOR ITS HEMOPROTEIN NATURE. The Journal of biological chemistry 239, 2370–2378. [PubMed] [Google Scholar]
- (36).Herron J, Hines KM, and Xu L (2018) Assessment of Altered Cholesterol Homeostasis by Xenobiotics Using Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry. Curr Protoc Toxicol 78, e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Fliesler SJ, Peachey NS, Herron J, Hines KM, Weinstock NI, Ramachandra Rao S, and Xu L (2018) Prevention of Retinal Degeneration in a Rat Model of Smith-Lemli-Opitz Syndrome. Sci Rep 8, 1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).von Bahr C, Spina E, Birgersson C, Ericsson O, Goransson M, Henthorn T, and Sjoqvist F (1985) Inhibition of desmethylimipramine 2-hydroxylation by drugs in human liver microsomes. Biochemical pharmacology 34, 2501–2505. [DOI] [PubMed] [Google Scholar]
- (39).Otton SV, Brinn RU, and Gram LF (1988) In vitro evidence against the oxidation of quinidine by the sparteine/debrisoquine monooxygenase of human liver. Drug metabolism and disposition: the biological fate of chemicals 16, 15–17. [PubMed] [Google Scholar]
- (40).Branch RA, Adedoyin A, Frye RF, Wilson JW, and Romkes M (2000) In vivo modulation of CYP enzymes by quinidine and rifampin. Clinical pharmacology and therapeutics 68, 401–411. [DOI] [PubMed] [Google Scholar]
- (41).Yamaori S, Araki N, Shionoiri M, Ikehata K, Kamijo S, Ohmori S, and Watanabe K (2018) A Specific Probe Substrate for Evaluation of CYP4A11 Activity in Human Tissue Microsomes and a Highly Selective CYP4A11 Inhibitor: Luciferin-4A and Epalrestat. The Journal of pharmacology and experimental therapeutics 366, 446–457. [DOI] [PubMed] [Google Scholar]
- (42).Johnson AL, Edson KZ, Totah RA, and Rettie AE (2015) Cytochrome P450 omega-Hydroxylases in Inflammation and Cancer. Advances in pharmacology (San Diego, Calif.) 74, 223–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Edson KZ, Prasad B, Unadkat JD, Suhara Y, Okano T, Guengerich FP, and Rettie AE (2013) Cytochrome P450-dependent catabolism of vitamin K: omega-hydroxylation catalyzed by human CYP4F2 and CYP4F11. Biochemistry 52, 8276–8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kotsuma M, Tokui T, Ishizuka-Ozeki T, Honda T, Iwabuchi H, Murai T, Ikeda T, and Saji H (2008) CYP2D6-Mediated metabolism of a novel acyl coenzyme A:cholesterol acyltransferase inhibitor, pactimibe, and its unique plasma metabolite, R-125528. Drug metabolism and disposition: the biological fate of chemicals 36, 529–534. [DOI] [PubMed] [Google Scholar]
- (45).Chauret N, Dobbs B, Lackman RL, Bateman K, Nicoll-Griffith DA, Stresser DM, Ackermann JM, Turner SD, Miller VP, and Crespi CL (2001) The use of 3-[2-(N,N-diethyl-N-methylammonium)ethyl]-7-methoxy-4-methylcoumarin (AMMC) as a specific CYP2D6 probe in human liver microsomes. Drug metabolism and disposition: the biological fate of chemicals 29, 1196–1200. [PubMed] [Google Scholar]
- (46).Kotsuma M, Hanzawa H, Iwata Y, Takahashi K, and Tokui T (2008) Novel binding mode of the acidic CYP2D6 substrates pactimibe and its metabolite R-125528. Drug metabolism and disposition: the biological fate of chemicals 36, 1938–1943. [DOI] [PubMed] [Google Scholar]
- (47).Henne KR, Kunze KL, Zheng YM, Christmas P, Soberman RJ, and Rettie AE (2001) Covalent linkage of prosthetic heme to CYP4 family P450 enzymes. Biochemistry 40, 12925–12931. [DOI] [PubMed] [Google Scholar]
- (48).Zheng YM, Baer BR, Kneller MB, Henne KR, Kunze KL, and Rettie AE (2003) Covalent heme binding to CYP4B1 via Glu310 and a carbocation porphyrin intermediate. Biochemistry 42, 4601–4606. [DOI] [PubMed] [Google Scholar]
- (49).Baer BR, Kunze KL, and Rettie AE (2007) Mechanism of formation of the ester linkage between heme and Glu310 of CYP4B1: 18O protein labeling studies. Biochemistry 46, 11598–11605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Hoch U, and Ortiz De Montellano PR (2001) Covalently linked heme in cytochrome p4504a fatty acid hydroxylases. The Journal of biological chemistry 276, 11339–11346. [DOI] [PubMed] [Google Scholar]
- (51).Edson KZ, and Rettie AE (2013) CYP4 enzymes as potential drug targets: focus on enzyme multiplicity, inducers and inhibitors, and therapeutic modulation of 20-hydroxyeicosatetraenoic acid (20-HETE) synthase and fatty acid omega-hydroxylase activities. Current topics in medicinal chemistry 13, 1429–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Zhou SF (2009) Polymorphism of human cytochrome P450 2D6 and its clinical significance: Part I. Clin Pharmacokinet 48, 689–723. [DOI] [PubMed] [Google Scholar]
- (53).McDonald MG, Rieder MJ, Nakano M, Hsia CK, and Rettie AE (2009) CYP4F2 is a vitamin K1 oxidase: An explanation for altered warfarin dose in carriers of the V433M variant. Molecular pharmacology 75, 1337–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Caldwell MD, Awad T, Johnson JA, Gage BF, Falkowski M, Gardina P, Hubbard J, Turpaz Y, Langaee TY, Eby C, King CR, Brower A, Schmelzer JR, Glurich I, Vidaillet HJ, Yale SH, Qi Zhang K, Berg RL, and Burmester JK (2008) CYP4F2 genetic variant alters required warfarin dose. Blood 111, 4106–4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Jarrar YB, and Lee SJ (2019) Molecular Functionality of Cytochrome P450 4 (CYP4) Genetic Polymorphisms and Their Clinical Implications. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Yi M, Cho SA, Min J, Kim DH, Shin JG, and Lee SJ (2017) Functional characterization of a common CYP4F11 genetic variant and identification of functionally defective CYP4F11 variants in erythromycin metabolism and 20-HETE synthesis. Arch Biochem Biophys 620, 43–51. [DOI] [PubMed] [Google Scholar]
- (57).Cauffiez C, Klinzig F, Rat E, Tournel G, Allorge D, Chevalier D, Pottier N, Lovecchio T, Colombel JF, Lhermitte M, Lo-Guidice JM, and Broly F (2004) Human CYP4F12 genetic polymorphism: identification and functional characterization of seven variant allozymes. Biochemical pharmacology 68, 2417–2425. [DOI] [PubMed] [Google Scholar]
- (58).Knights KM, Rowland A, and Miners JO (2013) Renal drug metabolism in humans: the potential for drug-endobiotic interactions involving cytochrome P450 (CYP) and UDP-glucuronosyltransferase (UGT). Br J Clin Pharmacol 76, 587–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.