

Abstract
Inflammation is involved in the pathogenesis of many disorders. However, the underlying mechanisms are often unknown. Here, we test whether cystinosin, the protein involved in cystinosis, is a critical regulator of galectin-3, a member of the β-galactosidase binding protein family, during inflammation. Cystinosis is a lysosomal storage disorder and, despite ubiquitous expression of cystinosin, the kidney is the primary organ impacted by the disease. Cystinosin was found to enhance lysosomal localization and degradation of galectin-3. In Ctns−/− mice, a mouse model of cystinosis, galectin-3 is overexpressed in the kidney. The absence of galectin-3 in cystinotic mice ameliorates pathologic renal function and structure and decreases macrophage/monocyte infiltration in the kidney of the Ctns−/− Gal3−/− mice compared to Ctns−/− mice. These data strongly suggest that galectin-3 mediates inflammation involved in kidney disease progression in cystinosis. Furthermore, galectin-3 was found to interact with the pro-inflammatory cytokine Monocyte Chemoattractant Protein-1, which stimulates the recruitment of monocytes/macrophages, and proved to be significantly increased in the serum of Ctns−/− mice and also patients with cystinosis. Thus, our findings highlight a new role for cystinosin and galectin-3 interaction in inflammation and provide an additional mechanistic explanation for the kidney disease of cystinosis. This may lead to the identification of new drug targets to delay cystinosis progression.
Keywords: chronic kidney disease, cystinosis, galectin-3, inflammation, monocyte chemoattractant protein–1
Inflammation is a normal acute response of an organism to injury or infection,1 but chronic inflammation is associated with tissue damage. In the case of chronic disease, such as diabetes, damaged tissues activate the immune system, leading to a continuous inflammatory response and, eventually, tissue degeneration.2 Cytokines are key modulators of inflammation and are capable of activating or resolving the inflammation process.3 In patients with chronic kidney disease (CKD), elevated plasma concentration of cytokines (interleukin [IL]-6 and tumor necrosis factor–α) and inflammation markers (C-reactive protein, IL-6, hyaluronan, and neopterin) are associated with progression to end-stage renal disease.4 Understanding the specific mediators that trigger inflammatory responses facilitates the discovery of appropriate drug targets to prevent degenerative damage.
Cystinosis is an autosomal-recessive metabolic disease that belongs to the lysosomal storage disorder family and is characterized by the accumulation of cystine within all organs.5 The gene defective in cystinosis, CTNS, encodes the lysosomal cystine/proton co-transporter, cystinosin.6,7 Although CTNS is ubiquitously expressed, the kidney is the first organ affected in persons with cystinosis. Patients typically present in their first year of life with Fanconi syndrome, characterized by severe fluid and electrolyte disturbances, poor growth, and rickets.8 CKD subsequently develops, which progresses to end-stage renal disease and requires kidney transplantation. Cystine accumulation in all tissues eventually leads to multiorgan dysfunction; patients experience photophobia and blindness, hypothyroidism, hypogonadism, diabetes, myopathy, and central nervous system deterioration.9 In the C57BL/6 background, a knockout mouse model (Ctns−/−)10 replicates the kidney phenotype of cystinotic patients,11 as well as deposition of cystine crystals in the cornea12 and thyroid dysfunction.13
Besides supportive therapy, the current treatment for cystinosis is the substrate depletion drug cysteamine, which delays the progression of cystinosis complications but has no effect on Fanconi syndrome and does not prevent end-stage renal disease.14–16 The specific sensitivity of the kidneys in persons with cystinosis is still not fully understood because cystine accumulates in all tissues.17 Recently, new cellular pathways were shown to be affected by the absence of cystinosin, in addition to cystine transport. As such, chaperone-mediated autophagy (CMA), a selective autophagy degrading KFERQ sequence-bearing proteins, is impaired, and lysosome-associated membrane protein 2, a main protagonist of CMA, is mislocalized in murine Ctns-deficient cells and tissues.18 Interaction of cystinosin with almost all components of vacuolar-type H+ adenosine triphosphatase, ragulator, and RagA/RagC was demonstrated, as well as defective lysosomal recruitment of mammalian target of rapamycin upon nutrient re-introduction after shortage in the absence of cystinosin.19 Expression of transcription factor EB, a regulator of lysosomal clearance, autophagy, and anabolism, also was shown to be impaired in cystinosin-deficient proximal tubular cells.20 A striking common property of these studies is that defects in mammalian target of rapamycin signaling, transcription factor EB expression, and CMA could not be corrected upon cystine clearance by cysteamine, showing that these cellular anomalies are due to the absence of cystinosin, besides cystine overload.18–20
In the present study, we reveal a new role for cystinosin in inflammation through its interaction with Gal-3. Gal-3 is a member of the lectin and β-galactoside–binding protein family 21 and is involved in multiple biologic functions, including inflammation.22 In the context of kidney diseases, Gal-3 inhibition was shown to reduce proinflammatory marker expression and renal fibrosis and prevent the decline of glomerular filtration rate in hyperaldosteronism, hypertension, or obesity models.23–26 Here we provide evidence that, in cystinosis, absence of cystinosin leads to Gal-3 overexpression in kidneys, enhancing macrophage infiltration and CKD progression. In addition, we identify monocyte chemoattractant protein–1 (MCP-1) as a potential mediator, revealing new gene targets for drug therapy.
RESULTS
Gal-3 interacts with cystinosin via its carbohydrate recognition domain
To identify specific interaction partners using quantitative mass spectrometry, Madin-Darby canine kidney (MDCK) cells were transduced with a lentiviral construct to stably express the cystinosin–green fluorescent protein (GFP) fusion protein. In addition to the 9 subunits of vacuolar-type H+ adenosine triphosphatase published previously,19 Gal-3 was identified as one of the proteins interacting with cystinosin (Figure 1a). This interaction was further confirmed by co-immunoprecipitation followed by Western blotting (Figure 1b and c). In addition, we showed that interaction between Gal-3 and cystinosin was mediated by Gal-3 carbohydrate recognition domain (CRD) localized in the C-terminal tail of the protein. Interaction was inhibited by thiodigalactoside, a potent inhibitor of galectin-carbohydrate interactions (Figure 1d). Like all galectins, Gal-3 has an affinity for glycosylated proteins,21 so it is likely that the interaction with Gal-3 occurs through the cystinosin glycosylated moiety located in the intralysosomal N-terminal tail of the protein.27
Figure 1 |. Galectin-3 (Gal-3) interacts with cystinosin–green fluorescent protein (GFP) via its carbohydrate recognition domain.

(a) Volcano plot representation of proteins immunoprecipitated by the anti-GFP antibody and identified by mass spectrometry (nanoRSLC-Q Exactive Plus MS) in Madin-Darby canine kidney (MDCK) cells overexpressing cystinosin-GFP versus nontransfected cells (4 independent experiments). Lysates of MDCK cells stably expressing cystinosin-GFP were immunoprecipitated with (b) anti-GFP or (c) anti–Gal-3 antibodies, and co-immunoprecipitated proteins were analyzed by Western blotting (n = 3). (d) Lysates of MDCK cells stably expressing cystinosin-GFP were treated or not treated for 30 minutes with 5 mM thiodigalactoside (TDG), a potent inhibitor of galectin-carbohydrate interactions. Lysates were then immunoprecipitated with anti-GFP antibodies and co-immunoprecipitated proteins were analyzed by Western blotting (n = 3). ATPase, adenosine triphosphatase; IP, immunoprecipitation. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Cystinosin enhances Gal-3 lysosomal localization and degradation
To verify the potential lysosomal localization of Gal-3 when interacting with cystinosin, we showed that Gal-3, like cathepsin D (an intralysosomal protease), was protected from digestion by proteinase K, whereas both proteins were digested when lysosomes were permeabilized with Triton X-100 (Figure 2a and Supplementary Figure S1). Similar data were obtained in lysosomes isolated from mouse liver, confirming lysosomal localization of Gal-3 in vivo (Figure 2b and c). Because of the absence of reliable antibody to detect cystinosin, immunostaining was performed on cystinosin-GFP–expressing MDCK cell lines with an anti–Gal-3 antibody. It confirmed the presence of Gal-3 in the lumen of the lysosomes, whereas cystinosin-GFP and Lamp-2 (a lysosomal transmembrane protein) were found only at the membrane of the vesicles (Figure 2d).
Figure 2 |. Galectin-3 (Gal-3) is localized into the lumen of late endosomes and lysosomes.

(a) Lysates of Madin-Darby canine kidney (MDCK) cells stably expressing cystinosin–green fluorescent protein (GFP) were treated or not treated with 2.5 µg/ml proteinase K for 30 minutes at 4 °C in the presence or absence of 2% Triton X-100 that permeabilized internal membranes. The samples were loaded on sodium dodecylsulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and processed for immunoblot with either anti-GFP (localized on the outer aspect of the lysosome), anti–cathepsin-D (intralysosomal hydrolase), or anti–Gal-3 antibodies. (b) Lysates (Ly) or lysosome-enriched fractions (LEF) from mouse liver were probed with the indicated antibodies. (c) Lysosome-enriched fractions were treated or not treated with 25 mg/ml proteinase K for 30 minutes at 4 °C in the presence or absence of 2% Triton X-100. The samples were loaded on SDS-PAGE and processed for immunoblot with anti–Gal-3 antibodies. (d) Immunostaining of late endosomes and lysosomes with lysosome-associated membrane protein 2 (Lamp-2) in cystinosin-GFP MDCK cell lines revealed a co-localization between cystinosin-GFP and Lamp-2 at the lysosomal membrane. Gal-3 is present within the vesicles, whereas cystinosin-GFP is located at the membrane of the vesicles. Bar = 10 µm. (e) Ctns−/− mouse embryonic fibroblasts expressing both Gal-3–GFP and cystinosin-DsRed were analyzed by total internal reflection fluorescence microscopy. Vesicular trafficking was monitored during 60 seconds and analyzed using ImageJ software. Magnifications of the selected areas are represented in the bottom panels and show true co-localization as determined by the spatiotemporal co-distribution of the proteins (indicated with arrows). A total of 9 cells were analyzed for each condition. Bar = 2 µm. The dynamics of the labeled vesicles and the spatiotemporal distribution of Gal-3 and cystinosin can be viewed in associated Supplementary Movies S1 and S2. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
To investigate if cystinosin was involved in Gal-3 trafficking, we analyzed the dynamics of both cystinosin and Gal-3–positive vesicles using dual-color live cell total internal reflection fluorescence microscopy in mouse embryonic fibroblasts (MEFs) from wild-type (WT) and Ctns−/− mice expressing both CTNS-DsRed and Gal-3–GFP. Our analysis confirmed that cystinosin and Gal-3 undergo true colocalization in Ctns−/− MEFs as validated by the similar spatiotemporal distribution of these molecules in the total internal reflection fluorescence microscopy zone (Figure 2e). Data in WT MEFs were similar (data not shown). Moreover, when MEFs transfected only with Gal-3–GFP were analyzed, Gal-3–GFP was found in the cytoplasm and no distinct vesicular structure was observed, contrary to the co-transfection with CTNS-DsRed (data not shown).
These data were confirmed in 293T cells, in which a strong GFP signal in the cytoplasm was observed in cells expressing only Gal-3–GFP, whereas in cells expressing both Gal-3–GFP and cystinosin-DsRed, Gal-3–GFP was mainly localized within cystinosin-DsRed–expressing lysosomes (Figure 3a). Moreover, quantification of Gal-3 protein expression in cells transfected with Gal-3–GFP alone or Gal-3–GFP and CTNS-DsRed, and Gal-3–GFP and DsRed as control, showed significantly less Gal-3–GFP proteins in cells co-transfected with CTNS-DsRed compared with cells transfected with Gal-3–GFP alone or co-transfected with DsRed (Figure 3b). Altogether, these results suggest that cystinosin enhances the lysosomal localization and degradation of Gal-3.
Figure 3 |. Cystinosin enhances Galectin-3 (Gal-3) lysosomal localization and degradation.

(a) Fluorescent images of large views of 293T cells transfected with Gal-3–green fluorescent protein (GFP) alone or with either DsRed or cystinosin-DsRed. Bar =50 µm. (b) Proteins were isolated from the transfected 293T cells, resolved on sodium dodecylsulfate–polyacrylamide gel electrophoresis, revealed with an anti–Gal-3 or anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody as reference, and quantified using ImageJ software, n = 3; statistical test used was 1-way analysis of variance followed by Tukey’s test. *P < 0.05; **P < 0.01. (c) Ctns−/− mouse embryonic fibroblasts were transfected with Gal-3–GFP expression vector and immunostained for endogenous Hsc70. Bar = 20 µm. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Cystinosin was shown to be involved in CMA.28 Heat shock 70 kDa protein (Hsc70) participates in CMA by aiding the unfolding and translocation of substrate proteins across the membrane into the lysosomal lumen in a LAMP2A-dependent manner.29,30 Because Gal-3 was proposed to be degraded by CMA,31 we investigated the localization of Gal-3 relative to Hsc70 in cystinotic MEFs. Confocal microscopy analysis confirmed the colocalization of Gal-3–GFP at Hsc70-positive structures in both WT (data not shown) and Ctns−/− cells (Figure 3c), supporting the co-transportation of Gal-3 with Hsc70 and suggesting that lysosomal internalization but not Gal-3 recognition by the chaperone is defective in cystinosis.
Gal-3 is involved in kidney inflammation in Ctns−/− mice
To study the role of Gal-3 in cystinosis in vivo, we generated a double knockout mouse model deficient in both cystinosin and Gal-3 (Ctns−/− Gal-3−/− mice). WT, Ctns−/−, and Gal-3−/− mice were used as control subjects. In the C57BL/6 Ctns−/− mice, renal anomalies appear around 10 months of age.11 Therefore, studies were performed at 8 to 9 months, when kidney anomalies start to be detected, and at 12 to 15 months, when kidney anomalies have progressed. Because cystine content was found to be different in male and female Ctns−/− kidneys, they were analyzed separately. If cystine content increases with age in both genotypes, no significant difference was observed in male and female kidneys between the Ctns−/− Gal-3−/− mice and Ctns−/− mice at 8 to 9 months and 12 to 15 months of age (Figure 4a).
Figure 4 |. Effect of the absence of Galectin-3 (Gal-3) on renal function and kidney structure in 12- to 15-month-old mice.
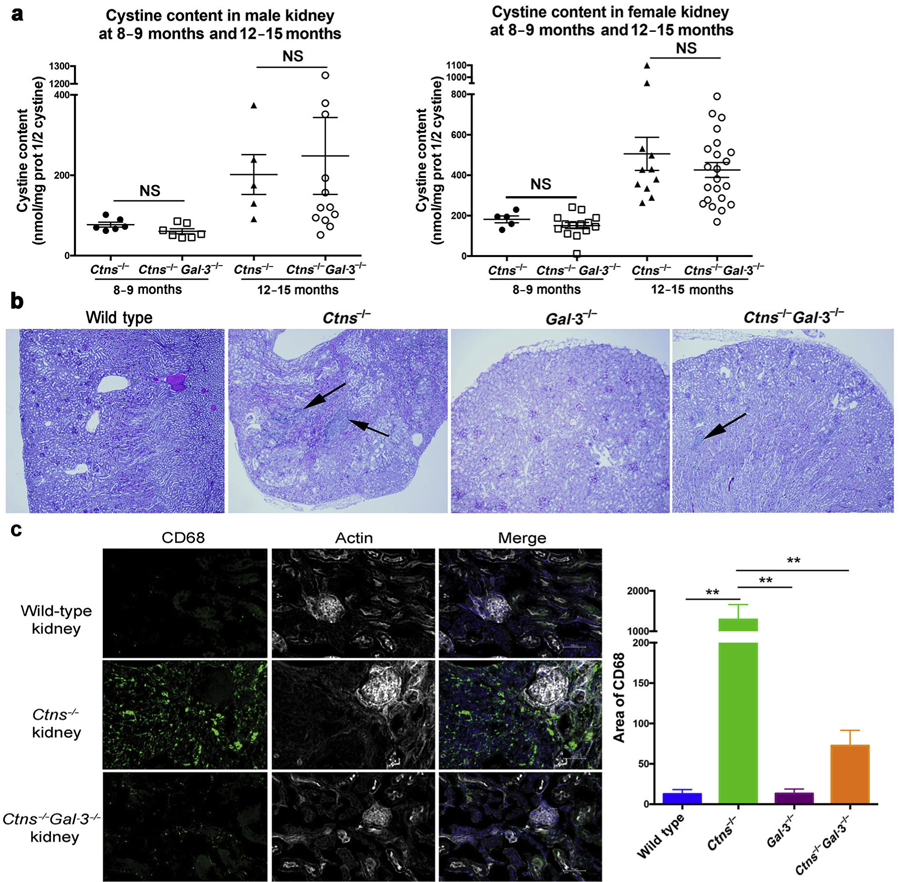
(a) Cystine content levels (nmol half cystine/mg protein) in the kidney of male (left panel) and female (right panel) Ctns−/− and Ctns−/− Gal-3−/− mice at 8 to 9 months (Ctns−/−: n = 11, 6 males and 5 females; Ctns−/− Gal-3−/−: n = 21, 7 males and 14 females) and 12 to 15 months (Ctns−/−: n = 16, 5 males and 11 females; Ctns−/− Gal-3−/−: n = 34, 12 males and 22 females). P values were determined using the unpaired 2-tailed Student t-test with Welch’s correction. (b) Histologic pictures of kidney sections from wild-type, Ctns−/−, Gal-3−/−, and Ctns−/− Gal-3−/− mice at 12 months of age stained with hematoxylin and eosin. Mononuclear infiltrates are indicated by arrows. (c) Kidney sections from wild-type, Ctns−/−, and Ctns−/− Gal-3−/− mice at 8 to 9 months of age were stained with anti-CD68 antibodies (green), phalloidin (white), and 4′,6- diamidino-2-phenylindole (blue). Bar = 50 µm. Quantification was then performed using ImageJ software (wild type: n = 3; Ctns−/− and Ctns−/−Gal-3−/−: n = 4). P values were determined using 1-way analysis of variance followed by Tukey’s test. **P < 0.01. NS, not significant. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Renal function was assessed in serum and urine and no significant difference was observed between WT and Ctns−/− mice at 8 to 9 months (data not shown), but at 12 to 15 months, Ctns−/− mice showed significantly higher serum creatinine and urea levels compared with WT mice. Interestingly, Ctns−/− Gal-3−/− mice exhibited improved renal function compared with Ctns−/− mice at 12 to 15 months, with lower serum creatinine (P < 0.05) and urea (P < 0.05; Table 1).
Table 1 |.
Serum and urine analyses for renal function of 12- to 15-month-old mice
| Wild type (n = 13) | Ctns−/− (n = 16) | Gal-3−/− (n = 5) | Ctns−/− Gal-3−/− (n = 34) | |
|---|---|---|---|---|
| Serum | ||||
| Creatinine (mg/dl) | 0.29 ± 0.02 | 0.41 ± 0.03a | 0.26 ± 0.02b | 0.31 ± 0.01b |
| Urea (mg/dl) | 48.95 ± 3.25 | 90.35 ± 3.34a | 52.68 ± 2.15b | 77.43 ± 2.49a,b,c |
| Phosphate (mg/dl) | 13.84 ± 1.07 | 12.87 ± 1.03 | 12.06 ± 0.28 | 12.03 ± 0.46 |
| Urine | ||||
| Phosphate (µmol/24 h) | 4.30 ± 0.62 | 5.56 ± 0.75 | 2.89 ± 0.81 | 6.06 T 0.81 |
| Protein (mg/24 h) | 12.36 ± 1.96 | 13.06 ± 2.04 | 14.69 ± 3.94 | 12.51 ± 1.42 |
| Volume (ml) | 0.85 ± 0.14 | 1.29 ± 0.14 | 0.89 ± 0.25 | 1.25 ± 0.12 |
Gal-3, Galectin-3.
P value was calculated by 1-way analysis of variance followed by Tukey’s test.
P < 0.05 compared with wild-type mice.
P < 0.05 compared with Ctns−/− mice.
P < 0.05 compared with Gal-3−/− mice.
Histologic studies of 8- to 9-month-old and 12- to 15-month-old mouse kidney sections revealed the presence of severe anomalies in Ctns−/− kidneys, including tubular atrophy, retracted glomeruli, and mononuclear infiltrates. Blind analysis of kidney sections ranged the extent of cortical damage from 1 (preserved tissue structure) to 6. The average score for Ctns−/− mice was 2.64 ± 0.36 at 9 months of age (n = 7) and 4.12 ± 0.37 at 12 to 15 months of age (n = 16). In the kidneys of Ctns−/− Gal-3−/− mice at the same age, these anomalies were significantly less extensive: 1.62 ± 0.11 at 9 months (n = 21; P < 0.05, Mann-Whitney t test) and 3.04 ± 0.22 at 12 to 15 months (n = 33; P < 0.05, Mann-Whitney t test) (Figure 4b and Supplementary Figure S2). Moreover, a percentage of tubular atrophy was assigned to every tissue, and the average for Ctns−/− mice and Ctns−/− Gal-3−/− mice were 66% and 42%, respectively, at 12 to 15 months (P = 0.01). Furthermore, a striking difference between Ctns – and Ctns−/− Gal-3−/− kidney sections was the presence of few mononuclear infiltrates in the Ctns−/− Gal-3−/− sections, whereas heavy infiltrates were consistently observed in the Ctns−/− kidney (Figure 4b).
We characterized the mononuclear infiltrates observed by histologic examination in 8- to 9-month-old Ctns−/− kidneys as macrophages/monocytes by co-immunostaining with CD68 and CD45 and with CD68 and major histocompatibility class II (Supplementary Figure S3), but not with CD68 and CD163, suggesting that these macrophages have a M1-like proinflammatory profile.32,33 Quantification of CD68-positive cells revealed significantly fewer macrophages/monocytes in WT, Gal-3−/−, and Ctns−/− Gal-3−/− kidneys compared with Ctns−/− kidneys (Figure 4c), confirming the histologic data (Figure 4b).
Altogether, these findings suggest that Gal-3 is involved in the recruitment of macrophages/monocytes in cystinotic kidneys and that its absence ameliorates kidney disease in Ctns−/− mice. Hence it suggests that inflammation is responsible, at least in part, for kidney deterioration in Ctns−/− mice.
Ctns-deficient kidneys exhibit Gal-3 overexpression
Using Western blot analysis, we found that Gal-3 expression was significantly increased in kidneys of 12-month-old Ctns−/− mice compared with WT mice (Figure 5a and b). To investigate if this increased expression of Gal-3 is specific to Ctns−/− kidneys, we quantified Gal-3 expression at the transcript and protein levels in kidneys from mice with CKD induced by standard subtotal 2-step nephrectomy operation and from the animals that underwent a sham operation. WT and Ctns−/− mice were used as control subjects. Gal-3 transcripts were significantly increased in Ctns−/− and sham kidneys but highly increased in CKD kidneys compared with WT mice (Figure 5c). In contrast, at the protein level, Gal-3 was detected only in Ctns−/− kidneys, strongly suggesting that only Ctns−/− kidneys were unable to degrade Gal-3 protein (Figure 5d). Immunofluorescent staining of Gal-3 in murine kidney at 8 to 9 months also demonstrated overexpression of Gal-3 in the Ctns−/− kidneys compared with WT kidneys (Figure 5e). Moreover, Gal-3 was expressed by CD68+ macrophages, as well as renal cells in Ctns−/− kidneys at 8 to 9 months (Supplementary Figure S4). Altogether, these data are consistent with decreased Gal-3 degradation in the absence of cystinosin as demonstrated in vitro (Figure 3a and b), leading to accumulation of Gal-3 protein in Ctns−/− kidneys.
Figure 5 |. Galectin-3 (Gal-3) expression is increased in the Ctns−/− mice.

Gal-3 expression in whole kidney homogenates of 12-month-old wild-type or Ctns−/− mice was evaluated by (a) Western blot and (b) quantified using ImageJ software (wild type: n = 6; Ctns−/−: n = 9). P values were determined using the unpaired 2-tailed Student t-test with Welch’s correction. **P < 0.01. mRNA and proteins were isolated from kidneys of sham-operated (n = 6) or 5/6 nephrectomized (chronic kidney disease; n = 6) animals, and Gal-3 expression was determined by droplet digital polymerase chain reaction (c) or Western blot (d). Protein lysates of kidney from WT and Ctns−/− mice at 12 months were used as negative and positive controls, respectively, for Gal-3 expression. P values were determined using 1-way analysis of variance followed by Tukey’s test. **P < 0.01. ***P < 0.0001. (e) Wild-type, Ctns−/−, and Ctns−/− Gal-3−/− kidneys were stained with anti–Gal-3 antibodies (green), phalloidin (white), and 4′,6-diamidino-2-phenylindole (blue). Bar = 50 µm. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Absence of cystinosin leads to increased serum MCP-1 via Gal-3 upregulation
Gal-3 regulates inflammation through different mechanisms.34 Thus, to gain further insights into the mechanism in cystinosis, we investigated the expression of different proinflammatory or antiinflammatory cytokines in the serum of 12- to 15-month-old WT, Ctns−/−, Gal-3−/−, and Ctns−/− Gal-3−/− mice. Six cytokine levels were measured by mouse cytometric bead array, MCP-1, interferon-γ, tumor necrosis factor, and IL-10, IL-6, and IL-12p70. Only MCP-1 expression was significantly increased in Ctns−/− mice serum compared with WT and Gal-3−/− mice and also to Ctns−/− Gal-3−/− mice, whose MCP-1 serum levels were comparable with WT mice (Figure 6a). MCP-1 is produced by different cell types, with the major source of MCP-1 being macrophages and monocytes,35 and is acting as a chemoattractant for monocytes and macrophages regulating their migration and infiltration. In contrast, at the same age, Gal-3 expression was found to be similar in the serum of Ctns−/− mice (44.33 ± 9.81 ng/ml; n = 5) and WT mice (40.86 ± 6.41 ng/ml; n = 7).
Figure 6 |. Galectin-3 (Gal-3) interacts with monocyte chemoattractant protein–1 (MCP-1), a macrophage/monocyte chemoattractant protein upregulated in cystinotic mice serum.

(a) A cytometric beads array has been used to determine MCP-1 concentration in the serum of wild-type (n = 7), Ctns−/− (n = 7), Gal-3−/− (n = 3), and Ctns−/− Gal-3−/− mice (n = 10). P values were determined using 1-way analysis of variance followed by Tukey’s test. *P < 0.05. (b) Lysates of 293T cells transfected with Gal-3–green fluorescent protein (GFP) and MCP-1–DsRed or Gal-3–GFP and CTNS-DsRed (control) were immunoprecipitated with anti-GFP antibody and co-immunoprecipitated proteins were analyzed by Western blotting. (c) The same lysates were treated or not treated with 5 mM of N-Acetyl-D-lactosamine, a potent inhibitor of Gal-3 carbohydrate interactions. Lysates were then immunoprecipitated with anti-GFP antibody and co-immunoprecipitated proteins were then analyzed by Western blotting. (d) An enzyme-linked immunosorbent assay (ELISA) directed against human MCP-1 has been used to determine MCP-1 expression in human serum samples from healthy donors (n = 10) and cystinotic patients (n = 19). P values were determined using the unpaired 2-tailed Student t-test. ***P < 0.001. (e) Using an ELISA assay, Gal-3 expression was determined in human serum samples from healthy donors (n = 10) and cystinotic patients (n = 17). P values were determined using the unpaired 2-tailed Student t-test. NS, not significant. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
The relationship between Gal-3 and MCP-1 was further investigated by co-immunoprecipitation using Gal-3–GFP and MCP-1–DsRed– or Gal-3–GFP and CTNS-DsRed– (as a positive control) expressing 293T cells. Interaction between Gal-3 and MCP-1 was observed (Figure 6b), suggesting a direct induction of MCP-1 by Gal-3. Incubation with N-Acetyl-D-lactosamine (LacNAc), an inhibitor of Gal-3 CRD, showed that, in contrast to cystinosin interaction, the CRD is not involved in the interaction between Gal-3 and MCP-1 (Figure 6c).
To assess whether this finding is relevant in humans, we measured Gal-3 and MCP-1 levels in patients with cystinosis who were not undergoing immunosuppressive therapy or taking antiinflammatory medications. Although the Gal-3 level was found to be slightly increased in the serum of patients with cystinosis compared with healthy donors, the difference was not significant (Figure 6e), confirming our results obtained in Ctns−/− mice. Only 2 patients had a high Gal-3 level (25.34 and 39.54 ng/ml); they were considered outliers and thus were not included in Figure 6e. In contrast, using an enzyme-linked immunosorbent assay directed against human MCP-1 (Figure 6d), serum MCP-1 levels were found to be significantly increased in patients with cystinosis (252.1 ± 18.4 pg/ml; n = 19; age range 1–23 years) despite cysteamine treatment compared with healthy control subjects (166.9 ± 13.5 pg/ml; n = 10; age range 8–63 years) (P < 0.001). No correlation has been found between age and MCP-1 level in both patients with cystinosis and healthy donors (data not shown).
DISCUSSION
Despite the fact that cystine accumulates in all tissues in patients affected with cystinosis, kidneys are the organs that are the most vulnerable to damage. Thus we believe cystine accumulation is not solely responsible for kidney degeneration. Herein, we describe a new role for cystinosin in the regulation of inflammation involved in the progression of chronic renal disease in cystinosis. This study may explain why patients evolve to end-stage renal failure despite undergoing cysteamine therapy.
The study of cystinosin’s molecular partners revealed a direct interaction with Gal-3, which can be inhibited by inhibitors of Gal-3 CRD. Recent studies showed that Gal-3 could interact with glycans located in the lysosomal lumen after lysosomal damage such as crystal buildup.36 In our study, the interaction between Gal-3 and cystinosin was studied in healthy cells expressing cystinosin and, therefore, not accumulating cystine or cystine crystals. In addition, overexpression of both Gal-3 and cystinosin in healthy cells modified the subcellular localization of Gal-3, which was then localized to the lysosomes, compared with overexpression of Gal-3 only, which was located in the cytoplasm. These results strongly suggest that interaction between Gal-3 and cystinosin is occurring independently of lysosomal damage and that cystinosin is actively responsible for Gal-3 lysosomal localization. Previously Gal-3 has been shown to be degraded through lysosomal-dependent proteolysis in absence of cAbl and Arg, 2 tyrosine kinases.31
In addition, we showed that cystinosin and Gal-3 co-localize at dynamic vesicular structures and are together mobilized in a spatiotemporal manner. Cystinosin previously has been associated with the trafficking mechanisms of the lysosomal LAMP2A, the only known CMA receptor, which is mislocalized in cystinosis.28 Gal-3 degradation previously has been shown to be mediated by CMA.31 Confocal microscopy analysis confirmed the colocalization of Gal-3 at Hsc70-positive structures in both Ctns−/− and WT cells, supporting the co-transportation of Gal-3 with Hsc70 for presentation at the lysosome and degradation by CMA as Hsc70 aids the translocation of substrate proteins across the membrane into the lysosomal lumen.29,30 A possible interpretation of our results is that cystinosin regulates the trafficking and delivery of Gal-3 to the CMA-active lysosomes for degradation.
This novel pathway of cystinosin-dependent Gal-3 lysosomal localization and degradation is supported in vivo as Ctns-deficient mice exhibit overexpression of Gal-3 within the kidney. Moreover, whereas increased Gal-3 mRNA was limited in Ctns−/− kidneys compared with another model of CKD, a large quantity of Gal-3 protein was detected only in Ctns−/− kidneys. These data strongly support the conclusion that cystinosin is involved in the degradation of Gal-3 protein, which can control overexpression of Gal-3 mRNA in stress conditions such as CKD.
Gal-3 has several biologic roles, including roles in acute and chronic inflammation by attracting monocytes and macrophages.37,38 In Ctns−/− mice, we observed heavy mononuclear infiltrates mainly in the kidney as previously described,11,39 which were characterized as monocytes/macrophages. In contrast, kidneys from the double knockout Ctns−/− Gal-3−/− mice exhibited very few monocyte/macrophage infiltrates, strongly suggesting that Gal-3 is involved in inflammation in the kidneys of Ctns−/− mice. In the context of repetitive tissue injury, Gal-3 may trigger the transition to chronic inflammation and fibrosis.34 Consistent with these findings, our data demonstrate the involvement of Gal-3 in the progression of the CKD in cystinosis. Indeed, the double knockout Ctns−/− Gal-3−/− mice exhibited better kidney function and structure compared with Ctns−/− mice. Similarly, Gal-3–deficient mice exhibit less renal fibrosis in the kidney compared with WT mice after unilateral ureteric obstruction,40 and Gal-3 knockout mice subjected to ischemia-reperfusion injury had a better renal function compared with WT mice subjected to ischemia-reperfusion injury.41
Although a correlation between levels of Gal-3 in plasma and the development of CKD was found,42 no difference of Gal-3 expression was observed in the serum of mice and humans affected by cystinosis and healthy control subjects. By measuring different cytokine levels in the serum, we found a significant increase of MCP-1 in Ctns−/− mice, whereas Ctns−/− Gal-3−/− mice had levels comparable with those of WT mice. MCP-1 is a cytokine that can be released in the serum and forms a gradient to attract monocytes and macrophages to the site of injury.35 The increase of MCP-1 in the sera of Ctns−/− mice compared with Ctns−/− Gal-3−/− mice was independent of cystine accumulation because kidney cystine content was similar in both genotypes. Furthermore, despite cysteamine treatment that allowed the exit of cystine out of the lysosomes, MCP-1 was found to be significantly increased in the sera of patients with cystinosis compared with healthy donors. These data strongly suggest that the absence of cystinosin, rather than cystine accumulation, is responsible for MCP-1 increase in serum. In addition, we showed a direct interaction between Gal-3 and MCP-1, which was recently suggested by Gordon-Alonso et al.43 but was not studied any further. The mechanism of Gal-3–mediated MCP-1 activation was not studied here. A hypothesis is the presence of a signal peptide in the human MCP-1, which can be cleaved to enhance its secretion.44 Moreover, plasmin can cleave the C-terminus of MCP-1, which increases its chemoattractant potency.45 In addition, correlating with our data, inhibition of MCP-1 in a mouse model of diabetic nephropathy prevented renal macrophage infiltration and improved renal function.46,47 In cystinosis, our data strongly suggest that the absence of cystinosin leads to the increase of Gal-3, which activates MCP-1 blood mobilization, enhancing renal inflammation and the progression of chronic kidney disease.
These findings open new perspectives in potential therapeutic targets that could limit or delay kidney degeneration in patients with cystinosis. Indeed, nonsteroidal antiinflammatory drugs such as aspirin and indomethacin have been found to inhibit Gal-3 expression by inhibiting its transcription.48 Indomethacin is actually used to regulate polyuria and polydipsia in cystinosis,49 and a recent study of 307 European patients with cystinosis showed an improved renal outcome in patients treated with indomethacin.50 In light of our study, the use of indomethacin or nonsteroidal antiinflammatory drugs for cystinosis patients may be reconsidered.
METHODS
Animal experiments
The C57BL/6 Ctns−/− mice were provided by Dr. Antignac (Inserm U1163, Paris, France). C57BL/6 galectin-3 null (Gal-3−/−) mice were provided by Dr. Fu-Tong Liu (University of California, Davis) and Dr. Jerrold M. Olefsky (University of California, San Diego). The breeding strategy to generate WT, Ctns−/−, Gal-3−/−, Ctns−/− and Gal-3−/− mice is described in Supplementary Methods.
Cell lines
MEFs were generated from newborn skin biopsies of WT and Ctns−/− mice. MEF, 293T, and MDCK type II cell lines (ATCC, Manassas, VA) were grown in Dulbecco’s modified Eagle medium containing 10% fetal calf serum or fetal bovine serum, 100 units/ml penicillin, 0.1 mg/ml streptomycin, and 2 mM L-glutamine.
Co-immunoprecipitation and mass spectrometry analysis
To study protein-protein interactions, MDCK cells were transduced using the lentiviral pRRL.SIN.cPPT.PGK/WPRE vector backbone to stably express cystinosin-GFP.51 Co-immunoprecipitation was performed as described in Supplementary Methods. Co-immunoprecipitated proteins were resolved by sodium dodecylsulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by mass spectrometry, LTQ Orbitrap as already described.19
293T cells expressing Gal-3–GFP and MCP-1–DsRed or Gal-3–GFP and CTNS-DsRed were used for co-immunoprecipitation as described in Supplementary Methods. Co-immunoprecipitated proteins were resolved by SDS-PAGE, and Gal-3–binding MCP-1 was detected using anti-DsRed antibody (Clontech Laboratories, Mountain View, CA).
Subcellular Fractionation
Eight livers were obtained from C57BL/6 mice and prepared as described previously.52 Conditions of the gradient were essentially the same as described in the original publication,52 except that Nycodenz was used instead of metrizamide.
Gal-3–inhibitor and proteinase K treatments
Lysates from cystinosin-GFP MDCK cells and 293T expressing Gal-3–GFP and CTNS-DsRed or Gal-3–GFP and MCP-1–DsRed were incubated with or without 5 mM thiodigalactoside or 5 mM of N-Acetyl-D-Lactosamine, respectively, 2 inhibitors of Gal-3 CRD. After 30 minutes of incubation at 4 °C with constant shaking, lysates were incubated with 50 µl anti-GFP microbeads and immunoprecipitation was performed as mentioned previously. Precipitated proteins were resolved by SDS-PAGE on 10% gel.
Lysates from cystinosin-GFP MDCK cells and mouse liver lysosome-enriched fractions were incubated with or without 2% Triton X-100 for 10 minutes at 4 °C and then incubated 30 minutes with or without 2.5 µg/ml and 25 µg/ml proteinase K, respectively. After enzyme inactivation with 1 mM phenylmethylsulfonyl fluoride for 5 minutes at 4 °C, proteins were resolved by SDS-PAGE on 10% gel.
Immunofluorescence analysis
Fixed MDCK cells were incubated with anti–Lamp-2 (OriGene Technologies, Rockville, MD) and anti–Gal-3 antibody (Cedarlane Laboratories, Burlington, Ontario, Canada) 2 hours at room temperature, followed by Alexa Fluor 555 secondary antibody (Invitrogen, Carlsbad, CA) for 1 hour at room temperature. Confocal images were taken using a Zeiss LSM 700 microscope (Carl Zeiss Microscopy, Jena, Germany).
293T cells transiently expressing Gal-3–GFP alone, Gal-3–GFP, and DsRed or Gal-3–GFP and cystinosin-DsRed were cultured on a coverslip before being mounted on a slide. Cells were imaged using a Keyence BZ-X700 instrument (Keyence Corp., Osaka, Japan).
Fixed kidney sections were incubated overnight at 4 °C with anti-CD68 (BioLegend, San Diego, CA) or anti–Gal-3 antibody (BioLegend), followed by Alexa Fluor 488 secondary antibody (Invitrogen), 1 hour at room temperature. Images were acquired using a Keyence BZ-X700 instrument. Quantification of CD68 expression is described in Supplementary Methods.
MEFs expressing Gal-3–GFP were stained using anti-Hsc70 antibody (Enzo Life Sciences, Farmingdale, NY) and imaged as described previously.53
Total internal reflection fluorescence microscopy
WT and Ctns−/− MEF expressing Gal-3–GFP and CTNS-DsRed were seeded on a 4-chamber 35- mm borosilicate bottom dish (Cellvis, Mountain View, CA). After 2 days in culture, MEFs were analyzed by total internal reflection fluorescence microscopy as described previously54 and in Supplementary Methods. Images were analyzed using ImageJ software.
Gal-3 expression analysis
293T cells expressing Gal-3–GFP, Gal-3–GFP and DsRed, or Gal-3– GFP and CTNS-DsRed and explanted kidneys were lysed in radio-immunoprecipitation assay buffer containing a proteinase inhibitor cocktail. Proteins were separated on a 4% to 15% gel and revealed using anti–Gal-3 (Abcam, Cambridge, UK) or anti–glyceraldehyde-3-phosphate dehydrogenase (Cell Signaling Technology, Danvers, MA) antibody.
Kidneys were homogenized in RLT buffer containing β-mercaptoethanol using Precellys 24 (Bertin Instruments, Montigny-le-Bretonneux, France). RNA was isolated and Gal-3– specific droplet digital polymerase chain reaction was performed as described in Supplementary Methods. Gal-3 transcript expression was expressed as a ratio compared with the endogenous control 18S.
An enzyme-linked immunosorbent assay (Abcam) was used to detect the Gal-3 level in mice and human sera, according to manufacturer’s instructions.
Renal function
Serum and urine phosphate, serum creatinine, and urea levels were estimated using the QuantiChrom Phosphate Assay Kit, Quanti-Chrom Creatinine Kit, and QuantiChrom Urea Assay Kit (Bioassay Systems, Hayward, CA). Protein levels in urine were measured using the Pierce BCA Protein Assay Kit (Rockford, IL).
Histology
At time the mice were killed, kidneys were collected, fixed in formalin, and embedded in paraffin. Sections stained with hematoxylin and eosin were reviewed in a blinded fashion by Dr. Marie-Claire Gubler as described in the Supplementary Methods.
Cystine content measurement
Tissue cystine measurement was performed by mass spectrometry (LC-ESI-MS/MS) as described previously.39
Standard nephrectomy and sham operation
Mice CKD was induced by the standard subtotal 2-stage nephrectomy operation as described previously.55,56 The sham group of mice underwent the same procedure but without cutting any kidney tissue.
Cytokine level in serum
To measure cytokine levels in mouse serum, a mouse inflammation kit cytometric bead array (BD Biosciences, San Jose, CA) was performed according to the manufacturer’s instructions. MCP-1 concentration in human serum was determined using an enzyme-linked immunosorbent assay directed against MCP-1 (Abcam, Cambridge, UK) according to the manufacturer’s instructions.
Statistical analysis
Values are expressed as mean ± SEM. The significance of the results was assessed by unpaired 2-tailed t-test or unpaired 2-tailed t-test with Welch’s correction. Group comparisons of 3 conditions or more were made with parametric analyses of variance, followed by Tukey’s multiple comparisons test for pairwise comparisons. Histologic scores were compared using the Mann-Whitney test. Analyses were performed using Prism 6 software (GraphPad, San Diego, CA). A P value less than 0.05 was considered significant.
Study approval
Mice experiments were conducted in compliance with Institutional Animal Use Committee protocols. Use of human tissue in this study was approved by the University of California, San Diego, Human Research Protections Program.
Supplementary Material
Movie S1. Visualization of the dynamics of vesicles carrying Galectin-3–GFP and cystinosin-DsRed in Ctns−/− fibroblasts using total internal reflection fluorescence microscopy.
Movie S2. Visualization of the dynamics of vesicles carrying Galectin-3–GFP and cystinosin-DsRed in Ctns−/− fibroblasts using total internal reflection fluorescence microscopy.
Figure S1. Galectin-3 (Gal-3) is localized in the lysosomes of wild-type Madin-Darby canine kidney (MDCK) cells. Lysate of wild-type MDCK cells was treated or not treated with proteinase K in the presence or absence of Triton X-100 that permeabilizes lysosomal membranes. Samples were then loaded on sodium dodecylsulfate–polyacrylamide gel electrophoresis and revealed with anti–gal-3 or anti–cathepsin-D (an intralysosomal hydrolase) antibody.
{kind=link}
Figure S3. Infiltration of CD68+, major histocompatibility class (MHC) class II+ and CD45+ macrophages in kidneys of 9-month-old Ctns−/− mice. Representative confocal microscopy pictures of immunostaining of cellular infiltrates in 9-month-old Ctns−/− kidneys are provided. The cells are macrophages that co-express CD68 (red) and CD45 (green; upper panel) or CD68 (red) and MHC class II (green; lower panel). Bar = 20 µm.
{kind=link}
Figure S2. Kidney structures are preserved in 12-month-old Ctns−/− Galectin-3 (Gal-3)−/− mice compared with Ctns−/− mice. Representative histologic pictures of kidney sections from 12-month-old Ctns−/− and Ctns−/− Gal-3−/− mice were stained with hematoxylin and eosin. More abundant cortical injury and mononuclear infiltrates (arrows) were observed in Ctns−/− kidneys than in Ctns−/− Gal-3−/− kidneys.
{kind=link}
Figure S4. Galectin-3 (Gal-3) is expressed by CD68+ macrophages and in renal cells in Ctns−/− kidneys. Representative confocal microscopy pictures of immunostaining of kidney sections from 9-month-old Ctns−/− mice with anti–Gal-3 antibody (green) and anti-CD68 antibody (red) shows partial co-localization of both proteins (arrows; upper panel), suggesting that CD68+ macrophages express Gal-3. In addition, kidney cells also can express Gal-3 (arrowheads; lower panel). Bar = 100 µm.
{kind=link}
Translational Statement.
Despite treatment, patients still progress to end-stage kidney failure. In this study we showed that absence of cystinosin results in impaired galectin-3 (Gal-3) lysosomal degradation, leading to inflammation and the progression of chronic kidney disease in cystinosis. These findings open new perspectives in potential therapeutic targets that could limit or delay kidney degeneration in patients with cystinosis. As such, antiinflammatory drugs and inhibitors of Gal-3 such as nonsteroidal antiinflammatory drugs or indomethacin may improve the renal pathogenesis in cystinosis.
ACKNOWLEDGMENTS
We acknowledge Dr. Fu-Tong Liu (University of California, Davis) and Dr. Jerrold M. Olefsky (University of California, San Diego) for providing the Gal-3−/− mice. We thank Lou Devanneaux and Nichole Flerchinger for their technical help and Elizabeth Souter for reviewing the manuscript. We acknowledge Christopher Alfonso from BD Bioscience for providing the mouse inflammation cytometric bead array and for his help with the interpretation of the results. This work was supported by National Institutes of Health grants RO1-DK090058 and R01-DK110162, the Cystinosis Research Foundation, and the California Institute of Regenerative Medicine (CIRM, CLIN-09230). TL is supported by a graduate fellowship from the Vaincre les Maladies Lysosomales. AB and JZ are supported by a fellowship from the Cystinosis Research Foundation. UCSD Neuroscience Microscopy Shared Facility was funded by National Institute of Neurological Disorder and Stroke (NINDS) grant P30-NS047101.
Footnotes
DISCLOSURE
SC is a Scientific Board member and member of the Board of Trustees of the Cystinosis Research Foundation. SC is a cofounder, shareholder, and member of both the scientific board and board of directors of GenStem Therapeutics Inc. The terms of this arrangement have been reviewed and approved by the University of California–San Diego in accordance with its conflict of interest policies. All the other authors declared no competing interests.
Supplementary material is linked to the online version of the paper at www.kidney-international.org.
REFERENCES
- 1.Medzhitov R Origin and physiological roles of inflammation. Nature. 2008;454:428–435. [DOI] [PubMed] [Google Scholar]
- 2.Donath MY. Targeting inflammation in the treatment of type 2 diabetes. Diabetes Obes Metab. 2013;15(suppl 3):193–196. [DOI] [PubMed] [Google Scholar]
- 3.Jaffer U, Wade RG, Gourlay T. Cytokines in the systemic inflammatory response syndrome: a review. HSR Proc Intensive Care Cardiovasc Anesth. 2010;2:161–175. [PMC free article] [PubMed] [Google Scholar]
- 4.Pecoits-Filho R, Heimburger O, Barany P, et al. Associations between circulating inflammatory markers and residual renal function in CRF patients. Am J Kidney Dis. 2003;41:1212–1218. [DOI] [PubMed] [Google Scholar]
- 5.Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. 2002;347: 111–121. [DOI] [PubMed] [Google Scholar]
- 6.Cherqui S, Kalatzis V, Trugnan G, Antignac C. The targeting of cystinosin to the lysosomal membrane requires a tyrosine-based signal and a novel sorting motif. J Biol Chem. 2001;276:13314–13321. [DOI] [PubMed] [Google Scholar]
- 7.Kalatzis V, Cherqui S, Antignac C, Gasnier B. Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. EMBO J. 2001;20:5940–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cherqui S, Courtoy PJ. The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives. Nat Rev Nephrol. 2017;13:115–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nesterova G, Gahl W. Nephropathic cystinosis: late complications of a multisystemic disease. Pediatr Nephrol. 2008;23:863–878. [DOI] [PubMed] [Google Scholar]
- 10.Cherqui S, Sevin C, Hamard G, et al. Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis. Mol Cell Biol. 2002;22:7622–7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nevo N, Chol M, Bailleux A, et al. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol Dial Transplant. 2010;25:1059–1066. [DOI] [PubMed] [Google Scholar]
- 12.Kalatzis V, Serratrice N, Hippert C, et al. The ocular anomalies in a cystinosis animal model mimic disease pathogenesis. Pediatr Res. 2007;62:156–162. [DOI] [PubMed] [Google Scholar]
- 13.Gaide Chevronnay HP, Janssens V, Van Der Smissen P, et al. A mouse model suggests two mechanisms for thyroid alterations in infantile cystinosis: decreased thyroglobulin synthesis due to endoplasmic reticulum stress/unfolded protein response and impaired lysosomal processing. Endocrinology. 2015;156:2349–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brodin-Sartorius A, Tete MJ, Niaudet P, et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 2012;81:179–189. [DOI] [PubMed] [Google Scholar]
- 15.Cherqui S Cysteamine therapy: a treatment for cystinosis, not a cure. Kidney Int. 2012;81:127–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gahl WA, Balog JZ, Kleta R. Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann Intern Med. 2007;147:242–250. [DOI] [PubMed] [Google Scholar]
- 17.Cherqui S, Courtoy PJ. The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives. Nat Rev Nephrol. 2017;13:115–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Napolitano G, Johnson JL, He J, et al. Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol Med. 2015;7:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrzejewska Z, Nevo N, Thomas L, et al. Cystinosin is a component of the vacuolar H+-ATPase-ragulator-rag complex controlling mammalian rarget of rapamycin complex 1 signaling. J Am Soc Nephrol. 2016;27: 1678–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rega LR, Polishchuk E, Montefusco S, et al. Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells. Kidney Int. 2016;89:862–873. [DOI] [PubMed] [Google Scholar]
- 21.Dumic J, Dabelic S, Flogel M. Galectin-3: an open-ended story. Biochim Biophys Acta. 2006;1760:616–635. [DOI] [PubMed] [Google Scholar]
- 22.Sciacchitano S, Lavra L, Morgante A, et al. Galectin-3: one molecule for an alphabet of diseases, from A to Z. Int J Mol Sci. 2018;19(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calvier L, Martinez-Martinez E, Miana M, et al. The impact of galectin-3 inhibition on aldosterone-induced cardiac and renal injuries. JACC Heart Fail. 2015;3:59–67. [DOI] [PubMed] [Google Scholar]
- 24.Frenay AR, Yu L, van der Velde AR, et al. Pharmacological inhibition of galectin-3 protects against hypertensive nephropathy. Am J Physiol Renal Physiol. 2015;308:F500–F509. [DOI] [PubMed] [Google Scholar]
- 25.Kolatsi-Joannou M, Price KL, Winyard PJ, Long DA. Modified citrus pectin reduces galectin-3 expression and disease severity in experimental acute kidney injury. PloS One. 2011;6:e18683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez-Martinez E, Ibarrola J, Calvier L, et al. Galectin-3 blockade reduces renal fibrosis in two normotensive experimental models of renal damage. PloS One. 2016;11:e0166272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nevo N, Thomas L, Chhuon C, et al. Impact of cystinosin glycosylation on protein stability by differential dynamic stable isotope labeling by amino acids in cell culture (SILAC). Mol Cell Proteomics. 2017;16:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Napolitano G, Johnson JL, He J, et al. Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol Med. 2015;7:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36:2435–2444. [DOI] [PubMed] [Google Scholar]
- 30.Xie W, Zhang L, Jiao H, et al. Chaperone-mediated autophagy prevents apoptosis by degrading BBC3/PUMA. Autophagy. 2015;11:1623–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Ma Q, Wang J, et al. c-Abl and Arg tyrosine kinases regulate lysosomal degradation of the oncoprotein Galectin-3. Cell Death Differ. 2010;17:1277–1287. [DOI] [PubMed] [Google Scholar]
- 32.Takeuchi H, Tanaka M, Tanaka A, et al. Predominance of M2-polarized macrophages in bladder cancer affects angiogenesis, tumor grade and invasiveness. Oncol Lett. 2016;11:3403–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chavez-Galan L, Olleros ML, Vesin D, Garcia I. Much more than M1 and M2 macrophages, there are also CD169(+) and TCR(+) macrophages. Front Immunol. 2015;6:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henderson NC, Sethi T. The regulation of inflammation by galectin-3. Immunologic Rev. 2009;230:160–171. [DOI] [PubMed] [Google Scholar]
- 35.Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skowyra ML, Schlesinger PH, Naismith TV, Hanson PI. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. 2018;360(6384). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacKinnon AC, Farnworth SL, Hodkinson PS, et al. Regulation of alternative macrophage activation by galectin-3. J Immunol. 2008;180: 2650–2658. [DOI] [PubMed] [Google Scholar]
- 38.Sano H, Hsu DK, Yu L, et al. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J Immunol. 2000;165:2156–2164. [DOI] [PubMed] [Google Scholar]
- 39.Yeagy BA, Harrison F, Gubler MC, et al. Kidney preservation by bone marrow cell transplantation in hereditary nephropathy. Kidney Int. 2011;79:1198–1206. [DOI] [PubMed] [Google Scholar]
- 40.Henderson NC, Mackinnon AC, Farnworth SL, et al. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am J Pathol. 2008;172:288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernandes Bertocchi AP, Campanhole G, Wang PH, et al. A role for galectin-3 in renal tissue damage triggered by ischemia and reperfusion injury. Transpl Int. 2008;21:999–1007. [DOI] [PubMed] [Google Scholar]
- 42.O’Seaghdha CM, Hwang SJ, Ho JE, et al. Elevated galectin-3 precedes the development of CKD. J Am Soc Nephrol. 2013;24:1470–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gordon-Alonso M, Hirsch T, Wildmann C, van der Bruggen P. Galectin-3 captures interferon-gamma in the tumor matrix reducing chemokine gradient production and T-cell tumor infiltration. Nat Commun. 2017;8:793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leonard EJ, Yoshimura T. Human monocyte chemoattractant protein-1 (MCP-1). Immunol Today. 1990;11:97–101. [DOI] [PubMed] [Google Scholar]
- 45.Sheehan JJ, Zhou C, Gravanis I, et al. Proteolytic activation of monocyte chemoattractant protein-1 by plasmin underlies excitotoxic neurodegeneration in mice. J Neurosci. 2007;27:1738–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chow FY, Nikolic-Paterson DJ, Ozols E, et al. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006;69:73–80. [DOI] [PubMed] [Google Scholar]
- 47.Seok SJ, Lee ES, Kim GT, et al. Blockade of CCL2/CCR2 signalling ameliorates diabetic nephropathy in db/db mice. Nephrol Dial Transplant. 2013;28:1700–1710. [DOI] [PubMed] [Google Scholar]
- 48.Dabelic S, Flogel M, Dumic J. Effects of aspirin and indomethacin on galectin-3. Croat Chem Acta. 2005;78:433–440. [Google Scholar]
- 49.Haycock GB, Al-Dahhan J, Mak RH, Chantler C. Effect of indomethacin on clinical progress and renal function in cystinosis. Arch Dis Child. 1982;57: 934–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emma FL, Ariceta E, Greco G, et al. Outcome and prognostic factors of nephropathic cystinosis: data from the Eunefron cohort [abstract]. Pediatr Nephrol. 2016:1748. [Google Scholar]
- 51.Dull T, Zufferey R, Kelly M, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wattiaux R, Wattiaux-De Coninck S, Ronveaux-dupal MF, Dubois F. Isolation of rat liver lysosomes by isopycnic centrifugation in a metrizamide gradient. J Cell Biol. 1978;78:349–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang J, Johnson JL, He J, et al. Cystinosin, the small GTPase Rab11, and the Rab7 effector RILP regulate intracellular trafficking of the chaperone-mediated autophagy receptor LAMP2A. J Biol Chem. 2017;292:10328–10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson JL, Napolitano G, Monfregola J, et al. Upregulation of the Rab27a-dependent trafficking and secretory mechanisms improves lysosomal transport, alleviates endoplasmic reticulum stress, and reduces lysosome overload in cystinosis. Mol Cell Biol. 2013;33:2950–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheung W, Yu PX, Little BM, et al. Role of leptin and melanocortin signaling in uremia-associated cachexia. J Clin Invest. 2005;115:1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheung WW, Kuo HJ, Markison S, et al. Peripheral administration of the melanocortin-4 receptor antagonist NBI-12i ameliorates uremia-associated cachexia in mice. J Am Soc Nephrol. 2007;18:2517–2524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Visualization of the dynamics of vesicles carrying Galectin-3–GFP and cystinosin-DsRed in Ctns−/− fibroblasts using total internal reflection fluorescence microscopy.
Movie S2. Visualization of the dynamics of vesicles carrying Galectin-3–GFP and cystinosin-DsRed in Ctns−/− fibroblasts using total internal reflection fluorescence microscopy.
Figure S1. Galectin-3 (Gal-3) is localized in the lysosomes of wild-type Madin-Darby canine kidney (MDCK) cells. Lysate of wild-type MDCK cells was treated or not treated with proteinase K in the presence or absence of Triton X-100 that permeabilizes lysosomal membranes. Samples were then loaded on sodium dodecylsulfate–polyacrylamide gel electrophoresis and revealed with anti–gal-3 or anti–cathepsin-D (an intralysosomal hydrolase) antibody.
Figure S3. Infiltration of CD68+, major histocompatibility class (MHC) class II+ and CD45+ macrophages in kidneys of 9-month-old Ctns−/− mice. Representative confocal microscopy pictures of immunostaining of cellular infiltrates in 9-month-old Ctns−/− kidneys are provided. The cells are macrophages that co-express CD68 (red) and CD45 (green; upper panel) or CD68 (red) and MHC class II (green; lower panel). Bar = 20 µm.
Figure S2. Kidney structures are preserved in 12-month-old Ctns−/− Galectin-3 (Gal-3)−/− mice compared with Ctns−/− mice. Representative histologic pictures of kidney sections from 12-month-old Ctns−/− and Ctns−/− Gal-3−/− mice were stained with hematoxylin and eosin. More abundant cortical injury and mononuclear infiltrates (arrows) were observed in Ctns−/− kidneys than in Ctns−/− Gal-3−/− kidneys.
Figure S4. Galectin-3 (Gal-3) is expressed by CD68+ macrophages and in renal cells in Ctns−/− kidneys. Representative confocal microscopy pictures of immunostaining of kidney sections from 9-month-old Ctns−/− mice with anti–Gal-3 antibody (green) and anti-CD68 antibody (red) shows partial co-localization of both proteins (arrows; upper panel), suggesting that CD68+ macrophages express Gal-3. In addition, kidney cells also can express Gal-3 (arrowheads; lower panel). Bar = 100 µm.