

FIV is a nonprimate lentivirus that infects domestic cats and causes a syndrome that is reminiscent of AIDS in humans. Based on its similarity to HIV with regard to different molecular and biochemical properties, FIV represents an attractive model for the development of strategies to prevent and/or treat HIV infection. Here, we show that the Bro1 domain of the human cellular protein Alix is sufficient to rescue the budding of FIV mutants devoid of canonical L-domains. Furthermore, we demonstrate that the integrity of the CCHC motifs in the Gag NC domain is dispensable for Alix-mediated rescue of virus budding, suggesting the involvement of other regions of the Gag viral protein. Our research is pertinent to the identification of a conserved yet mechanistically divergent ESCRT-mediated lentivirus budding process in general, and to the role of Alix in particular, which underlies the complex viral-cellular network of interactions that promote late steps of the retroviral life cycle.
KEYWORDS: Alix, late domain, nucleocapsid, feline immunodeficiency virus
ABSTRACT
The structural protein Gag is the only viral component required for retroviral budding from infected cells. Each of the three conserved domains—the matrix (MA), capsid (CA), and nucleocapsid (NC) domains—drives different phases of viral particle assembly and egress. Once virus assembly is complete, retroviruses, like most enveloped viruses, utilize host proteins to catalyze membrane fission and to free progeny virions. These proteins are members of the endosomal sorting complex required for transport (ESCRT), a cellular machinery that coats the inside of budding necks to perform membrane-modeling events necessary for particle abscission. The ESCRT is recruited through interactions with PTAP and LYPXnL, two highly conserved sequences named late (L) domains, which bind TSG101 and Alix, respectively. A TSG101-binding L-domain was identified in the p2 region of the feline immunodeficiency virus (FIV) Gag protein. Here, we show that the human protein Alix stimulates the release of virus from FIV-expressing human cells. Furthermore, we demonstrate that the Alix Bro1 domain rescues FIV mutants lacking a functional TSG101-interacting motif, independently of the entire p2 region and of the canonical Alix-binding L-domain(s) in FIV Gag. However, in contrast to the effect on human immunodeficiency virus type 1 (HIV-1), the C377,409S double mutation, which disrupts both CCHC zinc fingers in the NC domain, does not abrogate Alix-mediated virus rescue. These studies provide insight into conserved and divergent mechanisms of lentivirus-host interactions involved in virus budding.
IMPORTANCE FIV is a nonprimate lentivirus that infects domestic cats and causes a syndrome that is reminiscent of AIDS in humans. Based on its similarity to HIV with regard to different molecular and biochemical properties, FIV represents an attractive model for the development of strategies to prevent and/or treat HIV infection. Here, we show that the Bro1 domain of the human cellular protein Alix is sufficient to rescue the budding of FIV mutants devoid of canonical L-domains. Furthermore, we demonstrate that the integrity of the CCHC motifs in the Gag NC domain is dispensable for Alix-mediated rescue of virus budding, suggesting the involvement of other regions of the Gag viral protein. Our research is pertinent to the identification of a conserved yet mechanistically divergent ESCRT-mediated lentivirus budding process in general, and to the role of Alix in particular, which underlies the complex viral-cellular network of interactions that promote late steps of the retroviral life cycle.
INTRODUCTION
Feline immunodeficiency virus (FIV), a nonprimate lentivirus, causes an immunodeficiency syndrome in domestic cats that is strikingly similar to AIDS in humans. Furthermore, FIV shares with human immunodeficiency virus type 1 (HIV-1) several biological features in both feline and human cells (1). FIV and HIV-1, however, display important differences due to the fact that they have evolved alternative ways of overcoming specific replication challenges (2). Thus, it is important to conduct studies aimed at the identification of molecular and biological characteristics that are either common or significantly different between FIV and HIV-1 in order to clarify whether the domestic cat represents a potential nonprimate model for AIDS. Additionally, such studies are important for identifying the molecular elements needed for the development of efficient FIV-based vectors that can be used to safely deliver transgenes to human cells. Taking these considerations into account, detailed analysis and characterization of the interaction network between FIV and the host in human cells are particularly significant and could also be useful for studies of lentivirus phylogenesis and evolution.
Mutational and functional studies have shown that the C-terminal region of HIV-1 Gag, p6, whose disruption results in the arrest of viral budding, harbors L-domain determinants (3). To date, three different classes of L-domains have been well characterized (PT/SAP, PPXY, and LYPXnL) and are not unique features of retroviruses, since they are also present in both positive- and negative-stranded RNA enveloped viruses (4, 5), as well as in some DNA enveloped viruses (6–11). L-domains are docking sites for a set of host cell factors that are essential for the biogenesis of a cellular organelle, the multivesicular body (MVB), which is involved in the degradation of integral membrane proteins in all eukaryotic cells. The array of cellular proteins instrumental for the biogenesis of the MVB constitutes the endosomal sorting complex required for transport (ESCRT) machinery, organized into four main heteroprotein complexes: ESCRT-0, -I, -II, and -III. ESCRT factors are essential for additional important cellular processes, including cytokinesis and plasma membrane (12) and nuclear envelope (13, 14) repair. All these cellular events lead to the formation of narrow membrane necks whose interiors are contiguous with the cytosol. The ESCRT machinery, along with ESCRT-associated proteins and the AAA-ATPase vacuolar protein sorting-associated 4 (VPS4), drive the complex membrane remodeling that results in the scission of these necks. This process is also known as reverse-topology membrane scission and is topologically identical to the budding of viruses away from the cytosol (12, 15). Indeed, structural viral proteins, carrying L-domains, hijack members of the ESCRT machinery to execute their exit from infected cells (16). In particular, the PT/SAP L-domain interacts with the ESCRT-I component tumor susceptibility gene 101 (TSG101), a cellular protein that normally helps to sort proteins into MVB (17). The LYPXnL motif facilitates virus budding by binding the apoptosis-linked gene 2-interacting protein X (Alix) (18). Alix is an ESCRT-associated protein, which interacts with ESCRT-III via its component charged multivesicular body protein 4 (CHMP4) (19, 20). Alix is also one of the cellular proteins involved in cytokinesis (15). The PPXY sequence is a consensus binding site for members of the ubiquitin E3 ligase Nedd4 family. These ubiquitin ligases play essential roles in the MVB pathway, as well as in virus budding (21).
Like its retroviral counterparts, the FIV Gag protein can assemble and bud from cells in the absence of any other viral factors (22, 23). FIV Gag comprises three conserved domains, the matrix (MA), capsid (CA), and nucleocapsid (NC) domains (Fig. 1), which carry out different steps of viral particle assembly and egress (24). A short domain named p2, located within the carboxy-terminal region of Gag, contains a PSAP-type L-domain. Interestingly, the PT/SAP motif is present in all FIV sequences found not only in domestic cats but also in cougars and lions (25). We and others have demonstrated previously that the budding of FIV is strictly dependent on its PSAP L-domain (Fig. 1), irrespective of the presence or absence of a functional viral protease (26, 27). Moreover, we provided evidence that FIV egress involves Gag ubiquitination, which requires a functional L-domain (27). Finally, although FIV Gag does not contain a PPXY motif, we showed that the Nedd4-2s ubiquitin ligase enhances FIV Gag ubiquitination and robustly rescues viral mutants harboring a knocked-down L domain (27). Luttge and colleagues (28) reported that the expression of an N-terminal fragment of TSG101 containing the ubiquitin E2 variant (UEV) domain affects FIV egress from infected cells and that mutations of functional domains in the viral envelope glycoprotein negated this detrimental effect. Collectively, these data indicate that while FIV and HIV-1 share the ability to exploit the host cell ESCRT machinery to bud and release virus from infected cells, FIV-specific host-virus interactions have yet to be identified (29). Thus, in order to take advantage of FIV as a model for the study of HIV-1 production or transmission in vivo, or for the development and testing of anti-HIV interventions, further studies aimed at a full understanding of the molecular requirements for FIV replication in human cells are needed.
FIG 1.

Schematic representation of the HIV-1 and FIV Gag regions. Shown are the matrix (MA), capsid (CA), and nucleocapsid (NC) domains, as well as the HIV-1 p2, p1, and p6 peptides and the FIV p1 and p2 domains. The amino acid sequences of the HIV-1 and FIV NC domains are also shown. The HIV-1 and FIV regions containing the recognized viral L-domains (PTAP and YPLASL for HIV-1; PSAP for FIV) are underlined, as is the FIV LLDL motif. The HIV-1 NY5/BRU (LAV-1) recombinant clone pNL4-3 (GenBank accession no. M19921) and the FIV Petaluma strain, clone 34TF10 (GenBank accession no. M25381), were used as references.
It has been demonstrated previously that the budding of HIV-1 lacking a functional PT/SAP L domain can be rescued by the overexpression of Alix (30). Here, we set out to investigate whether Alix has a similar function in FIV budding from human cells and to identify the Gag domain(s) involved in this process. Our data provide a new example of conserved yet mechanistically divergent Alix-mediated lentivirus budding and uncover a complex virus-cell interplay involved in retroviral egress.
RESULTS AND DISCUSSION
In order to analyze the involvement of Alix in FIV egress from human cells, human embryonic kidney 293T cells were either transfected with a plasmid expressing hemagglutinin (HA)-tagged Alix (HA-Alix) (18) or mock transfected, in combination with constructs encoding either wild-type (WT) FIV Gag (here named PSAPx5LLDL) or FIV Gag carrying a replacement of both prolines in the PSAP motif with alanines (here named ASAAx5LLDL). Both constructs have been described elsewhere (named PSAP and ASAA, respectively, in reference 27). Of note, we showed previously that although the ASAAx5LLDL mutant is highly impaired in virus budding, it is assembly competent. Indeed, overexpression of the ubiquitin ligase Nedd4-2s, which is considered to restore only viruses that can complete virus assembly (31), efficiently rescued the budding of the ASAAx5LLDL mutant (27). Twenty-four hours posttransfection, virus particles released in the supernatants were analyzed by Western blotting, as described previously (32). As shown in Fig. 2A (virus-like particles [VLPs], fourth lane) and Fig. 2B, Alix clearly rescued the budding of the ASAAx5LLDL mutant. To investigate whether p2 sequences were involved in this process, a construct bearing a stop codon in place of the first amino acid of the Gag p2 domain (here named Δp2) was generated. Next, 293T cells were transfected with a plasmid encoding the Δp2 mutant with or without ectopic expression of HA-Alix. As shown in Fig. 2A (VLPs, fifth lane) and Fig. 2B, Δp2 mutations drastically affected virus egress. Once it was ascertained that Nedd4-2s overexpression could rescue this mutant (data not shown), we showed that Alix restored the release of virus particles by the Δp2 mutant (Fig. 2A, VLPs, sixth lane), thus excluding an overall role for the p2 domain in this Alix function. The Δp2 mutant lacks the highly conserved LLDL motif (Fig. 1), a sequence reminiscent of the YPDL Alix-binding site domain found in equine infectious anemia virus (EIAV) (18, 33–35). LXXL motifs, which are known to play a role in viral budding by interacting with different clathrin adaptor proteins and have been shown to play roles in Alix-mediated virus release (33, 34), are highly conserved in FIV p2 (25). As an exception, the FIV from cougars, PLV-14, displays a p2 Gag truncated before the LXXL motif, while it bears a canonical YXXL motif, equivalent to the EIAV L-domain, upstream of the PSAP sequence (25). In contrast to an initial report (36), Luttge and coworkers (26) showed that mutations of the LLDL motif had no detectable effect on virus particle production from either feline or human cells, a finding that we confirmed (data not shown). Based on these data, we generated an FIV mutant lacking a functional PSAP motif and with the EIAV YPDL L-domain replacing the LLDL sequence (ASAAx5YPDL). On the one hand, we found that the YPDL domain by itself does not rescue the budding of a PSAP-defective FIV mutant (Fig. 2A, VLPs, seventh lane). L-domains of different retroviruses usually act independently of their positions within Gag and are functionally interchangeable (4). However, the ASAAx5YPDL mutant shows a clear defect in budding, comparable to those observed with the ASAAx5LLDL and Δp2 mutants. This finding suggested that in the absence of a TSG101-interacting site, the presence of a canonical Alix-interacting domain, such as the YPDL sequence replacing the LLDL motif, is not sufficient to allow FIV VLP egress. Context-dependent functions of L-domains have been reported previously. For instance, different PPXY motifs have been shown to fail to mediate HIV-1 budding (37), demonstrating a certain degree of diversity in the mechanisms adopted by different viruses for the recruitment of the same host machinery. The budding defect of the ASAAx5YPDL mutant further supports previous observations, indicating that the PSAP motif is the dominant L-domain functioning in FIV budding (26, 27). On the other hand, Alix overexpression restored particle release for the ASAAx5YPDL mutant as efficiently as for the other p2 mutants (Fig. 2). Of note, for both the ASAAx5YPDL and Δp2 mutants, upon rescue with Alix, partially processed Gag forms appeared in the cell supernatants (Fig. 2A, arrows). Since no major defects in intracellular Gag processing were visible (Fig. 2A, Cells), this finding could be explained by defects of Gag polyprotein maturation in ASAAx5YPDL and Δp2 budding virions. Overall, our data strongly suggest that Alix functions in FIV egress independently of its canonical binding to L-domain sequences. This observation is consistent with previously published data showing that FIV budding is insensitive to the inhibitory effect of the Alix V-domain (26).
FIG 2.

Alix functions in FIV budding independently of the entire Gag p2 domain and independently of the presence of a binding motif. (A and B) 293T cells were transfected with plasmids expressing wild-type FIV Gag/Pol (PSAPx5LLDL) or an FIV Gag/Pol variant either with a mutation in the PSAP motif (ASAAx5LLDL), lacking the entire Gag p2 region (Δp2), or combining the PSAP inactivation with a YPDL motif replacing the conserved LLDL domain (ASAAx5YPDL). (A) Some of the samples (+) were also cotransfected with a construct expressing an HA-tagged version of Alix. Twenty-four hours posttransfection, VLPs were spun down by ultracentrifugation and were subjected to Western blot analysis by employing an anti-FIV capsid Gag antibody (p24). Cell lysates (Cells) were also assayed with α-p24 or with an antibody specific for the HA tag (α-HA). C– stands for mock-transfected cells. Asterisks point to capsid-containing intermediate products of the Gag polyprotein (Pr50) cleavage process (likely MA-CA-p1-NC, MA-CA-p1/CA-p1-NC-p2). The arrowhead in the lane for mock-transfected cells indicates a cross-reactive protein that is also visible in the Δp2 lanes. Arrows point to partially processed Gag intermediates present in the cell supernatants of the rescued Δp2 and ASAAx5YPDL mutants. Numbers indicate molecular size markers (in thousands). (B) Graph showing the relative virus release efficiency, calculated as the ratio of virion-associated Gag to total Gag (cells plus VLPs), normalized to FIV Gag/Pol PSAPx5LLDL release (26). Values are averages calculated from at least three independent experiments, and error bars indicate standard deviations. Band pixel intensity was quantified by using the software provided with the Alliance Q9 advanced chemiluminescence imager (Uvitec; Cleaver Scientific).
We have previously shown that the Alix Bro1 domain, and not the L-domain binding V-domain, is sufficient for the rescue of HIV-1 PT/SAP mutant budding (38). Interestingly, the phenylalanine of Bro1 (Phe105 in human Alix), which is essential for the ability of Alix to promote HIV-1 release (39), is conserved in feline Alix (Ensembl accession no. ENSFCAT00000030012 [https://useast.ensembl.org/Multi/Search/Results?q=ENSFCAT00000030012;site=ensembl]; NCBI accession no. XP_023116558), along with the surrounding residues, which have been shown to be important for this function, owing to the ability of the “Phe105 loop” to be inserted into cholesterol-rich membrane (40). Similarly, the Bro1 phenylalanine (F199 in human Alix), leucine (L216 in human Alix) (30), and isoleucine (I212 in human Alix) (39, 41) that are involved in the recruitment of the downstream-acting ESCRT-III component CHMP4B and in the rescue of the budding-defective HIV-1 ΔPTAP variant, are also conserved in feline Alix. Based on these domain and structural conservations, we decided to assess the ability of the human Alix Bro1 domain to rescue the budding of the FIV mutant lacking the p2 domain (Δp2); the VPRD C-terminal domain was used as a control (Fig. 3A). While the overexpression of VPRD had no effect on the budding of the Δp2 mutant, Alix Bro1 restored the release of Δp2 VLPs quite efficiently relative to restoration by WT Alix (Fig. 3B, VLPs).
FIG 3.

Overexpression of the Alix Bro1 domain is sufficient to rescue FIV Δp2 budding. (A) Schematic representation of the Alix mutants employed in this study. (B) 293T cells were transfected with the construct encoding FIV Δp2 (+) along with a plasmid expressing either wild-type Alix (WT) or the indicated mutant (Bro1 or VPRD), each fused to an HA tag. Twenty-four hours posttransfection, VLPs were spun down by ultracentrifugation and were analyzed by Western blotting with the α-p24 antibody (VLPs). Cell lysates (Cells) were also assayed with α-p24 (right) and α-HA (left) antibodies. C– stands for cells transfected with the corresponding empty vector (in place of an Alix-expressing plasmid) along with the construct encoding FIV Δp2. The results shown have been replicated in at least three independent experiments. Numbers indicate molecular weight markers (in thousands). (C) Graph showing the relative virus release efficiency, calculated as the ratio of virion-associated Gag to total Gag (cells plus VLPs), normalized to the release of FIV Δp2 plus WT Alix. Values are averages calculated from at least three independent experiments, and error bars indicate standard deviations. Band pixel intensity was quantified by using the software provided with the Alliance Q9 advanced chemiluminescence imager (Uvitec; Cleaver Scientific).
We and others have reported previously that the nucleocapsid (NC) region of HIV-1 Gag is involved in the ability of Alix Bro1 to restore the budding of mutant viruses lacking functional L-domains (35, 38, 42). Specifically, the cysteine (Cys) residues of each zinc finger (42) and the positively charged RNA-binding residues (arginines [R] and lysines [K]) of HIV-1 NC (43) are involved in this process. Furthermore, Popov and colleagues have clearly demonstrated that the substitution of the two terminal cysteine residues of each zinc (Zn) finger is sufficient to eliminate the ability of Alix to rescue the budding of the PTAP-devoid HIV L-domain mutant (42). Even though the overall structure of domestic cat FIV (FIVDC) NC is similar to that of HIV-1 NC, the latter has a long N-terminal tail rich in basic residues and a short C-terminal domain, while the opposite is seen in FIVDC NC (Fig. 1) (25). Nevertheless, the architecture of the tandem Zn fingers in the HIV and FIV NC proteins is conserved, with identical spacing of the cysteine and histidine residues involved in Zn coordination (Fig. 1) (23, 25), and a high degree of sequence similarity is observed, a common feature among all other lentiviruses. Thus, in order to investigate whether the ability of Alix to restore the budding of PSAP-devoid FIV involves NC Zn fingers, we first wanted to generate FIV mutants with no detectable assembly defects. We took in account the conservation between NC proteins described above and the knowledge that the first cysteine residue in the FIV NC N-terminal Zn finger, as well as the basic residues in the short sequence linking the two Zn fingers, is involved in nucleic acid binding and virus particle production (36). We therefore focused our attention on the two terminal cysteine residues of each Zn finger, also conserved between the FIV and HIV NC domains. Each of these cysteines was replaced with a serine (C390,409S/PSAP). It should be noted that the corresponding mutant in HIV-1 NC retained Gag-Gag interactions (44), further supporting our rationale for FIV NC residue selection as a target for mutagenesis. When transfected into 293T cells, FIV C390,409S/PSAP displayed a lower level of virus budding than that for the WT FIV construct. However, this defect was less drastic than the defect caused by the PSAP-to-ASAA mutation (compare Fig. 4C, left, with Fig. 2B). Next, the C390S and C409S mutations were combined with the functional inactivation of the PSAP L-domain (C390,409S/ASAA). 293T cells were transfected with the latter mutant or with the ASAAx5LLDL construct, along with or without a construct expressing HA-Alix. Figure 4B (right) and C (right) show that Alix rescued FIV C390,409S/ASAA budding as efficiently as FIV ASAAx5LLDL budding, thus indicating that, in contrast to what has been observed for HIV-1, the two cysteines in the N- and C-terminal FIV NC CCHC Zn fingers are not critical for this process.
FIG 4.

C390 and C409 are not involved in the ability of Alix to rescue the budding of PSAP-deficient FIV. 293T cells were transfected with the PSAPx5LLDL or C390,409S/PSAP construct (A) or with the ASAAx5LLDL or C390,409S/ASAA plasmid (B). In the experiment for which results are shown in panel B, cells were also transfected with a construct expressing HA-Alix (+) or with the corresponding empty vector (–). Twenty-four hours posttransfection, VLPs were spun down by ultracentrifugation and were analyzed by Western blotting by employing the α-p24 antibody. Cell lysates (Cells) were also assayed with α-p24 (A and B) and α-HA (B) antibodies. The results shown have been replicated in at least three independent experiments. Numbers indicate molecular weight markers (in thousands). (C) Graphs show the relative virus release efficiency, calculated as the ratio of virion-associated Gag to total Gag (cells plus VLPs), normalized to the release of FIV PSAPx5LLDL (left) or FIV ASAAx5LLDL (right). Values are averages calculated from at least three independent experiments, and error bars indicate standard deviations. Band pixel intensity was quantified by using the software provided with the Alliance Q9 advanced chemiluminescence imager (Uvitec; Cleaver Scientific).
Finally, we took advantage of a protease-defective FIV construct bearing a stop codon in place of amino acid 10 of the protease (PSAPx5LLDL/STOP), which we described previously (27). In this backbone, we combined the C390,409S mutations with the inactivation of either the PSAP domain or the LLDL motif (C390,409S/ASAA/STOP or C390,409S/PSAPx5AAAA/STOP, respectively). We showed that Alix is recruited in PSAPx5LLDL/STOP VLPs, while the mutant constructs displayed slightly less efficient Alix incorporation than the unmutated counterpart (Fig. 5). These findings indicate that NC Zn fingers might contribute to the efficiency of Alix incorporation without being a key determinant, pointing to a multifactor mechanism of Alix-FIV interaction that warrants further investigation.
FIG 5.
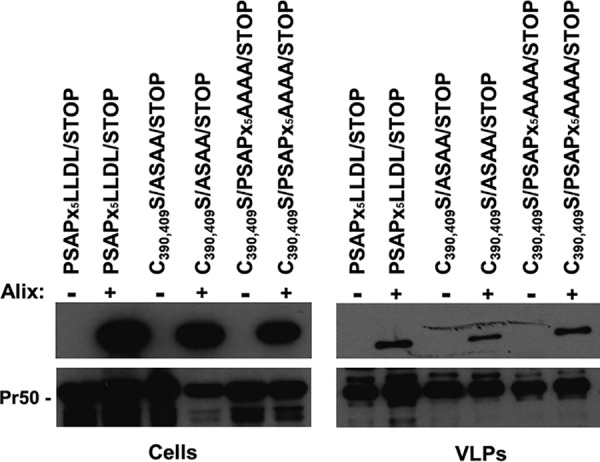
Neither C390 and C409 nor the PSAP motif nor the LLDL domain is the main determinant in the interaction of Alix with FIV Gag. 293T cells were transfected with constructs expressing either PSAPx5LLDL/STOP, C390,409S/ASAA/STOP, or C390,409S/PSAPx5AAAA/STOP along with a plasmid encoding HA-Alix (+) or the corresponding empty vector (–). Forty-eight hours posttransfection, similar amounts of VLPs were analyzed by Western blotting, using either the α-HA (upper panels) or the α-p24 (lower panels) antibody. Cell lysates (Cells) were also assayed.
Overall, our data present evidence that Alix Bro1 can rescue the release of a PSAP-defective FIV from human cells. Although this finding illustrates an additional host cell-virus interaction shared by FIV and HIV, it indicates that these two viruses present some significant differences in the mechanism by which Alix promotes this function. Indeed, the last two cysteines of each Zn finger, which are crucial for Alix recruitment to HIV NC (42), do not play a major role in the case of FIV. Furthermore, in the case of FIV, Alix function in particle budding is independent of known L-domains (results with the ASAAx5YPDL mutant here [Fig. 2A] and previous data [26]). Thus, additional studies are required to elucidate the region(s) within FIV Gag that plays a role in the interaction with Alix and the potential engagement of intermediary factors. In this respect, it is reasonable to assume that the C-terminal region of FIVDC might contribute to the recruitment of Alix Bro1. Indeed, basic residues within this region and the N-terminal portion of HIV-1 NC are functionally equivalent, at least as far as interaction with nucleic acids is concerned (45), and RNA is important for Alix Bro1–HIV-1 NC interactions (46). Finally, our studies cannot exclude the possibility that other domains or sequences within FIV Gag, in addition to NC and p2, might also take part in Alix binding and the catalysis of retrovirus budding, or that new molecular mechanisms are involved.
MATERIALS AND METHODS
Cells and viral constructs.
Human kidney cells (293T cells [ATCC CRL-11268]) were grown in Dulbecco’s modified Eagle’s medium (DMEM) with the addition of 10% heat-inactivated fetal calf serum.
Viral mutants were generated starting from the pΔenv1 construct, which contains Gag/Pol- and Rev-encoding sequences of the feline immunodeficiency virus (FIV), strain p34TF10 (GenBank accession no. NC_001482), as described previously (27). Briefly, an appropriate PCR-amplified region of the Gag-Pol-encoding sequence was cloned into the pCR2.1-Topo plasmid (Invitrogen). Mutations of the L-domain region and of the NC region were performed by using the QuikChange II site-directed mutagenesis kit (Stratagene). The mutated fragments were inserted back into the pΔenv1 plasmid.
Virion pelleting.
293T cells (1.5 × 106) were seeded into 25-cm2 tissue culture flasks. Twenty-four hours later, cells were transfected by the calcium phosphate method with 1.25 μg of the appropriate viral construct. In cotransfection experiments, along with the viral construct, 2 μg of the appropriate Alix-expressing vector was employed. When necessary, total DNA was brought to 3.25 μg with the corresponding empty vector. pBluescript KS(+) was used, when required, as a negative control for transfection. Twenty-four hours posttransfection, cell supernatants were harvested, and the cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (140 mM NaCl, 8 mM Na2HPO4, 2 mM NaH2PO4, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.05% sodium dodecyl sulfate [SDS]). Supernatants were clarified by low-speed centrifugation and were filtered through 0.45-μm-pore-size filters. Released VLPs were spun through a 20% (wt/vol) sucrose cushion for 2 h at 4°C and 27,000 rpm in a Beckman SW41 rotor. Pelleted VLPs were lysed in RIPA buffer, and viral proteins were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE), followed by Western blotting. Cell lysates were subjected to the same procedure in order to examine the intracellular Gag and Alix expression levels. In all gels, the PageRuler prestained protein ladder (Thermo Fisher Scientific) was used as a molecular weight marker.
In the experiments aimed at analyzing Alix recruitment, cells (4.5 × 106) were seeded into 75-cm2 tissue culture flasks, and 10 μg of the appropriate proviral construct, along with 5 μg of an Alix-expressing plasmid or empty vector, was transfected in triplicate as described above. Forty-eight hours posttransfection, cell supernatants were harvested, clarified by low-speed centrifugation, and filtered through 0.45-μm-pore-size filters. Released VLPs were spun through a 20% (wt/vol) sucrose cushion for 2 h at 4°C and 27,000 rpm. Pellets derived from three flasks were pooled and were lysed in RIPA buffer. The same volumes of viral proteins were analyzed by SDS-PAGE, followed by Western blotting. The pixel intensity of VLPs was quantified by using the software provided with the Alliance Q9 advanced chemiluminescence imager (Uvitec; Cleaver Scientific). Similar amounts of VLPs were then loaded onto SDS-PAGE gels, followed by Western blotting.
Western blot analysis.
For immunoblot analysis, aliquots of lysed VLPs and of the corresponding cell lysates were resolved by SDS-PAGE and electroblotted onto Hybond-C Extra membranes (Amersham Pharmacia). The membranes were incubated with the appropriate antibody, namely, a monoclonal anti-FIV capsid antiserum (anti-FIV-p24 Gag; Serotec) or a mouse monoclonal anti-HA antibody (Covance), followed by a peroxidase-conjugated anti-mouse IgG antibody (GE Healthcare). The blots were developed with enhanced chemiluminescence reagents (Amersham Pharmacia) as described elsewhere (47).
Data availability.
The raw data displayed in the Western blotting figures are available at the following links: https://doi.org/10.6084/m9.figshare.11822103 and https://doi.org/10.6084/m9.figshare.11828652. Data reported as “not shown” are available at https://doi.org/10.6084/m9.figshare.11828676.
ACKNOWLEDGMENTS
This work was supported by grants CPDA151030 and DOR (University of Padua) to A.C.; by the DIR, National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD (to F.B.); and by the ANRS (Agence Nationale de Recherche sur le Sida et les hépatites virales) (agreement 13018), the Istituto Superiore di Sanità, Rome, Italy (AIDS Project grant 40H98), and the Regione Veneto (Ricerca Sanitaria Finalizzata grant 312/10) (to C.P.).
We thank Francesca Spanevello for technical help in some experiments and Giulia Masi for assistance in the quantification of Western blots with the software provided with the Alliance Q9 advanced chemiluminescence imager (Uvitec; Cleaver Scientific).
We state that there are no conflicts of interest.
REFERENCES
- 1.Elder JH, Lin YC, Fink E, Grant CK. 2010. Feline immunodeficiency virus (FIV) as a model for study of lentivirus infections: parallels with HIV. Curr HIV Res 8:73–80. doi: 10.2174/157016210790416389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kenyon JC, Lever AM. 2011. The molecular biology of feline immunodeficiency virus (FIV). Viruses 3:2192–2213. doi: 10.3390/v3112192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Göttlinger HG, Dorfman T, Sodroski JG, Haseltine WA. 1991. Effect of mutations affecting the p6 Gag protein on human immunodeficiency virus particle release. Proc Natl Acad Sci U S A 88:3195–3199. doi: 10.1073/pnas.88.8.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calistri A, Salata C, Parolin C, Palù G. 2009. Role of multivesicular bodies and their components in the egress of enveloped RNA viruses. Rev Med Virol 19:31–45. doi: 10.1002/rmv.588. [DOI] [PubMed] [Google Scholar]
- 5.Votteler J, Sundquist WI. 2013. Virus budding and the ESCRT pathway. Cell Host Microbe 14:232–241. doi: 10.1016/j.chom.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calistri A, Sette P, Salata C, Cancellotti E, Forghieri C, Comin A, Göttlinger H, Campadelli-Fiume G, Palù G, Parolin C. 2007. Intracellular trafficking and maturation of herpes simplex virus type 1 gB and virus egress require functional biogenesis of multivesicular bodies. J Virol 81:11468–11478. doi: 10.1128/JVI.01364-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calistri A, Munegato D, Toffoletto M, Celestino M, Franchin E, Comin A, Sartori E, Salata C, Parolin C, Palù G. 2015. Functional interaction between the ESCRT-I component TSG101 and the HSV-1 tegument ubiquitin specific protease. J Cell Physiol 230:1794–1806. doi: 10.1002/jcp.24890. [DOI] [PubMed] [Google Scholar]
- 8.Crump CM, Yates C, Minson T. 2007. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J Virol 81:7380–7387. doi: 10.1128/JVI.00222-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee CP, Liu PT, Kung HN, Su MT, Chua HH, Chang YH, Chang CW, Tsai CH, Liu FT, Chen MR. 2012. The ESCRT machinery is recruited by the viral BFRF1 protein to the nucleus-associated membrane for the maturation of Epstein-Barr virus. PLoS Pathog 8:e1002904. doi: 10.1371/journal.ppat.1002904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pawliczek T, Crump CM. 2009. Herpes simplex virus type 1 production requires a functional ESCRT-III complex but is independent of TSG101 and Alix expression. J Virol 83:11254–11264. doi: 10.1128/JVI.00574-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stieler JT, Prange R. 2014. Involvement of ESCRT-II in hepatitis B virus morphogenesis. PLoS One 9:e91279. doi: 10.1371/journal.pone.0091279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurley JH. 2015. ESCRTs are everywhere. EMBO J 34:2398–2407. doi: 10.15252/embj.201592484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olmos Y, Hodgson L, Mantell J, Verkade P, Carlton JG. 2015. ESCRT-III controls nuclear envelope reformation. Nature 522:236–239. doi: 10.1038/nature14503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olmos Y, Carlton JG. 2016. The ESCRT machinery: new roles at new holes. Curr Opin Cell Biol 38:1–11. doi: 10.1016/j.ceb.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schöneberg J, Lee IH, Iwasa JH, Hurley JH. 2017. Reverse-topology membrane scission by the ESCRT proteins. Nat Rev Mol Cell Biol 18:5–17. doi: 10.1038/nrm.2016.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christ L, Raiborg C, Wenzel EM, Campsteijn C, Stenmark H. 2017. Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem Sci 42:42–56. doi: 10.1016/j.tibs.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 17.Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Côté M, Rich RL, Myszka DG, Sundquist WI. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 18.Strack B, Calistri A, Craig S, Popova E, Göttlinger HG. 2003. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114:689–699. doi: 10.1016/s0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- 19.Fisher RD, Chung HY, Zhai Q, Robinson H, Sundquist WI, Hill CP. 2007. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell 128:841–852. doi: 10.1016/j.cell.2007.01.035. [DOI] [PubMed] [Google Scholar]
- 20.Bissig C, Gruenberg J. 2014. ALIX and the multivesicular endosome: ALIX in Wonderland. Trends Cell Biol 24:19–25. doi: 10.1016/j.tcb.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 21.Morita E, Sundquist WI. 2004. Retrovirus budding. Annu Rev Cell Dev Biol 20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- 22.Adamson CS, Freed EO. 2007. Human immunodeficiency virus type 1 assembly, release, and maturation. Adv Pharmacol 55:347–387. doi: 10.1016/S1054-3589(07)55010-6. [DOI] [PubMed] [Google Scholar]
- 23.Luttge BG, Freed EO. 2010. FIV Gag: virus assembly and host-cell interactions. Vet Immunol Immunopathol 134:3–13. doi: 10.1016/j.vetimm.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bell NM, Lever AM. 2013. HIV Gag polyprotein: processing and early viral particle assembly. Trends Microbiol 21:136–144. doi: 10.1016/j.tim.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 25.Burkala E, Poss EM. 2007. Evolution of feline immunodeficiency virus Gag proteins. Virus Genes 35:251–264. doi: 10.1007/s11262-006-0058-8. [DOI] [PubMed] [Google Scholar]
- 26.Luttge BG, Shehu-Xhilaga M, Demirov DG, Adamson CS, Soheilian F, Nagashima K, Stephen AG, Fisher RJ, Freed EO. 2008. Molecular characterization of feline immunodeficiency virus budding. J Virol 82:2106–2119. doi: 10.1128/JVI.02337-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calistri A, Del Vecchio C, Salata C, Celestino M, Celegato M, Göttlinger H, Palù G, Parolin C. 2009. Role of the feline immunodeficiency virus L-domain in the presence or absence of Gag processing: involvement of ubiquitin and Nedd4-2s ligase in viral egress. J Cell Physiol 218:175–182. doi: 10.1002/jcp.21587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luttge BG, Panchal P, Puri V, Checkley MA, Freed EO. 2014. Mutations in the feline immunodeficiency virus envelope glycoprotein confer resistance to a dominant-negative fragment of Tsg101 by enhancing infectivity and cell-to-cell virus transmission. Biochim Biophys Acta 1838:1143–1152. doi: 10.1016/j.bbamem.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.González SA, Affranchino JL. 2018. Properties and functions of feline immunodeficiency virus Gag domains in virion assembly and budding. Viruses 10:E261. doi: 10.3390/v10050261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Usami Y, Popov S, Göttlinger HG. 2007. Potent rescue of human immunodeficiency virus type 1 late domain mutants by ALIX/AIP1 depends on its CHMP4 binding site. J Virol 81:6614–6622. doi: 10.1128/JVI.00314-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung HY, Morita E, von Schwedler U, Müller B, Kräusslich HG, Sundquist WI. 2008. NEDD4L overexpression rescues the release and infectivity of human immunodeficiency virus type 1 constructs lacking PTAP and YPXL late domains. J Virol 82:4884–4897. doi: 10.1128/JVI.02667-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Celestino M, Calistri A, Del Vecchio C, Salata C, Chiuppesi F, Pistello M, Borsetti A, Palù G, Parolin C. 2012. Feline tetherin is characterized by a short N-terminal region and is counteracted by the feline immunodeficiency virus envelope glycoprotein. J Virol 86:6688–6700. doi: 10.1128/JVI.07037-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee S, Joshi A, Nagashima K, Freed EO, Hurley JH. 2007. Structural basis for viral late-domain binding to Alix. Nat Struct Mol Biol 14:194–199. doi: 10.1038/nsmb1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhai Q, Fisher RD, Chung HY, Myszka DG, Sundquist WI, Hill CP. 2008. Structural and functional studies of ALIX interactions with YPX(n)L late domains of HIV-1 and EIAV. Nat Struct Mol Biol 15:43–49. doi: 10.1038/nsmb1319. [DOI] [PubMed] [Google Scholar]
- 35.Bello NF, Dussupt V, Sette P, Rudd V, Nagashima K, Bibollet-Ruche F, Chen C, Montelaro RC, Hahn BH, Bouamr F. 2012. Budding of retroviruses utilizing divergent L domains requires nucleocapsid. J Virol 86:4182–4193. doi: 10.1128/JVI.07105-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manrique ML, Raudd ML, González SA, Affranchino JL. 2004. Functional domains in the feline immunodeficiency virus nucleocapsid protein. Virology 327:83–92. doi: 10.1016/j.virol.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 37.Martin-Serrano J, Perez-Caballero D, Bieniasz PD. 2004. Context-dependent effects of L domains and ubiquitination on viral budding. J Virol 78:5554–5563. doi: 10.1128/JVI.78.11.5554-5563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dussupt V, Javid MP, Abou-Jaoudé G, Jadwin JA, de La Cruz J, Nagashima K, Bouamr F. 2009. The nucleocapsid region of HIV-1 Gag cooperates with the PTAP and LYPXnL late domains to recruit the cellular machinery necessary for viral budding. PLoS Pathog 5:e1000339. doi: 10.1371/journal.ppat.1000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sette P, Mu R, Dussupt V, Jiang J, Snyder G, Smith P, Xiao TS, Bouamr F. 2011. The Phe105 loop of Alix Bro1 domain plays a key role in HIV-1 release. Structure 19:1485–1495. doi: 10.1016/j.str.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bissig C, Lenoir M, Velluz MC, Kufareva I, Abagyan R, Overduin M, Gruenberg J. 2013. Viral infection controlled by a calcium-dependent lipid-binding module in ALIX. Dev Cell 25:364–373. doi: 10.1016/j.devcel.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCullough J, Fisher RD, Whitby FG, Sundquist WI, Hill CP. 2008. ALIX-CHMP4 interactions in the human ESCRT pathway. Proc Natl Acad Sci U S A 105:7687–7691. doi: 10.1073/pnas.0801567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Popov S, Popova E, Inoue M, Göttlinger HG. 2008. Human immunodeficiency virus type 1 Gag engages the Bro1 domain of ALIX/AIP1 through the nucleocapsid. J Virol 82:1389–1398. doi: 10.1128/JVI.01912-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dussupt V, Sette P, Bello NF, Javid MP, Nagashima K, Bouamr F. 2011. Basic residues in the nucleocapsid domain of Gag are critical for late events of HIV-1 budding. J Virol 85:2304–2315. doi: 10.1128/JVI.01562-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cimarelli A, Sandin S, Höglund S, Luban J. 2000. Rescue of multiple viral functions by a second-site suppressor of a human immunodeficiency virus type 1 nucleocapsid mutation. J Virol 74:4273–4283. doi: 10.1128/jvi.74.9.4273-4283.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu H, Wang W, Naiyer N, Fichtenbaum E, Qualley DF, McCauley MJ, Gorelick RJ, Rouzina I, Musier-Forsyth K, Williams MC. 2014. Single aromatic residue location alters nucleic acid binding and chaperone function of FIV nucleocapsid protein. Virus Res 193:39–51. doi: 10.1016/j.virusres.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sette P, Dussupt V, Bouamr F. 2012. Identification of the HIV-1 NC binding interface in Alix Bro1 reveals a role for RNA. J Virol 86:11608–11615. doi: 10.1128/JVI.01260-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Accola MA, Strack B, Göttlinger HG. 2000. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J Virol 74:5395–5402. doi: 10.1128/jvi.74.12.5395-5402.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data displayed in the Western blotting figures are available at the following links: https://doi.org/10.6084/m9.figshare.11822103 and https://doi.org/10.6084/m9.figshare.11828652. Data reported as “not shown” are available at https://doi.org/10.6084/m9.figshare.11828676.