

Human papillomaviruses are small, nonenveloped DNA viruses that infect skin and mucosa. The so-called high-risk HPVs (e.g., HPV16, HPV18, HPV31) have transforming potential and are associated with various anogenital and oropharyngeal tumors. These viruses enter host cells by a novel endocytic pathway with unknown cellular function. To date, it is unclear how endocytic vesicle formation occurs mechanistically. Here, we addressed the role of epidermal growth factor receptor signaling, which has previously been implicated in HPV16 endocytosis and identified the kinase Abl2 as a novel regulator of virus uptake. Since other viruses, such as influenza A virus and lymphocytic choriomeningitis virus, possibly make use of related mechanisms, our findings shed light on fundamental strategies of virus entry and may in turn help to develop new host cell-targeted antiviral strategies.
KEYWORDS: virus entry, endocytosis, HPV, papillomavirus, signaling
ABSTRACT
Human papillomavirus 16 (HPV16), the leading cause of cervical cancer, exploits a novel endocytic pathway during host cell entry. This mechanism shares many requirements with macropinocytosis but differs in the mode of vesicle formation. Previous work indicated a role of the epidermal growth factor receptor (EGFR) in HPV16 endocytosis. However, the functional outcome of EGFR signaling and its downstream targets during HPV16 uptake are not well characterized. Here, we analyzed the functional importance of signal transduction via EGFR and its downstream effectors for endocytosis of HPV16. Our findings indicate two phases of EGFR signaling as follows: a—likely dispensable—transient activation with or shortly after cell binding and signaling required throughout the process of asynchronous internalization of HPV16. Interestingly, EGFR inhibition interfered with virus internalization and strongly reduced the number of endocytic pits, suggesting a role for EGFR signaling in the induction of HPV16 endocytosis. Moreover, we identified the Src-related kinase Abl2 as a novel regulator of virus uptake. Inhibition of Abl2 resulted in an accumulation of misshaped endocytic pits, indicating Abl2’s importance for endocytic vesicle maturation. Since Abl2 rather than Src, a regulator of membrane ruffling during macropinocytosis, mediated downstream signaling of EGFR, we propose that the selective effector targeting downstream of EGFR determines whether HPV16 endocytosis or macropinocytosis is induced.
IMPORTANCE Human papillomaviruses are small, nonenveloped DNA viruses that infect skin and mucosa. The so-called high-risk HPVs (e.g., HPV16, HPV18, HPV31) have transforming potential and are associated with various anogenital and oropharyngeal tumors. These viruses enter host cells by a novel endocytic pathway with unknown cellular function. To date, it is unclear how endocytic vesicle formation occurs mechanistically. Here, we addressed the role of epidermal growth factor receptor signaling, which has previously been implicated in HPV16 endocytosis and identified the kinase Abl2 as a novel regulator of virus uptake. Since other viruses, such as influenza A virus and lymphocytic choriomeningitis virus, possibly make use of related mechanisms, our findings shed light on fundamental strategies of virus entry and may in turn help to develop new host cell-targeted antiviral strategies.
INTRODUCTION
Human papillomaviruses (HPVs) are a family of nonenveloped DNA viruses with transforming potential. In particular, so-called high-risk types are associated with various anogenital and oropharyngeal tumors. The most prevalent high-risk type, Human papillomavirus 16 (HPV16), is the leading cause of cervical cancer (1). While the mechanisms of transformation are rather well studied (2), the process of entry, i.e., the delivery of the viral genome into the nucleus for replication during initial infection, remains only partially understood (3, 4). This can be attributed to the complex life cycle of HPVs. Initial infection is established in basal keratinocytes of skin or mucosal epidermis, where maintenance replication also occurs (5, 6). Transformation, viral genome amplification, as well as assembly are tightly linked to tissue differentiation, and new particles are shed from granular/corneal cells of the epithelium (7–9). Thus, virus propagation is challenging, which can be overcome by the use of pseudovirions (PsVs) and other surrogate in vitro models. Similar to native HPV virions, PsVs consist of two structural proteins, L1 and L2, which self-assemble into viral capsids. Instead of the viral genome, PsVs incorporate a reporter plasmid as a pseudogenome. Reporter gene expression indicates successful entry upon infection of permissive cells. Importantly, PsVs are antigenetically indistinguishable from native HPVs (10–12).
The use of PsVs has allowed important insights into the early steps of HPV entry. For instance, we know that HPVs enter cells using endocytosis. More specifically, HPV16 and further high-risk types use a poorly characterized, novel endocytic mechanism (13, 14). This pathway is independent of major components of established endocytic mechanisms, such as clathrin, caveolin, flotillin, dynamin, and cholesterol. Instead, it relies on actin dynamics and signaling factors, such as the epidermal growth factor receptor (EGFR), phosphatidylinositol 3-kinase (PI3K), protein kinase C (PKC), and p21-activated kinase 1 (PAK1). This minimal “footprint” of requirements is mostly shared by macropinocytosis (15–17). Macropinocytosis is responsible for the uptake of large amounts of extracellular fluids and is often triggered by high amounts of growth factors. However, in contrast to macropinocytosis, HPV16 uptake is independent of cholesterol and Rho GTPase signaling (13). Moreover, the mode of vesicle formation is distinct. HPV16 enters cells in small inward budding pits, whereas macropinocytosis generates large outward protrusions that fold back to form vesicles. As a final distinction, HPV16 endocytosis occurs asynchronously over a protracted period of time, with a halftime of 10 to 12 h (13), while macropinocytosis occurs within about 20 min after induction and ceases thereafter (18). Since other viruses, such as influenza A virus (IAV) and lymphocytic choriomeningitis virus, can make use of similar mechanisms for entry, a better characterization of how endocytic vesicle formation is regulated and executed is of high interest (19–22).
Initiation of HPV16 endocytosis occurs after binding to heparan sulfate proteoglycans (HSPGs) on the cell surface (23, 24). This causes a first conformational change in the viral capsid, which is followed by cleavage of the major structural protein L1 by the extracellular protease kallikrein-8 (KLK8) (25, 26). Cyclophilins then aid to expose the N terminus of the minor capsid protein L2, which is subsequently cleaved by furin (27, 28). All of these changes reduce the affinity to HSPGs, allowing a transfer of the virus particle to an elusive secondary receptor for internalization (29, 30). Previous studies identified a variety of receptor candidates, such as the tetraspanins cluster of differentiation 151 (CD151) and CD63 (31, 32), EGFR (13), growth factor receptors (33, 34), integrin α6 (32, 35–37), and annexin A2 (38, 39). Hypothetically, these proteins may form a complex that serves as an entry platform and induces signaling, possibly via EGFR and/or integrin α6, for HPV16 uptake (3). Alternatively, Surviladze and colleagues propose that HPV16 capsids do not detach from HSPGs but are instead shed from the cell surface by matrix metalloproteinases releasing a complex of virus, HSPG ectodomain, and growth factors (33). This complex would then bind back to the cell surface and induce the activation of growth factor receptors and subsequent downstream signaling via extracellular signal-regulated kinase 1 (ERK1) and ERK2. A recent third addition to these models implicates a disintegrin and metalloproteinase 17 (ADAM17) (40), which supposedly facilitates the formation of CD151 containing entry platforms. In this model, ADAM17 mediates ectodomain shedding of membrane proteins and thereby releases growth factors, which in turn would activate EGFR and the downstream kinases ERK1 and 2. This ultimately triggers the formation of an entry platform, making the host cell susceptible for infection.
Despite their mechanistic differences, all of these models conclude that EGFR-derived signaling eventually triggers endocytic uptake. Although some evidence exists as to specific kinases that may facilitate HPV16 entry, the functional outcome of the signaling events remains speculative. Here, we analyzed the specificity and functional importance of EGFR and downstream effectors in signal transduction in light of the mechanistic differences between HPV16 endocytosis and macropinocytosis. Using phosphospecific Western blotting, small-molecule inhibitors, RNA interference (RNAi), knockout cell lines, and electron microscopy (EM) to study EGFR and different downstream signaling factors, we showed that EGFR-derived signaling is required for the initiation of endocytic pit formation. Moreover, we identified the kinase Abelson tyrosine-protein kinase 2 (Abl2) as a downstream target involved in endocytic pit maturation as a distinction from macropinocytosis.
RESULTS
EGFR signaling is required throughout HPV16 endocytosis.
Initially, we aimed to shed some light on whether EGFR signaling observed during or very shortly after binding facilitates HPV16 endocytosis (13, 33). Previous work indicates HPV16-induced phosphorylation of downstream EGFR targets such as ERK1 (Thr202/Tyr204)/ERK2 (Thr185/Tyr187) (referred to as pERK) as early as 5 min postinfection (p.i.) (33). As previously reported, HPV16 infection of serum-starved HaCaT cells led to detectable ERK phosphorylation (Fig. 1D and F) (33). In comparison to stimulation by epidermal growth factor (EGF) that served as a positive control and caused elevated pERK up to 30 min poststimulation, HPV16 binding led to levels of pERK above background that were short-lived and detected only at 10 but not 30 min p.i. (Fig. 1D to G). In line with previous hypotheses (3, 33, 40), this may suggest an indirect way of EGFR stimulation. Of note, pERK background signals were more prominent in solvent-treated control samples (mock) at 10 versus 30 min poststimulation (Fig. 1D to G). This elevated pERK background was likely due to mechanical stimulation during solvent addition as previously observed (41, 42). Notably, ERK activation downstream of HPV16 infection was detectable only upon high viral loads with varying levels of pERK, including experiments where pERK above background was not detectable. The cause of the variability could not be elucidated, suggesting that the degree of ERK activation even upon high viral loads was typically close to the detection limit. Taken together, we confirmed that EGFR signaling can occur with or shortly after virus binding. Corroborating the importance of EGFR signaling, the presence of the ATP competitive EGFR inhibitor Iressa (43) dose-dependently reduced HPV16 infection (Fig. 1A) (13). Moreover, we addressed the requirement of EGFR signaling for HPV16 endocytosis using a previously established assay to measure the internalization of infectious virus particles (13). At 12 h p.i., extracellular viruses were rendered noninfectious by a high-pH wash so that only already internalized virus was able to cause infection. The inhibitor was washed out at the time of pH wash, and infection was allowed to continue in the absence of the inhibitor, thus negating any effects of the inhibitor that may occur at later steps during the entry process. In the presence of Iressa, the endocytic uptake of infectious virions was reduced by about 70% (Fig. 1B). Overall, these experiments confirmed that EGFR signaling was important for HPV16 endocytosis and that EGFR signaling could be induced immediately after infection.
FIG 1.

EGFR signaling is essential for HPV16 endocytosis. (A) HeLa cells were pretreated with the EGFR inhibitor Iressa at the indicated concentrations for 30 min before they were infected with HPV16 PsVs. To reduce cytotoxicity, Iressa was exchanged for NH4Cl at 12 h p.i. Infection was scored by flow cytometry at 48 h p.i. Infection values were normalized to solvent controls and are depicted as the mean of three independent experiments ± standard deviation (SD). (B) HeLa cells were preincubated with 25 μM Iressa for 30 min and subsequently infected with HPV16 PsVs. At 12 h p.i., cells were either treated as in panel A (infection), or extracellular viruses were inactivated by treatment with a high pH buffer (infectious internalization). Infection was scored by flow cytometry and normalized to inhibitor reversibility. Depicted is the mean ± SD (n = 3). (C) HaCaT cells were infected with HPV16 PsVs at different MOIs (in particles/cell). Iressa (25 μM) was added at the indicated time points p.i., and infection was continued in the presence of the inhibitor. Infection was analyzed by flow cytometry at 48 h p.i., and infection values were normalized to solvent-treated controls. The area in gray indicates baseline infection levels upon Iressa treatment. Depicted is the mean of three independent experiments ± SD. (D, E) Serum-starved HaCaT cells were either incubated with PBS (mock), EGF, or different MOIs of HPV16, MCPyV, or HSV-1 for 10 (D) or 30 min (E). Cell lysates were first blotted against pERK and thereafter against ERK. (F, G) pERK levels were quantified and normalized to the respective mock samples and ERK (loading control). Depicted are the values from each experiment as fold activation of the background of n = 7 independent experiments (for HPV16). The individual experiments are indicated by individual geometric data points, whereas the data points are grouped by color according to the stimulus.
To test whether the signaling observed with or shortly after binding was indeed crucial for virus uptake, the EGFR inhibitor Iressa was added at various time points before and after infection. Addition of Iressa before and up to 4 h p.i. reduced infection by about 80 to 90% (Fig. 1C). At later time points, HPV16 infection increased to reach 56% ± 13%, when Iressa was added at 10 h p.i. (Fig. 1C). Since the time course resembled the known internalization kinetics (13), our data indicated that EGFR signaling was required throughout the asynchronous process of particle uptake. However, such EGFR signaling did not lead to a sustained and observable pERK signal (Fig. 1E and G). This may be due to the asynchronous events leading to infectious uptake so that pERK levels may only be raised in individual cells or locally and thereby may not rise above background within cell populations at these time points.
If EGFR signaling responsible for uptake needed to occur throughout internalization over a period of several hours, what role did the detectable ERK stimulation with or shortly after binding play? Detection of pERK within 10 min p.i. suggested that the coinciding HPV16 binding of HSPGs may be important. Since HSPGs also bind growth factors, which in turn interact with their receptors (44, 45), we hypothesized that HPV16-induced clustering of HSPGs may lead to subsequent clustering and activation of EGFR. To test this, we used herpes simplex virus 1 (HSV-1), an enveloped DNA virus, and Merkel cell polyomavirus (MCPyV), a nonenveloped polyomavirus, as controls. HSV-1 and MCPyV both bind to HSPGs (46–48) so that we expected EGFR stimulation upon their binding to cells. Like HPV16, HSV-1 stimulated pERK at 10 min p.i., whereas infection with MCPyV resulted in ERK phosphorylation only at 30 min p.i. (Fig. 1D to G). This correlates with different binding kinetics of these viruses (data not shown), resulting in the induction of pERK at different time points after virus addition. Since both viruses elicited pERK shortly after binding (Fig. 1D to G), and since neither HSV-1 nor MCPyV are known to interact with growth factor receptors, EGFR activation may in fact occur by indirect clustering of EGFR associated with HSPGs as has also been observed for other viruses (49, 50). Together with our inhibitor data, this suggested that EGFR signaling upon virus binding to HSPGs at high viral loads occurred coincidentally rather than functionally.
HPV16 infection is independent of an initial burst of EGFR signaling.
To investigate the functional importance of early EGFR signaling induced upon virus binding, we aimed to suppress early activation but not any signaling potentially occurring later. For this, Iressa was added prior to and during stimulation and washed out to allow signaling after binding of the stimulus. When Iressa was present during EGF stimulation, pERK as an indicator of signaling was suppressed, whereas EGF-stimulated signaling was observed again once the inhibitor was washed out (Fig. 2A). Thus, Iressa washout allowed reversible inhibition of EGFR-related signaling. However, when HPV16 was bound in the presence of Iressa and ERK activation was analyzed after subsequent washout of the inhibitor, no rise in pERK levels was detected, whereas EGF-derived pERK was still observable (Fig. 2B). This suggested that HPV16-derived detectable pERK only arose during binding to HSPGs but was not sustained through specific interactions of HPV16 with EGFR. Next, we asked whether HPV16-induced EGFR signaling during binding was crucial for HPV16 infection. Serum-starved HaCaT cells were infected in the presence of the EGFR inhibitor, which was subsequently removed at different time points p.i. Even an Iressa washout at 6 h p.i. only marginally reduced infection (Fig. 2C). This indicated that HPV16 infection of immortalized epithelial cells can occur independently of an initial strong pERK stimulation. Taken together, HPV16 binding to cells could activate EGFR signaling, but this early activation was dispensable for infection, whereas an asynchronous EGFR signaling was required throughout HPV16 internalization. The absence of observable activation of downstream targets at these times meant that studying any signaling cascades by this most typical approach (e.g., reviewed in reference 51) was not a feasible approach.
FIG 2.

Early ERK activation is dispensable for HPV16 infection. (A) Serum-starved HaCaT cells were either directly incubated with PBS (mock) or EGF for 10 min (left) or pretreated with 25 μM Iressa for 30 min and subsequently stimulated with EGF for 10 min (middle). To test for the reversibility of Iressa treatment, cells were preincubated with 25 μM Iressa for 30 min and stimulated with EGF after washout of the inhibitor for 10 min (right). Cell lysates were first blotted against pERK and thereafter against ERK. pERK levels were quantified and normalized to the mock of the untreated condition and ERK (loading control). (B) Serum-starved HaCaT cells were incubated with 25 μM Iressa for 30 min prior to binding of HPV16 PsVs or EGF for 1 h (mock = PBS). Subsequently, Iressa was washed out, and incubation was continued in the presence of HPV16 or EGF for the indicated time points. Lysates were processed as in panel A. pERK levels were quantified and normalized to the respective mock samples and ERK (loading control). (C) HaCaT cells were preincubated with 25 μM Iressa for 30 min and infected with HPV16 PsVs at different MOIs. At the indicated time points p.i., Iressa was washed out and infection was continued in the absence of the inhibitor. Cells were scored for infection 48 h p.i. by flow cytometry, and infection was normalized to the untreated control. Depicted is the mean of three experiments ± SD.
Abl2 is required for HPV16 endocytosis.
Small-compound inhibitor studies targeting potential EGFR downstream effectors during HPV16 endocytosis were employed as an alternative approach. Here, we focused on potential regulators of actin dynamics, as our previous work demonstrated that HPV16 uptake depends on actin polymerization, which is independent of classical Rho GTPase signaling (13). Sarcoma (Src) family kinases (SFK) are important regulators of actin polymerization during macropinocytosis (52), which shares similarities with HPV16 endocytosis (13). Moreover, the nonreceptor tyrosine kinase Src has previously been implicated in HPV16 infection (34, 39). To analyze the role of Src and the closely related and highly similar Abl kinases during HPV16 entry, the ATP competitive dual small-molecule inhibitor saracatinib was used (53). Saracatinib-mediated inhibition of Src and Abl kinases decreased HPV16 infection by 65% ± 9% (Fig. 3A). In contrast, uptake of vesicular stomatitis virus (VSV) by actin-dependent clathrin-mediated endocytosis (CME) (54) was unaffected (Fig. 3A). To more specifically address the involvement of Src, the highly selective inhibitor PP2 was utilized (55). PP2 modulates Src kinase-ligand interactions but not Abl activity (56, 57). However, PP2 treatment neither had an effect on HPV16 nor on VSV infection but reduced reovirus infection by 70% ± 14% (58), suggesting that Src was not required for HPV16 infection (Fig. 3B). Our evidence contrasted previous findings from Dziduszko and Ozbun that suggest the importance of Src kinases in HPV16 infection (39). Similar to our work, their studies relied on small-compound inhibitors, which need to be very well controlled due to pleiotropic effects on closely related kinases. To provide an alternative approach, we resorted to genetic experiments. In confirmation of the notion that Src family kinases are dispensable for entry, HPV16 infection of mouse embryonic fibroblasts (MEFs) lacking the three most ubiquitously expressed SFK members Src, Fyn, and Yes (59), was even increased (Fig. 3E). Infection with Semliki Forest virus (SFV), which enters cells via CME (60–63), was unaffected in these cells (Fig. 3E).
FIG 3.

HPV16 internalization depends on the tyrosine kinase Abl2. (A to D) HeLa cells were incubated with the small-compound inhibitors saracatinib (Src, Abl) (A), PP2 (Src) (B), nilotinib (Abl) (C), or imatinib (Abl) (D) at the indicated concentrations for 30 min. The cells were then either infected with HPV16 PsVs (black bars) for 48 h, with VSV-GFP for 5 h (white bars), or with reovirus for 16 h (gray bars). In the case of HPV16 infection, the inhibitor was exchanged by NH4Cl at 12 h p.i. to reduce cytotoxicity. Infection was scored by flow cytometry and normalized to solvent-treated control cells. (E) Mouse embryonic fibroblasts with a knockout of Src, Fyn, and Yes (SFY) as well as the littermate control cell line (Src+/+) were infected with HPV16 PsVs (black bars) for 48 h or SFV (white bars) for 5 h. Infection was scored by flow cytometry. (F) HeLa Kyoto cells were lysed 48 h after transfection with indicated siRNAs. Lysates were immunoblotted with Abl1- or Abl2-specific antibodies. (G) HeLa Kyoto cells were transfected with siRNAs against Abl1 and 2. At 48 h posttransfection, cells were infected with HPV16 PsVs for 48 h. Infection was analyzed by microscopy and automated cell scoring, and values were normalized to the control siRNA. (H) HeLa cells were preincubated with 25 μM Iressa, 25 μM imatinib, or 10 μM nilotinib for 30 min and subsequently infected with HPV16 PsVs. Cells were treated with a high-pH buffer at 12 h p.i. Infection was continued in the absence of the inhibitor. Infectious internalization was scored 48 h p.i. by flow cytometry and normalized to solvent-treated controls. All infection values are depicted as the mean of three independent experiments ± SD.
Next, we turned our attention to the Src-related kinases Abl1 and 2, as they are the most likely target for variable inhibition by Src family kinase inhibitors. Inhibition of Abl kinases by the ATP competitive inhibitors nilotinib (64) and imatinib (65) reduced HPV16 infection by 74% ± 4% and 79% ± 7%, respectively (Fig. 3C and D). At the same time, infection with VSV was unaffected (Fig. 3C and D), suggesting that HPV16 infection depended on Abl kinases. To further investigate whether HPV16 infection involves Abl kinases, we depleted cells of the two closely related kinases Abl1 or Abl2 by transfection of small interfering RNAs (siRNAs). All siRNAs led to an efficient depletion on the protein level (Fig. 3F). Interestingly, Abl1 knockdown did not affect HPV16 infection (Fig. 3G), whereas depletion of Abl2 reduced infection by 80% ± 15% (Fig. 3G). This indicated that Abl2 was required for HPV16 infection, while Abl1 seemed to be dispensable. To examine if Abl contributed to HPV16 endocytosis, we analyzed infectious internalization in the presence of the Abl inhibitors as described above. Both inhibitors reduced virus uptake, highlighting a role for Abl2 in the endocytic uptake of HPV16 (Fig. 3H). We concluded that Abl2, but not Src, was a crucial downstream target of EGFR required for HPV16 infection and internalization.
EGFR regulates pit induction, whereas Abl2 regulates pit maturation.
So far, we implicated EGFR and Abl2 as essential regulators of the novel endocytic pathway used for HPV16 internalization. To more specifically determine the regulatory role of both factors in endocytosis, quantitative ultrastructural analysis of endocytic pits in the presence or absence of either EGFR or Abl inhibitors was performed by ultrathin section transmission electron microscopy (TEM). Most virus containing pits (79%) in untreated cells were small, inward budding vesicles with diameters of about 60 to 80 nm (Fig. 4A). Each of these pits contained one virus particle and was termed a typical pit (Fig. 4A). Iressa-mediated EGFR inhibition resulted in a 64% decrease in the number of endocytic pits, with about half of the remaining pits appearing elongated and wider (about 130-nm diameter) compared to those of untreated cells (Fig. 4B and D). The reduction in pit number indicated that pit formation failed to initiate if EGFR signaling was suppressed. Interestingly, Abl inhibition by imatinib and nilotinib led to an enrichment of virus-containing pits with aberrant pit shapes by 275% and 195%, respectively (Fig. 4E). These pits were much larger (about 145 nm in diameter), contained most often several virus particles per pit, and were frequently associated with multiple budding structures that had a grape-like appearance (Fig. 4C). Importantly, only the number of endocytic pits containing virus particles changed, whereas upon visual inspection, the overall number of pits of various existing endocytic mechanisms did not (data not shown). This indicated that the treatments affected HPV16 endocytosis specifically. Since EGFR inhibition interfered with pit initiation and virus-filled pits did form upon Abl2 inhibition, Abl2 acted downstream of EGFR and was not involved in pit induction. Due to the observed change in pit size and shape, Abl2 signaling rather regulated pit dilation or maturation prior to scission.
FIG 4.
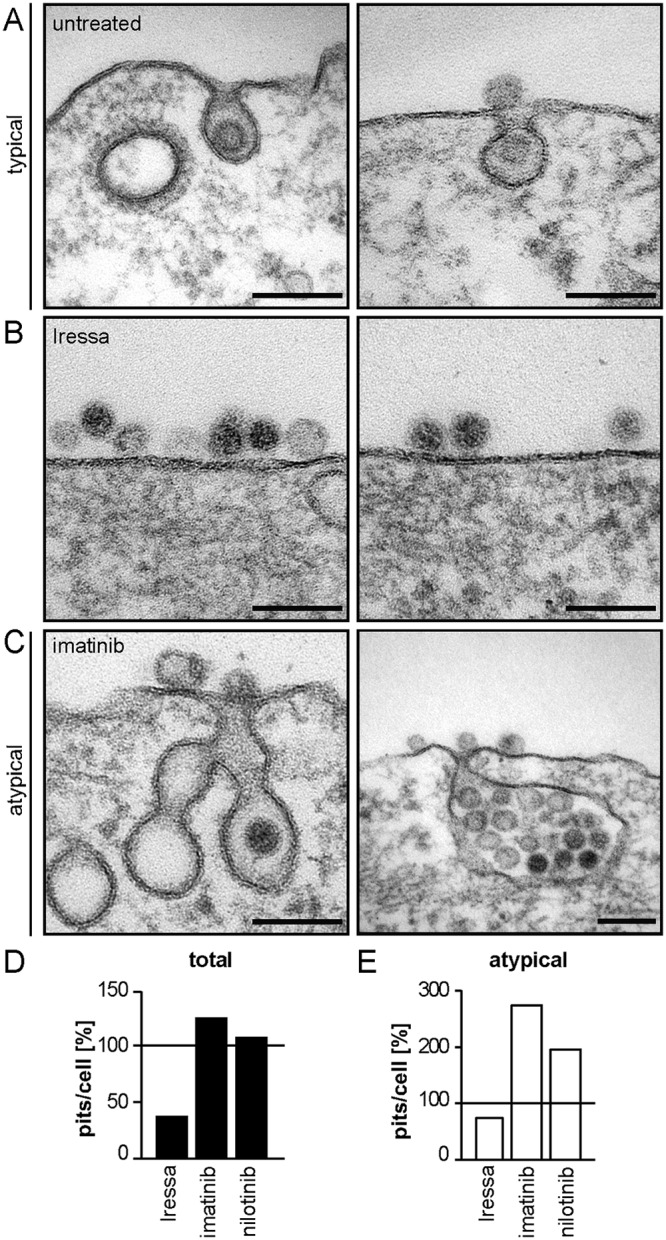
EGFR and Abl2 regulate pit induction and dilation/maturation, respectively. (A to C) HeLa ATCC cells were treated with 25 μM Iressa (B) or 25 μM imatinib (C) for 30 min or were left untreated (A) and subsequently infected with HPV16 PsVs. Cells were fixed at 6 h p.i., and endocytic pits were analyzed by ultrathin section TEM. (D) Virus-containing plasma membrane invaginations were counted and normalized to untreated control cells. At least 10 cells were counted per experiment in several independent experiments, excluding nilotinib where only one experiment was done. (E) The amount of virus-filled atypical pits per cell (e.g., in panel C) was determined and normalized to the amount of atypical pits in control cells.
DISCUSSION
Several studies have linked EGFR signaling to HPV16 endocytosis (13, 33, 34, 40). In this study, we aimed to further elucidate its role and potential downstream signaling events facilitating HPV16 endocytosis. Besides corroborating an essential role of EGFR for endocytosis, our results strongly indicated that EGFR signaling was crucial for the induction of endocytic pit formation. Moreover, we found that Abl2 kinase, a potential downstream target of EGFR signaling, regulated pit dilation or maturation during HPV16 endocytosis. In addition, biochemical assessment of phosphorylation dynamics combined with inhibitor studies determined that the burst of EGFR signaling observed during HPV16 binding was not required for infection of immortalized cells.
The observation of an early burst of EGFR signaling and the requirement of EGFR signaling throughout the asynchronous uptake of HPV16 that occurs over several hours poses a number of questions. First, are the two processes linked, and if not, how is signaling induced? Second, what is the potential role of initial EGFR signaling, as it was not required for endocytosis? Third, what is the detailed functional outcome of signaling and what are the effectors that are being targeted. Although at present in part speculative, we will discuss these questions in light of the existing experimental evidence.
Currently there are different models on how EGFR signaling is induced during HPV16 endocytosis. Raff et al. (3) propose that signaling is initiated upon transfer of terminally restructured virus particles to an entry platform containing the EGFR. An alternative model suggests that HPV16 bound to HSPG/growth factor complexes is shed from the cell surface (33). The decorated particle then induces signaling by binding to EGFR, which facilitates virus entry. A more recent model proposes that EGFR is activated through soluble growth factors shed from the cell surface by ADAM17. Signaling then triggers the formation of a virus entry platform over several hours (40). However, none of these models necessitates the early burst of EGFR signaling but rather throughout the asynchronous virus uptake, which may indicate that the two are not functionally linked.
But what causes the early burst of ERK activation? Viruses provide multiple receptor-binding sites and typically engage more than one receptor molecule. For instance, murine leukemia virus (MLV) clusters its receptor, mouse cationic amino acid transporter 1, upon binding (66). Interestingly, the HPV16 capsid displays multiple HSPG-binding sites (67) so that it likely clusters several HSPGs upon binding. Since HSPGs bind growth factors and thereby activate adjacent EGFR (44, 45, 68), HPV16 binding-induced HSPG-EGFR coclustering perhaps triggers EGFR signaling observed with or shortly after binding to cells. In support, our results demonstrated that binding of other HSPG-engaging viruses, namely, MCPyV and HSV-1, raised pERK levels briefly after binding comparable to those of HPV16 and EGF.
Our findings that early EGFR signaling was not required for HPV16 endocytosis and infection of immortalized cells do not rule out a putative importance for successful infection in vivo. Early EGFR signaling could prime host cells for viral gene expression as has, for example, been suggested for HIV-1 binding to chemokine receptors (69, 70), antagonize the host cell immune response (71, 72), or trigger proliferation of primary cells, which would be required for HPV16 nuclear import (73, 74).
Since our combined evidence from inhibitor kinetics and ultrathin section TEM indicated that EGFR activation was needed for induction of endocytic pit formation and occurred asynchronously over a period of several hours, EGFR activation for pit induction most likely occurred through a mechanism different from virus-induced HSPG-EGFR coclustering. This is supported by previous work that demonstrates infection of HSPG-deficient cells upon structural activation of the virus (25, 30, 33, 75). In the entry platform model, the endocytosis-facilitating microdomain is formed by the tetraspanin CD151 and contains EGFR, integrin α6, and further plasma membrane proteins (3). Since the ability of CD151 to form tetraspanin-enriched microdomains is essential for HPV16 infection (32), one possibility is that the CD151 microdomain associates with multiple EGFR molecules, thereby activating the receptor through transautophosphorylation (76). This is in analogy to processes described in cancer cells, where CD151 enhances growth factor receptor signaling through complex formation with integrins (77). Alternatively or in conjunction, ADAM17-dependent coclustering of preformed CD151-EGFR microdomains with HPV16 facilitates maturation of the virus entry platform and allows uptake (40).
The next step after initiation of endocytic pit formation via EGFR signaling would be pit dilation and maturation. Endocytic vesicle scission subsequently occurs by actin polymerization in a Rho GTPase-independent manner (13). In other endocytic mechanisms, these steps are carefully regulated by signaling events. Wondering whether downstream signaling of the EGFR would influence endocytosis in an additional manner to pit induction, we focused on Abl and Src kinases. These are prominent regulators of actin dynamics (52, 78) and could drive actin polymerization during HPV16 endocytosis and infection. By use of RNAi, inhibitors, and knockout cell lines, we identified the kinase Abl2 as a regulator of endocytic pit maturation, whereas Src was dispensable for HPV16 infection. While in line with the observations of Abban and Meneses that the Src inhibitor PP2 has no effect on HPV16 infection (79), the Ozbun lab observed an inhibition of infection upon PP2 treatment (34, 39). Including an inhibitor swap to NH4Cl after virus internalization (introduced in reference 13) likely avoided pleiotropic effects in our experiments and may thus explain the difference between the data presented here and previous work, where kinase inhibitors were kept throughout experimentation (34, 39). This may also mean that PP2-mediated inhibition of Src possibly affects infection at a later step of HPV16 entry.
The EM analysis implied that Abl2 was required for endocytic pit maturation since the amount of atypically formed pits increased upon Abl2 inhibition. A considerable fraction of atypical pits displayed multiple budding structures. A similar phenotype has previously been observed for caveolar endocytosis, where phosphatase inhibition leads to the formation of multicaveolar clusters (80, 81). Even though HPV16 uptake is independent of caveolin (13), the similarity in morphology may suggest a requirement of Abl2-regulated phosphatases for HPV16 endocytosis. In fact, we previously established a role of phosphatases during endocytosis but not binding (13). One candidate is SHIP2, which acts downstream of Abl kinases and controls phosphatidylinositol (3–5) trisphosphate and phosphatidylinositol (4, 5) bisphosphate levels, which are important regulators of plasma membrane trafficking (82–85). Thus, Abl2 may activate SHIP2 to drive phosphoinositide turnover and therefore pit maturation during HPV16 endocytosis. Additionally, we observed that, despite being misshaped upon Abl inhibition, many endocytic pits were already constricted at the vesicle neck. This suggests a perturbation of endocytic vesicle scission, which depends on actin polymerization (13). Abl2 has binding sites for actin (86, 87) and stimulates actin branching by interaction with Arp2/3 and cofilin, respectively (88). Thus, Abl2 may induce actin polymerization for endocytic vesicle scission. Since Abl2 had never been linked to endocytosis, our findings suggest a novel function of this kinase in membrane trafficking.
As discussed above, EGFR and Abl2 kinase signaling directly regulate different steps in the formation of the endocytic vesicle and must, therefore, occur subsequently with EGFR signaling occurring prior to Abl2 signaling. Given previous work implicating EGFR signaling in the activation of Abl kinases (89–91), it is likely that the two signaling events are part of a hierarchical cascade. However, without further experimental evidence, we cannot exclude that both events occur through different virus-cell interactions.
Taken together, we demonstrated that the initiation of HPV16 endocytosis depended on EGFR signaling but was independent of an initial signaling induced with or shortly after cell binding. In addition, we identified Abl2 as cellular kinase involved in endocytic pit maturation. The requirement of Abl2 kinase for HPV16 endocytic pit maturation demarcates an essential difference to macropinocytosis. While EGFR signaling is required for both macropinocytosis and HPV16 endocytosis (13, 33, 34, 40, 92), the induction kinetics are entirely different. A transient, strong stimulation in macropinocytosis is opposed by an asynchronous activation occurring over several hours in HPV16 endocytosis. This may be due to the mode of EGFR activation and/or downstream regulatory factors. Macropinocytosis depends on Src activation and signaling, which leads to recruitment and activation of cortactin to drive membrane ruffling (52, 93), macropinosome formation, and maturation (94, 95), whereas HPV16 endocytosis needs the Src-like kinase Abl2 to drive vesicle maturation. Thus, it is tempting to speculate that the involvement of Abl2 instead of Src together with the sustained low activation of EGFR is one of the causes for differences between the modes of vesicle formation, which is the most striking divergence between the pathways.
MATERIALS AND METHODS
Cells, viruses, pharmacological inhibitors, ligands, siRNAs, and antibodies.
HeLa (ATCC [96]), HeLa Kyoto (kind gift from L. Pelkmans, ETH Zürich Switzerland [97]), HaCaT (kind gift from J. T. Schiller, NCl, Bethesda, MD, USA [98]), and mouse embryonic fibroblasts (Src/Fyn/Yes [SFY]) and its littermate control Src+/+ (kind gift from R. Mancini and L. Pelkmans, ETH, Zürich, Switzerland [59]) were cultivated in Dulbecco modified Eagle medium (DMEM) high glucose (Sigma) supplemented with 10% fetal calf serum (Biochrom). Recombinant HPV16 PsVs containing green fluorescent protein (GFP) reporter plasmids were produced as previously described using the plasmids pClneo-GFP and p16sheLL (11). VSV-GFP (Indiana), SFV, MCPyV, and HSV-1 were generated as described earlier (60, 99–101). Reovirus (T3D [102]) was a kind gift from S. Boulant (DKFZ/Heidelberg University, Germany). Small-compound inhibitors in this study included the following: Iressa and PP2 from Tocris Bioscience as well as saracatinib, nilotinib, and imatinib from LC Laboratories. The ligand EGF was from Sigma. The siRNAs against Abl1 (ABL1_5, AAAGGTGAAAAGCTCCGGGTC; ABL1_11, CCAGTGGAGATAACACTCTAA) and Abl2 (ABL2_6, ATCAAGCATCCTAATCTGGTA; ABL2_8, AACCCTGTCCTTAATAACTTA) and the negative-control siRNA (AllStarNeg) were purchased from Qiagen. Antibodies in this study were the following: Lady Di (against SFV glycoproteins, ETH Zürich, Switzerland [13]), ERK-2 (Santa Cruz [33]), pERK (NEB [33]), Abl1 (Millipore), Abl2 (Abcam), α-tubulin (Sigma-Aldrich), and μNS (kind gift from S. Boulant, DKFZ/Heidelberg University, Germany).
Infection and infectious internalization studies.
HeLa ATCC, HaCaT, or mouse embryonic fibroblasts seeded in 12-well plates (5 × 104 cells/well) were preincubated with small-compound inhibitors at the indicated concentrations for 30 min. Cells were subsequently infected with HPV16 PsVs, VSV-GFP, or SFV in the presence of the inhibitor to result in about 20% infection of the unperturbed control. In case of infection assays with HPV16 PsV, the medium was exchanged by 10 mM NH4Cl/10 mM HEPES at 12 h p.i. to reduce cytotoxicity (13). For infectious internalization assays, cells were washed twice with phosphate-buffered saline (PBS) at 12 h p.i. and incubated with high-pH buffer (0.1 mM N-cyclohexyl-3-aminopropanesulfonic acid [CAPS] buffer, pH 10.5) for 90 s to inactivate extracellular virions (13). Then, cells were washed twice with PBS and further cultivated in complete growth medium to allow infection by already internalized virus. For all assays, cells were trypsinized (Sigma) and fixed in 4% (vol/wt) paraformaldehyde (PFA) either at 5 h p.i. for SFV, 6 h p.i. for VSV-GFP, or 48 h p.i. for HPV16 PsV. In the case of SFV, immunostaining against the glycoproteins followed by a secondary antibody staining using Alexa Flour 488 (Molecular Probes) was performed. Cells were analyzed for infection (level of GFP or Alexa Flour 488 staining) by flow cytometry (FACSCalibur; Becton, Dickinson). The relative number of infected cells was normalized to the solvent control. For infectious internalization assays, infection was normalized to inhibitor reversibility as previously described (13). For reovirus experiments, 6,000 HeLa ATCC cells were seeded in optical bottom 96-well plates (Greiner Bio-One). Cells were preincubated with PP2 at indicated concentrations and subsequently infected with reovirus in the presence of PP2 to result in about 20% infection in the solvent treated control. At 16 h p.i., cells were fixed with 4% PFA and immunostained against the nonstructural μNS protein followed by secondary antibody staining. Nuclei were stained with RedDot 2 (VWR/Biotium), and infection was analyzed microscopically (as described in RNAi). Cell number and infection were determined using CellProfiler (version 2.1.1). Nuclei were segmented, and a mask covering the nuclei and 10 adjacent pixels in the perinuclear area was transferred to the respective images with virus signal (μNS staining). Cells were classified as infected or uninfected based on the virus intensity in the masked area. The threshold was set based on the intensity of uninfected samples. Readout was the average percentage of infected cells per sample.
RNAi.
A total of 2,000 HeLa Kyoto cells in 96-well optical bottom plates were reverse transfected with 20 nM siRNA using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer’s instructions. Cells were infected with HPV16 PsVs at 48 h posttransfection to result in about 20% infection in AllStarNeg control cells. Cells were fixed with 4% PFA at 48 h p.i., and nuclei were stained with RedDot 2. Infection was analyzed on a Zeiss Axiovert Z.1 microscope equipped with a Yokogawa CSU22 spinning disc module. Images were acquired with a 20× objective using a CoolSnap HQ camera (Visitron Systems). Cell number and infection were determined using a MATLAB-based infection scoring procedure (MathWorks [13, 97, 103]). Raw infection values were normalized to the control siRNA. To analyze knockdown efficiencies, 2 × 104 HeLa Kyoto cells in 12-well plates were reverse transfected with 20 nM siRNA. Cell lysates were prepared 48 h posttransfection. Lysates from two wells per condition were combined and prepared for SDS-PAGE and Western blotting by addition of 2× SDS sample buffer (4% SDS, 100 mM Tris-HCl, pH 6.8, 20% glycerol, 8 mM dithiothreitol [DTT], 4% bromphenol blue) and incubation for 5 min at 95°C for protein denaturation. The samples were analyzed for Abl1 and Abl2 expression, and α-tubulin was used as a loading control.
ERK activation studies.
Two days prior to stimulation, 3 × 105 HaCaT cells were seeded in 6-well plates. The following day, cells were serum starved for 16 h. Then, cells were exposed to PBS (mock), 30 ng EGF and HPV16 PsV, MCPyV, or HSV-1 at multiplicities of infection (MOI) of 100, 1,000, or 10,000 particles/cell for the indicated time points. Alternatively, cells were preincubated with a small-compound inhibitor at the designated concentrations for 30 min prior to infection. The cells were lysed on ice by incubation with radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 7.4) supplemented with protease inhibitors (Roche complete mini) for 5 min. Nuclei were sedimented by centrifugation and protein concentrations were determined with a bicinchoninic acid (BCA) test kit (Thermo Scientific). Then, the lysates were incubated with 2× SDS sample buffer as above, and pERK levels were determined by Western blotting analysis. Thereafter, the nitrocellulose membrane was stripped, twice with elution buffer I (50 mM Tris-HCl, pH 7.5, 5 mM EDTA, 150 mM NaCl) for 2 min, once with elution buffer II (5 mM DTT, 200 mM glycine-HCl, pH 2.8) for 2 min and reprobed against ERK as a loading control. Relative signal intensity of pERK was determined using ImageJ and normalized to the signal intensity of the loading control ERK before normalization to the indicated mock sample.
Electron microscopy.
A total of 1 × 105 HeLa ATCC cells seeded in 3-cm dishes were preincubated with 25 μM Iressa, 10 μM nilotinib, or 25 μM imatinib for 30 min and infected with HPV16 PsVs (10,000 particles/cell). Cells were fixed at 6 h p.i. in 2.5% glutaraldehyde (GA) in Dulbecco's phosphate-buffered saline (D-PBS), postfixed in 1% OsO4, block stained with 0.5% uranylacetate (UAC), dehydrated, and embedded in Epon. Ultrathin sections (60 nm) were cut and counterstained with uranyl acetate and lead. Samples were analyzed at 80 kV on a FEI Tecnai 12 electron microscope (FEI, Eindhoven, Netherlands). Images of selected areas were documented with an Olympus Veleta 4k charge-coupled-device (CCD) camera or with Ditabis imaging plates (Ditabis, Pforzheim, Germany). The total number of pits/cell as well as the number of atypical pits/cell (elongated, multiple particles and/or pits) was determined for at least 10 cells per experiment in several independent experiments, excluding nilotinib, where only one experiment was performed. Since HPV16 endocytic pits are not easily distinguishable from caveolae or uncoated pits from other endocytic mechanisms without further staining, only pits containing virus(es) were counted. Pit numbers were normalized to untreated controls.
ACKNOWLEDGMENTS
We thank I. Fels (Institute of Cellular Virology, Münster, Germany) for technical support during virus production. Thanks also to members of the Schelhaas laboratory for helpful comments on this manuscript.
This work was supported by funding to M.S. by the German Research Foundation (grants SCHE 1552 6-2 and INST211/817A09).
REFERENCES
- 1.Zur Hausen H. 2002. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 2.Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, Stanley MA. 2012. The biology and life-cycle of human papillomaviruses. Vaccine 30(Suppl):F55–F70. doi: 10.1016/j.vaccine.2012.06.083. [DOI] [PubMed] [Google Scholar]
- 3.Raff AB, Woodham AW, Raff LM, Skeate JG, Yan L, Da Silva DM, Schelhaas M, Kast WM. 2013. The evolving field of human papillomavirus receptor research: a review of binding and entry. J Virol 87:6062–6072. doi: 10.1128/JVI.00330-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mikuličić S, Florin L. 2019. The endocytic trafficking pathway of oncogenic papillomaviruses. Papillomavirus Res 7:135–137. doi: 10.1016/j.pvr.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmitt A, Zeltner R, Iftner T, Rochat A, Barrandon Y, Borenstein L, Wettstein FO. 1996. The primary target cells of the high-risk cottontail rabbit papillomavirus colocalize with hair follicle stem cells. J Virol 70:1912–1922. doi: 10.1128/JVI.70.3.1912-1922.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McBride AA. 2008. Replication and partitioning of papillomavirus genomes. Adv Virus Res 72:155–205. doi: 10.1016/S0065-3527(08)00404-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doorbar J. 2005. The papillomavirus life cycle. J Clin Virol 32(Suppl):S7–S15. doi: 10.1016/j.jcv.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 8.Schiller JT, Day PM, Kines RC. 2010. Current understanding of the mechanism of HPV infection. Gynecol Oncol 118:S12–S17. doi: 10.1016/j.ygyno.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kajitani N, Satsuka A, Kawate A, Sakai H. 2012. Productive lifecycle of human papillomaviruses that depends upon squamous epithelial differentiation. Front Microbiol 3:152. doi: 10.3389/fmicb.2012.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT. 1992. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc Natl Acad Sci U S A 89:12180–12184. doi: 10.1073/pnas.89.24.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buck CB, Pastrana DV, Lowy DR, Schiller JT. 2004. Efficient intracellular assembly of papillomaviral vectors. J Virol 78:751–757. doi: 10.1128/jvi.78.2.751-757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buck CB, Pastrana DV, Lowy DR, Schiller JT. 2005. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol Med 119:445–462. doi: 10.1385/1-59259-982-6:445. [DOI] [PubMed] [Google Scholar]
- 13.Schelhaas M, Shah B, Holzer M, Blattmann P, Kühling L, Day PM, Schiller JT, Helenius A. 2012. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog 8:e1002657. doi: 10.1371/journal.ppat.1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spoden G, Kühling L, Cordes N, Frenzel B, Sapp M, Boller K, Florin L, Schelhaas M. 2013. Human papillomavirus types 16, 18, and 31 share similar endocytic requirements for entry. J Virol 87:7765–7773. doi: 10.1128/JVI.00370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schelhaas M. 2010. Come in and take your coat off—how host cells provide endocytosis for virus entry. Cell Microbiol 12:1378–1388. doi: 10.1111/j.1462-5822.2010.01510.x. [DOI] [PubMed] [Google Scholar]
- 16.Mercer J, Schelhaas M, Helenius A. 2010. Virus entry by endocytosis. Annu Rev Biochem 79:803–833. doi: 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- 17.Doherty GJ, McMahon HT. 2009. Mechanisms of endocytosis. Annu Rev Biochem 78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 18.Kerr MC, Lindsay MR, Luetterforst R, Hamilton N, Simpson F, Parton RG, Gleeson PA, Teasdale RD. 2006. Visualisation of macropinosome maturation by the recruitment of sorting nexins. J Cell Sci 119:3967–3980. doi: 10.1242/jcs.03167. [DOI] [PubMed] [Google Scholar]
- 19.Sieczkarski SB, Whittaker GR. 2002. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol 76:10455–10464. doi: 10.1128/jvi.76.20.10455-10464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quirin K, Eschli B, Scheu I, Poort L, Kartenbeck J, Helenius A. 2008. Lymphocytic choriomeningitis virus uses a novel endocytic pathway for infectious entry via late endosomes. Virology 378:21–33. doi: 10.1016/j.virol.2008.04.046. [DOI] [PubMed] [Google Scholar]
- 21.Rojek JM, Perez M, Kunz S. 2008. Cellular entry of lymphocytic choriomeningitis virus. J Virol 82:1505–1517. doi: 10.1128/JVI.01331-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Vries E, Tscherne DM, Wienholts MJ, Cobos-Jiménez V, Scholte F, García-Sastre A, Rottier PJM, de Haan C. 2011. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog 7:e1001329. doi: 10.1371/journal.ppat.1001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joyce JG, Tung JS, Przysiecki CT, Cook JC, Lehman ED, Sands JA, Jansen KU, Keller PM. 1999. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J Biol Chem 274:5810–5822. doi: 10.1074/jbc.274.9.5810. [DOI] [PubMed] [Google Scholar]
- 24.Giroglou T, Florin L, Schafer F, Streeck RE, Sapp M. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J Virol 75:1565–1570. doi: 10.1128/JVI.75.3.1565-1570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cerqueira C, Liu Y, Kühling L, Chai W, Hafezi W, van Kuppevelt TH, Kühn JE, Feizi T, Schelhaas M. 2013. Heparin increases the infectivity of human papillomavirus type 16 independent of cell surface proteoglycans and induces L1 epitope exposure. Cell Microbiol 15:1818–1836. doi: 10.1111/cmi.12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cerqueira C, Samperio Ventayol P, Vogeley C, Schelhaas M. 2015. Kallikrein-8 proteolytically processes human papillomaviruses in the extracellular space to facilitate entry into host cells. J Virol 89:7038–7052. doi: 10.1128/JVI.00234-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bienkowska-Haba M, Patel HD, Sapp M. 2009. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog 5:e1000524. doi: 10.1371/journal.ppat.1000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richards RM, Lowy DR, Schiller JT, Day PM. 2006. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc Natl Acad Sci U S A 103:1522–1527. doi: 10.1073/pnas.0508815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Selinka H-C, Florin L, Patel HD, Freitag K, Schmidtke M, Makarov VA, Sapp M. 2007. Inhibition of transfer to secondary receptors by heparan sulfate-binding drug or antibody induces noninfectious uptake of human papillomavirus. J Virol 81:10970–10980. doi: 10.1128/JVI.00998-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Becker M, Greune L, Schmidt MA, Schelhaas M. 2018. Extracellular conformational changes in the capsid of human papillomaviruses contribute to asynchronous uptake into host cells. J Virol 92:e02106-17. doi: 10.1128/JVI.02106-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spoden G, Freitag K, Husmann M, Boller K, Sapp M, Lambert C, Florin L. 2008. Clathrin- and caveolin-independent entry of human papillomavirus type 16–involvement of tetraspanin-enriched microdomains (TEMs). PLoS One 3:e3313. doi: 10.1371/journal.pone.0003313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheffer KD, Gawlitza A, Spoden GA, Zhang XA, Lambert C, Berditchevski F, Florin L. 2013. Tetraspanin CD151 mediates papillomavirus type 16 endocytosis. J Virol 87:3435–3446. doi: 10.1128/JVI.02906-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Surviladze Z, Dziduszko A, Ozbun MA. 2012. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog 8:e1002519. doi: 10.1371/journal.ppat.1002519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Surviladze Z, Sterk RT, DeHaro SA, Ozbun MA. 2013. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J Virol 87:2508–2517. doi: 10.1128/JVI.02319-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evander M, Frazer IH, Payne E, Qi YM, Hengst K, McMillan NA. 1997. Identification of the alpha6 integrin as a candidate receptor for papillomaviruses. J Virol 71:2449–2456. doi: 10.1128/JVI.71.3.2449-2456.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McMillan NAJ, Payne E, Frazer IH, Evander M. 1999. Expression of the α6 integrin confers papillomavirus binding upon receptor-negative B-cells. Virology 261:271–279. doi: 10.1006/viro.1999.9825. [DOI] [PubMed] [Google Scholar]
- 37.Yoon C-S, Kim K-D, Park S-N, Cheong S-W. 2001. α6 Integrin is the main receptor of human papillomavirus type 16 VLP. Biochem Biophys Res Commun 283:668–673. doi: 10.1006/bbrc.2001.4838. [DOI] [PubMed] [Google Scholar]
- 38.Woodham AW, Da Silva DM, Skeate JG, Raff AB, Ambroso MR, Brand HE, Isas JM, Langen R, Kast WM. 2012. The S100A10 subunit of the annexin A2 heterotetramer facilitates L2-mediated human papillomavirus infection. PLoS One 7:e43519. doi: 10.1371/journal.pone.0043519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dziduszko A, Ozbun MA. 2013. Annexin A2 and S100A10 regulate human papillomavirus type 16 entry and intracellular trafficking in human keratinocytes. J Virol 87:7502–7515. doi: 10.1128/JVI.00519-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mikuličić S, Finke J, Boukhallouk F, Wüstenhagen E, Sons D, Homsi Y, Reiss K, Lang T, Florin L. 2019. ADAM17-dependent signaling is required for oncogenic human papillomavirus entry platform assembly. Elife 8:e44345. doi: 10.7554/eLife.44345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fukada T, Sakajiri H, Kuroda M, Kioka N, Sugimoto K. 2017. Fluid shear stress applied by orbital shaking induces MG-63 osteosarcoma cells to activate ERK in two phases through distinct signaling pathways. Biochem Biophys Rep 9:257–265. doi: 10.1016/j.bbrep.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jo H, Sipos K, Go YM, Law R, Rong J, McDonald JM. 1997. Differential effect of shear stress on extracellular signal-regulated kinase and N-terminal jun kinase in endothelial cells: Gi2- and Gβ/γ- dependent signaling pathways. J Biol Chem 272:1395–1401. doi: 10.1074/jbc.272.2.1395. [DOI] [PubMed] [Google Scholar]
- 43.Ciardiello F, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, Bianco AR, Tortora G. 2000. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res 6:2053–2063. [PubMed] [Google Scholar]
- 44.Rapraeger AC, Krufka A, Olwin BB. 1991. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science 252:1705–1708. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- 45.Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. 1991. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 64:841–848. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]
- 46.Lycke E, Johansson M, Svennerholm B, Lindahl U. 1991. Binding of herpes simplex virus to cellular heparan sulphate, an initial step in the adsorption process. J Gen Virol 72:1131–1137. doi: 10.1099/0022-1317-72-5-1131. [DOI] [PubMed] [Google Scholar]
- 47.Shieh MT, WuDunn D, Montgomery RI, Esko JD, Spear PG. 1992. Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J Cell Biol 116:1273–1281. doi: 10.1083/jcb.116.5.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schowalter RM, Pastrana DV, Buck CB. 2011. Glycosaminoglycans and sialylated glycans sequentially facilitate Merkel cell polyomavirus infectious entry. PLoS Pathog 7:e1002161. doi: 10.1371/journal.ppat.1002161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diao J, Pantua H, Ngu H, Komuves L, Diehl L, Schaefer G, Kapadia SB. 2012. Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry. J Virol 86:10935–10949. doi: 10.1128/JVI.00750-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eierhoff T, Hrincius ER, Rescher U, Ludwig S, Ehrhardt C. 2010. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog 6:e1001099. doi: 10.1371/journal.ppat.1001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Day EK, Sosale NG, Lazzara MJ. 2016. Cell signaling regulation by protein phosphorylation: a multivariate, heterogeneous, and context-dependent process. Curr Opin Biotechnol 40:185–192. doi: 10.1016/j.copbio.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mettlen M, Platek A, Van Der Smissen P, Carpentier S, Amyere M, Lanzetti L, de Diesbach P, Tyteca D, Courtoy PJ. 2006. Src triggers circular ruffling and macropinocytosis at the apical surface of polarized MDCK cells. Traffic 7:589–603. doi: 10.1111/j.1600-0854.2006.00412.x. [DOI] [PubMed] [Google Scholar]
- 53.Hennequin LF, Allen J, Breed J, Curwen J, Fennell M, Green TP, Lambert-Van Der Brempt C, Morgentin R, Norman RA, Olivier A, Otterbein L, Plé PA, Warin N, Costello G. 2006. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2–(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem 49:6465–6488. doi: 10.1021/jm060434q. [DOI] [PubMed] [Google Scholar]
- 54.Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, Whelan S. 2009. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog 5:e1000394. doi: 10.1371/journal.ppat.1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. 1996. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor: Study of Lck- and FynT-dependent T cell activation. J Biol Chem 271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 56.Zhu X, Kim JL, Newcomb JR, Rose PE, Stover DR, Toledo LM, Zhao H, Morgenstern KA. 1999. Structural analysis of the lymphocyte-specific kinase Lck in complex with non-selective and Src family selective kinase inhibitors. Structure 7:651–661. doi: 10.1016/s0969-2126(99)80086-0. [DOI] [PubMed] [Google Scholar]
- 57.Brandvold KR, Steffey ME, Fox CC, Soellner MB. 2012. Development of a highly selective c-Src kinase inhibitor. ACS Chem Biol 7:1393–1398. doi: 10.1021/cb300172e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mainou BA, Dermody TS. 2011. Src kinase mediates productive endocytic sorting of reovirus during cell entry. J Virol 85:3203–3213. doi: 10.1128/JVI.02056-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klinghoffer RA, Sachsenmaier C, Cooper JA, Soriano P. 1999. Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J 18:2459–2471. doi: 10.1093/emboj/18.9.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marsh M, Helenius A. 1980. Adsorptive endocytosis of Semliki Forest virus. J Mol Biol 142:439–454. doi: 10.1016/0022-2836(80)90281-8. [DOI] [PubMed] [Google Scholar]
- 61.Helenius A, Kartenbeck J, Simons K, Fries E. 1980. On the entry of Semliki Forest virus into BHK-21 cells. J Cell Biol 84:404–420. doi: 10.1083/jcb.84.2.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.White J, Kartenbeck J, Helenius A. 1980. Fusion of Semliki Forest virus with the plasma membrane can be induced by low pH. J Cell Biol 87:264–272. doi: 10.1083/jcb.87.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marsh M, Kielian MC, Helenius A. 1984. Semliki Forest virus entry and the endocytic pathway. Biochem Soc Trans 12:981–983. doi: 10.1042/bst0120981. [DOI] [PubMed] [Google Scholar]
- 64.Weisberg E, Manley PW, Breitenstein W, Brüggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E, Kung AL, Mestan J, Daley GQ, Callahan L, Catley L, Cavazza C, Azam M, Mohammed A, Neuberg D, Wright RD, Gilliland DG, Griffin JD. 2005. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 7:129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 65.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. 2000. Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science 289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 66.Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W. 2005. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J Cell Biol 170:317–325. doi: 10.1083/jcb.200503059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dasgupta J, Bienkowska-Haba M, Ortega ME, Patel HD, Bodevin S, Spillmann D, Bishop B, Sapp M, Chen XS. 2011. Structural basis of oligosaccharide receptor recognition by human papillomavirus. J Biol Chem 286:2617–2624. doi: 10.1074/jbc.M110.160184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aviezer D, Yayon A. 1994. Heparin-dependent binding and autophosphorylation of epidermal growth factor (EGF) receptor by heparin-binding EGF-like growth factor but not by EGF. Proc Natl Acad Sci U S A 91:12173–12177. doi: 10.1073/pnas.91.25.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wojcechowskyj JA, Didigu CA, Lee JY, Parrish NF, Sinha R, Hahn BH, Bushman FD, Jensen ST, Seeholzer SH, Doms RW. 2013. Quantitative phosphoproteomics reveals extensive cellular reprogramming during HIV-1 entry. Cell Host Microbe 13:613–623. doi: 10.1016/j.chom.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. 2006. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 71.Ueki IF, Min-Oo G, Kalinowski A, Ballon-Landa E, Lanier LL, Nadel JA, Koff JL. 2013. Respiratory virus-induced EGFR activation suppresses IRF1-dependent interferon λ and antiviral defense in airway epithelium. J Exp Med 210:1929–1936. doi: 10.1084/jem.20121401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zheng K, Kitazato K, Wang Y. 2014. Viruses exploit the function of epidermal growth factor receptor. Rev Med Virol 24:274–286. doi: 10.1002/rmv.1796. [DOI] [PubMed] [Google Scholar]
- 73.Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PF. 2009. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog 5:e1000318. doi: 10.1371/journal.ppat.1000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aydin I, Weber S, Snijder B, Samperio Ventayol P, Kühbacher A, Becker M, Day PM, Schiller JT, Kann M, Pelkmans L, Helenius A, Schelhaas M. 2014. Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathog 10:e1004162. doi: 10.1371/journal.ppat.1004162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Day PM, Lowy DR, Schiller JT. 2008. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J Virol 82:12565–12568. doi: 10.1128/JVI.01631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Böni-Schnetzler M, Pilch PF. 1987. Mechanism of epidermal growth factor receptor autophosphorylation and high-affinity binding. Proc Natl Acad Sci U S A 84:7832–7836. doi: 10.1073/pnas.84.22.7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Klosek SK, Nakashiro KI, Hara S, Shintani S, Hasegawa H, Hamakawa H. 2005. CD151 forms a functional complex with c-Met in human salivary gland cancer cells. Biochem Biophys Res Commun 336:408–416. doi: 10.1016/j.bbrc.2005.08.106. [DOI] [PubMed] [Google Scholar]
- 78.Stuart JR, Gonzalez FH, Kawai H, Yuan ZM. 2006. c-Abl interacts with the WAVE2 signaling complex to induce membrane ruffling and cell spreading. J Biol Chem 281:31290–31297. doi: 10.1074/jbc.M602389200. [DOI] [PubMed] [Google Scholar]
- 79.Abban CY, Meneses PI. 2010. Usage of heparan sulfate, integrins, and FAK in HPV16 infection. Virology 403:1–16. doi: 10.1016/j.virol.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Parton RG, Joggerst B, Simons K. 1994. Regulated internalization of caveolae. J Cell Biol 127:1199–1215. doi: 10.1083/jcb.127.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kiss AL, Botos E. 2009. Ocadaic acid retains caveolae in multicaveolar clusters. Pathol Oncol Res 15:479–486. doi: 10.1007/s12253-008-9139-4. [DOI] [PubMed] [Google Scholar]
- 82.Colicelli J. 2010. ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci Signal 3:re6. doi: 10.1126/scisignal.3139re6 (Erratum, 4:er4, 2011.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Edimo WE, Ghosh S, Derua R, Janssens V, Waelkens E, Vanderwinden JM, Robe P, Erneux C. 2016. SHIP2 controls plasma membrane PI(4,5)P2 thereby participating in the control of cell migration in 1321 N1 glioblastoma cells. J Cell Sci 129:1101–1114. doi: 10.1242/jcs.179663. [DOI] [PubMed] [Google Scholar]
- 84.Liang X, Hajivandi M, Veach D, Wisniewski D, Clarkson B, Resh MD, Pope RM. 2006. Quantification of change in phosphorylation of BCR-ABL kinase and its substrates in response to Imatinib treatment in human chronic myelogenous leukemia cells. Proteomics 6:4554–4564. doi: 10.1002/pmic.200600109. [DOI] [PubMed] [Google Scholar]
- 85.Plattner R, Irvin BJ, Guo S, Blackburn K, Kazlauskas A, Abraham RT, York JD, Pendergast AM. 2003. A new link between the c-Abl tyrosine kinase and phosphoinositide signalling through PLC-γ1. Nat Cell Biol 5:309–319. doi: 10.1038/ncb949. [DOI] [PubMed] [Google Scholar]
- 86.Liu G, Huang YJ, Xiao R, Wang D, Acton TB, Montelione GT. 2010. NMR structure of F-actin-binding domain of Arg/AbI2 from Homo sapiens. Proteins Struct Funct Bioinform 78:1326–1330. doi: 10.1002/prot.22656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang Y, Miller AL, Mooseker MS, Koleske AJ. 2001. The Abl-related gene (Arg) nonreceptor tyrosine kinase uses two F-actin-binding domains to bundle F-actin. Proc Natl Acad Sci U S A 98:14865–14870. doi: 10.1073/pnas.251249298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Courtemanche N, Gifford SM, Simpson MA, Pollard TD, Koleske AJ. 2015. Abl2/Abl-related gene stabilizes actin filaments, stimulates actin branching by actin-related protein 2/3 complex, and promotes actin filament severing by cofilin. J Biol Chem 290:4038–4046. doi: 10.1074/jbc.M114.608117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Plattner R, Kadlec L, DeMali KA, Kazlauskas A, Pendergast AM. 1999. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev 13:2400–2411. doi: 10.1101/gad.13.18.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jones R, Gordus A, Krall J, Macbeath G. 2006. A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature 439:168–174. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- 91.Sini P, Cannas A, Koleske AJ, Di Fiore PP, Scita G. 2004. Abl-dependent tyrosine phosphorylation of Sos-1 mediates growth-factor-induced Rac activation. Nat Cell Biol 6:268–274. doi: 10.1038/ncb1096. [DOI] [PubMed] [Google Scholar]
- 92.Haigler HT, McKanna JA, Cohen S. 1979. Rapid stimulation of pinocytosis in human carcinoma cells A-431 by epidermal growth factor. J Cell Biol 83:82–90. doi: 10.1083/jcb.83.1.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bougnères L, Girardin SE, Weed SA, Karginov AV, Olivo-Marin JC, Parsons JT, Sansonetti PJ, Van Nhieu GT. 2004. Cortactin and Crk cooperate to trigger actin polymerization during Shigella invasion of epithelial cells. J Cell Biol 166:225–235. doi: 10.1083/jcb.200402073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kasahara K, Nakayama Y, Sato I, Ikeda K, Hoshino M, Endo T, Yamaguchi N. 2007. Role of Src-family kinases in formation and trafficking of macropinosomes. J Cell Physiol 211:220–232. doi: 10.1002/jcp.20931. [DOI] [PubMed] [Google Scholar]
- 95.Donepudi M, Resh MD. 2008. c-Src trafficking and co-localization with the EGF receptor promotes EGF ligand-independent EGF receptor activation and signaling. Cell Signal 20:1359–1367. doi: 10.1016/j.cellsig.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Scherer WF, Syverton JT, Gey GO. 1953. Studies on the propagation in vitro of poliomyelitis viruses. J Exp Med 97:695–710. doi: 10.1084/jem.97.5.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Snijder B, Sacher R, Rämö P, Liberali P, Mench K, Wolfrum N, Burleigh L, Scott CC, Verheije MH, Mercer J, Moese S, Heger T, Theusner K, Jurgeit A, Lamparter D, Balistreri G, Schelhaas M, De Haan CAM, Marjomäki V, Hyypiä T, Rottier PJM, Sodeik B, Marsh M, Gruenberg J, Amara A, Greber U, Helenius A, Pelkmans L. 2012. Single-cell analysis of population context advances RNAi screening at multiple levels. Mol Syst Biol 8:579. doi: 10.1038/msb.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. 1988. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Johannsdottir HK, Mancini R, Kartenbeck J, Amato L, Helenius A. 2009. Host cell factors and functions involved in vesicular stomatitis virus entry. J Virol 83:440–453. doi: 10.1128/JVI.01864-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schowalter RM, Pastrana DV, Pumphrey KA, Moyer AL, Buck CB. 2010. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe 7:509–515. doi: 10.1016/j.chom.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hafezi W, Lorentzen EU, Eing BR, Müller M, King NJC, Klupp B, Mettenleiter TC, Kühn JE. 2012. Entry of herpes simplex virus type 1 (HSV-1) into the distal axons of trigeminal neurons favors the onset of nonproductive, silent infection. PLoS Pathog 8:e1002679. doi: 10.1371/journal.ppat.1002679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Berard A, Coombs KM. 2009. Mammalian reoviruses: propagation, quantification, and storage. Curr Protoc Microbiol 14:15C.1.1–15C.1.18. doi: 10.1002/9780471729259.mc15c01s14. [DOI] [PubMed] [Google Scholar]
- 103.Engel S, Heger T, Mancini R, Herzog F, Kartenbeck J, Hayer A, Helenius A. 2011. Role of endosomes in simian virus 40 entry and infection. J Virol 85:4198–4211. doi: 10.1128/JVI.02179-10. [DOI] [PMC free article] [PubMed] [Google Scholar]