

Klebsiella pneumoniae strains that produce extended-spectrum beta lactamases (ESBLs) are a persistent public health threat. There are relatively few therapeutic options, and there is undue reliance on carbapenems. Alternative therapeutic options are urgently required. A combination of cefepime and the novel beta lactamase inhibitor enmetazobactam is being developed for the treatment of serious infections caused by ESBL-producing organisms. The pharmacokinetics-pharmacodynamics (PK-PD) of cefepime-enmetazobactam against ESBL-producing K. pneumoniae was studied in a neutropenic murine pneumonia model.
KEYWORDS: ESBL, Klebsiella, beta-lactamases, cefepime, enmetazobactam, epithelial lining fluid, multidrug resistance, pharmacodynamics, pharmacokinetics, pneumonia
ABSTRACT
Klebsiella pneumoniae strains that produce extended-spectrum beta lactamases (ESBLs) are a persistent public health threat. There are relatively few therapeutic options, and there is undue reliance on carbapenems. Alternative therapeutic options are urgently required. A combination of cefepime and the novel beta lactamase inhibitor enmetazobactam is being developed for the treatment of serious infections caused by ESBL-producing organisms. The pharmacokinetics-pharmacodynamics (PK-PD) of cefepime-enmetazobactam against ESBL-producing K. pneumoniae was studied in a neutropenic murine pneumonia model. Dose-ranging studies were performed. Dose fractionation studies were performed to define the relevant PD index for the inhibitor. The partitioning of cefepime and enmetazobactam into the lung was determined by comparing the area under the concentration-time curve (AUC) in plasma and epithelial lining fluid. The magnitude of drug exposure for cefepime-enmetazobactam required for logarithmic killing in the lung was defined using 3 ESBL-producing strains. Cefepime, given as 100 mg/kg of body weight every 8 h intravenously (q8h i.v.), had minimal antimicrobial effect. When this background regimen of cefepime was combined with enmetazobactam, a half-maximal effect was induced with enmetazobactam at 4.71 mg/kg q8h i.v. The dose fractionation study suggested both fT > threshold and fAUC:MIC are relevant PD indices. The AUCELF:AUCplasma ratio for cefepime and enmetazobactam was 73.4% and 61.5%, respectively. A ≥2-log kill in the lung was achieved with a plasma and ELF cefepime fT > MIC of ≥20% and enmetazobactam fT > 2 mg/liter of ≥20% of the dosing interval. These data and analyses provide the underpinning evidence for the combined use of cefepime and enmetazobactam for nosocomial pneumonia.
INTRODUCTION
Enterobacteriaceae that produce extended-spectrum beta lactamases (ESBL) are endemic in many health care settings (1–3). Infections caused by these pathogens are expensive, compromise clinical outcomes, and force an ever-increasing upward spiral of the use of broad-spectrum antimicrobial agents. Quinolones, carbapenems, and aminoglycosides are the antibiotics of choice for the treatment of infections caused by ESBL-producing organisms. However, the coexpression of quinolone and aminoglycoside resistance mechanisms and/or concerns about toxicity with these agents (4, 5) means the carbapenems are often the agents of choice (6). Newer β-lactam–β-lactamase inhibitor combinations, such as ceftazidime-avibactam (7) and meropenem-vaborbactam (8), are active against ESBL-producing Enterobacteriaceae but have been developed primarily for infections caused by carbapenemase-producing organisms. Carbapenem resistance is becoming increasingly prevalent. The loss of this class, because of inappropriate and excessive use, will be nothing short of catastrophic for the provision of modern health care. Hence, new options for the treatment of infections caused by ESBL-producing pathogens that can spare the use of carbapenems are urgently required (9).
Enmetazobactam (formerly AAI101; Allecra Therapeutics, Saint-Louis, France) is a novel penicillanic acid sulfone beta lactamase inhibitor (BLI) that is structurally similar to tazobactam. Enmetazobactam is active against a wide range of class A β-lactamases, including ESBLs (e.g., TEM, SHV, and CTX-M) as well as some class C and class D β-lactamases (10, 11). Enmetazobactam is being developed in combination with cefepime to protect the latter against hydrolysis by Ambler class A enzymes. The potential efficacy of cefepime-enmetazobactam against class C and class D OXA-48 enzymes is conferred by the intrinsic stability of cefepime, while the combination is required for those organisms that coproduce class A and C enzymes. The efficacy of cefepime-enmetazobactam is currently being compared with that of piperacillin-tazobactam in a phase III clinical trial of patients with complicated urinary tract infection and/or acute pyelonephritis. The potential utility of cefepime-enmetazobactam for other diseases remains to be determined.
Here, we describe the pharmacokinetics and pharmacodynamics of the combination of cefepime-enmetazobactam with the principal aim of establishing the utility of cefepime-enmetazobactam for nosocomial pneumonia (i.e., hospital-acquired and ventilator-acquired pneumonia [HAP-VAP]). The studies were designed to provide a pharmacodynamic rationale for dosage selection for HAP-VAP as the current clinical program moves forwards from initial clinical studies in complicated urinary tract infection to other more complex nosocomial infections.
RESULTS
In vitro susceptibility and genotype.
The MICs and genotypes of challenge strains used in the study are summarized in Table 1. All strains were obtained from IHMA. The genotype was determined by multiplex PCR (12). MICs were determined in the presence of enmetazobactam at a fixed concentration of 4 and 8 mg/liter using Clinical and Laboratory Standards Institute methodology and the quality control strains Escherichia coli ATCC 25922 and E. coli NCTC 13353 to ensure appropriate assay performance (13). MICs were determined over the course of five experiments, and the modal value was used for the pharmacodynamic analyses.
TABLE 1.
Details of the study strains
| Isolate | Resistance mechanism(s) | MICa for: |
|||
|---|---|---|---|---|---|
| Cefepime [mg/liter (range)] | Cefepime + enmetazobactam [4 mg/liter (range)] | Cefepime + enmetazobactam [8 mg/liter (range)] | Meropenem [mg/liter (range)] | ||
| K. pneumoniae 1280740 | CTX-M-15, DHA-1 | 128 (128–>128) | 0.12 (0.06–0.12) | 0.06 (0.06–0.06) | 0.12 (0.06–0.12) |
| K. pneumoniae 1256506 | CTX-M-2, CMY-2 | 128 (64–>128) | 0.25 (0.12–0.5) | 0.12 (0.12–0.12) | 0.06 (0.06–0.06) |
| K. pneumoniae 1091463 | CTX-M-3 | >128 | 4 (4–16) | 2 (1–2) | 8 (4–8) |
The modal MIC is reported and the range determined from 5 independently conducted experiments.
Murine pharmacokinetics of cefepime and enmetazobactam (in combination).
The pharmacokinetics of cefepime and enmetazobactam were determined in combination and are shown in Fig. 1. The pharmacokinetics of both agents were linear. Total drug concentrations were determined; however, the plasma protein binding for both compounds in mice is negligible (14) and was assumed to be zero for the PK-PD calculations.
FIG 1.

Plasma PK profile for cefepime and enmetazobactam. (A and B) Total drug concentration-time profiles for cefepime (A) and enmetazobactam (B). Both drugs were coadministered i.v. at 0, 8, and 16 h with sampling in the 1st and 3rd intervals. A destructive design was used, with groups of mice at each dose-time point being serially sacrificed. Data are means ± standard deviations from 3 mice.
The fit of a standard two-compartment PK structural model with time-delineated zero-order intravenous input of drug (i.e., i.v. injection into the tail vein) and first-order clearance of drug from the central compartment was satisfactory for both cefepime and enmetazobactam. However, closer inspection of the cefepime PK data suggested the presence of a prolonged terminal gamma phase of drug elimination with plasma concentrations that approximated the MIC and, hence, were likely biologically relevant (Fig. 1 and 2). A two-compartment model fit to the cefepime data resulted in significant underprediction of these later time points (i.e., 4 and 8 h), and simulations from this model resulted in the underestimation of the fT > MIC, especially for strains with lower MICs. Hence, for cefepime, a third compartment was added. This enabled the gamma phase and the later time points to be better described by the pharmacokinetic model and more accurate estimates of fT > MIC. The parameter estimates for the final cefepime and enmetazobactam models are summarized in Table 2. The half-life of the terminal gamma phase of elimination for cefepime was approximately 1 h.
FIG 2.

Intrapulmonary pharmacokinetics of cefepime and enmetazobactam. Cefepime (A to C) and enmetazobactam (D to F) were coadministered as 25 and 6.25 mg/kg, 50 and 25 mg/kg, and 200 and 200 mg/kg once. Plasma and epithelial lining fluid (ELF) were sampled using a destructive design from 0 to 8 h postdose. Data are means ± standard deviations from n = 3 mice. Samples of cefepime and enmetazobactam in ELF in the latter part of the dosing interval in panels A, B, D, and E are not shown because they were beneath the limit of detection, which was likely related to the dilution effect from the bronchoalveolar lavage.
TABLE 2.
Parameter estimates from the population PK models fit to total drug concentration-time profiles for cefepime and enmetazobactama
| Parameterb | Cefepime |
Enmetazobactam |
||||
|---|---|---|---|---|---|---|
| Mean | Median | SD | Mean | Median | SD | |
| SCL (liters/h) | 0.043 | 0.043 | 0.005 | 0.040 | 0.040 | 0.007 |
| V (liters) | 0.007 | 0.007 | 0.000 | 0.009 | 0.010 | 0.003 |
| kcp (h−1) | 4.046 | 3.654 | 1.510 | 0.868 | 0.639 | 0.499 |
| kpc (h−1) | 28.185 | 29.607 | 3.205 | 0.731 | 0.559 | 0.370 |
| kcd (h−1) | 0.350 | 0.325 | 0.082 | |||
| kdc (h−1) | 0.837 | 0.797 | 0.278 | |||
The mean and median parameter estimates, and their standard deviations, obtained from the population PK models fit to cefepime and enmetazobactam total drug concentration-time profiles. The point estimates of the population median parameter values for both cefepime and enmetazobactam were used for the PK modeling and estimation of drug exposure.
SCL is the first-order clearance of enmetazobactam from the central compartment; V is the volume of the central compartment; kcp, kpc, kcd, and kdc are the first-order intercompartmental rate constants.
Intrapulmonary PK and partitioning of cefepime and enmetazobactam into the murine lung.
Cefepime and enmetazobactam partitioned readily into the epithelial lining fluid (ELF) of mice (Fig. 2). There was no evidence of system hysteresis for either agent. The AUCELF:AUCplasma ratio was 73.4% and 61.5% for cefepime and enmetazobactam, respectively, when assessed from time 0 to 8 h after drug coadministration. These estimates were calculated using a population PK model fit to each agent separately. Protein binding in ELF was assumed to be negligible. A summary of the parameter estimates from the population PK model is provided in Table 3.
TABLE 3.
Summary of the parameter estimates from the population PK modela
| Parameterb | Cefepime |
Enmetazobactam |
||||
|---|---|---|---|---|---|---|
| Mean | Median | SD | Mean | Median | SD | |
| SCL (liters/h) | 0.027 | 0.022 | 0.009 | 0.026 | 0.021 | 0.016 |
| V (liters) | 0.002 | 0.002 | 0.001 | 0.005 | 0.002 | 0.008 |
| VELF (liters) | 0.006 | 0.008 | 0.004 | 0.013 | 0.003 | 0.027 |
| kcp (h−1) | 24.173 | 25.495 | 2.996 | 18.301 | 16.924 | 7.589 |
| kpc (h−1) | 28.521 | 29.633 | 2.129 | 8.825 | 7.296 | 5.476 |
| kcl (h−1) | 26.542 | 27.930 | 2.872 | 15.761 | 16.954 | 5.084 |
| klc (h−1) | 13.138 | 13.429 | 2.876 | 17.652 | 17.349 | 6.863 |
| kcd (h−1) | 1.343 | 1.350 | 0.434 | |||
| kdc (h−1) | 0.838 | 0.901 | 0.186 | |||
The mean and median parameter estimates, and their standard deviations, obtained from the population PK models fitted to cefepime and enmetazobactam total drug concentration-time profiles in plasma and lung. The point estimates of the population median parameter values for both cefepime and enmetazobactam were used for the PK modeling and estimation of drug exposure.
SCL is the first-order clearance of enmetazobactam from the central compartment; V is the volume of the central compartment; kcp, kpc, kcl, klc, kcd, and kdc are first-order intercompartmental rate constants connecting the central, peripheral, deep, and lung compartments.
Murine dose-response relationships for cefepime-enmetazobactam.
Multiple preliminary experiments were conducted to define the optimum background regimen of cefepime that enabled the elucidation of the pharmacodynamics of the combination of cefepime-enmetazobactam. A number of combinations of dosages and schedules were studied to identify regimens that produced a reproducible submaximal response. This approach provided the necessary “headroom” to enable the investigation of the pharmacodynamics of enmetazobactam (i.e., if there was too little cefepime, there is nothing that enmetazobactam can protect, and if there is too much, a near-maximal response is induced and there is no ability to elucidate the pharmacodynamics of enmetazobactam). As a result, a half-maximal effect (E50) was targeted. The challenge strain used for these experiments was Klebsiella pneumoniae 1280740, with a cefepime MIC of 128 mg/liter, and cefepime-enmetazobactam MICs determined in the presence of 4 and 8 mg/liter enmetazobactam were 0.125 and 0.06 mg/liter, respectively (Table 1).
A backbone of 100 mg/kg of body weight cefepime every 8 h intravenously (q8h i.v.) was selected, as it characteristically had no effect on reducing the bacterial burden in the lung after 24 h of treatment (Fig. 3 and 4, where the data from cefepime-treated mice essentially overlays that from vehicle-treated controls). This regimen produced an fT > 128 mg/liter and fT > 0.06 mg/liter for 2% and 65%, respectively, of the dosing interval. Both drugs were administered as an i.v. combination on a q8h schedule. The dose-response relationship is shown in Fig. 3. The dose of enmetazobactam that produced a half-maximal effect in the presence of 100 mg/kg cefepime q8h was 4.71 mg/kg q8h, or approximately 15 mg/kg/day.
FIG 3.

Exposure-response relationship for cefepime-enmetazobactam against Klebsiella pneumoniae. Pharmacodynamics of cefepime-enmetazobactam in a murine lung model with Klebsiella pneumoniae 1280740 as the challenge strain. Data are the means and standard deviations from n = 3 mice. The solid blue squares are the pretreatment and end-of-therapy vehicle-treated controls, with expansion of approximately 1-log over a 24-h period. The solid red line is the fit of an inhibitory sigmoid Emax model to the data. The broken blue line is the stasis line (i.e., the pretreatment control obtained 2 h postinoculation). Half-maximal activity is achieved with a regimen of 100 mg/kg cefepime q8h plus 4.71 mg/kg enmetazobactam q8h.
FIG 4.

Dose fractionation study of cefepime in combination with enmetazobactam against Klebsiella pneumoniae. In panel A, the data from vehicle-treated controls (solid black squares) are shown, and these data were overlaid with the data from a cohort of mice receiving 100 mg/kg cefepime q8h i.v. (solid red squares) obtained from the same experiment. Mice shown in panels B to D all received 100 mg/kg cefepime q8h i.v. and a total daily dosage of 15 mg/kg/day enmetazobactam. Enmetazobactam was administered as 7.5 mg/kg q12h (B), 5 mg/kg q8h (C), and 2.5 mg/kg q4h (D). The fit of a PK-PD model to the data is shown, with the plasma concentrations and bacterial density represented by the red and black solid lines, respectively. The PK was used in the fitting but was not specifically measured in this experiment. The pharmacodynamic data (solid black squares) are means ± standard deviations from n = 4 mice.
Murine dose fractionation experiments.
To determine the relevant pharmacodynamic index for enmetazobactam, a fixed backbone of 100 mg/kg cefepime q8h was used. The dose-finding experiments (described above) were used to design the dose fractionation experiment. While a number of different designs were possible, we ultimately chose an isodose experiment where the time course of antibacterial activity of candidate regimens comprising the same total daily dosage was assessed. A dosage of 5 mg/kg/day q8h resulted in (approximately) half-maximal effect. Hence, a total daily dosage of 15 mg/kg/day was fractionated, and the following regimens were used: (i) 7.5 mg/kg q12h; (ii) 5 mg/kg q8h; and (iii) 2.5 mg/kg q4h.
The fractionation study enabled the antibacterial activity of enmetazobactam to be established. There were statistically significant differences in overall changes in log10(CFU/g) with time between cefepime alone and 100 mg/kg cefepime q8h plus 2.5 mg/kg enmetazobactam q4h (P = 0.009) as well as cefepime alone and 100 mg/kg cefepime q8h plus 5 mg/kg enmetazobactam q8h (P = 0.06). However, there was no difference in cefepime alone and cefepime plus 7.5 mg/kg enmetazobactam q12h (P = 0.58).
Visual inspection of the observed data and the fits of the mathematical model (Table 4) suggested fT > threshold as the relevant pharmacodynamic index. More fractionated regimens resulted in (visually) greater logarithmic killing. However, these differences were not statistically significant when the different schedules were compared using the overall pairwise comparisons of log10(CFU/g) from estimated overall mean effects from the linear mixed-effects modeling. There were no differences between the effect of 100 mg/kg cefepime q8h plus 2.5 mg/kg enmetazobactam q4h versus 100 mg/kg cefepime q8h plus 5 mg/kg enmetazobactam q8h (P = 0.99); 100 mg/kg cefepime q8h plus 2.5 mg/kg enmetazobactam q4h versus 100 mg/kg cefepime q8h plus 7.5 mg/kg enmetazobactam q12h (P = 0.92); and 100 mg/kg cefepime q8h plus 5 mg/kg enmetazobactam q8h versus 100 mg/kg cefepime q8h plus 7.5 mg/kg enmetazobactam q12h (P = 0.99). Hence, there were no statistically significant differences in any of the enmetazobactam schedules. Regardless of the schedule, the population PK model predicted that bacterial regrowth occurred when enmetazobactam plasma concentrations were <0.07 mg/liter (Fig. 5).
TABLE 4.
Parameters estimates from the population PK-PD model fit to the dose fractionation experiment
| Parametera (unit) | Mean | Median | SD |
|---|---|---|---|
| SCL (liters/h) | 0.037 | 0.038 | 0.005 |
| V (liters) | 0.008 | 0.011 | 0.003 |
| kcp (h−1) | 0.872 | 0.736 | 0.723 |
| kpc (h−1) | 0.685 | 0.546 | 0.394 |
| kgmax (log10 CFU/g/h) | 0.126 | 0.145 | 0.036 |
| Popmax (CFU/g) | 6,593,420,133 | 5,636,594,684 | 3,022,060,598 |
| kkmax (log10 CFU/g/h) | 0.679 | 0.746 | 0.132 |
| Hk | 3.236 | 2.932 | 1.125 |
| C50k (mg/liter) | 0.112 | 0.118 | 0.033 |
| Initial condition (CFU/g) | 37,240,976.245 | 37,241,784.148 | 1,415,354.042 |
SCL is the first-order clearance of enmetazobactam from the central compartment; V is the volume of the central compartment; kcp and kpc are the first-order intercompartmental rate constants; kgmax is the maximum rate of bacterial growth; popmax is the maximum theoretical bacterial density in the thigh; kkmax is the maximum rate if drug-induced bacterial killing; Hk is the slope function; C50k is the concentration of enmetazobactam where the rate of killing is half maximal; initial condition is the estimated bacterial density in the thigh immediately postinoculation.
FIG 5.
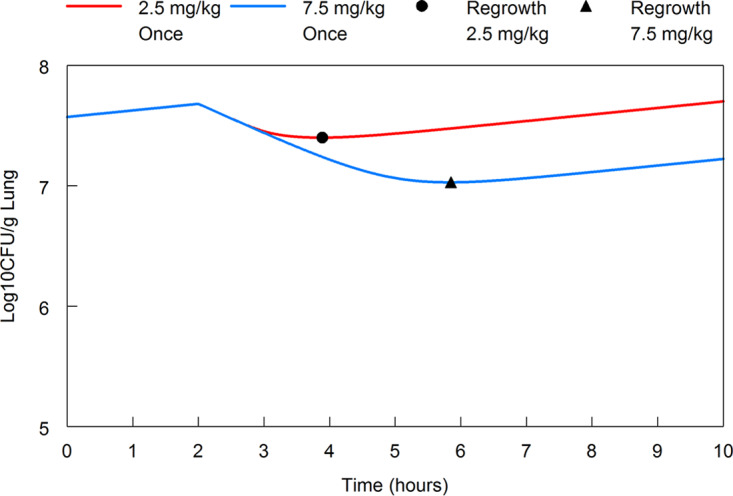
Threshold plasma concentration of enmetazobactam where regrowth is observed. The modeled enmetazobactam plasma concentration that allows regrowth is 0.07 mg/liter. The 2.5-mg/kg enmetazobactam regimen (solid red line) administered on a q4h schedule results in killing for approximately 47% of the dosing interval. The solid black circle indicates regrowth at 1.89 h postdose for the 2.5-mg/kg q4h schedule. The 7.5-mg/kg enmetazobactam regimen (solid blue line) administered on a q12h schedule results in killing for approximately 32% of the dosing interval. The solid black triangle indicates regrowth at 3.84 h postdose for the 7.5-mg/kg q12h schedule.
To further assess the impact of fractionation on the pharmacodynamics of cefepime-enmetazobactam, a bootstrap analysis was performed using the mathematical model fit to the PK-PD data set. There were no differences in the maximum rate of enmetazobactam-induced killing (kkmax) or the concentration of enmetazobactam that induced a half-maximal rate of killing (i.e., C50k), further supporting AUC:threshold as the relevant pharmacodynamic index.
In summary, both the AUC and time > threshold can be used to account for the experimental data. For dose identification, we chose to use fT > threshold, since it is potentially a more conservative measure for in vivo clinical bridging studies. The time-dependent pharmacodynamics of enmetazobactam was described using a threshold of 2 mg/liter. This value was estimated in a hollow-fiber infection model using ESBL-producing Enterobacteriaceae. Concentrations of enmetazobactam of >2 mg/liter infused on a background of concentrations similar to those achieved with the administration of 2 g cefepime q8h i.v. suppressed the emergence of resistance (data not shown).
Pharmacodynamics of cefepime-enmetazobactam against Klebsiella pneumoniae.
A 4-by-4 matrix design was used to elucidate the pharmacodynamics of the combination against three strains of Klebsiella pneumoniae. One reason for choosing this design was an observation from a previous study (15) that the amount of inhibitor required to achieve a given endpoint was dependent on the concentration of beta-lactam. The lack of intrinsic in vivo activity of enmetazobactam was confirmed in a single experiment using strain 1280740. The pharmacodynamics of strains 1256506 and 1091463 were determined using a 4-by-3 design; a cohort receiving enmetazobactam monotherapy was not further used once futility had been established. An interaction surface was fit to the entire data set using the relevant pharmacodynamic index for each agent. The interaction surface was steep, with negligible activity of cefepime alone and no intrinsic activity of enmetazobactam (Fig. 6).
FIG 6.

Pharmacodynamics of cefepime-enmetazobactam against ESBL-producing strains of Klebsiella pneumoniae. A matrix design was used throughout, with the same dosages of cefepime and enmetazobactam on a mg/kg basis, using 1280740 (MIC, 0.06 mg/liter), 1256506 (MIC, 0.125 mg/liter), and 1091463 (MIC 2 mg/liter). The fitted combination surface is shown for drug exposure in plasma (A and B) and epithelial lining fluid (ELF) (C and D). Panels B and D show rotated views of panels A and C, respectively. For plasma, a ≥2-log kill was achieved with a cefepime fT > MIC of ≥20% and enmetazobactam fT > 2 mg/liter of ≥20% of the dosing interval. For a ≥3-log kill plasma cefepime fT > MIC of ≥30% and enmetazobactam fT > 2 mg/liter of ≥50% of the dosing interval was required. When drug exposure in ELF is considered, a ≥2-log kill was achieved with a cefepime fT > MIC in ELF of ≥40% and enmetazobactam fT > 2 mg/liter of ≥10% of the dosing interval or a cefepime fT > MIC and enmetazobactam fT > 2 mg/liter in ELF of ≥20% for both. A 3-log kill was achieved with a cefepime fT > MIC in ELF of ≥30% and enmetazobactam fT > 2 mg/liter of ≥50% of the dosing interval.
Drug exposure targets for cefepime and enmetazobactam for pneumonia.
The pharmacodynamic models provide drug exposure targets in plasma and ELF that were predicted to achieve various orders of logarithmic killing (Fig. 7). For plasma, those targets were cefepime fT > MIC of ≥20% and enmetazobactam fT > 2 mg/liter of ≥20% of the dosing interval. For a ≥3-log kill, cefepime fT > MIC of ≥30% and enmetazobactam fT > 2 mg/liter of ≥50% of the dosing interval was required. When drug exposure in ELF was considered, a ≥2-log kill was achieved with a cefepime fT > MIC in ELF of ≥40% and enmetazobactam fT > 2 mg/liter of ≥10% of the dosing interval or a cefepime fT > MIC and enmetazobactam fT > 2 mg/liter in ELF of ≥20% for both. A 3-log kill was achieved with a cefepime fT > MIC in ELF of ≥30% and enmetazobactam fT > 2 mg/liter of ≥40% of the dosing interval.
FIG 7.

Graphical summary of the extent of antibacterial killing of cefepime and enmetazobactam, quantified in terms of the relevant pharmacodynamic indices for both agents. Drug exposure targets for plasma and epithelial lining fluid (ELF) are shown in the left and right panels, respectively. The panels provide a way of identifying pairs of drug exposure targets for cefepime and enmetazobactam in plasma and ELF associated with various degrees of logarithmic killing in the lung.
DISCUSSION
Cefepime-enmetazobactam has the potential to provide an alternative to the carbapenems for the treatment of infections caused by ESBL-producing Enterobacteriaceae. Such an addition would be clinically useful (9). There has been much recent debate about the role of piperacillin-tazobactam in the treatment of ESBLs, with conflicting data. A recent clinical trial suggested that patients receiving piperacillin-tazobactam as initial therapy have a 30-day mortality inferior to that of patients receiving meropenem (16). The reason for this remains unclear, especially since tazobactam inhibits the majority of ESBL enzymes. The intrinsic stability of cefepime to class D enzymes is further buttressed by at least some activity of enmetazobactam against some enzymes in this class. Hence, cefepime-enmetazobactam has broad-spectrum activity against Ambler class A, C, and D enzymes and, therefore, may be a suitable alternative for the treatment of a wide range of nosocomial pathogens harboring a variety of resistance mechanisms.
Cefepime and enmetazobactam behave in a highly predictable manner in mice. The pharmacokinetics of both agents are linear, with similar partitioning into plasma and epithelial lining fluid. For all β-lactam–β-lactamase inhibitor combinations, numerous preliminary studies are required to define the background of beta-lactam that enables the pharmacodynamics of inhibitors to be elucidated; a regimen of β-lactam needs to be selected to enable on-scale readouts when combined with the β-lactamase inhibitor. There are some subtleties related to cefepime that deserve emphasis. The half-life of cefepime in mice is often cited as being circa 10 to 12 min, which means a q8h regimen would not be predicted to be effective, since nearly all of the drug would be cleared after 5 half-lives (i.e., approximately 1 h). This assumption was not consistent with any of our experimental observations shown in Fig. 1 and 2. The PK data suggested there is a prolonged gamma elimination phase. Minimum concentrations (Cmin) concentrations approximated the potentiated MIC for many of the murine regimens. Therefore, the pharmacodynamics of enmetazobactam can be elucidated on a background of cefepime that is only relatively infrequently administered (q8h in this study). At least in our hands, the use of a q2h schedule (as described by others [15]) is not sustainable for repeated experimentation and raises significant ethical questions from an animal welfare perspective.
The pharmacodynamics of cefepime-enmetazobactam does not unequivocally fall into any one of the traditional pharmacodynamic indices, i.e., fT > threshold, fAUC:MIC, or fpeak:MIC. The dose fractionation study suggested both fT > threshold and fAUC:MIC are relevant and can be used to explain the data. This is similar to early descriptions of the pharmacodynamics of ceftazidime-avibactam, where some experiments and analyses suggested time-dependent behavior while others were more suggestive of concentration-dependent activity (15). The flip-flopping between time- and concentration-dependent antibacterial activity is somewhat puzzling and not entirely satisfactory given the field’s unyielding quest for certainty. The short duration of these studies means the emergence of resistance is an unlikely explanation for our findings. The factors that may favor AUC as the dynamically linked index are the slow off-rate of the inhibitor following the formation of a noncovalent adduct with the enzyme (10), intrinsically low rates of enzyme synthesis, enzymes that are readily inhibited by enmetazobactam (i.e., enzymes with low 50% inhibitory concentration values), and reduced expression of beta lactamases in some tissue sites, such as the lung (15). Conversely, more fractionated schedules may be more relevant for strains where there is hyperproduction of beta-lactamases or under circumstances where there is heightened expression of beta lactamase (high inoculum or propitious in vivo conditions). Irrespective of the reason, it is likely that the conclusions as to whether the pharmacodynamics of the inhibitor is time or concentration dependent are extremely sensitive to experimental design and the nature of the challenge strains that are used.
These analyses provide the preclinical foundation for identifying clinical regimens for cefepime-enmetazobactam for HAP/VAP. The design of the interaction matrix and fitting of a surface that describes the combined effect of cefepime and enmetazobactam enables multiple joint pharmacodynamic targets in both plasma and ELF to be delineated. Figures 6 and 7 show that much of the interaction is flat to achieve a ≥2-log effect. The highly concordant plasma-ELF concentration-time profiles and relatively high partitioning of both cefepime and enmetazobactam means that the targets for plasma and ELF are comparable. Additional considerations for defining a candidate regimen for HAP/VAP include a deep understanding of the in vitro susceptibility patterns of causative pathogens, the extent of partitioning into the epithelial lining fluid of volunteers and patients, and the impact of pharmacokinetic variability in critically ill patients on achieving drug exposure targets that are predicted to be safe and effective (17).
MATERIALS AND METHODS
Challenge strains, genotype, and in vitro susceptibility testing.
MICs were estimated using Clinical and Laboratory Standards Institute (CLSI) methodology (18) and determined in at least 5 independently conducted experiments. The mode was used for the pharmacodynamic analyses.
Drugs.
The clinical formulation of cefepime (Sandoz, Germany) and enmetazobactam was stored as a powder in the dark at –20°C. Cefepime and enmetazobactam powder both were reconstituted in 0.9% sodium chloride (Baxter Healthcare, Ltd., UK). Both reconstituted agents produced final solutions of 100 mg/ml. Enmetazobactam and cefepime were further diluted for use by preparing twice the intended final concentration to allow for coadministration at a 1:1 ratio. Solutions were stored at 4°C for the duration of the experiment (i.e., no longer than 24 h).
Murine model of pneumonia.
All experiments were conducted under UK Home Office project license PAC022930 and approved by the University of Liverpool’s Animal Welfare and Ethics Review Committee. Mice were supplied by Charles River (Margate, UK). Male CD-1 mice were used for all experiments. Mice were acclimatized 7 days prior to experimentation. Mice were rendered neutropenic with intraperitoneal injection of 150 mg/kg cyclophosphamide (Baxter Healthcare, Ltd., UK) on day −4 and 100 mg/kg on day −1 prior to lung inoculation.
Pneumonia was induced via the intranasal inoculation of a bacterial suspension. Mice were anesthetized with 2% isoflurane, and 25 μl of 1 × 108, 5 × 108, or 1 × 109 CFU/ml for strains 1256506, 1280740, and 1091463, respectively, was instilled into each nare. These inocula were chosen to produce a persistent nonlethal infection and were defined from preliminary inoculum-finding experiments (data not shown). Mice were sacrificed using 120 to 180 mg/kg pentobarbitone injected intraperitoneally at 2 (pretreatment controls) and 26 h postinoculation.
The lungs were removed en bloc, placed in 1 ml phosphate-buffered saline (PBS; VWR, Leicester, UK), weighed, and homogenized. Homogenates were serially diluted 10-fold in PBS and plated to Mueller-Hinton agar (Oxoid and Remel, Basingstoke, UK). Plates were incubated at 37°C for 24 h, after which the bacterial density per gram of lung was enumerated.
Dose-finding experiments.
The challenge strain for these experiments was K. pneumoniae 1280740. Preliminary dose-finding studies were conducted over the course of multiple independently conducted experiments to define the optimal dosage and schedule of cefepime as a fixed background that subsequently enabled the pharmacodynamics of enmetazobactam to be explored. The experiments with fixed cefepime dose and various doses of enmetazobactam were performed using the following combinations: 25 mg/kg cefepime q8h i.v. coadministered with 0.1, 1, 3.125, or 25 mg/kg enmetazobactam q8h i.v.; 50 mg/kg cefepime q8h i.v. coadministered with 0.01, 0.1, 0.5, 1, 1.5, 3.125, 6.25, or 25 mg/kg enmetazobactam q8h i.v.; 75 mg/kg cefepime q8h i.v. coadministered with 0.01, 0.1, 0.5, 1, 1.5, 3.125, 6.25, 12.5, 25 or 50 mg/kg enmetazobactam q8h i.v.; 100 mg/kg cefepime q8h i.v. coadministered with 0.01, 0.1, 0.5, 1, 1.5, 3.125, 6.25, 12.5, 25 or 50 mg/kg enmetazobactam q8h i.v.; and 200 mg/kg cefepime q8h i.v. coadministered with 6.25, 12.5, 25, 50, 100 or 200 mg/kg enmetazobactam q8h i.v.
Plasma pharmacokinetics.
The plasma pharmacokinetics of cefepime and enmetazobactam were determined by coadministering cefepime/enmetazobactam at 25/6.25 mg/kg q8h, 50/25 mg/kg q8h, 100/75 mg/kg q8h, and 200/200 mg/kg q8h to mice infected intranasally with K. pneumoniae 1280740. A destructive design was used where groups of n = 3 mice from each dosage-time point were sacrificed at 0.083, 0.25, 0.5, 1, 2, 4, 8, 16, 16.083, 16.25, 16.5, 17, 18, 20, and 24 h after initial drug administration.
Cardiac bleeds were performed under deep anesthesia induced with 5% isoflurane, followed by sacrifice with cervical dislocation. Whole blood was collected using heparinized syringes before being placed in Eppendorf tubes and centrifuged at 19,000 × g for 2 min. The plasma was removed and then stored at −80°C until bioanalysis.
Intrapulmonary pharmacokinetics.
A separate study was performed to estimate the intrapulmonary pharmacokinetics of cefepime and enmetazobactam. This experiment was performed in infected mice with Klebsiella pneumoniae 1280740. Groups of mice (n = 3 per dosage-time point) received cefepime-enmetazobactam in the following combinations: 25/6.25, 50/25, and 200/200 mg/kg q8h i.v.
Samples to estimate plasma and ELF concentrations of cefepime and enmetazobactam were obtained at 0 min, 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, and 8 h postdose. Both plasma and epithelial lining fluid were collected once the chest cavity was opened to ensure the lung was not pierced at the time of cardiac puncture. Bronchoalveolar lavage was performed by instilling two sequential aliquots of 0.5 ml PBS into the lung via the trachea, which then were combined prior to storage and subsequent bioanalysis.
Measurement of cefepime and enmetazobactam by LC-MS/MS.
Concentrations of cefepime and enmetazobactam in plasma and epithelial lining fluid were measured using a Waters I-class ultrahigh-performance liquid chromatography (UPLC) system coupled with a Xevo TQ-S in tandem mass spectrometry mode (LC-MS/MS). Samples were extracted by protein crash using acetonitrile as the precipitating agent, and chromatography was performed on a BEH HILIC (50 by 2.1 mm; 1.7 μm; Waters). For cefepime, the standard curve and the quality control samples were prepared in blank mouse plasma, and the internal standard was [13C2H3]cefepime; the mobile phase was 20 mM ammonium formate-acetonitrile in an isocratic composition of 17:83, vol/vol. The multiple reaction monitoring (MRM) transition values for cefepime and the internal standard were m/z 481→125 and m/z 485→125, respectively. The same LC-MS/MS conditions were used to measure cefepime in epithelial lining fluid using PBS plus 1% bovine serum albumin as the surrogate matrix for the construction of the standard curve and of the quality controls. The assay for plasma samples was linear over the range of 0.01 to 10 mg/ml, the within-run precision was ≤3%, and accuracy was 97.3% to 104.4%. The assay for epithelial lining fluid samples was linear over the range of 0.01 to 10 mg/liter, the within-run precision was ≤4.5%, and accuracy was 99% to 102.4%.
The same approach was adopted for enmetazobactam quantification, using 2H3-AAI101 as an internal standard. The MRM transition values were m/z 315→84 and m/z 318→87 for enmetazobactam and the internal standard, respectively. The dynamic range of the plasma sample assay was 0.01 to 10 mg/liter, the within-run precision was ≤2.4%, and accuracy was between 98% and 101.1%. The dynamic range of the epithelial lining fluid assay was 0.01 to 10 mg/liter, the within-run precision was ≤0.8%, and accuracy was between 100.6% and 101.7%. The same mobile phase as that for cefepime was used; however, the isocratic composition of 20 mM ammonium formate-acetonitrile was 15:85, vol/vol.
Measurement of urea by LC-MS/MS.
Urea concentrations were measured in plasma and epithelial lining fluid to enable correction for the dilution effect from the bronchoalveolar lavage. Calibration curve and quality control samples were prepared in the proper matrix. Sample preparation was a protein crash with acetonitrile, followed by a derivatization with camphanic chloride. [13C15N2]urea was used as the internal standard. The MRM transition values were m/z 241→109 and m/z 244→109 for urea and the internal standard, respectively. The dynamic range of the assay for plasma samples was 50 to 750 mg/liter, within-run precision was ≤1.5%, and accuracy was 98.2% to 99.2%. The dynamic range of the epithelial lining fluid assay was 1 to 250 mg/liter, within-run precision was ≤3.3%, and accuracy was between 95.9% and 97.9%. Quantification was performed using the same equipment as that for cefepime and enmetazobactam. Chromatographic separation was carried out on an HSS T3 (50 by 2.1 mm; 1.8 μm; Waters), and the mobile phase was an isocratic composition of 10 mM ammonium formate (pH 3.0) and acetonitrile in a ratio of 75:25, vol/vol.
Dose fractionation experiments.
Dose fractionation experiments were conducted to identify the relevant pharmacodynamic index that best links cefepime-enmetazobactam with the observed antibacterial effect. The dosage of enmetazobactam that induces half-maximal reduction in bacterial density was estimated from the inhibitory sigmoid Emax model, and the total daily dosage was administered on a number of different schedules (i.e., q4h, q8h, and q12h).
Pharmacokinetic modeling.
The pharmacokinetics of cefepime and enmetazobactam were modeled using a population methodology with the program Pmetrics (19). The mean observation from n = 3 mice collected from each dosage-time point combination was used as the observation. For cefepime, a 3-compartment model consisting of central, peripheral, and deep-tissue compartments was used. The central compartment was measured in volume, V (liters). The clearance of cefepime from cefepime was modeled as a first-order process. Similarly, the intercompartmental transfers of drug were modeled as first-order processes. To adequately describe the plasma concentrations at the end of the dosing interval (i.e., 8 h postdose), a deep-tissue compartment was required.
A similar approach was used for enmetazobactam. However, a deep compartment was not required to account for terminal plasma concentrations. A standard open 2-compartment model was used with zero-order input of drug into the central compartment via i.v. injection and first-order clearance from the central compartment, which had volume, V (liters).
Integrative measures of drug exposure following administration of cefepime and enmetazobactam (i.e., fT > MIC and fT > threshold) were calculated using ADAPT 5 (20).
Pharmacokinetic-pharmacodynamic modeling.
All PK modeling was performed with the population PK program Pmetrics (19) and ADAPT 5 (20). The dose fractionation data set was modeled using the following three inhomogeneous differential equations.
where RATEIV(1) is the zero-order intravenous injection of enmetazobactam, SCL is the first-order clearance of enmetazobactam from the central compartment, Vc is the volume of the central compartment, k12 and k21 are the first-order intercompartmental rate constants, kgmax is the maximum rate of bacterial growth, popmax is the maximum theoretical bacterial density in the thigh, and Hk is the slope function.
Output equations were
The means and standard deviations from each group of three mice were used for the PK. For the pharmacodynamics, an “individual” consisted of a cohort of mice.
To compare regimens from the dose fractionation studies, a bootstrap analysis was used. Pmetrics (19) was used to generate 1,000 simulated subjects for every individual in the original analysis (an individual consisted of a cohort of mice receiving a given regimen). Thus, for the pharmacodynamic analyses, the bootstrap included 1,000-subject parameter values for each of the 100 mg/kg cefepime q8h, 100 mg/kg cefepime q8h plus 7.5 mg/kg enmetazobactam q12h, 100 mg/kg cefepime q8h plus 5 mg/kg enmetazobactam q8h, and 100 mg/kg cefepime q8h plus 2.5 mg/kg enmetazobactam q4h groups. The parameters from these sets of 1,000-subject simulations then were collated and compared.
Statistical modeling.
Linear mixed-effect modeling was used to compare the changes of log10(CFU/g) over time for different schedules of 100 mg/kg cefepime q8h plus 2.5 enmetazobactam mg/kg q4h, 5 mg/kg enmetazobactam q8h, or 7.5 mg/kg enmetazobactam q12h against cefepime alone. Accounting for random variations across different cages, the fitted model took the form
where U0 is the random intercept for each cage and ϵ is the model error term. The estimated means of log10(CFU/g) between different schedules were compared in a post hoc analysis using Tukey’s test, and the reported P values were corrected for multiple comparisons.
Pharmacodynamic interaction modeling.
The interactions of cefepime and enmetazobactam were modeled so that the addition of the latter had an impact upon the maximum effect of cefepime (i.e., made it larger) and reduced the exposure of cefepime that resulted in half-maximal effect (i.e., made it smaller). In this process, a standard inhibitory sigmoid, Emax, was used that took the form
where Econ is the bacterial density in cefepime-treated mice, Emax is the maximum effect induced by cefepime (and modified by enmetazobactam), fT > MIC is the fraction of the dosing interval where cefepime concentrations are above the MIC, E50 is the fT > MIC at which there is half-maximal effect, and H is the slope function.
Enmetazobactam was allowed to affect Emax using the following function:
where A and B are constants, with A being the Emax of cefepime in the absence of enmetazobactam and B the additional activity delivered by the inhibitor that is described using a Hill-like function. The drug exposure of the inhibitor is expressed in terms of the relevant pharmacodynamic index (i.e., AUC). E50ant1 is the exposure of the inhibitor that results in half-maximal activity, and H1 is the slope function for the inhibitor’s effect on the Emax.
Enmetazobactam was allowed to affect E50 using the following function:
where A1 and B1 are constants, with A being the E50 of cefepime in the absence of enmetazobactam and B1 the additional activity delivered by the inhibitor that is described using a Hill-like function. The drug exposure of the inhibitor is expressed in terms of the relevant pharmacodynamic index (i.e., AUC). E50ant2 is the exposure of the inhibitor that results in half-maximal activity, and H2 is the slope function for the inhibitor’s effect on E50.
ACKNOWLEDGMENTS
This work was supported by a research grant from Allecra.
William Hope holds or has recently held research grants with F2G, Astellas Pharma, Spero Therapeutics, Antabio, Allecra, Bugworks, and NAEJA-RGM. He holds awards from the Medical Research Council, National Institutes of Health Research, FDA, and the European Commission. William Hope has received personal fees in his capacity as a consultant for F2G, Amplyx, Ausperix, Spero Therapeutics, VenatoRx, Pfizer, and BLC/TAZ.
Footnotes
For a companion article on this topic, see https://doi.org/10.1128/AAC.00078-20.
REFERENCES
- 1.Arnaud I, Maugat S, Jarlier V, Astagneau P, for the National Early Warning, Investigation and Surveillance of Healthcare-Associated Infections Network (RAISIN)/multidrug resistance study group . 2015. Ongoing increasing temporal and geographical trends of the incidence of extended-spectrum beta-lactamase producing Enterobacteriaceae infections in France, 2009 to 2013. Euro Surveill 20:pii=30014. doi: 10.2807/1560-7917.ES.2015.20.36.30014. [DOI] [PubMed] [Google Scholar]
- 2.McNulty CAM, Lecky DM, Xu-McCrae L, Nakiboneka-Ssenabulya D, Chung KT, Nichols T, Thomas HL, Thomas M, Alvarez-Buylla A, Turner K, Shabir S, Manzoor S, Smith S, Crocker L, Hawkey PM. 2018. CTX-M ESBL-producing Enterobacteriaceae: estimated prevalence in adults in England in 2014. J Antimicrob Chemother 73:1368–1388. doi: 10.1093/jac/dky007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McDanel J, Schweizer M, Crabb V, Nelson R, Samore M, Khader K, Blevins AE, Diekema D, Chiang HY, Nair R, Perencevich E. 2017. Incidence of extended-spectrum β-Lactamase (ESBL)-producing Escherichia coli and Klebsiella infections in the United States: a systematic literature review. Infect Control Hosp Epidemiol 38:1209–1215. doi: 10.1017/ice.2017.156. [DOI] [PubMed] [Google Scholar]
- 4.Bennett AC, Bennett CL, Witherspoon BJ, Knopf KB. 2019. An evaluation of reports of ciprofloxacin, levofloxacin, and moxifloxacin-association neuropsychiatric toxicities, long-term disability, and aortic aneurysms/dissections disseminated by the Food and Drug Administration and the European Medicines Agency. Expert Opin Drug Saf 18:1055–1063. doi: 10.1080/14740338.2019.1665022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drusano GL, Ambrose PG, Bhavnani SM, Bertino JS, Nafziger AN, Louie A. 2007. Back to the future: using aminoglycosides again and how to dose them optimally. Clin Infect Dis 45:753–760. doi: 10.1086/520991. [DOI] [PubMed] [Google Scholar]
- 6.Gudiol C, Cuervo G, Carratalà J. 2019. Optimizing therapy of bloodstream infection due to extended-spectrum β-lactamase-producing Enterobacteriaceae. Curr Opin Crit Care 25:438–448. doi: 10.1097/MCC.0000000000000646. [DOI] [PubMed] [Google Scholar]
- 7.Hackel M, Kazmierczak KM, Hoban DJ, Biedenbach DJ, Bouchillon SK, De Jonge BLM, Stone GG. 2016. Assessment of the in vitro activity of ceftazidime-avibactam against multidrug-resistant Klebsiella spp. collected in the INFORM Global Surveillance Study, 2012 to 2014. Antimicrob Agents Chemother 60:4677–4683. doi: 10.1128/AAC.02841-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lomovskaya O, Sun D, Rubio-Aparicio D, Nelson K, Tsivkovski R, Griffith DC, Dudley MN. 2017. Vaborbactam: spectrum of beta-lactamase inhibition and impact of resistance mechanisms on activity in enterobacteriaceae. Antimicrob Agents Chemother 61:e01443-17. doi: 10.1128/AAC.01443-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karaiskos I, Giamarellou H. 2020. Carbapenem-sparing strategies for ESBL producers: when and how. Antibiotics 9:61. doi: 10.3390/antibiotics9020061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papp-Wallace KM, Bethel CR, Caillon J, Barnes MD, Potel G, Bajaksouzian S, Rutter JD, Reghal A, Shapiro S, Taracila MA, Jacobs MR, Bonomo RA, Jacqueline C. 2019. Beyond piperacillin-tazobactam: cefepime and AAI101 as a potent β-lactam−β-lactamase inhibitor combination. Antimicrob Agents Chemother 63:e00105-19. doi: 10.1128/AAC.00105-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrissey I, Magnet S, Hawser S, Shapiro S, Knechtle P. 2019. In vitro activity of cefepime-enmetazobactam against Gram-negative isolates collected from U.S. and European hospitals during 2014–2015. Antimicrob Agents Chemother 63:e00514-19. doi: 10.1128/AAC.00514-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lob SH, Kazmierczak KM, Badal RE, Hackel MA, Bouchillon SK, Biedenbach DJ, Sahm DF. 2015. Trends in susceptibility of Escherichia coli from intra-abdominal infections to ertapenem and comparators in the United States according to data from the SMART program, 2009 to 2013. Antimicrob Agents Chemother 59:3606–3610. doi: 10.1128/AAC.05186-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belley A, Huband MD, Fedler KA, Watters AA, Flamm RK, Shapiro S, Knechtle P. 2019. Development of broth microdilution MIC and disk diffusion antimicrobial susceptibility test quality control ranges for the combination of cefepime and the novel β-lactamase inhibitor enmetazobactam. J Clin Microbiol 57:e00607-19. doi: 10.1128/JCM.00607-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crandon JL, Nicolau DP. 2015. In vivo activities of simulated human doses of cefepime and cefepime-AAI101 against multidrug-resistant Gram-negative Enterobacteriaceae. Antimicrob Agents Chemother 59:2688–2694. doi: 10.1128/AAC.00033-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berkhout J, Melchers MJ, Van Mil AC, Seyedmousavi S, Lagarde CM, Schuck VJ, Nichols WW, Mouton JW. 2016. Pharmacodynamics of ceftazidime and avibactam in neutropenic mice with thigh or lung infection. Antimicrob Agents Chemother 60:368–375. doi: 10.1128/AAC.01269-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris PNA, Tambyah PA, Lye DC, Mo Y, Lee TH, Yilmaz M, Alenazi TH, Arabi Y, Falcone M, Bassetti M, Righi E, Rogers BA, Kanj S, Bhally H, Iredell J, Mendelson M, Boyles TH, Looke D, Miyakis S, Walls G, Al Khamis M, Zikri A, Crowe A, Ingram P, Daneman N, Griffin P, Athan E, Lorenc P, Baker P, Roberts L, Beatson SA, Peleg AY, Harris-Brown T, Paterson DL, MERINO Trial Investigators and the Australasian Society for Infectious Disease Clinical Research Network (ASID-CRN) . 2018. Effect of piperacillin-tazobactam vs meropenem on 30-day mortality for patients with E coli or Klebsiella pneumoniae bloodstream infection and ceftriaxone resistance: a randomized clinical trial. JAMA 320:984–994. doi: 10.1001/jama.2018.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodvold KA, Hope W, Boyd S. 2017. Considerations for effect site pharmacokinetics to estimate drug exposure: concentrations of antibiotics in the lung. Curr Opin Pharmacol 36:114–123. doi: 10.1016/j.coph.2017.09.019. [DOI] [PubMed] [Google Scholar]
- 18.Clinical and Laboratory Standards Institute. 2015. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 19.Neely MN, van Guilder MG, Yamada WM, Schumitzky A, Jelliffe RW. 2012. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit 34:467–476. doi: 10.1097/FTD.0b013e31825c4ba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Argenio DZ, Schumitzky A, Wang X. 2009. ADAPT 5 user’s guide: pharmacokinetic/pharmacodynamic systems analysis software. Biomedical Simulations Resource, Los Angeles, CA. [Google Scholar]