

Abstract
Immune response to therapeutic enzymes poses a detriment to patient safety and treatment outcome. Enzyme replacement therapy (ERT) is a standard therapeutic option for some types of mucopolysaccharidoses, including Morquio A syndrome caused by N-acetylgalactosamine-6-sulfate sulfatase (GALNS) deficiency. Current protocols tolerize patients using cytotoxic immunosuppressives, which can cause adverse effects. Here we show development of tolerance in Morquio A mice via oral delivery of peptide or GALNS for 10 days prior to ERT. Our results show that using an immunodominant peptide (I10) or the complete GALNS enzyme to orally induce tolerance to GALNS prior to ERT resulted in several improvements to ERT in mice: (a) decreased splenocyte proliferation after in vitro GALNS stimulation, (b) modulation of the cytokine secretion profile, (c) decrease in GALNS-specific IgG or IgE in plasma, (d) decreased GAG storage in liver, and (e) fewer circulating immune complexes in plasma. This model could be extrapolated to other lysosomal storage disorders in which immune response hinders ERT.
Keywords: Genetics, Immunology
Keywords: Lysosomes, Mouse models, Tolerance

Introduction
Mucopolysaccharidosis type IVA (MPS IVA) or Morquio A syndrome (MIM ID #253000) is an autosomal recessive disorder caused by the deficiency of N-acetylgalactosamine-6-sulfate sulfatase (GALNS: E.C.3.1.6.4), resulting in accumulation of the glycosaminoglycans (GAGs) keratan sulfate (KS) and chondroitin-6-sulfate in lysosomes (1, 2). GAG accumulation leads to chronic and progressive deterioration of affected cells, tissues, and organs. Clinical manifestations include bone abnormalities, dysostosis multiplex, joint pathology, organomegaly, short stature, pulmonary compromise, and valvular heart disease (3, 4). Heterogeneity in patients’ phenotype is confirmed by more than 250 mutations reported to date (5–9).
There is no cure for Morquio A, and for many years only palliative treatments have been available (3). Currently, hematopoietic stem cell transplantation (HSCT) and enzyme replacement therapy (ERT) have been clinically evaluated for Morquio A. HSCT has had limited effectiveness due to graft-versus-host disease, risk of infections, and high mortality rates (10, 11). ERT with human recombinant GALNS enzyme or elosulfase alfa (Vimizim) was approved by the Food and Drug Administration (FDA) in 2014 (12). Although ERT is known to ease MPS complications, it fails to adequately treat particular tissues, especially the skeletal and cardiovascular systems (13–17). In addition, the major reason behind partial effectiveness of ERT is that the immune system makes antibodies against the infused recombinant protein that neutralize enzymatic activity (18–21). In Morquio A clinical trials, study drug–related adverse events were present in 73% to 100% of the patients, and all patients tested positive for anti–elosulfase alfa antibody by weeks 4 to 6 until the end of the study (17, 22–24). In phase III clinical trials (MOR-004, MOR-005), all patients developed anti-drug antibodies, nearly all developed neutralizing antibodies, and 9.7% of patients developed drug-specific IgE antibodies. Only 2 patients developed study drug–related serious adverse events (anaphylaxis and hematuria) (17, 25, 26).
These antibodies can cause (a) treatment resistance, (b) type III hypersensitivity reactions, and (c) glomerulonephritis due to depositions of immune complexes in the kidney (27). Immune response may depend on factors such as the nature of the replaced protein, patient genetic background, structural differences between the infused and the defective protein, and presence or absence of the residual mutant protein or cross-reactive immunologic material (CRIM) (28). CRIM-negative patients do not express any residual protein due to large deletions or nonsense mutations, among others. Meanwhile, CRIM-positive patients present mutations that result in circulation of an inactive protein. Therefore, there is a relationship between CRIM status and development of antibodies against the therapeutic protein. It has been shown that CRIM-negative patients could develop a stronger immune response to the protein than CRIM-positive patients (20, 29). To control the potential impact in these patients, immunosuppressive drugs such as cyclosporine A, azathioprine, and methotrexate have been evaluated (30–32). Although nonspecific immunosuppressive protocols have demonstrated good results inhibiting immune responses to the infused protein, side effects include damage to bone marrow, gastrointestinal tract, liver, and kidney, and risk of bacterial and viral infections, among others (33–35). The current challenge is to replace chronic treatments of nonspecific immunosuppression and their toxicities with new therapies that safely induce specific immune tolerance (36).
To establish new strategies to modulate immune responses to ERT, we evaluated GALNS antigenicity by selecting the most immunogenic epitopes in the protein. In this study, we have developed a protein- or a peptide-based oral immunotherapy to induce specific tolerance to GALNS in Morquio A mice treated with ERT. Oral immunotherapy was chosen as the approach because it is antigen-specific, nontoxic, has a long-lasting effect, and has demonstrated potential in allergy research. Beyond inducing tolerance to ERT, the treatment improved outcomes, reducing GAG accumulation in the liver and reducing circulating immune complexes.
Results
Ten regions in the GALNS enzyme were predicted as highly immunogenic in silico.
To comprehensively profile the most immunogenic regions in the GALNS enzyme, we used algorithms designed to predict B and T cell epitopes (37, 38). Ten 20-mer peptides containing the immunogenic regions in the GALNS enzyme were selected: 5 peptides (A2, B3, G6, H7, and J1) were predicted to contain B cell epitopes and 3 peptides (D9, E8, and I10) were predicted as regions containing T cell epitopes. In addition, 2 peptide sequences (C4 and F5) were predicted to contain both B and T cell epitopes (Table 1). We also predicted the human HLA binding sites within the sequences of the 10 peptides (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI125607DS1). The locations of the predicted peptides are displayed within the structural model of GALNS (Figure 1A), and the presence or absence of structural domains was confirmed (Table 1) (2, 39, 40).
Table 1. Amino acid sequences of predicted GALNS synthetic peptides.

Figure 1. Selection of immunodominant peptides within the GALNS enzyme.

(A) Location of the synthetic peptides in the 3D structure of GALNS enzyme. Peptides A2 (blue), B3 (green), C4 (magenta), D9 (yellow), E8 (violet), F5 (orange), G6 (cyan), H7 (light blue), I10 (purple), and J1 (light green) are highlighted. The active site of the protein (C79) is shown in red. (B–F) Selection of immunodominant peptides after in vitro stimulation of splenocytes. MKC mice were treated with 16 intravenous weekly infusions of human GALNS (E, black dots) or PBS (P, red dots). Ten days after the last infusion, mice were euthanized and splenocytes were in vitro stimulated with GALNS or a single peptide. The background levels from unstimulated cells were subtracted. (B) Levels of splenocyte proliferation (n = 6 measurements from 2 different mice) and secretion levels of cytokines (C) IFN-γ, (D) IL-4, (E) IL-5, and (F) IL-13 (n = 9 measurements from 3 different mice). Data are shown as scatter plots with mean ± 95% CI. *P < 0.05; **P < 0.01; ***P < 0.001 represent statistically significant differences between treated and untreated mice as determined by 2-tailed paired t test.
Immunological evaluation of GALNS and synthetic peptides after ERT showed preferential immunogenicity.
The combination of in silico predictions of immunogenic regions of GALNS with in vitro and in vivo experiments was aimed to select the best peptides as representative epitopes within GALNS. Here, we used a knockout Morquio A mouse model (MKC; Galns–/–), which corresponds to CRIM-negative patients having the strongest humoral and cellular responses against the therapy. MKC mice were treated with 16 intravenous weekly infusions of recombinant human GALNS or PBS. Cellular responses were measured by assessing splenocyte proliferation and secretion of proinflammatory cytokines after in vitro stimulation of isolated splenocytes with GALNS or individual peptides (Figure 1, B–F). Splenocyte proliferation in ERT-treated MKC mice was significantly higher than in PBS-treated control mice after in vitro stimulation of isolated splenocytes with peptide C4, E8, I10, or the GALNS enzyme (Figure 1B). In immunotolerance studies, it has been demonstrated that after the antigen is taken up in the gut by dendritic cells, and in the presence of tolerization, Th1 (IFN-γ) and Th2 (IL-4, IL-5, and IL-13) cytokine production is decreased. To differentiate the peptides’ capacity to modulate a Th1 or Th2 response, a profile of cytokines was characterized. IFN-γ was measured to define the Th1 response, and IL-4, IL-5, and IL-13 for the Th2 response (41). Levels of IFN-γ in GALNS-stimulated cells of ERT-treated MKC mice were significantly higher than PBS controls for peptides C4, E8, I10, and for GALNS (Figure 1C). High IL-4 secretion was observed with the 3 peptides (C4, E8, and I10) or with GALNS in ERT-treated mice. The differences were statistically significant when compared with PBS controls. Higher IL-4 secretion was detected after stimulation with GALNS or peptide I10 (Figure 1D). Only peptides C4 and I10 and GALNS protein induced relatively high secretion of IL-5 after the in vitro stimulation in ERT-treated mice compared with the PBS controls (Figure 1E). A significant increase in IL-13 secretion was observed exclusively with peptide I10 and GALNS after the in vitro stimulation (Figure 1F). As in proliferation assays, splenocytes secreted significantly more cytokines after in vitro stimulation only with peptides C4, E8, I10, or with GALNS in the ERT-treated MKC mice when compared with PBS control mice (Figure 1, C–F). In summary, peptide C4 induced the most IFN-γ secretion among the 10, even when compared with GALNS upon the in vitro stimulation (Figure 1C). These results suggested that peptides C4, E8, and I10 were the most immunogenic of the 10 examined.
Oral tolerance reduces cellular and humoral responses to GALNS ERT.
We selected peptide I10 and the GALNS enzyme to induce oral tolerance, owing to the similarity of the in vitro responses to both. MKC mice received either GALNS or peptide I10 via oral gavage at 3 different doses (50, 100, or 500 μg) prior to the regular intravenous ERT regimen (tolerized group). Two control groups were given PBS (nontolerized groups). The positive control group was treated with ERT (nontolerized, treated) and the negative control group received intravenous PBS (nontolerized, untreated group) (Table 2). The effect of inducing GALNS tolerance was evaluated by measuring cellular and humoral responses in the mice treated with ERT. Cellular responses were evaluated by assessing splenocyte proliferation and secreted cytokines (IFN-γ, IL-4, IL-5, IL-10, and IL-13) after GALNS in vitro stimulation in the tolerized, nontolerized, and untreated groups. Mice fed with peptide I10 showed significantly less splenocyte proliferation after GALNS in vitro stimulation in 2 groups (50 or 500 μg) than nontolerized mice. The 3 groups of mice that received GALNS orally (50, 100, or 500 μg) presented a statistically significant decrease in splenocyte proliferation (Figure 2A). These results demonstrated that oral administration of peptides affected the cytokine profile in treated mice. Less IFN-γ was secreted in mice treated orally with peptide I10. The IFN-γ decrease was statistically significant (50 or 500 μg of peptide I10) when compared with the nontolerized group, suggesting modulation of the Th1 response. Interestingly, mice that received GALNS orally did not show any modulation in IFN-γ secretion after splenocytes’ in vitro stimulation with GALNS (Figure 2B).
Table 2. Groups of mice for the oral tolerance induction.
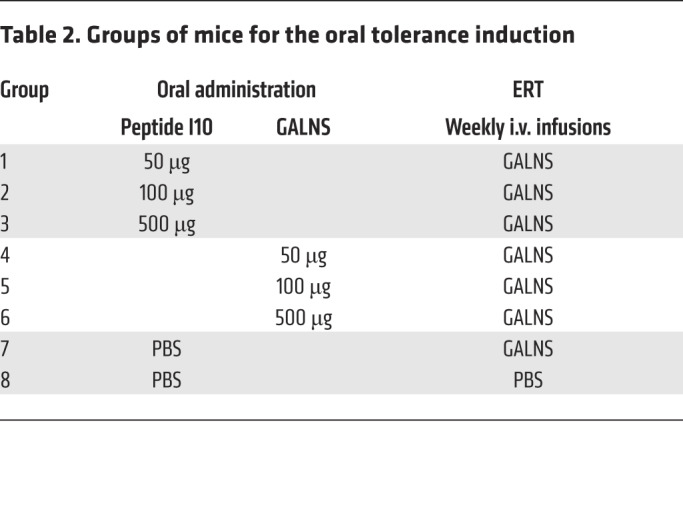
Figure 2. Effect of tolerance induction on cellular response after in vitro GALNS stimulation of splenocytes.

Oral tolerance (OT) was induced by feeding MKC mice with 50, 100, or 500 μg of peptide I10 (red) or GALNS enzyme (blue). Control groups were fed with PBS (gray and purple). One week after the last oral dose, mice received 16 weekly intravenous infusions of human GALNS (red, blue, and gray) or PBS (purple). Ten days after the last infusion, mice were euthanized and splenocytes were in vitro stimulated with GALNS. The background levels from unstimulated cells were subtracted. The induction of tolerance was evaluated by (A) levels of splenocyte proliferation (data are shown as scatter plots with mean ± 95% CI. Each scatter plot represents the average of 3 measurements for each mouse, n = 3 mice per group), and secretion levels of (B) IFN-γ, (C) IL-4, (D) IL-5, (E) IL-10, and (F) IL-13. Quantitative data are represented as a box-and-whisker plot, with bounds from 25th to 75th percentile, median line, and whiskers ranging from 5th and 95th percentile values of the average of 2 measurements for each mouse, n = 3 mice per group. *Benjamini and Hochberg–adjusted P values represent statistically significant differences between tolerized and nontolerized (PBS-ERT) mice: (A) P < 0.04, (B) P < 0.025, (C) P < 0.04, (D) P = not significant (NS), (E) P < 0.03, and (F) P = NS. §Benjamini and Hochberg–adjusted P values represent statistically significant differences between ERT-treated mice and untreated (PBS-PBS) mice: (A) P < 0.03, (B) P < 0.008, (C) P < 0.04, (D) P = 0.04, (E) P < 0.02, and (F) P = 0.02.
IL-4 (Th2-biased cytokine) production by splenocytes after in vitro stimulation with GALNS was evaluated. The results showed a statistically significant decrease in the secreted IL-4 by splenocytes of the tolerized groups with 50 or 500 μg peptide I10, and with 100 or 500 μg GALNS when compared with the nontolerized treated mice. The difference in the IL-4 levels of the nontolerized untreated mice was statistically significant when compared with all groups except mice treated with 100 μg peptide I10 (Figure 2C). Inducing tolerance did not affect Th2 cytokine levels (IL-5 and IL-13). All tolerized groups showed elevated IL-5 and IL-13, but this was not significantly different from nontolerized mice (Figure 2, D and F). IL-10 is an immunoregulatory cytokine required to induce tolerance. IL-10 did not increase in some tolerized groups that showed inhibition in the GALNS-specific splenocyte proliferation or proinflammatory cytokine secretion. In contrast, significant IL-10 downregulation was observed in groups treated with 50 μg peptide I10 and 500 μg GALNS, compared with nontolerized mice. This divergent result may relate to the experimental system used (time of detection), or the observed induction of tolerance was IL-10 independent (42). Higher IL-10 in nontolerized mice could be a mechanism to counteract the inflammation from Th1 and Th2 responses induced after in vitro stimulation with GALNS (Figure 2, B and E).
The effect of oral tolerance on humoral response was examined by measuring the levels of specific IgG and IgE against GALNS. Plasma levels of GALNS-specific IgG were significantly lower in mice fed with 50 μg peptide I10 or 500 μg GALNS than in nontolerized mice (Figure 3A). GALNS-specific IgE in plasma was also reduced for mice treated with 50 μg peptide I10 or with 500 μg GALNS when compared with the nontolerized group (Figure 3B). In summary, we conclude that tolerization with either GALNS or peptide I10 could protect against harmful formation of both the enzyme-specific neutralizing IgG and anaphylaxis-inducing IgE antibodies during ERT, improving treatment efficiency.
Figure 3. Effect of tolerance induction on humoral response to GALNS.

Oral tolerance (OT) was induced by feeding MKC mice with 50, 100, or 500 μg of peptide I10 (red) or GALNS enzyme (blue). Control groups were fed with PBS (gray and purple). One week after the last oral dose, mice received 16 weekly intravenous infusions of human GALNS (red, blue, and gray) or PBS (purple). The induction of tolerance was evaluated in plasma samples after 16 intravenous infusions by (A) levels of IgG specific for GALNS and (B) levels of IgE specific for GALNS. Quantitative data are represented as a box-and-whisker plot, with bounds from 25th to 75th percentile, median line, and whiskers ranging from 5th and 95th percentile values of the average of 2 measurements for each mouse, n = 3 mice per treatment group. *P < 0.01, Benjamini and Hochberg adjusted, for differences between tolerized and nontolerized (PBS-ERT) mice. §P < 0.02, Benjamini and Hochberg adjusted, for differences between ERT-treated mice and untreated (PBS-PBS) mice.
Altogether, these studies indicate that peptide I10 at a concentration of 50 μg and GALNS protein at a concentration of 500 μg have the strongest effect in ameliorating the immune responses to ERT.
Mesenteric lymph nodes play a role in oral tolerance to GALNS.
To elucidate the tolerogenic role of gut-associated lymphoid tissue (GALT), we measured forkhead box P3 (Foxp3) and transforming growth factor β1 (Tgfb1) gene expression in mesenteric lymph nodes (MLNs) and Peyer’s patches (PPs) of tolerized and nontolerized mice. We detected upregulation of Foxp3 and Tgfb1 (molecules associated with immune suppression) in MLN samples, confirming that oral tolerance is GALT originated and requires MLNs (Figure 4, A and C). In contrast, Foxp3 and Tgfb1 did not show any change in expression in PPs of tolerized and nontolerized mice (Figure 4, B and D). This suggests that PPs are not required for oral tolerance (43).
Figure 4. Mesenteric lymph nodes play a role in oral tolerance to GALNS.

RNA from MLNs and PPs of tolerized and nontolerized mice were used to perform quantitative RT-PCR analysis. Expression levels of Foxp3 were measured in (A) MLNs and (B) PPs. Expression levels of Tgfb1 were measured in (C) MLNs and (D) PPs. Fold change indicates the change in RNA transcripts of tolerized mice and nontolerized mice normalized by untreated (PBS-PBS) mice. Quantitative data are represented as a box-and-whisker plot, with bounds from 25th to 75th percentile, median line, and whiskers ranging from 5th and 95th percentile values of 9 measurements from 3 mice. *P < 0.025, Benjamini and Hochberg adjusted, for differences between tolerized and nontolerized (PBS-ERT) mice. The dotted horizontal line indicates 1-fold change.
Decreased circulating immune complexes after oral tolerance.
Antibodies against proteins used for ERT often cause glomerulonephritis due to immune-complex deposits in the kidney. We measured plasma levels of circulating immune complexes (CICs) to evaluate whether oral tolerance had reduced neutralizing antibodies. As expected, CIC plasma levels were higher in the group that underwent ERT without tolerization (nontolerized treated mice, PBS-ERT). In contrast, the lowest levels were found in mice treated with vehicle (PBS) and in naive animals. Plasma CICs were reduced significantly in groups treated orally with 50 and 100 μg peptide I10 when compared with PBS-ERT mice (Figure 5). Our results show that oral tolerance induced with GALNS or peptide I10 can be used to prevent the development of type III hypersensitivity reactions in MKC mice treated with ERT.
Figure 5. Circulating immune complexes in plasma.

Quantities of M complement proteins C1q, C3, C4, and C5 bound to circulating immune complexes (CICs) were measured using a Preceptor CIC ELISA kit. Oral tolerance was induced by feeding MKC mice with 50, 100, or 500 μg of peptide I10 (red) or GALNS enzyme (blue). Control groups were fed with PBS (gray and purple). One week after the last oral dose, mice received 16 weekly intravenous infusions of human GALNS (red, blue, and gray) or PBS (purple). Blood samples were obtained 10 days after the last infusion. Control naive MKC mice did not undergo any treatment (orange). Data are shown as scatter plots with mean ± 95% CI. Each scatter plot represents the average of 10 measurements from 5 mice. *P < 0.05, Benjamini and Hochberg adjusted, for differences between tolerized and nontolerized (PBS-ERT) mice. §P < 0.007, Benjamini and Hochberg adjusted, for differences between ERT-treated mice and untreated (PBS-PBS) mice.
Vacuole reduction in liver samples of tolerized Morquio A mice.
To measure the effect of the oral tolerization protocol in the treated mice, we performed a pathological evaluation of GAGs in liver tissues. Sections were stained with toluidine blue and evaluated by light microscopy. The number of cells with cytoplasmic vacuoles and the total number of vacuoles were counted in 8 random microscopic high-power fields, as described in the Methods section (Figure 6, A–H). Vacuoles were observed in endothelial and Kupffer cells. The highest GAG score and vacuole number were in nontolerized, untreated control mice (PBS-PBS). Although PBS-ERT mice showed decreases in GAG score and vacuole number, their GAG levels were significantly higher than those in the tolerized mice. All tolerized groups showed a significantly lower GAG accumulation than the nontolerized or the untreated group. Improvements were observed in both GAG score (Figure 6I) and GAG vacuole number (Figure 6J). These results were confirmed by electron microscopy studies. At the ultrastructural level, sinusoidal endothelial cells and Kupffer cells demonstrated cytoplasmic vacuolar accumulation, with finely granular material consistent with GAG storage. Cells with the highest number of vacuoles were present in the untreated and nontolerized groups. No significant changes were seen in cellular organelles or lipid content (Figure 7).
Figure 6. Glycosaminoglycan (GAG) accumulation in liver.

Light microscopic findings of toluidine blue–stained, 1-μm-thick sections of mouse liver from (A) PBS control group, (B) ERT nontolerized group, and groups tolerized with peptide I10 (C, 50 μg; D, 100 μg; and E, 500 μg) or GALNS (F, 50 μg; G, 100 μg; and H, 500 μg) prior to ERT. The red arrows and insets show GAG aggregates. Accumulation of GAGs was significantly higher in control groups (A and B) and much less in treated groups (D and H). Wild-type mouse cells do not have any GAG accumulation because they have a functional GALNS enzyme. Original magnification, ×100 and ×400 (insets). The number of cells with cytoplasmic vacuoles (I) as well as total number of vacuoles (J) were counted in 8 random microscopic high-power fields (×100). A score from 0 to 5 (0 = none, 1 = 1, 2 = 2–3, 3 = 4–5, 4 = 6–7, and 5 = 7) was assigned according to the number of cells containing vacuoles per high-power field. Quantitative data are represented as a box-and-whisker plot, with bounds from 25th to 75th percentile, median line, and whiskers ranging from 5th and 95th percentile values. *P < 0.05, Benjamini and Hochberg adjusted, for differences between tolerized and nontolerized (PBS-ERT) mice. §P < 0.05, Benjamini and Hochberg adjusted, for differences between ERT-treated mice and untreated (PBS-PBS) mice.
Figure 7. Ultrastructural demonstration of glycosaminoglycan (GAG) accumulation in liver.

Electron microscopic findings of mouse liver from (A) PBS control group, (B) ERT nontolerized group, and groups tolerized with peptide I10 (C, 50 μg; D, 100 μg; and E, 500 μg) or GALNS (F, 50 μg; G, 100 μg; and H, 500 μg). The red arrows show GAG aggregates and distended lysosomal vacuoles. Accumulation of GAGs was significantly higher in control groups (A and B) and much less in treated groups (D and H). Original magnification, ×15,000. RBC, red blood cells; N, nucleus; L, lipid vacuole.
Reduction of KS in plasma samples of tolerized Morquio A mice.
The primary biomarker of Morquio A treatment outcomes is KS. We measured plasma levels of KS to evaluate whether oral tolerance affected GAG levels in circulation. We found significantly higher plasma KS levels in the PBS-ERT group compared with each of the tolerized groups (Figure 8). The highest levels of KS were present in untreated mice (PBS-PBS).
Figure 8. Keratan sulfate (KS) levels in plasma.

Quantities of KS in plasma were measured using a high-sensitivity KS ELISA kit. Oral tolerance was induced by feeding MKC mice with 50, 100, or 500 μg of peptide I10 (pink dots) or GALNS enzyme (blue dots). Control groups were fed with PBS (green and purple dots). One week after the last oral dose, mice received 16 weekly intravenous infusions of human GALNS (pink, blue, and green dots) or PBS (purple dots). Blood samples were obtained 10 days after the last infusion. Data are shown as scatter plots with mean ± 95% CI (n = 2–3 measurements for each mouse, n = 3 mice per group). *P < 0.05, Benjamini and Hochberg adjusted, for differences between tolerized and nontolerized (PBS-ERT) mice. §P < 0.05, Benjamini and Hochberg adjusted, for differences between ERT-treated mice and untreated (PBS-PBS) mice.
Altogether, these results suggest tolerance achieved by this protocol could reach improved therapeutic levels when compared with regular ERT.
Discussion
Oral tolerance is induced mainly in the GALT and in other mucosal surfaces such as the respiratory tract. It has been defined as the specific suppression of B and T cell responses to an antigen administered orally (44, 45). Antigenic immune response represents a significant obstacle to ERT efficacy (17–20, 46, 47). Oral tolerance constitutes an ideal approach to prevent immune responses and to eliminate side effects of immunosuppressive drugs.
In the present study, we report what we believe is a novel approach to improve ERT efficacy while reducing the immune response by using a protein- or peptide-based oral immunotherapy to induce specific tolerance to GALNS in Morquio A mice treated with ERT. The results provide evidence that oral tolerance can decrease immune responses and improve ERT outcomes. In particular, we show that an immunodominant peptide (I10) or the complete enzyme (GALNS) was used to orally induce tolerance to GALNS prior to ERT. The experiment resulted in several improvements to ERT in mice, including: (a) decreased splenocyte proliferation after in vitro GALNS stimulation, (b) modulation of the cytokine secretion profile, (c) decrease in GALNS-specific IgG or IgE in plasma, (d) decreased GAG storage in liver, and (e) fewer circulating immune complexes in plasma.
Immunodominant epitopes are specific subunits of the antigenic regions easily recognized by the immune system. The identification of immunodominant epitopes in a specific protein is based on the synthesis of overlapping peptides that results in hundreds of molecules to evaluate (48). In this study, the combination of in silico prediction of immunogenic regions of GALNS with in vitro experiments aimed to speed up the screening of immunodominant epitopes in GALNS. Ten peptides were selected by the predicted presence of T and B cell epitopes in the GALNS sequence.
Th1-driven responses are regarded mainly as cell-mediated immune responses, whereas Th2-type cytokines are predominantly related to antibody- and/or immune complex–mediated responses (49). Although there is not a clear characterization of the type of response that develops in patients undergoing ERT, immune response to infused enzymes is recognized as one of the major complications for treatment success (18, 19, 21, 32, 50). The degree of immune response is related to (a) the presence or absence of residual mutant protein in the individual and (b) structural differences between the infused and defective proteins.
CRIM-negative patients cannot produce any precursor form of the native enzyme, or at least it cannot be detected in circulation. CRIM-positive patients produce some precursor forms of abnormal enzyme in circulation (20, 47, 51). CRIM status could predict immune response. A strong immune response against the therapy could be expected in CRIM-negative patients, if the immune system recognizes the infused protein as a foreign molecule. This activates strong inflammation, predicting poor clinical outcome. CRIM-positive patients could present a more heterogeneous response against the therapy (52). This would depend mainly on structural differences between the abnormal and native proteins. Other factors could include the surrounding environment, patient genetics, route of administration, dosage, frequency, and/or duration of the treatment (19, 32).
Because the mechanisms of oral tolerance are determined by the dose of antigen fed (44), we used low and high doses (50, 100, or 500 μg) of peptide I10 or GALNS enzyme for tolerization. At any given concentration, the molarity of peptide I10 is 25.5-fold higher than that of GALNS because of the difference in molecular weight. Oral administration of peptide I10 or GALNS enzyme prior to ERT promoted a significant reduction in splenocyte proliferation after in vitro GALNS stimulation (except for mice fed 100 μg peptide I10; Figure 2A). These results could be explained by induction of T cell anergy, a condition in which T cells exposed to a specific antigen fail to proliferate upon restimulation with the same specific antigen (53).
Modulation of cytokine secretion varied by type of oral antigen. Secretion of IFN-γ after in vitro splenocyte stimulation with GALNS was dramatically lower for mice fed peptide I10 (50 and 500 μg) than the nontolerized group (Figure 2B). Additionally, a statistically significant decrease in IL-4 secretion was observed in the same groups of mice (Figure 2C). In these groups, there was clear suppression of the Th1-biased (IFN-γ) and Th2-biased (IL-4) responses. Mice fed 100 μg peptide I10 did not show modulation of these cytokines.
Although mice fed GALNS presented a reduction in splenocyte proliferation, they did not show a significant decrease in IFN-γ secretion (Figure 2B). In contrast, decreased IL-4 was detected in mice fed with GALNS (100 and 500 μg) (Figure 2C). These observations indicated a predominantly Th1-biased (IFN-γ) response that counteracts the Th2-biased (IL-4) response. IFN-γ is a versatile cytokine in the immune system and could play a role in inflammation and tolerance as well. The immunomodulatory enzyme indoleamine 2,3-dioxygenase (IDO) is mainly induced by IFN-γ to establish immune tolerance or to induce T cell unresponsiveness (54). In tolerance to allografts, there is evidence that IDO induction is mediated by CD8+ Treg secretion of IFN-γ (55). In models of induction of tolerance to alloantigens, it was reported that CD4+ Tregs require secretion of IFN-γ for immune regulation (56). Induction of IFN-γ has been reported in other models of oral tolerance (57). Therefore, it could be suggested that immune modulation in mice treated with GALNS orally may be mediated by IFN-γ and activation of IDO.
Secretion of IL-5 or IL-13 was not downregulated in any groups tolerized orally. Their levels did not differ from the nontolerized group. In mice treated orally with GALNS, increased levels of IFN-γ may suppress and counteract IL-4 but not IL-5 and IL-13 (Figure 2, D and F).
IL-10 is an important cytokine for tolerance induction. Surprisingly, IL-10 secretion after splenocyte stimulation was markedly reduced, especially for the groups fed 50 μg peptide I10 or 500 μg GALNS, compared with the nontolerized group (Figure 2E). This divergent result may be related to the experimental system used (time of detection), or it could be speculated that induction of tolerance was IL-10 independent (42). Higher IL-10 levels in nontolerized mice could be a mechanism to counteract inflammation due to the higher levels of Th1- and Th2-type cytokines induced after in vitro stimulation with GALNS. Interestingly, although splenocytes of GALNS-fed mice produced higher levels of IFN-γ, no IL-10 increase was detected.
The effect of oral tolerance induction on humoral response was determined. As expected, and in accord with the cellular response profile, plasma levels of GALNS-specific IgG in mice fed 50 μg peptide I10 or 500 μg GALNS were significantly lower than in nontolerized mice (Figure 3A). Cytokines like IL-4 and IFN-γ are important in B cell biology and in mediating regulation of antibody isotypes by B cells. Previous reports demonstrate that IL-4 secretion is critical for the optimal maturation and selection of high-affinity IgG1 antibodies (58). GALNS-specific IgE plasma levels significantly decreased in mice treated with 50 μg peptide I10 or with 500 μg GALNS when compared with the nontolerized group (Figure 3B). Instant hypersensitivity reactions (anaphylaxis, urticarial, allergic asthma) are mediated by IgE antibodies, and infiltration of eosinophils directs chronic inflammation (59). Also, IL-4 signaling regulates immunoglobulin class switching to IgE through STAT6 activation (60, 61), whereas IFN-γ is mainly associated with switching to IgG2a (62). Therefore, downregulation of IL-4 and IFN-γ in mice treated orally reduces GALNS-specific IgG and IgE in plasma. Although mice fed GALNS secreted higher levels of IFN-γ after GALNS in vitro stimulation, levels of IgG and IgE were significantly lower than the nontolerized mice. This result agrees with our hypothesis that in mice treated orally with GALNS, IFN-γ could be tolerogenic.
We could speculate that induction of tolerance in groups of mice fed with peptide I10 is regulated by a different mechanism than that in mice fed with the complete enzyme. Induction of anergy and downregulation of the Th1/Th2 response in mice treated with peptide I10 could be mediated by induction of CTLA-4. On the other hand, it seems that induction of tolerance in mice treated by GALNS orally could be mediated by IFN-γ. Interestingly, both mechanisms may induce the immunomodulatory enzyme IDO (54, 63–65). IDO is a cytosolic enzyme that catalyzes the first step of oxidative tryptophan catabolism. Tryptophan metabolism suppresses effector T cells (65, 66). However, confirming this hypothesis requires additional evaluations of regulatory markers.
We measured Foxp3 expression to confirm the hypothesis that MLNs support the generation of Foxp3+ Tregs in tolerized animals (67–69). We observed high Foxp3 expression in MLNs and not in PPs. These findings confirm that MLNs are indispensable to induce oral tolerance. On the other hand, it is possible that PPs are important to transport the antigen from the gut lumen to the lymphoid areas of the GALT (68, 69) but are not required to induce oral tolerance to GALNS. We found changes in Foxp3 expression in MLNs, which could be related to the dose of antigen used (70) (Figure 4).
We also evaluated how oral tolerance affected pathological improvement in mice treated with ERT. CICs in plasma and GAG accumulation in liver were evaluated in tolerized and nontolerized mice. Glomerulonephritis has been reported in ERT patients due to deposits of immune complexes in the kidney as a secondary effect of the immune response (21, 27). Inducing tolerance to GALNS used in ERT reduced immune-complex deposits in the glomeruli (data not shown) and in plasma (Figure 5). There was significant reduction of CICs in mice fed with 50 and 100 μg of I10, reaching levels of nontolerized, untreated mice (PBS-PBS). This result presents a clinical impact — the potential to avoid nephrotic syndrome in ERT patients.
There was a significant reduction in liver GAGs in all groups tolerized with I10 (Figure 6, C–E) and GALNS (Figure 6, F–H) when compared with the nontolerized, ERT-treated group (Figure 6B) and untreated mice (Figure 6A). Measuring GAGs by a scoring system or by counting vacuoles confirmed these results (Figure 6, I and J). In addition, there was a significant reduction in plasma KS in all tolerized groups compared with the nontolerized group (Figure 8). These results suggest that buildup of tolerance to GALNS can even further decrease GAG levels in liver and in circulation when compared with regular ERT.
Conclusions.
Our present study established a protocol to evaluate immunogenicity of a lysosomal enzyme, namely GALNS. This evaluation was based on the combination of in silico predictions with in vitro and in vivo studies. The main goal was to induce oral tolerance to GALNS prior to ERT. We demonstrated that repeated administration of GALNS immunogenic peptides or the complete enzyme decreased the response to GALNS in mice treated with ERT. Beyond inducing tolerance to ERT, the treatment improved outcomes, reducing GAG accumulation in the liver and circulation and reducing CICs. To the best of our knowledge this is the first immunogenicity evaluation of the GALNS enzyme and characterization of immune response to ERT in Morquio A mouse models. We also believe it is the first report that demonstrates induction of oral tolerance to a lysosomal enzyme used for ERT with a significant improvement.
Methods
Production and purification of recombinant human GALNS enzyme.
Supernatants of Chinese hamster ovary (CHO) cells overexpressing human GALNS were cultured in protein-free media and purified by a 2-column procedure, as described previously (71). Enzymatic activity of GALNS was determined by using 4-methylumbelliferyl-β-D-galactopyranoside-6-sulfate as substrate (72). One unit of GALNS enzymatic activity is defined as the amount of enzyme that catalyzes the conversion of 1 nmol of 4 methylumbelliferyl-β-D-galactopyranoside-6-sulfate per hour.
Prediction of GALNS epitopes.
The human GALNS sequence is available in the NCBI protein database (www.ncbi.nlm.nih.gov). Signal peptide prediction was determined using the proteomics and sequence analysis tools of the ExPASy Proteomics Server (http://au.expasy.org) to ensure that the epitopes were not located in the signal peptide region. To predict the immunogenic peptides in GALNS we used a combination of 2 computational algorithms to predict potential T and B cell epitopes: the Immune Epitope Database and Analysis Resource (IEBD, www.immuneepitope.org) and RANKPEP (http://bio.dfci.harvard.edu/RANKPEP/) (37, 38). Predictions of B cell epitopes were evaluated by the IEDB analysis resource. This algorithm is based on the predictions of surface accessibility and flexibility of the molecule, and the presence of β-turns and linear epitopes. MHC-II epitopes (H2-IAb) were selected according to IC50 values (nanomolar concentration of peptide that inhibits binding of a standard peptide by 50%) and binding potential. HLA binding sites were predicted by using the IEBD analysis resource. We selected 10 candidate peptides (Table 1), which were synthesized by Biomatik Corporation.
Computational model of GALNS and peptide location.
The structural model of GALNS was previously constructed based on the homology and x-ray crystal structures of N-acetylgalactosamine-4-sulfatase (4S) and arylsulfatase A (ASA) (2). The I-TASSER server was used to generate a computational model of the GALNS structure (http://zhanglab.ccmb.med.umich.edu/I-TASSER/). The Rasmol program was used for molecular visualization and peptide location.
In vitro evaluation of immunodominant peptides.
The selected peptides were evaluated in a Morquio A mouse model (knockout mice, Galns–/–, MKC). This model was produced by targeted disruption of the murine Galns gene. MKC mice do not express Galns mRNA, and no enzymatic activity of GALNS is detected (73). Morquio A mice were treated with ERT, receiving 16 weekly infusions of human GALNS at a concentration of 250 U/g of body weight through the tail vein. A control group received PBS (74). Ten days after the last infusion, mice were euthanized and their spleens were aseptically removed. The tissues were homogenized with a syringe plunger in complete RPMI 1640 medium (10% fetal bovine serum, 2 μM glutamine, 50 U/mL penicillin, 50 μg/mL streptomycin, 100 μM nonessential amino acids, 50 μM 2-mercaptoethanol). The suspensions were centrifuged at approximately 120 g for 10 minutes. The red blood cells were lysed using Red Blood Cell Lysis Buffer (MilliporeSigma). The specificity of cellular response against the peptides or the complete enzyme in the in vitro stimulation was determined by splenocyte proliferation and cytokine secretion.
Splenocyte proliferation.
In a 96-well plate, 5 × 105 splenocytes/well were stimulated with the individual peptides (100 μg/mL), GALNS (150 μg/mL), concanavalin A (Con A) (3 μg/mL), or media in triplicate for 72 hours at 37°C, saturated humidity, and 5% CO2. Cells were pulsed with 1 μCi of radioactive thymidine for the last 18 hours of incubation. Thymidine incorporation was measured by beta scintillation counter (Trilux Microbeta Counter) (75).
Secreted cytokines.
Cytokines were determined in the cell culture supernatants. In a 96-well plate, 1 × 106 splenocytes/well were stimulated with the individual peptides (100 μg/mL), the complete GALNS enzyme (150 μg/mL), Con A (3 μg/mL), or media in triplicate for 72 hours at 37°C, saturated humidity, and 5% CO2. Cells were centrifuged at approximately 120 g for 10 minutes. Secreted cytokines (IL-4, IL-5, IL-13, and IFN-γ) in the collected supernatants were detected by LUMINEX technology, using a Millipore Milliplex kit according to the manufacturer’s instructions.
Detection of anti-GALNS IgG and IgE antibodies by ELISA.
An indirect ELISA technique was used to detect plasma levels of IgG and IgE antibodies against GALNS in treated and untreated mice. Ninety-six-well polystyrene microplates were coated with 2 μg/mL of GALNS enzyme in coating buffer (15 mM Na2CO3, 35 mM NaHCO3, 0.021% NaN3, pH 9.6) and incubated overnight at 4°C in a wet chamber. The plates were blocked with 3% casein in PBS for 1 hour at room temperature in a wet chamber and washed twice, first with TTBS (10 mM Tris, 150 mM NaCl, 0.05% Tween 20, pH 7.5), and then with TBS (10 mM Tris, 150 mM NaCl, 0.05%, pH 7.5). Mouse plasma samples (100 μL) were diluted (1:500 for IgE or 1:1,000 for IgG) in TTBS, added to the plate, and incubated 2 hours at 37°C in a wet chamber. Four washes with TTBS were performed, after which 100 μL of anti–mouse IgE–HRP (1:1,000, Thermo Fisher Scientific) in TTBS or anti–mouse IgG–HRP (1:5,000, MilliporeSigma) in TTBS was applied. After 3 washes with TTBS followed by 1 wash with TBS, the signals were developed with the substrate TMB (3,3′,5,5′-tetramethylbenzidine, MilliporeSigma). The enzymatic reaction was stopped with 1N HCl and the absorbance was measured at 450 nm in a Multiskan EL800 (Bio-Tek Instruments). Plasma concentrations of anti-GALNS IgG antibodies were derived by extrapolation of the absorbance values from a calibration curve using a monoclonal anti-GALNS antibody (19B2) (76).
Induction of oral tolerance.
Six-week-old MKC mice (Galns–/–) were divided into 8 groups (n = 3). Oral tolerance was induced by feeding mice with 50, 100, or 500 μg of peptide I10 or GALNS enzyme. Two control groups received PBS alone (Table 2). The mice were fed by oral gavage with ether peptide I10 or GALNS enzyme every other day over a period of 9 days (in total 5 times). One week after the last oral administration, mice were treated by weekly intravenous infusions of GALNS enzyme at a dose of 250 U/kg of body weight or PBS for the control group. The specificity of cellular response against GALNS was determined by splenocyte proliferation and cytokine secretion as described above.
Quantitative real-time PCR for Foxp3 and Tgfb1.
Total RNA was isolated from MLNs or PPs of tolerized and nontolerized mice using the Qiagen RNeasy Micro Kit according to the manufacturer’s instructions. First-strand complementary DNA synthesis was performed by using the iScript Advanced cDNA Synthesis Kit for RT-qPCR (Bio-Rad). cDNA levels of Foxp3 and Tgfb1 were quantified by qPCR using the Prime PCR SYBR Green Assay (Bio-Rad) in a Bio-Rad CFX Connect Real-Time PCR system. Both negative and positive controls were included in each plate. Each study sample was analyzed with 3 technical replicates. Amplification conditions used for qPCR were 95°C for 2 minutes, followed by 40 cycles of denaturation and annealing/extension cycles at 95°C for 5 seconds and 60°C for 30 seconds. Actb and Gapdh were used as normalization controls, with 3 values averaged for each sample to compare fold changes in regulation of the 2 experimental genes. Fold change was determined by the ΔΔCt method. Samples were handled per the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines.
Determination of immune complexes in plasma.
A Proceptor CIC ELISA kit (ProGen Biologics) was used to detect mouse CICs in mouse plasma samples. The assay was performed according to the manufacturer’s instructions. Briefly, 100 μL of 20-fold-diluted mouse plasma sample was placed in an antibody-coated 96-well microplate and incubated for 90 minutes at 25°C. After 3 washes, 100 μL of HRP-conjugated antibody was added to the plate and incubated for 1 hour at 25°C. Three additional washes were performed and 100 μL of substrate solution was used for color development. After 5 minutes of incubation at room temperature, 50 μL of stop solution was used. Absorbance was measured at 450 nm in a microplate reader.
Determination and quantification of GAG accumulation.
Liver tissues from 24 mice used in the oral tolerance protocol were evaluated for GAG storage. Tissues were fixed in 4% paraformaldehyde/2% glutaraldehyde and embedded in Spurr’s resin. Assessment of GAG accumulation was performed on toluidine-stained 1-μm sections using light microscopy. Both the number of cells with cytoplasmic vacuoles as well as total number of vacuoles were counted in 8 random microscopic high-power fields (×100 oil immersion). A score from 0 to 5 (0 = none, 1 = 1, 2 = 2–3, 3 = 4–5, 4 = 6–7, and 5 = 7) was assigned according to the number of cells containing vacuoles per high-power field. The total number of vacuoles per high-power field was also counted in each of the 8 random microscopic fields. Data were obtained and tabulated blindly. The means of both, number of cells with vacuoles and total number of vacuoles, was calculated and compared among the different groups. Tolerized and nontolerized MKC mice treated with ERT were compared.
Quantification of KS levels in plasma.
A high-sensitivity Keratan Sulfate ELISA kit (Seikagaku Biobusiness) was used to detect KS levels in mouse plasma samples. The assay was performed per the manufacturer’s instructions. Briefly, 20 μL of 5-fold-diluted mouse plasma sample was placed in an antibody-coated 96-well microplate and incubated for 1 hour at room temperature. After 5 washes, 100 μL of HRP-conjugated antibody was added to the plate and incubated for 1 hour at room temperature. Five additional washes were performed and 100 μL of substrate solution was used for color development. After 30 minutes of incubation at room temperature, 100 μL of stop solution was used. The absorbance was measured at 450 nm in a microplate reader.
Statistics.
The results are expressed as mean ± SD for each group of mice. Statistical analyses were performed with IBM SPSS Statistics 23.0. A 2-tailed paired t test was used to compare the differences between groups. P values were adjusted for multiple comparisons by controlling the false discovery rate using the Benjamini and Hochberg method (77).
Study approval.
All mice were housed in a pathogen-free environment with normal diet. All procedures were in accordance with Institutional Animal Care and Use Committee (IACUC) guidelines under approved protocols at Saint Louis University and followed the NIH’s Guide for the Care and Use of Laboratory Animals (8th ed., National Academies Press, 2011).
Author contributions
ACS and AMM conceived and designed the study. ACS, AMM, BK, and QG performed experiments. APK, CJB, and AKC provided additional technical guidance and expertise. MAG conducted the pathological evaluation. EA performed statistical analyses. LAB and ST assisted in manuscript preparation and discussions. AMM supervised the study. ACS and AMM wrote the manuscript, with input from all authors.
Supplementary Material
Acknowledgments
This work was funded by the NIH/Eunice Kennedy Shriver National Institute of Child Health & Human Development grant R03HD064749 (to AMM and ST), Saint Louis University Department of Pediatrics in association with the Fleur de Lis Organization and the Cardinal Glennon Foundation (to AMM), Pontificia Universidad Javeriana grant PS3887 (to ACS), and Convocatoria Doctorados Nacionales – Departamento Administrativo para la Ciencia y la Educación – COLCIENCIAS (to ACS). Disclaimer: The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health & Human Development or the NIH. We are grateful to M. Marcinkowski for editorial assistance and S. Gonzalo for critical reading of the manuscript.
Version 1. 11/19/2019
In-Press Preview
Version 2. 01/27/2020
Electronic publication
Version 3. 03/02/2020
Print issue publication
Footnotes
Conflict of interest: ACS, APK, CJB, LAB, ST, and AMM have filed US patent application 15/876,892. This patent application covers the underlying concept of oral tolerance to treat mucopolysaccharidosis IVA described in this manuscript.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(3):1288–1300.https://doi.org/10.1172/JCI125607.
Contributor Information
Angela C. Sosa, Email: cataluna2881@gmail.com.
Barbara Kariuki, Email: barbara.kariuki@health.slu.edu.
Qi Gan, Email: qi.gan@health.slu.edu.
Alan P. Knutsen, Email: knutsenm@slu.edu.
Clifford J. Bellone, Email: clifford.bellone@slu.edu.
Miguel A. Guzmán, Email: mguzman4@slu.edu.
Luis A. Barrera, Email: abarrera@javeriana.edu.co.
Shunji Tomatsu, Email: stomatsu@nemours.org.
Anil K. Chauhan, Email: chauhanakc0@gmail.com.
Eric Armbrecht, Email: eric.armbrecht@slu.edu.
Adriana M. Montaño, Email: adriana.montano@health.slu.edu.
References
- 1. Neufeld EF, Muenzer, J. The mucopolysaccharidoses. In: Valle D, Antonarakis S, Ballabio A, Beaudet A, Mitchell GA, eds. The Metabolic and Molecular Bases of Inherited Diseases. New York, NY: McGraw Hill; 2001. [Google Scholar]
- 2.Sukegawa K, et al. Biochemical and structural analysis of missense mutations in N-acetylgalactosamine-6-sulfate sulfatase causing mucopolysaccharidosis IVA phenotypes. Hum Mol Genet. 2000;9(9):1283–1290. doi: 10.1093/hmg/9.9.1283. [DOI] [PubMed] [Google Scholar]
- 3.Montaño AM, Tomatsu S, Gottesman GS, Smith M, Orii T. International Morquio A Registry: clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis. 2007;30(2):165–174. doi: 10.1007/s10545-007-0529-7. [DOI] [PubMed] [Google Scholar]
- 4.Tomatsu S, et al. Mucopolysaccharidosis type IVA (Morquio A disease): clinical review and current treatment. Curr Pharm Biotechnol. 2011;12(6):931–945. doi: 10.2174/138920111795542615. [DOI] [PubMed] [Google Scholar]
- 5.Morrone A, et al. Morquio A syndrome-associated mutations: a review of alterations in the GALNS gene and a new locus-specific database. Hum Mutat. 2014;35(11):1271–1279. doi: 10.1002/humu.22635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomatsu S, et al. Mucopolysaccharidosis IVA: identification of mutations and methylation study in GALNS gene. J Med Genet. 2004;41(7):e98. doi: 10.1136/jmg.2003.018010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fateen EM, El Mawgoud HA, Eissa NR, Ibrahim MM, Aglan MS, Essawi ML. Four novel mutations in the N-acetylgalactosamine-6-sulfate sulfatase gene among Egyptian patients with Morquio A disease. Gene. 2017;600:48–54. doi: 10.1016/j.gene.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Bidchol AM, et al. GALNS mutations in Indian patients with mucopolysaccharidosis IVA. Am J Med Genet A. 2014;164A(11):2793–2801. doi: 10.1002/ajmg.a.36735. [DOI] [PubMed] [Google Scholar]
- 9.Wood TC, et al. Diagnosing mucopolysaccharidosis IVA. J Inherit Metab Dis. 2013;36(2):293–307. doi: 10.1007/s10545-013-9587-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomatsu S, et al. Impact of enzyme replacement therapy and hematopoietic stem cell transplantation in patients with Morquio A syndrome. Drug Des Devel Ther. 2015;9:1937–1953. doi: 10.2147/DDDT.S68562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tomatsu S, Montaño AM, Nishioka T, Orii T. Mucopolysaccharidosis IV (Morquio syndrome). In: Barranger JA, Cabrera-Salazar MA, eds. Lysosomal Storage Disorders. Boston, MA;Springer; 2007:433–445. [Google Scholar]
- 12.Sanford M, Lo JH. Elosulfase alfa: first global approval. Drugs. 2014;74(6):713–718. doi: 10.1007/s40265-014-0210-z. [DOI] [PubMed] [Google Scholar]
- 13.Mercimek-Mahmutoglu S, Reilly C, Human D, Waters PJ, Stoeckler-Ipsiroglu S. Progression of organ manifestations upon enzyme replacement therapy in a patient with mucopolysaccharidosis type I/Hurler. World J Pediatr. 2009;5(4):319–321. doi: 10.1007/s12519-009-0062-x. [DOI] [PubMed] [Google Scholar]
- 14.Braunlin EA, Berry JM, Whitley CB. Cardiac findings after enzyme replacement therapy for mucopolysaccharidosis type I. Am J Cardiol. 2006;98(3):416–418. doi: 10.1016/j.amjcard.2006.02.047. [DOI] [PubMed] [Google Scholar]
- 15.Yano S, Moseley K, Pavlova Z. Postmortem studies on a patient with mucopolysaccharidosis type I: histopathological findings after one year of enzyme replacement therapy. J Inherit Metab Dis. 2009;32 Suppl 1:S53–S57. doi: 10.1007/s10545-009-1057-4. [DOI] [PubMed] [Google Scholar]
- 16.van den Broek L, et al. Fatal coronary artery disease in an infant with severe mucopolysaccharidosis type I. Pediatrics. 2011;127(5):e1343–e1346. doi: 10.1542/peds.2009-2047. [DOI] [PubMed] [Google Scholar]
- 17.Puckett Y, Mulister H, Montaño AM. Enzyme replacement therapy for mucopolysaccharidosis IVA (Morquio A syndrome): Milestones and challenges. Expert Opin Orphan Drugs. 2017;5:741–752. [Google Scholar]
- 18.Ponder KP. Immune response hinders therapy for lysosomal storage diseases. J Clin Invest. 2008;118(8):2686–2689. doi: 10.1172/JCI36521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brooks DA, Kakavanos R, Hopwood JJ. Significance of immune response to enzyme-replacement therapy for patients with a lysosomal storage disorder. Trends Mol Med. 2003;9(10):450–453. doi: 10.1016/j.molmed.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, et al. Neutralizing antibodies to therapeutic enzymes: considerations for testing, prevention and treatment. Nat Biotechnol. 2008;26(8):901–908. doi: 10.1038/nbt.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matzner U, et al. Non-inhibitory antibodies impede lysosomal storage reduction during enzyme replacement therapy of a lysosomal storage disease. J Mol Med. 2008;86(4):433–442. doi: 10.1007/s00109-008-0309-3. [DOI] [PubMed] [Google Scholar]
- 22.Harmatz PR, et al. Impact of elosulfase alfa in patients with morquio A syndrome who have limited ambulation: An open-label, phase 2 study. Am J Med Genet A. 2017;173(2):375–383. doi: 10.1002/ajmg.a.38014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones SA, et al. Safety and clinical activity of elosulfase alfa in pediatric patients with Morquio A syndrome (mucopolysaccharidosis IVA) less than 5 y. Pediatr Res. 2015;78(6):717–722. doi: 10.1038/pr.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burton BK, et al. Safety and physiological effects of two different doses of elosulfase alfa in patients with morquio a syndrome: A randomized, double-blind, pilot study. Am J Med Genet A. 2015;167A(10):2272–2281. doi: 10.1002/ajmg.a.37172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hendriksz CJ, et al. Long-term endurance and safety of elosulfase alfa enzyme replacement therapy in patients with Morquio A syndrome. Mol Genet Metab. 2016;119(1–2):131–143. doi: 10.1016/j.ymgme.2016.05.018. [DOI] [PubMed] [Google Scholar]
- 26.Hendriksz CJ, et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J Inherit Metab Dis. 2014;37(6):979–990. doi: 10.1007/s10545-014-9715-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hunley TE, et al. Nephrotic syndrome complicating alpha-glucosidase replacement therapy for Pompe disease. Pediatrics. 2004;114(4):e532–e535. doi: 10.1542/peds.2003-0988-L. [DOI] [PubMed] [Google Scholar]
- 28.Bali DS, et al. Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: lessons learned from 10 years of clinical laboratory testing experience. Am J Med Genet C Semin Med Genet. 2012;160C(1):40–49. doi: 10.1002/ajmg.c.31319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kishnani PS, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99(1):26–33. doi: 10.1016/j.ymgme.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banugaria SG, et al. Persistence of high sustained antibodies to enzyme replacement therapy despite extensive immunomodulatory therapy in an infant with Pompe disease: need for agents to target antibody-secreting plasma cells. Mol Genet Metab. 2012;105(4):677–680. doi: 10.1016/j.ymgme.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Messinger YH, et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet Med. 2012;14(1):135–142. doi: 10.1038/gim.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kishnani PS, et al. Immune response to enzyme replacement therapies in lysosomal storage diseases and the role of immune tolerance induction. Mol Genet Metab. 2016;117(2):66–83. doi: 10.1016/j.ymgme.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 33.Lim KB, Schiano TD. Long-term outcome after liver transplantation. Mt Sinai J Med. 2012;79(2):169–189. doi: 10.1002/msj.21302. [DOI] [PubMed] [Google Scholar]
- 34.Briggs JD. A critical review of immunosuppressive therapy. Immunol Lett. 1991;29(1–2):89–94. doi: 10.1016/0165-2478(91)90206-p. [DOI] [PubMed] [Google Scholar]
- 35.Ponticelli C, Campise MR. Neurological complications in kidney transplant recipients. J Nephrol. 2005;18(5):521–528. [PubMed] [Google Scholar]
- 36.Bluestone JA, Auchincloss H, Nepom GT, Rotrosen D, St Clair EW, Turka LA. The Immune Tolerance Network at 10 years: tolerance research at the bedside. Nat Rev Immunol. 2010;10(11):797–803. doi: 10.1038/nri2869. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Q, et al. Immune epitope database analysis resource (IEDB-AR) Nucleic Acids Res. 2008;36(Web Server issue):W513–W518. doi: 10.1093/nar/gkn254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reche PA, Glutting JP, Zhang H, Reinherz EL. Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics. 2004;56(6):405–419. doi: 10.1007/s00251-004-0709-7. [DOI] [PubMed] [Google Scholar]
- 39.Tomatsu S, et al. Determinant factors of spectrum of missense variants in mucopolysaccharidosis IVA gene. Mol Genet Metab. 2006;89(1–2):139–149. doi: 10.1016/j.ymgme.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 40.Rivera-Colón Y, Schutsky EK, Kita AZ, Garman SC. The structure of human GALNS reveals the molecular basis for mucopolysaccharidosis IV A. J Mol Biol. 2012;423(5):736–751. doi: 10.1016/j.jmb.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amsen D, Spilianakis CG, Flavell RA. How are T(H)1 and T(H)2 effector cells made? Curr Opin Immunol. 2009;21(2):153–160. doi: 10.1016/j.coi.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dubois B, Chapat L, Goubier A, Papiernik M, Nicolas JF, Kaiserlian D. Innate CD4+CD25+ regulatory T cells are required for oral tolerance and inhibition of CD8+ T cells mediating skin inflammation. Blood. 2003;102(9):3295–3301. doi: 10.1182/blood-2003-03-0727. [DOI] [PubMed] [Google Scholar]
- 43.Worbs T, et al. Oral tolerance originates in the intestinal immune system and relies on antigen carriage by dendritic cells. J Exp Med. 2006;203(3):519–527. doi: 10.1084/jem.20052016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faria AM, Weiner HL. Oral tolerance. Immunol Rev. 2005;206:232–259. doi: 10.1111/j.0105-2896.2005.00280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, et al. Mechanism of oral tolerance induction to therapeutic proteins. Adv Drug Deliv Rev. 2013;65(6):759–773. doi: 10.1016/j.addr.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brooks DA. Immune response to enzyme replacement therapy in lysosomal storage disorder patients and animal models. Mol Genet Metab. 1999;68(2):268–275. doi: 10.1006/mgme.1999.2894. [DOI] [PubMed] [Google Scholar]
- 47.Doerfler PA, Nayak S, Corti M, Morel L, Herzog RW, Byrne BJ. Targeted approaches to induce immune tolerance for Pompe disease therapy. Mol Ther Methods Clin Dev. 2016;3:15053. doi: 10.1038/mtm.2015.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Díaz I, et al. In silico prediction and ex vivo evaluation of potential T-cell epitopes in glycoproteins 4 and 5 and nucleocapsid protein of genotype-I (European) of porcine reproductive and respiratory syndrome virus. Vaccine. 2009;27(41):5603–5611. doi: 10.1016/j.vaccine.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 49.Moudgil KD, Choubey D. Cytokines in autoimmunity: role in induction, regulation, and treatment. J Interferon Cytokine Res. 2011;31(10):695–703. doi: 10.1089/jir.2011.0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ensina LF, et al. Laronidase hypersensitivity and desensitization in type I mucopolysaccharidosis: a case report. Pediatr Allergy Immunol. 2014;25(5):498–499. doi: 10.1111/pai.12209. [DOI] [PubMed] [Google Scholar]
- 51.Rohrbach M, et al. CRIM-negative infantile Pompe disease: 42-month treatment outcome. J Inherit Metab Dis. 2010;33(6):751–757. doi: 10.1007/s10545-010-9209-0. [DOI] [PubMed] [Google Scholar]
- 52.Banugaria SG, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med. 2011;13(8):729–736. doi: 10.1097/GIM.0b013e3182174703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwartz RH. T cell anergy. Annu Rev Immunol. 2003;21:305–334. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- 54.Brandacher G, Margreiter R, Fuchs D. Implications of IFN-gamma-mediated tryptophan catabolism on solid organ transplantation. Curr Drug Metab. 2007;8(3):273–282. doi: 10.2174/138920007780362536. [DOI] [PubMed] [Google Scholar]
- 55.Guillonneau C, et al. CD40Ig treatment results in allograft acceptance mediated by CD8CD45RC T cells, IFN-gamma, and indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117(4):1096–1106. doi: 10.1172/JCI28801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sawitzki B, Kingsley CI, Oliveira V, Karim M, Herber M, Wood KJ. IFN-gamma production by alloantigen-reactive regulatory T cells is important for their regulatory function in vivo. J Exp Med. 2005;201(12):1925–1935. doi: 10.1084/jem.20050419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang M, Yang C, Mine Y. Multiple T cell epitope peptides suppress allergic responses in an egg allergy mouse model by the elicitation of forkhead box transcription factor 3- and transforming growth factor-beta-associated mechanisms. Clin Exp Allergy. 2010;40(4):668–678. doi: 10.1111/j.1365-2222.2009.03442.x. [DOI] [PubMed] [Google Scholar]
- 58.Stockinger B, Veldhoen M, Martin B. Th17 T cells: linking innate and adaptive immunity. Semin Immunol. 2007;19(6):353–361. doi: 10.1016/j.smim.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 59.Averbeck M, Gebhardt C, Emmrich F, Treudler R, Simon JC. Immunologic principles of allergic disease. J Dtsch Dermatol Ges. 2007;5(11):1015–1028. doi: 10.1111/j.1610-0387.2007.06538.x. [DOI] [PubMed] [Google Scholar]
- 60.Kashiwada M, et al. IL-4-induced transcription factor NFIL3/E4BP4 controls IgE class switching. Proc Natl Acad Sci U SA. 2010;107(2):821–826. doi: 10.1073/pnas.0909235107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rothman PB. The transcriptional regulator NFIL3 controls IgE production. Trans Am Clin Climatol Assoc. 2010;121:156–171; discussion 171. [PMC free article] [PubMed] [Google Scholar]
- 62.Chang JH, Cha HR, Chang SY, Ko HJ, Seo SU, Kweon MN. IFN-gamma secreted by CD103+ dendritic cells leads to IgG generation in the mesenteric lymph node in the absence of vitamin A. J Immunol. 2011;186(12):6999–7005. doi: 10.4049/jimmunol.1003484. [DOI] [PubMed] [Google Scholar]
- 63.von Boehmer H. Mechanisms of suppression by suppressor T cells. Nat Immunol. 2005;6(4):338–344. doi: 10.1038/ni1180. [DOI] [PubMed] [Google Scholar]
- 64.Johnson BA, Baban B, Mellor AL. Targeting the immunoregulatory indoleamine 2,3 dioxygenase pathway in immunotherapy. Immunotherapy. 2009;1(4):645–661. doi: 10.2217/IMT.09.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4(10):762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 66.Huang L, Baban B, Johnson BA, Mellor AL. Dendritic cells, indoleamine 2,3 dioxygenase and acquired immune privilege. Int Rev Immunol. 2010;29(2):133–155. doi: 10.3109/08830180903349669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spahn TW, et al. Mesenteric lymph nodes are critical for the induction of high-dose oral tolerance in the absence of Peyer’s patches. Eur J Immunol. 2002;32(4):1109–1113. doi: 10.1002/1521-4141(200204)32:4<1109::AID-IMMU1109>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 68.Pabst O, Mowat AM. Oral tolerance to food protein. Mucosal Immunol. 2012;5(3):232–239. doi: 10.1038/mi.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wawrzyniak M, O’Mahony L, Akdis M. Role of regulatory cells in oral tolerance. Allergy Asthma Immunol Res. 2017;9(2):107–115. doi: 10.4168/aair.2017.9.2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Siewert C, et al. Experience-driven development: effector/memory-like alphaE+Foxp3+ regulatory T cells originate from both naive T cells and naturally occurring naive-like regulatory T cells. J Immunol. 2008;180(1):146–155. doi: 10.4049/jimmunol.180.1.146. [DOI] [PubMed] [Google Scholar]
- 71.Tomatsu S, et al. Characterization and pharmacokinetic study of recombinant human N-acetylgalactosamine-6-sulfate sulfatase. Mol Genet Metab. 2007;91(1):69–78. doi: 10.1016/j.ymgme.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 72.van Diggelen OP, et al. A fluorimetric enzyme assay for the diagnosis of Morquio disease type A (MPS IV A) Clin Chim Acta. 1990;187(2):131–139. doi: 10.1016/0009-8981(90)90339-T. [DOI] [PubMed] [Google Scholar]
- 73.Tomatsu S, et al. Mouse model of N-acetylgalactosamine-6-sulfate sulfatase deficiency (Galns-/-) produced by targeted disruption of the gene defective in Morquio A disease. Hum Mol Genet. 2003;12(24):3349–3358. doi: 10.1093/hmg/ddg366. [DOI] [PubMed] [Google Scholar]
- 74.Tomatsu S, et al. Enzyme replacement therapy in a murine model of Morquio A syndrome. Hum Mol Genet. 2008;17(6):815–824. doi: 10.1093/hmg/ddm353. [DOI] [PubMed] [Google Scholar]
- 75.Mirano-Bascos D, Steede NK, Robinson JE, Landry SJ. Influence of disulfide-stabilized structure on the specificity of helper T-cell and antibody responses to HIV envelope glycoprotein gp120. J Virol. 2010;84(7):3303–3311. doi: 10.1128/JVI.02242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tomatsu S, et al. Development of MPS IVA mouse (Galnstm(hC79S.mC76S)slu) tolerant to human N-acetylgalactosamine-6-sulfate sulfatase. Hum Mol Genet. 2005;14(22):3321–3335. doi: 10.1093/hmg/ddi364. [DOI] [PubMed] [Google Scholar]
- 77.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57(1):289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.