

Abstract
Fibroblast growth factor 23 (FGF23) is a bone-derived hormone that controls blood phosphate levels by increasing renal phosphate excretion and reducing 1,25-dihydroxyvitamin D3 [1,25(OH)2D] production. Disorders of FGF23 homeostasis are associated with significant morbidity and mortality, but a fundamental understanding of what regulates FGF23 production is lacking. Because the kidney is the major end organ of FGF23 action, we hypothesized that it releases a factor that regulates FGF23 synthesis. Using aptamer-based proteomics and liquid chromatography–mass spectrometry–based (LC-MS–based) metabolomics, we profiled more than 1600 molecules in renal venous plasma obtained from human subjects. Renal vein glycerol-3-phosphate (G-3-P) had the strongest correlation with circulating FGF23. In mice, exogenous G-3-P stimulated bone and bone marrow FGF23 production through local G-3-P acyltransferase–mediated (GPAT-mediated) lysophosphatidic acid (LPA) synthesis. Further, the stimulatory effect of G-3-P and LPA on FGF23 required LPA receptor 1 (LPAR1). Acute kidney injury (AKI), which increases FGF23 levels, rapidly increased circulating G-3-P in humans and mice, and the effect of AKI on FGF23 was abrogated by GPAT inhibition or Lpar1 deletion. Together, our findings establish a role for kidney-derived G-3-P in mineral metabolism and outline potential targets to modulate FGF23 production during kidney injury.
Keywords: Bone Biology, Nephrology
Keywords: Bone disease, Chronic kidney disease, Homeostasis

Introduction
Fibroblast growth factor-23 (FGF23) levels increase with both acute and chronic kidney disease (CKD) (1) and are a strong independent risk factor for adverse renal outcomes and mortality in these contexts (2–4). For example, higher FGF23 levels prior to cardiac surgery or upon arrival to the intensive care unit correlate with subsequent development of acute kidney injury (AKI). Once AKI is established, increased FGF23 levels are associated with a requirement for renal replacement therapy and mortality risk (5, 6). A growing body of evidence shows that high FGF23 levels may contribute directly to adverse outcomes in AKI, through profibrotic effects on the kidneys (7), impairment of immune function (8, 9), impairment of erythropoiesis (10), and toxic effects on the endothelium and cardiovascular system (11–13).
Over the past decade, significant progress has been made in understanding how FGF23 acts in the kidney to enhance phosphate excretion and decrease 1,25-dihydroxyvitamin D3 [1,25(OH)2D] production, including the requirement for signaling through the binary complex of an FGF receptor and α-klotho (14). By contrast, the upstream mechanisms that regulate FGF23 production in the setting of renal injury remain poorly understood. Known factors such as 1,25(OH)2D and parathyroid hormone (PTH) do not appear to drive FGF23 production in AKI, as mice lacking the vitamin D receptor (VDR) or the PTH receptor in osteocytes show robust increases in FGF23 with experimental kidney injury (15). In addition, FGF23 levels increase in AKI before or in the absence of changes in blood phosphate or calcium.
To address this gap in knowledge, we performed a proteomic and metabolomic screen of a unique renal arteriovenous sample set, with the goal of identifying potential kidney-derived mediators of FGF23 production. Using a range of genetic tools, including mice and cultured bone cells with targeted gene deletions, in conjunction with liquid chromatography–mass spectrometry–based (LC-MS–based) biochemical assays, we identified a kidney-to-bone metabolic signaling axis. Finally, we examined mice that underwent renal ischemia reperfusion injury (IRI), as well as humans who did or did not develop AKI following cardiac surgery, to demonstrate the disease relevance of our findings.
Results
A proteomic and metabolomic screen highlights an association between renal venous G-3-P and arterial FGF23 levels.
The kidney is an endocrine organ that synthesizes and releases both proteins (renin and erythropoietin) and a metabolite [1,25(OH)2D] with key systemic roles. To identify factors released by the kidney that might stimulate FGF23 production, we performed a screen of proteins and metabolites in aortic and renal venous samples from 17 human subjects with a mean estimated glomerular filtration rate (eGFR) of 66.6 mL/min/1.73 m2 (Table 1 and ref. 16). We quantitated 1317 proteins on an aptamer-based platform, and 300 known metabolites and 3612 unknown peaks were quantitated by LC-MS (Figure 1A). The levels of known markers of glomerular filtration — cystatin C (measured by aptamer) and creatinine (measured by LC-MS) — decreased from the artery to the renal vein in all subjects (Figure 1, B and C), confirming appropriate blood sampling. Next, we examined the correlations between all molecules measured in the renal vein with arterial intact FGF23 (iFGF23) levels (Figure 1D and Supplemental Tables 1 and 2). The top hit, and the only molecule that achieved statistical significance adjusting for the total 5229 measurements, was the metabolite G-3-P (r2 = 0.76, P = 5.2 × 10–6).
Table 1. Clinical characteristics of the human renal arteriovenous sampling cohort.
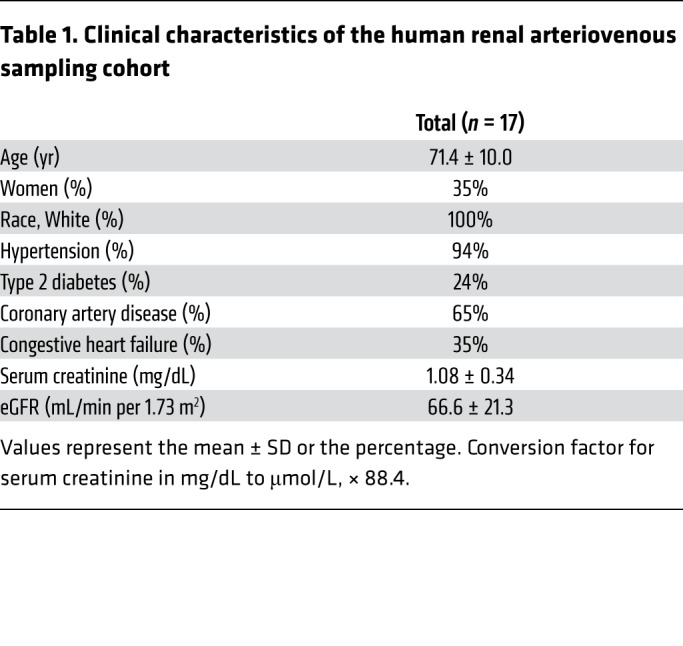
Figure 1. Screen of human renal venous plasma highlights the association between G-3-P and arterial FGF23.

(A) Study overview demonstrating arterial and renal venous plasma sampling with subsequent measurement of iFGF23, creatinine, and phosphate as well as proteomic and metabolomic profiling. (B and C) Cystatin C measured by aptamer (B) and creatinine measured by LC-MS (C) in the artery (A) and renal vein (V) of all individuals (n = 17). (D) Scatter plot with each molecule measured by aptamer or LC-MS in the renal vein samples (proteins in blue, metabolites in red, unknown LC-MS peaks, no color). Molecules are plotted along the y axis based on r2 with arterial iFGF23 and along the x axis based on the median V/A ratio of the molecule. Dotted lines show the median V/A ratio for cystatin C and creatinine. (E) Graph showing the V/A ratio for select proteins and metabolites, with each individual depicted by a separate vertical line. Median V/A ratios for the select proteins and metabolites are shown below the graph. P values were determined by 2-sided, paired t test. Epo, erythropoietin. (F–H) Scatter plots showing correlations between arterial iFGF23 and renal vein G-3-P (F), arterial phosphate (G), and eGFR (H).
G-3-P is a polar small molecule that would be expected to undergo glomerular filtration, but its median renal venous/arterial ratio was 1.0, rather than 0.87 or 0.83 for the glomerular filtration markers cystatin C or creatinine, respectively (Figure 1D). This median renal venous/arterial ratio is consistent with other renal anabolic products such as renin, erythropoietin, and 1,25(OH)2D (Figure 1E). The correlation between renal venous G-3-P and arterial iFGF23 (Figure 1F) was stronger than the correlations for either one with arterial phosphate or eGFR (Figure 1, G and H, and Supplemental Table 3; supplemental material available online with this article; https://doi.org/10.1172/JCI131190DS1). Because it was not measured with the aptamer-based platform, we also assessed renal venous levels of α-klotho by ELISA and found no correlation between renal venous α-klotho and renal venous G-3-P (r2 = 0.01) or between renal venous α-klotho and arterial iFGF23 (r2 = 0.06, Supplemental Table 1).
G-3-P administration increases FGF23 synthesis in bone and bone marrow.
In C57Bl/6J mice, plasma iFGF23 and C-terminal FGF23 (cFGF23) levels demonstrated a dose-dependent increase following a single dose of G-3-P, with no change in the cFGF23/iFGF23 ratio (Figure 2, A and B, and Supplemental Figure 1A). A single dose of similar molecules such as glycerol-2-phosphate (G-2-P) and glycerol, or the cytokine IL-6 had no effect on plasma FGF23 levels (Supplemental Figure 1, B and C). G-3-P administration also increased the urinary phosphate/creatinine ratio (Figure 2C) and decreased plasma 1,25(OH)2D levels (Figure 2D) but did not alter PTH levels or renal function (Figure 2, E and F). Although we observed no effect with a single dose of G-3-P, we found that blood phosphate levels were significantly reduced following G-3-P dosing over consecutive days (Figure 2, G–I). In contrast to G-2-P, we found that G-3-P also had no effect on bone mineralization in vitro (Figure 2J). Thus, the impact of exogenous G-3-P is specific and stimulates FGF23 and its downstream physiologic actions. To test whether FGF23 can feed back and directly modulate G-3-P production, we treated proximal tubular cells with FGF23 or vehicle and found no difference in G-3-P levels at 2 hours and 24 hours (Supplemental Figure 1D).
Figure 2. G-3-P increases FGF23 and its downstream physiologic actions.

(A) Plasma iFGF23 and cFGF23 levels after i.p. injection of G-3-P, sodium phosphate, or vehicle (Veh) (n = 4–10 per group). (B) Dose effect of i.p. G-3-P on plasma iFGF23 and cFGF23 levels (n = 3–6 per group). (C–G) Urinary phosphate/creatinine (phos/Cr) ratio (C), plasma 1,25(OH)2D (D), PTH (E), serum creatinine (sCr) (F), and blood phosphate (Phos) (G) after i.p. injection of G-3-P or vehicle (n = 4–10 per group). (H and I) Blood phosphate (H) and iFGF23 and cFGF23 levels (I) after 6 consecutive daily doses of G-3-P or vehicle (n = 4–5 per group). Doses for i.p. injections were 300 mg/kg, and all measurements were made 24 hours after i.p. injection unless otherwise specified. Data represent the mean ± SEM. *P < 0.05 , by 1-way ANOVA followed by Tukey’s multiple comparisons test (A and B) or 2-sided Student’s t test (C–I). (J) Mineralization (alizarin red staining) and CFU (alkaline phosphatase staining) of primary stromal progenitor cells treated for 3 weeks with ascorbic acid (50 mM), dexamethasone (0.1 mM), and G-3-P (10 μM), G-2-P (10 μM), or vehicle. The mean ± SD is shown for the percentage of area with positive staining. Scale bars: 100 μm.
To assess whether the G-3-P effect on FGF23 was specific to bone, we used mice in which Fgf23 had been deleted in osteocytes (Fgf23fl/fl Dmp1-Cre+) by crossing them with the 10-kb dentin matrix protein-1 promoter-Cre (DMP1-Cre) line (Figure 3, A and B, and ref. 17). Interestingly, G-3-P significantly increased FGF23 levels in Fgf23fl/fl Dmp1-Cre+ mice to an extent similar to that detected in Fgf23fl/fl controls (Figure 3C). We confirmed that G-3-P treatment of Fgf23fl/fl Dmp1-Cre+ mice had no effect on Fgf23 expression in bone (undetectable) but found a significant increase in Fgf23 expression in bone marrow (Figure 3D). To assess the specificity of this finding, we collected mRNA from various tissues 24 hours after G-3-P treatment of WT C57Bl/6J mice and found increased Fgf23 expression only in bone (~4.5-fold, P < 0.0001) and bone marrow (~5-fold, P < 0.0001), with a trend toward increased expression in spleen (~2.1-fold, P = 0.07) and no change in muscle, kidney, liver, heart, or lung (Figure 3, E and F). Next, we collected bone marrow cells from femurs and tibiae of WT C57Bl/6J mice following treatment with G-3-P or vehicle and isolated purified stromal and hematopoietic cells by flow cytometry (Figure 3G). Following G-3-P treatment, an increase in Fgf23 expression was confined to the nonhematopoietic stromal compartment of bone marrow (Figure 3H).
Figure 3. G-3-P stimulates FGF23 production in bone and bone marrow.

(A) Schema of gene-targeting vector (top), floxed Fgf23–exon 1 (E1) allele (middle), and ΔFgf23–exon 1 allele (bottom). (B) PCR of genomic DNA from tissues from Fgf23fl/fl Dmp1-Cre+ mice to amplify sequences with Fgf23 (ΔFgf23-E1) or without (Fl-Fgf23-E1) exon 1 deletion. Shown is a representative gel from 1 of 4 independent experiments. S. muscle, skeletal muscle. (C) Plasma iFGF23 levels in Fgf23fl/fl Dmp1-Cre+ and Fgf23fl/fl mice after i.p. G-3-P or vehicle (n = 5 per group). (D) Bone and bone marrow Fgf23 expression in Fgf23fl/fl Dmp1-Cre+ mice after i.p. administration of G-3-P or vehicle (n = 3–5 per group). ND, not detected. (E) Tissue Fgf23 expression in C57Bl/6J mice after i.p. administration of G-3-P or vehicle (n = 3–4). (F) FGF23 immunoblots of bone and bone marrow from C57Bl/6J mice treated with G-3-P or vehicle. Shown are representative blots from 1 of 2 independent experiments. (G) FACS strategy for isolation of bone marrow CD45+ mononuclear cells and CD45– stromal cells. Prior to sorting, samples destined for purification of stromal cells underwent negative selection (see Methods). All cells were fluorescently labeled with CD45.2 (APC) and TER119 (PE) antibodies. The viability dye 7AAD was included for the removal of dead cells. Cells were gated by size (top left), and doublets were excluded by forward scatter (top middle) and side scatter (top right). Live cells (bottom left, 7AAD–) were then examined for CD45.2 and TER119 staining (bottom middle). TER119+ (RBCs) were excluded, and CD45+ and CD45– cells were sorted for RNA isolation. FSC-A, forward scatter area; SSC-A, side scatter area; FSC-W, forward scatter pulse width; SSC-W, side scatter pulse width. (H) Fgf23 expression in stromal and hematopoietic bone marrow compartments after i.p. administration of G-3-P or vehicle (n = 4). The G-3-P injection dose was 300 mg/kg. All measurements were made 24 hours after i.p. injection. Data represent the mean ± SEM. *P < 0.05 and ***P < 0.0001, by 2-sided Student’s t test (C–E and H).
G-3-P increases FGF23 production via local GPAT-mediated LPA synthesis.
G-3-P is a biosynthetic precursor of LPA via the action of G-3-P acyltransferases (GPATs) (Figure 4A and ref. 18). Because our original profiling methods did not measure LPA, we re-assayed the renal venous samples obtained from human subjects and found that, unlike G-3-P, renal vein LPA did not correlate with circulating iFGF23 (Supplemental Figure 2A). Therefore, we turned our attention to the impact of G-3-P on local LPA production, focusing specifically on LPA 18:1, which is an excellent proxy for total LPA (Supplemental Figure 2B) and is the specific molecule we subsequently tested for functional effects. As with G-3-P, a single dose of LPA 18:1 significantly increased plasma iFGF23 and cFGF23 levels in C57Bl/6J mice, with no change in the cFGF23/iFGF23 ratio (Figure 4B and Supplemental Figure 2C). G-3-P injection resulted in a significant increase in LPA 18:1 levels in bone marrow, but not plasma (Figure 4C), an effect that was blocked when G-3-P was coadministered with the GPAT inhibitor FSG67 (Figure 4D and refs. 19, 20). In turn, GPAT inhibition blocked the stimulatory effect of G-3-P on plasma iFGF23 and cFGF23 levels and FGF23 expression in bone and bone marrow (Figure 4, E–G). Downstream of GPAT, exogenous LPA 18:1 significantly increased plasma iFGF23 and cFGF23 levels, despite coadministration with FSG67 (Figure 4E). Thus, G-3-P increased bone and bone marrow FGF23 expression in vivo through a local increase in LPA catalyzed by GPAT.
Figure 4. G-3-P increases FGF23 production via local GPAT-mediated LPA synthesis.

(A) Schema of GPAT-mediated conversion of G-3-P to LPA. (B) Plasma iFGF23 and cFGF23 levels after i.p. injection of LPA 18:1 or vehicle (n = 6–10 per group). (C and D) LPA 18:1 levels in bone marrow and plasma after i.p. injection of G-3-P or vehicle (C), and in bone marrow with or without the GPAT inhibitor (inh) FSG67 (D) (n = 3–5 per group). (E) Plasma iFGF23 and cFGF23 levels after i.p. injection of G-3-P, LPA 18:1, or vehicle, with or without FSG67 (n = 3–8 per group). (F and G) Bone Fgf23 mRNA levels (n = 4–5 per group) (F) and immunohistochemistry for FGF23 in femurs from mice treated with G-3-P or vehicle, with or without FSG67 (G). Shown are representative images from 1 of 2 independent experiments. Scale bars: 100 μm. (H and I) Fgf23 mRNA levels (n = 2–3 per group) (H) and FGF23 immunoblot (I) (blot is a representative gel from 1 of 2 independent experiments) of primary osteoblasts treated with Gpat2 or scrambled shRNA (scr), with or without G-3-P (10 μM). Doses for i.p. injections were G-3-P (300 mg/kg), LPA 18:1 (50 mg/kg), and FSG67 (5 mg/kg). All measurements were made 24 hours after i.p. injection. Data represent the mean ± SEM. *P < 0.05, by 2-sided Student’s t test (B, C, and H), 1-way ANOVA followed by Tukey’s multiple comparisons test (D and F), or 2-way ANOVA followed by Tukey’s multiple comparisons test (E).
Four GPAT isoforms have been identified. GPAT2 is known to be expressed in human bone (21). Because of our data highlighting Fgf23 expression in response to G-3-P in the stromal compartment of bone marrow, we isolated and differentiated primary osteoblasts from bone marrow for further analysis. We found that Gpat2 was the predominant Gpat isoform expressed in primary osteoblasts and bone marrow and that its expression increased following G-3-P treatment (Supplemental Figure 3, A and B). Knockdown of Gpat2 in primary osteoblasts resulted in reduced enzyme expression (Supplemental Figure 3, B–D) and reduced LPA 18:1 synthesis (Supplemental Figure 3E). In turn, knockdown by shRNA of Gpat2 — but not Gpat1, Gpat 3, or Gpat 4 — significantly attenuated the FGF23 response to G-3-P treatment (Figure 4, H and I, and Supplemental Figure 3, B and F). These results highlight GPAT2 as the GPAT isoform that may be responsible for the effect of G-3-P on bone and bone marrow LPA synthesis.
LPAR1 mediates the effect of G-3-P and LPA on FGF23 production.
To further explore how LPA regulates FGF23, we tested the effects of LPA on Ocy454 cells, which are conditionally immortalized osteocytes (22, 23). LPA administration triggered a dose-responsive increase in FGF23 levels that increased up to 24 hours after treatment, an effect that required coadministration of 1,25(OH)2D (Figure 5A and Supplemental Figure 4, A–C). LPA receptors are GPCRs known to be expressed in bone, with highest levels demonstrated for LPAR1 and LPAR4, a finding we confirmed in Ocy454 cells (Figure 5B). CRISPR/Cas9-mediated deletion of Lpar1, Lpar4, and Vdr (Figure 5C) resulted in reduced expression of FGF23 following treatment with LPA and 1,25(OH)2D, with the largest effect noted in Lpar1-deficient cells (Figure 5D). Further, ChIP at a putative VDR target site showed increased VDR occupancy at the Fgf23 promoter in response to LPA and 1,25(OH)2D treatment in WT cells, but not in Lpar1- or Vdr-deficient cells (Figure 5E, Supplemental Figure 4D, and ref. 24).
Figure 5. LPAR1 mediates the effect of G-3-P and LPA on FGF23 production.

(A) Fgf23 expression in Ocy454 cells treated with vehicle, 10 μM LPA, 10 nM 1,25(OH)2D (1,25D), or both (n = 3–6 per group). (B) Expression of LPA receptors in Ocy454 cells (n = 2). (C and D) Immunoblots of primary osteoblasts following stable deletion of Lpar1, Lpar4, or Vdr, probing for targeted gene products (C) or FGF23 following treatment with LPA (10 μM) and 1,25(OH)2D (10 nM) (D). Shown are representative gels from 1 of 2 independent experiments. (E) ChIP of the Fgf23 promoter following LPA 18:1 and 1,25(OH)2D treatment in WT or Lpar1- or Vdr-deficient cells. Plot shows relative enrichment of the putative VDR target site pulled down by an antibody against VDR compared with IgG. (F and G) Body weights (n = 5 mice per group) (F) and femur length (G) of Lpar1–/– mice and Lpar+/+ littermates. (H) Plasma iFGF23 and cFGF23 levels after i.p. injection of G-3-P (300 mg/kg), LPA 18:1 (50 mg/kg), or vehicle for Lpar1–/– mice and Lpar1+/+ littermates (n = 3–9 per group). Data represent the mean ± SEM. *P < 0.05 and ***P < 0.0001, by 1-way ANOVA followed by Tukey’s multiple comparisons test (A) or 2-sided Student’s t test (F and H).
Notably, a prior study has implicated LPAR1 in osteoblast development, showing that Lpar1–/– mice develop osteoporosis in trabecular and cortical bone (25). After backcrossing these animals with CD-1 mice, we established a colony of Lpar1–/– mice that were of equal weight and had similar bone lengths relative to their Lpar1+/+ littermates (Supplemental Figure 4E and Figure 5, F and G). Consistent with this prior study, Lpar1–/– mice had the same baseline plasma levels of iFGF23 and cFGF23 compared with levels in Lpar1+/+ littermates. However, G-3-P and LPA administration elicited a further increase in plasma iFGF23 and cFGF23 levels in Lpar1+/+ mice only (Figure 5H). These studies show that LPA signals through LPAR1 to increase FGF23 production in vitro and in vivo, cooperating with 1,25(OH)2D at the gene expression level.
Kidney-derived G-3-P increases FGF23 via GPAT and LPAR1 during AKI.
To model AKI in C57Bl/6J mice, we performed bilateral IRI. Relative to sham surgery, we found that IRI was associated with a significant increase in kidney tissue G-3-P levels (Figure 6A), consistent with the known glycolytic shift induced by ischemic AKI (26). Likewise, mouse renal epithelial cells (inner medullary collecting duct [IMCD] cells) treated with rotenone, an inhibitor of mitochondrial respiration that enhances glycolytic flux, also significantly increased G-3-P production (Supplemental Figure 5A). Next, we tested the impact of IRI-mediated AKI in Lpar1–/– mice and their Lpar1+/+ littermates. We observed no difference in the severity of AKI between groups (Figure 6B). Plasma G-3-P levels, in conjunction with several glycolytic intermediates, rose significantly within 4 hours of IRI (Figure 6C and Supplemental Figure 5B). Bone marrow G-3-P levels also increased significantly (Supplemental Figure 5C), along with bone marrow but not plasma LPA 18:1 levels (Figure 6D and Supplemental Figure 5D). Despite similar increases in circulating G-3-P and bone marrow LPA in Lpar1–/– and Lpar1+/+ mice, only Lpar1+/+ mice had a significant increase in plasma iFGF23 and cFGF23 levels (with no change in the cFGF23/iFGF23 ratio) and bone FGF23 expression (Figure 6, E and F, and Supplemental Figure 5E). Addition of the nonspecific LPA receptor inhibitor LPA-bromophosphonate did not further decrease iFGF23 levels in Lpar1–/– mice (Supplemental Figure 5F and refs. 27, 28). Furthermore, administration of FSG67 to WT C57Bl/6J mice at the time of IRI surgery significantly lowered bone marrow LPA levels and lessened the subsequent increase in plasma iFGF23 levels (Figure 6, G and H). In the context of AKI, increased blood phosphate levels were noted in one Lpar1–/– animal and one C57Bl/6J animal treated with the GPAT inhibitor FSG67 (Supplemental Figure 5, G and H). Collectively, these data demonstrate that kidney-derived G-3-P goes to the bone marrow and increases FGF23 production, and that this increase in FGF23 with AKI is dependent on the conversion of G-3-P to LPA and signaling through LPAR1 (Figure 6I).
Figure 6. AKI increases FGF23 production through G-3-P and LPA signaling.

(A) Kidney G-3-P levels 10 minutes after IRI or sham surgery (n = 6 per group). (B–E) Serum creatinine (sCr) (B), plasma G-3-P (C), bone marrow LPA (D), and plasma iFGF23 and cFGF23 (E) levels in Lpar1–/– mice and Lpar1+/+ littermates subjected to IRI. All measurements were made after 24 hours, except for G-3-P levels (4 hours). n = 2–6 per group. (F) Immunohistochemistry for FGF23 in femurs from Lpar1–/– mice and Lpar1+/+ littermates subjected to IRI. Shown are representative images from 1 of 2 independent experiments. Scale bars: 100 μm. (G and H) Bone marrow LPA (G) and plasma iFGF23 and cFGF23 (H) levels in C57Bl/6J mice subjected to IRI with or without FSG67 (0.25 mg/kg). n = 4 per group. Data represent the mean ± SEM. *P < 0.05, by 2-sided Student’s t test (A), 2-way ANOVA followed by Tukey’s multiple comparisons test (B, C, and E), or 1-way ANOVA followed by Tukey’s multiple comparisons test (D, G, and H). (I) Schema depicting the action of kidney-derived G-3-P on bone FGF23 production, whereby local GPAT mediates conversion of G-3-P to LPA, which subsequently signals through LPAR1 to stimulate FGF23 expression.
G-3-P increases in the circulation with human AKI and correlates with FGF23 levels.
FGF23 levels increase rapidly in patients who develop AKI. We measured plasma G-3-P levels in 26 individuals immediately before and after cardiac surgery, including individuals who did (AKI) or did not (controls) subsequently develop AKI, as defined by a doubling of serum creatinine or need for renal replacement therapy. Patients with AKI and controls were matched by age, sex, and baseline eGFR (Table 2). There was no baseline difference in plasma G-3-P levels, whereas G-3-P increased significantly more in patients with AKI than in controls immediately following surgery (Figure 7A). This immediate postoperative rise in G-3-P occurred before a change in blood creatinine levels (Figure 7B). Furthermore, immediate postoperative plasma G-3-P levels correlated with the subsequent rise in plasma iFGF23 levels on postoperative day 1 in patients with AKI (r2 = 0.31, Figure 7C), whereas postoperative levels of plasma PTH, phosphate, 1,25(OH)2D, and creatinine had no association with the increase in plasma iFGF23 among these patients (r2 ≤ 0.05 for all, Figure 7, D–G). Among AKI patients, cFGF23 levels increased even more than iFGF23 on postoperative day 1 (Supplemental Figure 6A), and immediate postoperative plasma G-3-P levels had a correlation with the subsequent rise in cFGF23 similar to that seen with iFGF23 (r2 = 0.39, Supplemental Figure 6B)
Table 2. Baseline characteristics of AKI case-control study subjects.
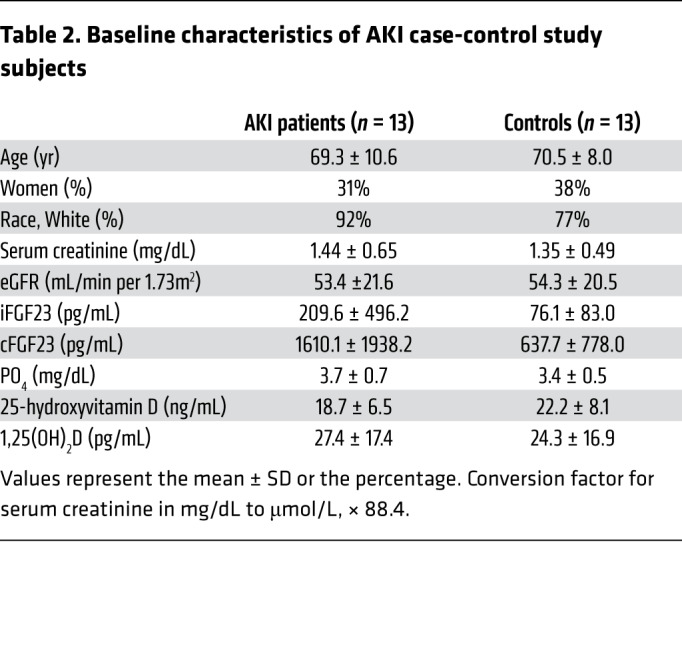
Figure 7. G-3-P increases immediately with human AKI and correlates with subsequent iFGF23 elevation.

(A and B) Plasma G-3-P (A) and creatinine (B) levels before (Preop) and immediately after (Postop) cardiac surgery for patients with AKI and control subjects (n = 13 for each group). Data represent the mean ± SEM. *P < 0.05, by 2-way ANOVA followed by Tukey’s multiple comparisons test. (C) Scatter plots of immediate postoperative plasma G-3-P and iFGF23 levels 24 hours after surgery in patients with AKI and control subjects. (D–G) Scatter plots of immediate postoperative PTH (D), phosphate (E), 1,25(OH)2D (F), and creatinine (G) levels and iFGF23 levels 24 hours after surgery for patients with AKI.
Discussion
Tools in systems biology such as proteomics and metabolomics enable an unbiased examination of interorgan physiology. Recent studies have revealed previously unanticipated functions for several circulating small molecules, including citric acid cycle intermediates, short-chain fatty acids, and bile acids, that coordinate metabolism, inflammation, and other systemic processes across tissues, often acting through cognate cell-surface or nuclear receptors (29–33). With the hypothesis that the kidney may release a factor that modulates FGF23 production, we performed a screen of renal venous plasma, followed by an extensive mechanistic interrogation. Spanning basic and clinical investigation, these studies identify a role for kidney-derived G-3-P in FGF23 homeostasis and demonstrate relevance to AKI.
G-3-P is a downstream product of glycolysis, a ubiquitous metabolic process. To our knowledge, it is not possible to label kidney-specific glycolysis by isotope or fluorescence. For example, if isotope-labeled glucose were injected directly into the renal artery, only a small fraction would be metabolized within the organ, with the rest returned to the systemic circulation, such that the tissue of origin for any isotope-labeled G-3-P in bone or bone marrow would be uncertain. To circumvent this challenge, we focused on a kidney-specific intervention that involved stimulation of kidney G-3-P production by clamping the renal arteries to induce kidney ischemia, using sham surgery as a control. Using this approach, we show that kidney-specific ischemia increased G-3-P within kidney tissue, with downstream G-3-P accumulation in plasma and bone marrow. In turn, this kidney-derived G-3-P increased bone marrow LPA, with no change in circulating LPA, findings that recapitulated the effect of exogenous G-3-P injection. Finally, the impact of circulating G-3-P on FGF23 production was specific to bone and bone marrow, with no increase in Fgf23 expression in the other organs tested. Together, these findings identify a kidney–to–bone and bone marrow signaling axis mediated by G-3-P and LPA.
Critically, the stimulatory effect of both IRI and G-3-P injection on both local LPA and FGF23 production was abrogated by GPAT inhibition. Because the conversion of G-3-P to LPA is the initial step in the synthesis of triglycerides, the contribution of GPATs to lipid metabolism has been closely examined (18, 34). GPAT1 and GPAT4 are important regulators of triglyceride synthesis, particularly in the liver, whereas GPAT3 plays a major role in adipose tissue. GPAT2 is believed to play a minor role in initiating triglyceride synthesis, as its presence in the liver does not prevent the substantial triglyceride depletion observed in Gpat1–/– mice (35); further, Gpat2 expression is not altered by fasting or refeeding, as observed for other lipogenic enzymes (36). Here, we found GPAT2 to be the major GPAT isoform expressed in bone and bone marrow and that GPAT2 knockdown in primary osteoblasts blocked the effect of G-3-P treatment on LPA and FGF23 production. Thus, we believe that GPAT2 is a strong candidate for converting circulating G-3-P to LPA in bone and bone marrow in vivo, but we acknowledge that additional genetic and specific pharmacologic approaches are required to confirm this possibility.
Once G-3-P was converted locally to LPA, our results showed that LPA acted through LPAR1 to stimulate FGF23 expression during AKI. However, LPAR1 was not the sole determinant of FGF23 production, as Lpar1 deletion in mice did not alter basal FGF23 or phosphate levels. In cultured osteocytes, we noted that Lpar4 deletion also reduced LPA-triggered FGF23 expression, raising the possibility of some redundancy in LPAR signaling (although we did not observe a further decrement in FGF23 levels in Lpar1–/– mice treated with a nonspecific LPAR inhibitor). Signaling through VDR is a known stimulus for FGF23 production, and, indeed, we found that Vdr deletion in osteocytes also reduced LPA-triggered FGF23 expression. Furthermore, we observed in vitro that the stimulatory impact of LPA required coadministration with 1,25(OH)2D (vitamin D was not required for in vivo experiments, presumably because of endogenous sources). Our ChIP experiments showed that LPA signaling through LPAR1 increased VDR occupancy at the Fgf23 promoter, placing 1,25(OH)2D action downstream of LPA/LPAR1. Understanding how LPA signaling through LPAR1 subsequently modulates VDR action — e.g., via expression, stability, phosphorylation, heterodimerization, nuclear localization, etc. — is an important area for future investigation. Interestingly, prior publications have shown that the long-recognized impact of 1,25(OH)2D on osteoblast differentiation is in fact dependent on the presence of LPA in culture media, a finding that has probably been missed because of the presence of LPA in commercially available sera used for cell culture (37).
In addition to highlighting G-3-P, LPA, and LPAR1, our findings underscore the important contribution that bone marrow makes to FGF23 production during AKI. Other groups have already shown that bone marrow is an important source of FGF23 in response to acute blood loss and erythropoietin treatment (38, 39). White’s group also used DMP1-Cre to knock out Fgf23 in bone, crossing Dmp1-Cre+ mice with Fgf23-Δ/floxed mice (40). High-phosphate challenge triggered a similar relative fold change in FGF23 expression in bone Fgf23-KO mice relative to levels in WT littermates, but from a lower baseline. In this same study, the authors also deleted Fgf23 using Col2.3-Cre and found that high-phosphate challenge did not elicit a significant increase in FGF23 in these KO mice. Whereas DMP1 is expressed in late osteoblasts/early osteocytes, Col2.3 is expressed in early osteoblasts and osteocytes. Thus, it is necessary to delete Fgf23 in earlier osteoblast precursors to abrogate FGF23 responsiveness, which corroborates our data on G-3-P and Fgf23 expression in flow-sorted bone marrow stromal precursor cells.
Although we emphasize the specific action of kidney-derived G-3-P, we acknowledge that other tissues subjected to ischemia can also release G-3-P. In fact, one study that examined humans undergoing planned ablation of myocardial tissue demonstrated an approximately 40% increase in coronary venous G-3-P levels within 10 minutes of injury, with a subsequent increase of approximately 20%–25% in peripheral vein G-3-P levels (41). Although lesser in magnitude than the more than 100% rise we observed with AKI, it is thus possible that other ischemic tissues could also drive FGF23 production by releasing G-3-P. The literature also shows that additional factors such as iron deficiency and inflammation can increase FGF23 levels. Whether these effects intersect with G-3-P or LPA metabolism requires further study (42, 43).
To date, strategies for counteracting excess FGF23 production have focused on the administration of neutralizing antibodies against FGF23 (44, 45). However, this treatment requires careful monitoring to avoid functional FGF23 deficiency with resultant hyperphosphatemia and soft tissue mineralization; it also requires s.c. injections and is costly. The identification of upstream regulators of FGF23 production amenable to small-molecule antagonism could facilitate the development of FGF23 modulators that are easier to titrate and deliver, although our results show these approaches can still cause hyperphosphatemia. Both GPAT and LPAR inhibitors have been developed, but to our knowledge, no inhibitors specific to GPAT2 or LPAR1 are yet clinically available. In our studies, the increase in FGF23 levels, but not serum creatinine, was markedly attenuated in Lpar1–/– mice, suggesting that FGF23 may not have a direct impact on AKI severity. An important caveat is that our experiments examined creatinine levels very shortly (24 hours) after IRI. Because our work focuses on the physiology of FGF23 production rather than its downstream effects, we did not extend follow up to assess for differences in renal, cardiac, or mortality outcomes in Lpar1–/– mice subjected to AKI.
This study has several strengths. First, we started with an unbiased screen of over 5000 measurements, including more than 1600 known proteins and metabolites in a unique human renal arteriovenous sample set, selecting the top hit for subsequent characterization. Second, we used a combination of biochemical and genetic approaches to outline the sequential action of G-3-P, LPA, and LPAR1 on FGF23 expression both in vitro and in vivo. Third, we extended the findings of the existing literature reporting bone marrow Fgf23 expression, as we identified the specific contribution of nonhematopoietic stromal precursor cells. Fourth, we used kidney IRI to demonstrate that kidney-derived G-3-P and its subsequent enzymatic conversion to LPA and signaling through LPAR1 are all necessary to stimulate FGF23 production during AKI. Fifth, we showed that G-3-P levels rise rapidly in humans who develop AKI and that they correlate better with the subsequent rise in FGF23 than creatinine or other mineral markers.
Several limitations also warrant mention. We note that the human renal arteriovenous cohort had a mean eGFR of 66.6 mL/min per 1.73 m2, along with a high prevalence of hypertension and other kidney disease risk factors, perhaps enriched for individuals with abnormal kidney metabolism, e.g., enhanced glycolysis and G-3-P production. Sampling in healthier cohorts will be required to assess the relationship between renal venous G-3-P and circulating FGF23 among individuals with normal kidney function. As noted, more work is also required to confirm a specific role for GPAT2 in vivo, as well as to further characterize the cooperation between LPA and VDR signaling. Additional experiments are also required to understand the exact mechanisms by which G-3-P is released from the kidney and taken up in bone; organic anion transporters, which share topology with an established bacterial G-3-P transporter, are attractive candidates for further study (46). Finally, future studies will need to examine whether enhanced renal release of G-3-P is implicated in the dramatic FGF23 increases observed in advanced CKD.
Ultimately, these studies are motivated by the strong association between elevated FGF23 levels and adverse outcomes in kidney disease, with emerging data ascribing direct cardiovascular and other end-organ toxicities to FGF23 in this context (8, 13, 47). Because metabolites and the enzymes and receptors that mediate their biologic actions may all be amenable to pharmacologic modulation, we believe our results identify new opportunities for therapeutic targeting in AKI, as well as other disorders of FGF23 excess.
Methods
Human renal arteriovenous sampling.
Details on this protocol have been published previously (16). We recruited consecutive patients referred to the Massachusetts General Hospital Cardiac Catheterization Laboratory for right and left heart catheterization. During the procedure, plasma was sampled from catheters placed in the ostium of a renal vein and the abdominal aorta. All patients were fasting, and all samples were obtained prior to coronary artery catheterization and administration of iodinated contrast dye. eGFR for study participants was calculated using the Modification of Diet in Renal Disease (MDRD) equation (48). Arterial and renal venous plasma samples from 17 individuals were used for metabolomic profiling and measurement of iFGF23; proteomics analysis was performed on samples obtained from 16 of the 17 study subjects.
Proteomics.
Proteomic profiling was performed with 50 μL plasma as previously described using the SOMAscan single-stranded DNA aptamer–based platform (1.3 k assay; SomaLogic) (49). All assays were performed using SOMAscan reagents (SomaLogic) according to the manufacturer’s detailed protocol. The median intra-assay coefficients of variation (CV) were calculated from inclusion of replicated pooled plasma calibrator samples on each assay plate, and was 4% and 2.5% across the 2 plates (96-well format) used for this study. The median inter-assay CV were calculated using replicate quality control plasma samples included across the 2 plates, and was 3%.
Metabolomics.
Metabolites were assayed as previously described (50). In brief, sugars, sugar phosphates, organic acids, bile acids, nucleotides, and other anionic polar metabolites including G-3-P were measured in 30 μL plasma (or tissue homogenate) using hydrophilic interaction liquid chromatography (LC) and multiple reaction monitoring in the negative ion mode on a 5500 QTRAP MS (SCIEX). Amino acids, amines, acylcarnitines, nucleotides, and other cationic polar metabolites were measured in 10 μL plasma using hydrophilic interaction LC coupled with nontargeted, positive ion mode MS analysis on an Exactive Plus Orbitrap MS (Thermo Fisher Scientific). Lipids were measured in 10 μL plasma using C8 chromatography and nontargeted, positive ion mode MS analysis on an Exactive Plus Orbitrap Mass Spectrometer. Identification of known metabolites was achieved by matching retention times and mass/charge ratio (m/z) to synthetic mixtures of reference compounds and characterized pooled plasma reference samples.
LPA measurement in human plasma.
LPA for renal arteriovenous samples was measured in 50 μL plasma by the Mass Spectrometry Laboratory at National Jewish Health (Denver, Colorado, USA) (51). In brief, LC-MS was performed with a 6500 QTRAP (SCIEX) hybrid triple quadrupole/ion trap mass spectrometer in the negative ion mode, coupled with chromatography on an Ascentis Express C8 column (75 × 2.1 mm, 2.7 μm; MilliporeSigma).
Human AKI case-control study.
We analyzed samples from a case-control study of 26 individuals who underwent cardiac surgery, with patients and controls matched by age (±10 years), sex, and baseline eGFR (±5 mL/min/1.73 m2). AKI was defined as a doubling of serum creatinine or need for renal replacement therapy. Patients were recruited from Brigham and Women’s Hospital between August 2007 and March 2012. In brief, the inclusion and exclusion criteria were chosen to capture patients at high risk of AKI and other adverse outcomes after cardiac surgery. Inclusion criteria were a baseline eGFR of 30 mL/min or less per 1.73 m2 or any 2 of the following: a baseline eGFR of 31–60 mL/min per 1.73 m2, diabetes mellitus, left ventricular ejection fraction of 40% or less, previous cardiac surgery, combined coronary artery bypass/valve procedure, urgent procedure, and preoperative intra-aortic balloon pump implantation. Exclusion criteria were preoperative AKI (defined as a 0.3 mg/dL rise in serum creatinine over 24 hours or a 0.5 mg/dL rise over 48 hours), recent aminoglycoside use, serum creatinine level above 4.5 mg/dL, end-stage renal disease requiring dialysis, renal transplantation, and pregnancy. Plasma aliquots were collected and stored at –80°C within 2 hours of collection. G-3-P was measured in plasma samples obtained preoperatively and immediately postoperatively (at the end of cardiopulmonary bypass); iFGF23 was additionally measured on postoperative day 1 (~24 hours after cardiopulmonary bypass).
Mice.
C57Bl/6J and CD-1 mice were purchased from The Jackson Laboratory. Lpar1–/+ mice were generated in-house (52, 53); because of low fertility, these mice were backcrossed for 5 generations with CD-1 mice before in-house generation of Lpar1–/– mice. Fgf23fl/fl Dmp1-Cre+ mice were generated by breeding DMP1-Cre mice (17) with floxed Fgf23 mice carrying loxP sequences flanking exon 1 of the Fgf23 gene. The floxed Fgf23 gene allele was generated by using a gene-targeting vector on the pNT1.1 plasmid backbone (GenBank: JN935771.1) that included a 9.7-kb target region around exons 1 and 2 of the Fgf23 gene (BAC clone RP23-8L20) along with 2 loxP sites flanking exon 1, an insertion of a 1.9-kb flippase recognition target–flanked (FRT-flanked) neomycin (Neo) cassette immediately at the 5′-end of the second loxP site (loxP no. 2), and a thymidine kinase (TK) cassette at the 3′-end of exon 2. After linearization by I-Ceu1 restriction enzyme digestion, the targeting vector was injected into 129S6/B6 hybrid G4 embryonic stem (ES) cells by members of the Transgenic Resources Program at the Department of Comparative Medicine of the University of Washington. Positive ES cell clones with homologous recombination events within their Fgf23 locus were injected into 129S blastocysts to produce chimeric mice. These mice were then bred with ROSA26/FLPe-transgenic mice (C57BL/6 FLP, stock 009086; The Jackson Laboratory) to remove the FRT-Neo cassette from the integrated gene allele and produce mice solely carrying the floxed Fgf23-E1 gene allele. The latter mice were backcrossed with C57BL/6 mice for 5 generations before being bred with DMP1-Cre mice. Mice (8–24 weeks of age) used in the experiments were sex- and age-matched and maintained in a specific pathogen–free environment at 25°C on a 12-hour light/12-hour dark cycle.
Animal experiments.
G-3-P (≤300 mg/kg; MilliporeSigma), LPA (50 mg/kg; Santa Cruz Biotechnology), sodium phosphate dibasic (300 mg/kg; MilliporeSigma), G-2-P (300 mg/kg; MilliporeSigma), glycerol (300 mg/kg; MilliporeSigma), 1,25(OH)2D (100 ng/kg or 6 ng/g; Enzo Life Sciences), and IL-6 (1.3 μg/kg; R&D Systems) were i.p. injected into 3–10 mice per group. G-3-P dosing was substantially lower than that of the inorganic phosphate used to stimulate FGF23 production in vivo. For a mouse weighing approximately 25 g, 300 mg/kg G-3-P is approximately 7.5 mg, or approximately 39 μmol. By comparison, one study found no increase in FGF23 following a single i.v. injection of 500 μmol NaPi (54), and a separate study noted a less than 2-fold increase in FGF23 following a 1-day infusion of 960 μmol NaPi (55). The GPAT inhibitor FSG67 (Focus Biomolecules) was injected i.p. 30 minutes prior to administration of G-3-P, LPA, or vehicle at a dose of 5 mg/kg (except for IRI experiments; see below). Unless otherwise specified, the mice were sacrificed 24 hours after i.p. injection, and blood, femurs, bone marrow, and other tissues were collected for further analysis. We used a bilateral IRI model for AKI, with the control animals undergoing sham surgery (56). Under inhaled anesthesia on a temperature-controlled heating blanket, both renal pedicles were clamped for 30 minutes with vascular clips (Roboz), and then incisions were closed in 2 layers after clamp removal. For mice that underwent IRI or sham surgery, FSG67 and LPA bromophosphonate (Echelon Biosciences) were injected i.p. 30 minutes prior to surgery at a dose of 0.25 mg/kg and 50 mg/kg, respectively. Unless otherwise specified, the mice were sacrificed 24 hours after surgery, and blood, femurs, and bone marrow were collected for further analysis.
Mouse creatinine, blood urea nitrogen, and phosphate measurements.
Creatinine was measured by alkaline picrate reaction and urease reaction at the Massachusetts General Hospital Animal Clinical Pathology Laboratory. Phosphate was measured by colorimetric assay (ab65622; Abcam).
Histology and immunohistochemistry.
Femur histology was examined on 3-mm formalin-fixed, paraffin-embedded sections stained with hematoxylin & eosin. FGF23 antibody (Immutopics, 186-206, 30-6310) was used to perform FGF23 staining of 2 consecutive femur sections.
LC-MS.
G-3-P and other glycolytic metabolites were measured in mouse plasma and cell culture media using the metabolomics method for human samples described above. For mouse kidney tissue, kidney was homogenized in water (1 mg:4 μL ratio), and for mouse bone marrow, the entire femoral bone marrow was homogenized in 200 μL water; homogenates were then treated as plasma for LC-MS analyses. For LPA measurement of mouse samples, mouse plasma was extracted with 100% methanol at a 1:5 ratio, and bone marrow (prepared by cutting femoral epiphyses and flushing with ice-cold PBS) was centrifuged and reconstituted in 1 mL 100% methanol. For LPA measurement of cell culture samples, cells were extracted with 800 μL 100% methanol per 6-well plate. Mouse and cell culture sample methanol extracts were then evaporated and reconstituted in 50 μL 100% methanol, and 10 μL of this reconstituted sample was separated using a 100 × 2.1 mm Acquity UPLC C8 column, and the peak for LPA 18:1 (transition 435.1/152.9) was monitored in the negative ion mode on a TSQ Quantiva Triple Quadrupole Mass Spectrometer (Thermo Fisher Scientific). For creatinine measurement in the human AKI cohort, 10 μL human plasma was extracted with 90 μL 80% methanol containing creatinine-d3 (MilliporeSigma) and separated using a 150 × 2 mm and 3-μm Atlantis HILIC Column (Waters), and the peaks for creatinine (transition 114.1/44) and creatinine-d3 (transition 117.1/47) were monitored in the positive ion mode on TSQ Quantiva Triple Quadrupole Mass Spectrometer.
Cell lines and experiments.
Primary stromal progenitor cells were isolated from the bone marrow of femurs from C57Bl/6J mice and then cultured as previously described (57). A purified population of stromal progenitor cells was obtained 3 weeks after the initiation of culture and differentiated toward osteoblasts using 0.1 mM dexamethasone and 50 mM ascorbate acid with 10% FBS and 1% antibiotic for 2 weeks (58). Primary osteoblasts, with or without Gpat 1-4 knockdowns, were treated with 10 μM G-3-P (MilliporeSigma) and 10 nM 1,25(OH)2D (Enzo Life Sciences) for 24 hours prior to analysis. For differentiation experiments, primary stromal progenitor cells were treated with either 10 μM G-2-P or G-3-P in addition to 0.1 mM dexamethasone and 50 mM ascorbic acid for a total of 3 weeks, and colonies were stained with alkaline phosphatase (MilliporeSigma) to detect CFU, or alizarin red (MilliporeSigma) to detect mineralization, as previously described (59, 60). Ocy454 cells were grown from a single subclone (22, 61). Cells were passaged in α-MEM supplemented with 10% heat-inactivated FBS and 1% antibiotics (penicillin-streptomycin; Fungizone) at 33°C in 5% CO2. Cells were plated at 50,000 cells/mL and allowed to reach confluency at 33°C (typically in 2–3 days) prior to transfer to a 37°C environment. Experiments were performed on cells cultured at 37°C for 7 days. Ocy454 cells, with or without gene deletions, were treated with 1, 10, and 100 μM LPA (Santa Cruz Biotechnology) and 10 nM 1,25(OH)2D for 24 hours (unless otherwise specified) prior to analysis. All cells were switched to 2% FBS media 24 hours prior to treatments. IMCD3 cells (American Type Culture Collection [ATCC]) were seeded at 200,000 cells per well in 6-well plates and incubated overnight. The following day, IMCD3 cells were washed once with 2 mL pyruvate-free DMEM (D5671; MilliporeSigma), and then the media were changed to 2 mL pyruvate-free DMEM containing 10% dialyzed FBS, penicillin-streptomycin, and 2 mM l-glutamine premixed with 200 nM rotenone or 0.1% DMSO. Media were collected for analysis after 24 hours. Opossum kidney (OK) cells (ATCC) were seeded at 100,000 cells per well in a 6-well plate and incubated overnight. The following day, cells were treated with 10 ng/mL FGF23 (R&D Systems) for 2 or 24 hours, and media were collected for analysis.
Flow cytometric analysis and sorting.
Bone marrow cells were collected from the femurs and tibiae of C57Bl/6J mice 24 hours after treatment with G-3-P or vehicle. Total bone marrow cells were isolated by crushing the femurs and tibiae and filtering cells through a 40-μm nylon mesh. Samples destined for the purification of hematopoietic cells (CD45+TER119–) were processed by density gradient centrifugation (Ficoll-Paque Plus; GE Healthcare) to isolate mononuclear cells. Samples destined for the purification of nonhematopoietic stromal cells (CD45–TER119–) were enriched for nonhematopoietic cells via negative selection. Here, total cells were labeled with biotinylated CD45.2 (clone 104) and TER119 (clone TER119) antibodies (BioLegend), which were then coupled to streptavidin-coated microbeads (Miltenyi Biotec) and passed over a magnetic column (LD; Miltenyi Biotec) for removal of labeled cells. The cells from both samples were fluorescence labeled (anti–CD45.2-APC and anti–Ter119-PE antibodies; BioLegend). Purified hematopoietic cells and purified stromal cells were isolated on a BD FACSAria 3. Flow cytometric analysis was performed using FlowJo software. Total RNA was isolated from sorted cells (RNAeasy Micro; QIAGEN), reverse transcribed into cDNA (SuperScript IV; Invitrogen, Thermo Fisher Scientific), and then used for quantitative PCR (qPCR).
GPAT knockdown.
shRNA lentivirus particles for Gpat1-4 were purchased from Origene (catalogs TL500848V, TL517838V, TL506938V, and TL512820V) and were transduced in HEK293T cells on a pLKO.1-puro (Addgene, plasmid 8453) backbone. Viral packaging was performed in 293T cells using standard protocols (http://www.broadinstitute.org/rnai/public/resources/protocols). Primary osteoblasts were exposed to lentivirus particles (MOI = 1) overnight in the presence of polybrene (5 μg/mL). Cells were subsequently selected with puromycin (2 μg/mL) and maintained in selection medium throughout the duration of the experiment.
Lpar1, Lpar4, and Vdr deletion.
CRISPR/Cas9-mediated gene deletions were performed using the sgRNA targeting sequences listed in Supplemental Table 4. First, Ocy454 cells were stably transduced with a blasticidin resistance–conferring Cas9-expressing lentivirus. sgRNA sequences were subcloned into lentiGuide-Puro (Addgene plasmid 52963) (62). Ocy454 cells were transfected with this plasmid using Fugene HD (Promega; 1 μg plasmid per well of a 6-well plate). Lentivirus particles were produced in HEK293T cells as described above. Cells were exposed to lentivirus particles (MOI = 1) overnight at 33°C in the presence of polybrene (5 μg/mL). Media were then changed, and puromycin (2 μg/mL) and blasticidin (4 μg/mL) were added. Cells were maintained in selection medium throughout the duration of the experiment.
FGF23 and klotho ELISA.
For the renal arteriovenous samples, plasma iFGF23 levels were measured by the Kainos ELISA, and plasma klotho levels were measured by DuoSet ELISA (R&D Systems) according to the manufacturer’s instructions. All other plasma and cell lysate iFGF23 and cFGF23 levels were measured by the respective Immutopics ELISAs according to the manufacturer’s instructions.
qPCR.
Total RNA was extracted from cultured cells using RNeasy (QIAGEN) following the manufacturer’s instructions. For long bone RNA isolation, mice were euthanized, and femurs were rapidly dissected on ice. Soft tissue was removed and epiphyses cut. Bone marrow cells were then removed by serial flushing with ice-cold PBS. TRIzol (Life Technologies, Thermo Fisher Scientific) was added, and samples were frozen at –80°C and then homogenized. For bone marrow RNA, flushed bone marrow was spun down and homogenized with TRIzol. RNA was then extracted according to the manufacturer’s instructions. For cDNA synthesis, 1 μg RNA was used for the synthesis reactions according to the manufacturer’s instructions (PrimeScript RT; Takara). SYBR Green–based qPCR detection was performed using FastStart Universal SYBR Green (Roche) on a StepOne Plus Thermocycler (Applied Biosystems). The PCR primer sequences are listed in Supplemental Table 4.
Immunoblotting.
Whole-cell lysates were prepared using RIPA buffer with protease inhibitors (Pierce, Thermo Fisher Scientific). Bone marrow from femurs and tibiae of C57Bl/6J mice was flushed in RIPA buffer with protease inhibitors. Bone marrow–free femurs and tibiae were homogenized into a fine powder in liquid nitrogen using a porcelain mortar and pestle and then transferred into RIPA buffer with protease inhibitors. After three 30-second sonications, the samples were centrifuged at 13,000 × g for 10 minutes, and the supernatants were collected. Lysates (15–20 μg cellular protein) were separated by SDS-PAGE, and proteins were transferred onto nitrocellulose membranes. Membranes were blocked with 5% milk in TBST and incubated with FGF23 (186-206, 21-6310; Immutopics), LPAR1 (PA1-1041; Thermo Fisher Scientific), LPAR4 (ab183076; Abcam), VDR (ab8756; Abcam), or GPAT2 (10299-378; VWR International) antibodies overnight at 4°C. The next day, the membranes were washed and incubated with the appropriate HRP-coupled secondary antibodies, and signals were detected with ECL (Pierce, Thermo Fisher Scientific). All immunoblots were repeated at least twice, with comparable results.
ChIP analysis.
ChIP analysis of 107 Ocy454 cells was performed with a Magna Chip Kit (MilliporeSigma) using a VDR antibody (Abcam) and IgG. The primer sequences are listed in Supplemental Table 4. qPCR was performed to amplify immunoprecipitated genomic fragments, and data are presented as the fold enrichment relative to IgG-associated DNA.Statistics. For the initial screen of renal venous plasma samples, Pearson’s correlation coefficients between venous metabolites and proteins and arterial iFGF23 were calculated on log-transformed data, because the raw data did not show normal distributions. Statistical significance was set at a P value of less than 9.6 × 10–6, adjusting for the total 5229 measurements (1317 proteins, 300 known metabolites, and 3612 unknown peaks by LC-MS). For other analyses, we performed an unpaired, 2-tailed Student’s t test or 1- or 2-way ANOVA, depending on the number of groups analyzed, with significance defined as a P value of less than 0.05.
Study approval.
The human renal arteriovenous sampling study protocol was approved by the IRB of the Massachusetts General Hospital and adhered to Declaration of Helsinki principles, and all participants provided written informed consent prior to inclusion in the study. The human AKI case-control study protocol was approved by the IRB of Brigham and Women’s Hospital and adhered to Declaration of Helsinki principles, and all participants provided written informed consent prior to inclusion in the study. Mouse studies were approved by the IACUC of Massachusetts General Hospital, UCSF, and the San Francisco Veterans Affair Health Care System and were conducted under their guidelines.
Author contributions
PS and WZ performed animal experiments with input from WDK, JC, AMT, WC, DBS, and MNW. PS, WDK, NG, NBA, and MNW performed cellular experiments with input from PDP. SE, REG, BRK, IHDB, and EPR designed and conducted arterial and renal venous sampling and measurement of mineral markers from patients undergoing invasive catheterization. WDK, WZ, KAP, CBC, and EPR generated metabolomics and other LC-MS data. DN and REG generated proteomics data. WC and ZC generated and studied Fgf23fl/fl Dmp1-Cre+ mice with input from MC. DEL and SSW conducted the AKI case-control study of patients undergoing cardiac surgery. PS, RIT, HJ, MNW, and EPR designed the experiments and analyzed results. PS and EPR wrote the manuscript with input from all authors.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01-NR-017399 and U01-DK-106981 (to EPR); K08-AR-067285 and R01-DK-116716 (to MNW); P01-DK011794 (subproject 3, to HJ); R01-AR-067291 (to WC); and R01-HL-133870, R01-HL-132320, and R01-DK-081572 ( to REG); and by a grant from the Extramural Grant Program of Satellite Healthcare, a not-for-profit renal care provider (to EPR).
Version 1. 02/17/2020
Electronic publication
Version 2. 03/02/2020
Print issue publication
Footnotes
Conflict of interest: DBS is a cofounder of and holds equity in Clear Creek Bio. SE received research grants from Edwards Lifesciences and Svelte Medical and consulting fees from Medtronic and AstraZeneca. JC has received compensation and/or research support from Arena, Celgene, Novartis, and Ono Pharmaceuticals and is a named coinventor on patents describing the sequence for novel EDG-5 receptor homologs (US patent 6,057,126), cloned lysophosphatidic receptors (US patent 6,140,060), methods for promoting survival of myelin-producing cells (US patent 6,150,345), and the use of mosaic aneuploid stem cells (European patent EP1951037A4). DEL received research support from BioPorto Diagnostics. SSW serves on a GlaxoSmithKline steering committee of phase III trials for daprodustat and has provided expert witness testimony for the Public Health Advocacy Institute and GE Healthcare. HJ is a named coinventor on a patent describing the measurement of FGF23 (US patent 7,094,551 B2). MNW is a coinventor on a pending patent (US patent application 16/333,546) regarding the use of SIK inhibitors for osteoporosis and receives research support from Radius Health and Galapagos NV. EPR received research support from Elysium Pharmaceuticals.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(3):1513–1526.https://doi.org/10.1172/JCI131190.
See the related Commentary at Kidney to bone via bedside to bench…and back?.
Contributor Information
Petra Simic, Email: psimic@partners.org.
Wondong Kim, Email: wdongkim@gmail.com.
Wen Zhou, Email: wzhou0@mgh.harvard.edu.
Wenhan Chang, Email: wenhan.chang@ucsf.edu.
David B. Sykes, Email: dbsykes@mgh.harvard.edu.
Sammy Elmariah, Email: SELMARIAH@mgh.harvard.edu.
Debby Ngo, Email: dngo@bidmc.harvard.edu.
Paola Divieti Pajevic, Email: pdivieti@bu.edu.
Nicolas Govea, Email: nicolas.govea@gmail.com.
Ian H. de Boer, Email: IDeBoer@Nephrology.washington.edu.
Zhiqiang Cheng, Email: Zhiqiang.Cheng@va.gov.
Marta Christov, Email: Marta_Christov@nymc.edu.
Jerold Chun, Email: jchun@sbpdiscovery.org.
Sushrut S. Waikar, Email: swaikar@partners.org.
Robert E. Gerszten, Email: rgerszte@bidmc.harvard.edu.
Clary B. Clish, Email: clary@broadinstitute.org.
Harald Jüppner, Email: hjueppner@partners.org.
Marc N. Wein, Email: MNWEIN@partners.org.
References
- 1.Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev. 2012;92(1):131–155. doi: 10.1152/physrev.00002.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leaf DE, et al. Fibroblast growth factor 23 levels are elevated and associated with severe acute kidney injury and death following cardiac surgery. Kidney Int. 2016;89(4):939–948. doi: 10.1016/j.kint.2015.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leaf DE, et al. Fibroblast growth factor 23 levels associate with AKI and death in critical illness. J Am Soc Nephrol. 2017;28(6):1877–1885. doi: 10.1681/ASN.2016080836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gutiérrez OM, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359(6):584–592. doi: 10.1056/NEJMoa0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christov M, Neyra JA, Gupta S, Leaf DE. Fibroblast growth factor 23 and klotho in AKI. Semin Nephrol. 2019;39(1):57–75. doi: 10.1016/j.semnephrol.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Leaf DE, et al. Fibroblast Growth Factor 23 Associates with Death in Critically Ill Patients. Clin J Am Soc Nephrol. 2018;13(4):531–541. doi: 10.2215/CJN.10810917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith ER, Tan SJ, Holt SG, Hewitson TD. FGF23 is synthesised locally by renal tubules and activates injury-primed fibroblasts. Sci Rep. 2017;7(1):3345. doi: 10.1038/s41598-017-02709-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rossaint J, et al. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J Clin Invest. 2016;126(3):962–974. doi: 10.1172/JCI83470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bacchetta J, et al. Fibroblast growth factor 23 inhibits extrarenal synthesis of 1,25-dihydroxyvitamin D in human monocytes. J Bone Miner Res. 2013;28(1):46–55. doi: 10.1002/jbmr.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coe LM, Madathil SV, Casu C, Lanske B, Rivella S, Sitara D. FGF-23 is a negative regulator of prenatal and postnatal erythropoiesis. J Biol Chem. 2014;289(14):9795–9810. doi: 10.1074/jbc.M113.527150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silswal N, et al. FGF23 directly impairs endothelium-dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am J Physiol Endocrinol Metab. 2014;307(5):E426–E436. doi: 10.1152/ajpendo.00264.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faul C, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121(11):4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grabner A, et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 2015;22(6):1020–1032. doi: 10.1016/j.cmet.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen G, et al. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature. 2018;553(7689):461–466. doi: 10.1038/nature25451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christov M, et al. Plasma FGF23 levels increase rapidly after acute kidney injury. Kidney Int. 2013;84(4):776–785. doi: 10.1038/ki.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhee EP, et al. A combined epidemiologic and metabolomic approach improves CKD prediction. J Am Soc Nephrol. 2013;24(8):1330–1338. doi: 10.1681/ASN.2012101006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu Y, Xie Y, Zhang S, Dusevich V, Bonewald LF, Feng JQ. DMP1-targeted Cre expression in odontoblasts and osteocytes. J Dent Res. 2007;86(4):320–325. doi: 10.1177/154405910708600404. [DOI] [PubMed] [Google Scholar]
- 18.Wendel AA, Lewin TM, Coleman RA. Glycerol-3-phosphate acyltransferases: rate limiting enzymes of triacylglycerol biosynthesis. Biochim Biophys Acta. 2009;1791(6):501–506. doi: 10.1016/j.bbalip.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wydysh EA, Medghalchi SM, Vadlamudi A, Townsend CA. Design and synthesis of small molecule glycerol 3-phosphate acyltransferase inhibitors. J Med Chem. 2009;52(10):3317–3327. doi: 10.1021/jm900251a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuhajda FP, et al. Pharmacological glycerol-3-phosphate acyltransferase inhibition decreases food intake and adiposity and increases insulin sensitivity in diet-induced obesity. Am J Physiol Regul Integr Comp Physiol. 2011;301(1):R116–R130. doi: 10.1152/ajpregu.00147.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilhelm M, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509(7502):582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 22.Spatz JM, et al. The Wnt inhibitor sclerostin is up-regulated by mechanical unloading in osteocytes in vitro. J Biol Chem. 2015;290(27):16744–16758. doi: 10.1074/jbc.M114.628313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wein MN, et al. HDAC5 controls MEF2C-driven sclerostin expression in osteocytes. J Bone Miner Res. 2015;30(3):400–411. doi: 10.1002/jbmr.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaneko I, Saini RK, Griffin KP, Whitfield GK, Haussler MR, Jurutka PW. FGF23 gene regulation by 1,25-dihydroxyvitamin D: opposing effects in adipocytes and osteocytes. J Endocrinol. 2015;226(3):155–166. doi: 10.1530/JOE-15-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gennero I, et al. Absence of the lysophosphatidic acid receptor LPA1 results in abnormal bone development and decreased bone mass. Bone. 2011;49(3):395–403. doi: 10.1016/j.bone.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lan R, et al. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol. 2016;27(11):3356–3367. doi: 10.1681/ASN.2015020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang G, et al. Alpha-substituted phosphonate analogues of lysophosphatidic acid (LPA) selectively inhibit production and action of LPA. ChemMedChem. 2007;2(5):679–690. doi: 10.1002/cmdc.200600280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nikitopoulou I, et al. A metabolically-stabilized phosphonate analog of lysophosphatidic acid attenuates collagen-induced arthritis. PLoS ONE. 2013;8(7):e70941. doi: 10.1371/journal.pone.0070941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toma I, et al. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J Clin Invest. 2008;118(7):2526–2534. doi: 10.1172/JCI33293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He W, et al. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature. 2004;429(6988):188–193. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 31.Pluznick JL, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci USA. 2013;110(11):4410–4415. doi: 10.1073/pnas.1215927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe M, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439(7075):484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 33.Ryan KK, et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature. 2014;509(7499):183–188. doi: 10.1038/nature13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamashita A, et al. Glycerophosphate/acylglycerophosphate acyltransferases. Biology (Basel) 2014;3(4):801–830. doi: 10.3390/biology3040801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hammond LE, et al. Mitochondrial glycerol-3-phosphate acyltransferase-deficient mice have reduced weight and liver triacylglycerol content and altered glycerolipid fatty acid composition. Mol Cell Biol. 2002;22(23):8204–8214. doi: 10.1128/MCB.22.23.8204-8214.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S, et al. Cloning and functional characterization of a novel mitochondrial N-ethylmaleimide-sensitive glycerol-3-phosphate acyltransferase (GPAT2) Arch Biochem Biophys. 2007;465(2):347–358. doi: 10.1016/j.abb.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mansell JP, Blackburn J. Lysophosphatidic acid, human osteoblast formation, maturation and the role of 1α,25-Dihydroxyvitamin D3 (calcitriol) Biochim Biophys Acta. 2013;1831(1):105–108. doi: 10.1016/j.bbalip.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 38.Rabadi S, Udo I, Leaf DE, Waikar SS, Christov M. Acute blood loss stimulates fibroblast growth factor 23 production. Am J Physiol Renal Physiol. 2018;314(1):F132–F139. doi: 10.1152/ajprenal.00081.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toro L, et al. Erythropoietin induces bone marrow and plasma fibroblast growth factor 23 during acute kidney injury. Kidney Int. 2018;93(5):1131–1141. doi: 10.1016/j.kint.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 40.Clinkenbeard EL, et al. Conditional deletion of murine Fgf23: interruption of the normal skeletal responses to phosphate challenge and rescue of genetic hypophosphatemia. J Bone Miner Res. 2016;31(6):1247–1257. doi: 10.1002/jbmr.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis GD, et al. Metabolite profiling of blood from individuals undergoing planned myocardial infarction reveals early markers of myocardial injury. J Clin Invest. 2008;118(10):3503–3512. doi: 10.1172/JCI35111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.David V, et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016;89(1):135–146. doi: 10.1038/ki.2015.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Onal M, et al. A novel distal enhancer mediates inflammation-, PTH-, and early onset murine kidney disease-induced expression of the mouse Fgf23 gene. JBMR Plus. 2018;2(1):32–47. doi: 10.1002/jbm4.10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shalhoub V, et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest. 2012;122(7):2543–2553. doi: 10.1172/JCI61405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carpenter TO, et al. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest. 2014;124(4):1587–1597. doi: 10.1172/JCI72829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 2003;301(5633):616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- 47.Singh S, et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016;90(5):985–996. doi: 10.1016/j.kint.2016.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130(6):461–470. doi: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
- 49.Ngo D, et al. Aptamer-based proteomic profiling reveals novel candidate biomarkers and pathways in cardiovascular disease. Circulation. 2016;134(4):270–285. doi: 10.1161/CIRCULATIONAHA.116.021803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paynter NP, et al. Metabolic predictors of incident coronary heart disease in women. Circulation. 2018;137(8):841–853. doi: 10.1161/CIRCULATIONAHA.117.029468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Montesi SB, et al. Docosatetraenoyl LPA is elevated in exhaled breath condensate in idiopathic pulmonary fibrosis. BMC Pulm Med. 2014;14:5. doi: 10.1186/1471-2466-14-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Contos JJ, Fukushima N, Weiner JA, Kaushal D, Chun J. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc Natl Acad Sci U S A. 2000;97(24):13384–13389. doi: 10.1073/pnas.97.24.13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tager AM, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14(1):45–54. doi: 10.1038/nm1685. [DOI] [PubMed] [Google Scholar]
- 54.Thomas L, Bettoni C, Knöpfel T, Hernando N, Biber J, Wagner CA. Acute adaption to oral or intravenous phosphate requires parathyroid hormone. J Am Soc Nephrol. 2017;28(3):903–914. doi: 10.1681/ASN.2016010082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arai-Nunota N, et al. Intravenous phosphate loading increases fibroblast growth factor 23 in uremic rats. PLoS ONE. 2014;9(3):e91096. doi: 10.1371/journal.pone.0091096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16(5):535–543, 1p following 143. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Soleimani M, Nadri S. A protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. Nat Protoc. 2009;4(1):102–106. doi: 10.1038/nprot.2008.221. [DOI] [PubMed] [Google Scholar]
- 58.Zuk PA, et al. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001;7(2):211–228. doi: 10.1089/107632701300062859. [DOI] [PubMed] [Google Scholar]
- 59.Simic P, et al. SIRT1 regulates differentiation of mesenchymal stem cells by deacetylating β-catenin. EMBO Mol Med. 2013;5(3):430–440. doi: 10.1002/emmm.201201606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schipani E, et al. Targeted expression of constitutively active receptors for parathyroid hormone and parathyroid hormone-related peptide delays endochondral bone formation and rescues mice that lack parathyroid hormone-related peptide. Proc Natl Acad Sci U S A. 1997;94(25):13689–13694. doi: 10.1073/pnas.94.25.13689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wein MN, et al. SIKs control osteocyte responses to parathyroid hormone. Nat Commun. 2016;7:13176. doi: 10.1038/ncomms13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.