

A new approach is developed for generating site-specific antibody-drug conjugates for targeted therapy.
Abstract
Most of the current antibody-drug conjugates (ADCs) in clinic are heterogeneous mixtures. To produce homogeneous ADCs, established procedures often require multiple steps or long reaction times. The introduced mutations or foreign sequences may cause high immunogenicity. Here, we explore a new concept of transforming CD38 enzymatic activity into a facile approach for generating site-specific ADCs. This was achieved through coupling bifunctional antibody-CD38 fusion proteins with designer dinucleotide-based covalent inhibitors with stably attached payloads. The resulting adenosine diphosphate–ribosyl cyclase–enabled ADC (ARC-ADC) with a drug-to-antibody ratio of 2 could be rapidly generated through single-step conjugation. The generated ARC-ADC targeting human epidermal growth factor receptor 2 (HER2) displays excellent stability and potency against HER2-positive breast cancer both in vitro and in vivo. This proof-of-concept study demonstrates a new strategy for production of site-specific ADCs. It may provide a general approach for the development of a novel class of ADCs with potentially enhanced properties.
INTRODUCTION
Antibody-drug conjugates (ADCs) enable targeted delivery of small-molecule drugs, providing significantly improved therapeutic index. Owing to their outstanding potency and selectivity, ADCs hold great promise for the treatment of a variety of human diseases (1–3). Most of the current ADCs in clinical use or development are generated through nonspecific conjugation to surface cysteine or lysine residues, resulting in heterogeneous ADCs with varied drug-to-antibody ratios (DARs) and distinct pharmacological properties (2, 4, 5). ADCs with site-specific conjugations show increased stability, pharmacokinetics, and safety profiles (6). Using engineered amino acids, carbohydrates, or unnatural amino acids, homogeneous ADCs could be generated (7–15). But the conjugation processes often require multiple steps or long reaction times due to inefficient chemistries. Although genetic fusions of peptide motifs or engineered enzymes allow more efficient production of site-specific ADCs (16–22), the introduced mutations or non–human-derived sequences may raise considerable immunogenicity. Moreover, despite successes of several types of linkers for attachments and release of cytotoxic payloads, optimal drug linkers remain limited for applying the ADC modality to non-oncology areas.
CD38, a type II transmembrane protein, belongs to the adenosine diphosphate (ADP)–ribosyl cyclase family. Its extracellular domain catalyzes the formation of cyclic ADP-ribose and ADP-ribose from nicotinamide adenine dinucleotide (NAD+). 2′-F-arabinose nicotinamide mononucleotide (2′-F-araNMN) and 2′-F-araNAD+ are potent covalent inhibitors of CD38 by rapidly forming a stable arabinosyl-ester bond with the catalytic glutamate 226 (E226) residue (23–28). These important findings raise the question of whether CD38 covalent inhibitors could be harnessed for the development of site-specific ADCs. Here, we explore this new concept through design, generation, and characterization of an innovative class of site-specific ADCs, termed as ADP-ribosyl cyclase–enabled ADCs (ARC-ADCs). Using a rationally designed bifunctional antibody-CD38 fusion protein targeting human epidermal growth factor receptor 2 (HER2) coupled with a novel CD38 covalent inhibitor, an anti-HER2 ARC-ADC with a defined DAR of 2 was rapidly generated through single-step conjugation. The resulting anti-HER2 ARC-ADC displays excellent cytotoxicity and specificity for HER2-expressing breast cancer cells and potent antitumor activity in mouse xenograft models. As a proof of concept, ARC-ADCs demonstrate a new strategy for facile production of homogeneous ADCs and may provide a general approach for the development of a novel class of ADCs with potentially improved properties.
RESULTS
To create ARC-ADCs, we used full-length anti-human HER2 antibody Herceptin as a model antibody. On the basis of x-ray structures of Herceptin antigen-binding fragment, immunoglobulin G (IgG) fragment crystallizable (Fc) region, and CD38 extracellular domain (29–31), we reasoned that genetic fusion of CD38 catalytic domain to the light chain N terminus or heavy chain C terminus of Herceptin may result in bifunctional fusion proteins with retained HER2-binding affinity and CD38 enzymatic activity. To this end, a flexible GGS linker was genetically inserted between CD38 catalytic domain and Herceptin light/heavy chain. The designed CD38 fusion proteins designated as CD38 N-fusion IgG and CD38 C-fusion IgG (Fig. 1A) together with Herceptin IgG were expressed in mammalian cells and purified to homogeneity (fig. S1A). The yields are ~12 mg/liter for CD38 N-fusion IgG and 9.5 mg/liter for CD38 C-fusion IgG, slightly lower than that of Herceptin (15 mg/liter). Their binding affinity to HER2 receptor was then examined by enzyme-linked immunosorbent assay (ELISA). Compared with CD38 N-fusion IgG with a half-maximal effective concentration (EC50) of 8.32 ± 0.99 nM for recombinant HER2, CD38 C-fusion IgG displays higher binding affinity (EC50 = 1.02 ± 0.47 nM), comparable to that of Herceptin (EC50 = 0.80 ± 0.10 nM) (Fig. 1B). Thus, CD38 C-fusion IgG was chosen for analysis of its CD38 enzymatic activity. Fluorescence-based activity assays revealed that, in contrast to CD38 catalytic domain, CD38 C-fusion IgG exhibits significantly higher activity for nicotinamide guanine dinucleotide (NGD+) (Fig. 1C). These results indicate that genetic fusion of CD38 catalytic domain to Herceptin has little effects on antibody binding and its enzymatic activity.
Fig. 1. Characterization of CD38-antibody fusions and 2′-Cl-araNAD+-N3.
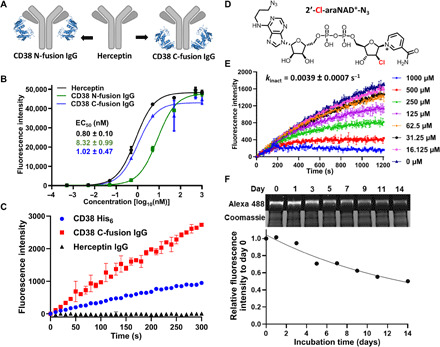
(A) Schematic of designed CD38 N-fusion IgG and CD38 C-fusion IgG. (B) Binding to recombinant HER2 extracellular domain by Herceptin and CD38 N- and C-fusion IgGs as analyzed by ELISA. (C) Enzymatic activity of CD38 catalytic domain, CD38 C-fusion IgG, and Herceptin. CD38-His6 (20 nM), CD38 C-fusion IgG (10 nM), and Herceptin (10 nM) were incubated with NGD+ (100 μM) in PBS. The CD38 cyclase activity was monitored on the basis of the formation of fluorescent cGDPR as measured at 410 nm. (D) Chemical structure of 2′-Cl-araNAD+-N3. (E) Inactivation of CD38 C-fusion IgG by 2′-Cl-araNAD+-N3. CD38 C-fusion IgG (2 nM) was incubated with NGD+ (100 μM) in PBS in the presence of various concentrations of 2′-Cl-araNAD+-N3. The enzymatic activity was measured using cGDPR-based fluorescence assays. (F) Stability of Alexa Fluor 488–conjugated CD38 C-fusion IgG in mouse plasma. Using 2′-Cl-araNAD+-N3, CD38 C-fusion IgG was labeled with Alexa Fluor 488 and incubated in mouse plasma at 37°C for up to 14 days, followed by in-gel fluorescence imaging and Coomassie staining. The quantified fluorescence intensities for intact fusion proteins are shown at the bottom.
To create functionalized CD38 covalent inhibitors as drug linkers for ARC-ADCs, we envisioned that relative to 2′-F-araNAD+–derived analogs, 2′-Cl–substituted NAD+ analogs may act as CD38 covalent inhibitors with improved stability for the arabinosyl-ester bonds formed with CD38 E226 residue. Moreover, structural analysis of CD38 indicated that the adenine moiety of NAD+ is largely solvent-exposed and has limited interactions with surrounding residues (30). In addition, the pyrophosphate diester bond within NAD+ revealed excellent plasma stability and rapid release of payloads upon cellular internalization for ADCs (32). We thus designed and synthesized a novel 2′-Cl-araNAD+ analog with an azido group at N6 of adenine (2′-Cl-araNAD+-N3) (Fig. 1D and fig. S1, B and C). The stereochemistry of the intermediate O-benzoyl protected 2′-Cl-arabinose nicotinamide riboside was determined as β-isomer based on 1H-1H correlation spectroscopy (COSY) and subsequent nuclear Overhauser effect spectroscopy (NOESY) experiments (fig. S1, D to F).
Fluorescence-based activity assays indicated that 2′-Cl-araNAD+-N3 could inactivate CD38 C-fusion IgG with a kinact of 0.0039 ± 0.0007 s−1 (Fig. 1E). To confirm that CD38 C-fusion IgG is covalently labeled at E226 by 2′-Cl-araNAD+-N3, the E226 residue was mutated to glutamine, which was shown to abolish catalytic activity of CD38 (33). CD38 C-fusion IgG, its mutant (E226), and Herceptin were then incubated without and with 2′-Cl-araNAD+-N3, followed by conjugation with Alexa Fluor 488 DBCO via click chemistry. In-gel fluorescence imaging and Coomassie-stained SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gels revealed that only the heavy chain of wild-type CD38 C-fusion IgG was labeled with fluorescent dyes (fig. S2A). These results suggest that E226 of CD38 C-fusion IgG is the conjugation site.
Using Alexa Fluor 488–conjugated CD38 C-fusion IgG, cellular uptake assays were performed. Confocal microscopy indicated that fluorescently labeled CD38 C-fusion IgG could be selectively internalized into HER2-positive HCC1954 cells, whereas HER2-negative MDA-MB-468 cells showed no detectable uptake (fig. S2B). Notably, in-gel fluorescence imaging revealed that a substantial portion of Alexa Fluor 488–conjugated CD38 C-fusion IgG remained intact after 14-day incubation in mouse plasma at 37°C (Fig. 1F and fig. S3), supporting excellent stability for the 2′-Cl-araNAD+-N3–based linker.
Next, the 2′-Cl-araNAD+-N3 linker was attached with a model payload, tubulin inhibitor monomethyl auristatin F (MMAF), through click chemistry (Fig. 2A and fig. S4, A and B). The resulting 2′-Cl-araNAD+–MMAF conjugate was incubated with CD38 C-fusion IgG at a molar ratio of 100 for various amounts of time. On the basis of residual enzymatic activity, 2′-Cl-araNAD+–MMAF could rapidly inactivate CD38 C-fusion IgG with a kobs of 0.140 ± 0.046 min−1 (Fig. 2B). Nearly more than 90% of initial CD38 activity was lost after the 20-min incubation. The anti-HER2 ARC-ADC was then prepared by incubating CD38 C-fusion IgG with the 2′-Cl-araNAD+–MMAF (molar ratio, 1:100) for overnight on ice, followed by removal of the free drug-linker conjugate. Mass spectrometry (MS) analysis of CD38 C-fusion IgG and ARC-ADC indicated no mass shifts for the light chain of ARC-ADC but an increase of 1410 Da for the heavy chain of ARC-ADC, matching the calculated addition for a 2′-Cl-arabinose–ADP–MMAF moiety (Fig. 2, C and D). No unmodified heavy chain was observed for ARC-ADC, consistent with its undetectable CD38 enzymatic activity. These results demonstrate the facile production of ARC-ADC with a defined DAR of 2. Furthermore, the CD38 C-fusion IgG E226Q mutant was incubated with 2′-Cl-araNAD+–MMAF (molar ratio, 1:100) for overnight on ice. MS indicated that no mass shifts for both light and heavy chains of the CD38 C-fusion IgG E226Q mutant underwent conjugation treatment (fig. S4, C to F). Liquid chromatography–MS (LC-MS) analysis of CD38 C-fusion IgG and anti-HER2 ARC-ADC treated by trypsin revealed a unique peptide for ARC-ADC, which matches an E226-containing peptide attached with 2′-Cl-arabinose–ADP–MMAF (fig. S4G). Moreover, we crystallized human CD38 catalytic domain with covalently attached 2′-Cl-araNAD+. The solved high-resolution (1.5 Å) x-ray structure revealed that 2′-Cl-arabinose–ADP forms an arabinosyl-ester bond with E226 residue at the active site of CD38 (Fig. 2E and table S1). Together with the fluorescent dye conjugation analysis (fig. S2A), these results support catalytic E226 residue as the conjugation site for ARC-ADC.
Fig. 2. Generation and in vitro evaluation of anti-HER2 ARC-ADC.
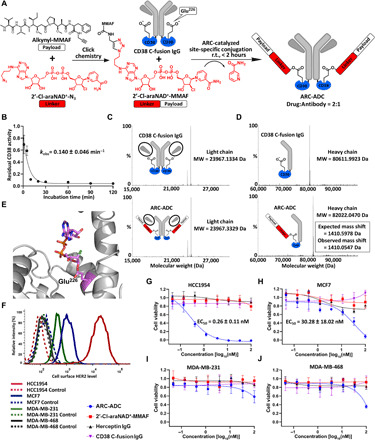
(A) Scheme for generating anti-HER2 ARC-ADC. r.t., room temperature. (B) Conjugation kinetics of drug linker to CD38 C-fusion IgG. 2′-Cl-araNAD+–MMAF (1 mM) was incubated with CD38 C-fusion IgG (10 μM) in 50 mM tris buffer (pH 8.5) for various amounts of time on ice. The residual enzymatic activity determined by fluorescence-based activity assays was plotted as a function of incubation time. (C and D) Mass spectra of light chains (C) and heavy chains (D) for CD38 C-fusion IgG and anti-HER2 ARC-ADC. MW, molecular weight. (E) X-ray structure of human CD38 catalytic domain with 2′-Cl-araNAD+ covalently attached to Glu226 residue. (F) Flow cytometric analysis of HER2 expression for four breast cancer cell lines. (G to J) In vitro cytotoxicity of anti-HER2 ARC-ADC. HCC1954 (G), MCF7 (H), MDA-MB-231 (I), and MDA-MB-468 (J) cells with varied levels of HER2 expression were incubated for 72 hours at 37°C with 5% CO2 in the presence of various concentrations of ARC-ADC, 2′-Cl-araNAD+–MMAF, Herceptin, and CD38 C-fusion IgG. Cell viability was measured by MTT assays. Cells treated with culture media and 5 μM paclitaxel were included as 100% viability and 0% viability controls, respectively.
In vitro cytotoxicity was then evaluated for the anti-HER2 ARC-ADC using four breast cancer cell lines with various levels of HER2 expression (Fig. 2F). The ARC-ADC could potently suppress proliferation of HCC1954 cells with an EC50 of 0.26 ± 0.11 nM. Its cytotoxicity positively correlates with levels of HER2 expression (Fig. 2, G to J). In contrast, 2′-Cl-araNAD+–MMAF, CD38 C-fusion IgG, and Herceptin displayed no cytotoxicity for these cell lines under the same conditions. These results indicate excellent in vitro potency and specificity for the anti-HER2 ARC-ADC. In addition, anti-HER2 ARC-ADC was incubated with mouse plasma for 3 days at 37°C. There was no significant difference (P = 0.5187 for HCC1954 cells and P = 0.5198 for MDA-MB-468 cells) in cytotoxicity for fresh and plasma-incubated ARC-ADCs (fig. S5). These results are consistent with plasma stability of Alexa Fluor 488–conjugated CD38 C-fusion IgG (Fig. 1F) and support high stability of 2′-Cl-araNAD+-N3 linker and its mediated covalent attachments to CD38 C-fusion IgG.
To investigate payload release of ARC-ADC upon cellular internalization, we first incubated 2′-Cl-araNAD+–MMAF with rat liver lysosomal lysates at 37°C for various amounts of time. On the basis of high-performance liquid chromatography (HPLC) retention times with reference to synthesized standards and MS analysis (fig. S6), 2′-Cl-araNAD+–MMAF could be rapidly degraded into 6-adenosine-MMAF in the lysosomal environment. Subsequent treatment of the lysosomal reaction mixture by HCC1954 cell lysates led to full conversion into 6-adenine-MMAF as revealed by HPLC and MS analysis (fig. S7). These results suggest that 6-adenine-MMAF may be the major form of MMAF released from anti-HER2 ARC-ADC inside target cells.
Pharmacokinetics of CD38 C-fusion IgG was then examined in mice. Two sandwich ELISAs using different detection antibodies revealed comparable half-lives (37.41 ± 16.63 hours by anti-κ light chain and 33.14 ± 19.49 hours by anti-CD38) for intravenously administered CD38 C-fusion IgG in mice (Fig. 3A). Next, in vivo biodistribution and efficacy were evaluated for the anti-HER2 ARC-ADC using NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice bearing tumors derived from HER2-expressing HCC1954 cells. Near-infrared fluorescence–based imaging revealed the implanted human tumors as major locations for IRDye-labeled anti-HER2 ARC-ADC post-intravenous injections (Fig. 3B and fig. S8). In contrast to the phosphate-buffered saline (PBS)–treated group with rapidly growing tumors, mice treated with anti-HER2 ARC-ADC showed tumor shrinkage upon treatment initiation and significant inhibition of tumor growth (Fig. 3C). These results demonstrate excellent in vivo efficacy of the anti-HER2 ARC-ADC for established tumors in mice. No loss of body weight or major organ weights or overt toxicity was observed for both groups during the study (Fig. 3, D and E). In comparison to the median survival of PBS-treated group (35 days), the median survival of ARC-ADC–treated group (74 days) was increased by more than 110% (Fig. 3F).
Fig. 3. Pharmacokinetics and pharmacodynamics in mice.
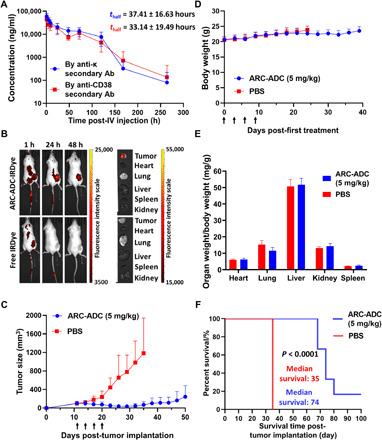
(A) Pharmacokinetics of CD38 C-fusion IgG in mice. A single dose (3 mg/kg) of CD38 C-fusion IgG was administered by intravenous (IV) injection into CD-1 mice (n = 5). Plasma concentrations of CD38 C-fusion IgG were determined by two sandwich ELISAs using the same capture antibody (Ab) [anti-human IgG (H+L)] but different detection antibodies (anti-κ light chain or anti-CD38). (B) Biodistribution of anti-HER2 ARC-ADC in mice. HCC1954 cells were subcutaneously implanted into the flank of female NSG mice. IRDye-labeled anti-HER2 ARC-ADC (5 mg/kg) or free IRDye at the same molar concentration was administered intravenously through tail vein 1 week after tumor implantation. Mice were then imaged at 1, 24, and 48 hours after injection, followed by euthanasia and imaging of harvested tumors and major organs. (C) In vivo efficacy of anti-HER2 ARC-ADC. HCC1954 cells were subcutaneously implanted into the flank of female NSG mice. Once the tumor sizes reached 100 mm3, mice (n = 6) were treated with PBS or ARC-ADC (5 mg/kg) by intravenous injection (black arrows) every 3 days for a total of four times. (D) Body weights of mice during the in vivo efficacy study. (E) Ratios of major organ weight to body weight of mice at the end of in vivo efficacy study. (F) Kaplan-Meier survival curve for PBS- and ARC-ADC–treated groups.
DISCUSSION
This study demonstrates the concept of transforming CD38 and its covalent inhibitors into a facile, single-step approach for generation of site-specific ADCs. It was achieved through coupling bifunctional antibody-CD38 fusion proteins with the designer covalent inhibitors with stably attached payloads. It may provide a general approach for production of homogeneous ADCs with defined DARs and can be extended to generate a variety of ADCs with distinct targeting antibodies and payloads. In addition, the success of ARC-ADC supports extension of CD38 fusion to other peptides and proteins for site-specific conjugations for biomedical applications, similar to other enzymatic conjugation strategies like Halo-tag and CLIP-tag (34, 35).
Four distinct linker designs have been established for current ADCs with cytotoxic payloads, including disulfide bonds (reduction by thiol groups), hydrazones (cleavage at acidic pH), cathepsin B cleavable dipeptides, and noncleavable linkers. Optimal linkers are still limited for generating ADCs with noncytotoxic payloads. The 2′-Cl-araNAD+-N3 linker displays considerable stability and efficiency for payload release, which provides a new linker design for the development of ADCs with potentially improved physicochemical and pharmacological properties through increasing solubility, tunability, and efficiency for the release of payloads. The 2′-Cl-araNAD+-N3 linker may add new and more effective strategies for the development of ADCs with noncytotoxic payloads for non-oncological indications.
In addition, this study used a native human CD38 enzyme for generating site-specific ADCs. In silico prediction of immunogenic peptides of CD38 C-fusion IgG heavy chain was carried out by using the NetMHCIIpan 3.2 server (36) and selecting common subtypes of human major histocompatibility complex (MHC) class II among major populations. No peptides within the junction region of CD38 C-fusion IgG heavy chain were predicted to have high binding affinity to the selected representative MHC class II molecules (table S2). In addition to its excellent catalytic efficiency for rapid and single-step conjugation, the incorporation of catalytic domain of human CD38 may prevent immunogenicity that could possibly occur for mutated, engineered, or non–human-derived therapeutic proteins. This CD38-based site-specific ADCs may lay the foundation for the development of innovative ADCs with improved efficacy and safety.
While this proof-of-concept study revealed excellent therapeutic efficacy and lack of apparent toxicity for ARC-ADCs in rodents, further detailed in vivo studies are needed to demonstrate superiority and sufficient biosafety of ARC-ADCs by including additional control groups such as 2′-Cl-araNAD+–MMAF, Herceptin, CD38 C-fusion IgG, and an approved ADC (e.g., ado-trastuzumab emtansine) for evaluation of antitumor efficacy, toxicity to major organs and tissues, and safety profiles. In addition, future studies include 2′-Cl-araNAD+-N3 linker–mediated payload release, in vivo mechanisms of action and immunogenicity of ARC-ADCs, and generation of ARC-ADCs targeting other diseases-associated antigens. The payload release of ARC-ADC in lysosomal and cytosol environments needs to be quantitatively analyzed. Further mechanistic studies are required for understanding their action mechanisms. The immunogenicity of ARC-ADCs could be examined using surrogate mouse fusion proteins in immunocompetent mice. Human CD38 catalytic domain can be genetically fused with light or heavy chain of humanized antibodies specific for antigens associated with oncologic or non-oncological indications. In addition to MMAF, other cytotoxic or noncytotoxic payloads can be attached with 2′-Cl-araNAD+-N3 for generation of site-specific ARC-ADCs.
In conclusion, genetically fused CD38 together with 2′-Cl-araNAD+-N3 covalent inhibitor enables facile production of site-specific ADCs. The resulting anti-HER2 ARC-ADC exhibits excellent stability and efficacy against HER2-positive breast cancer both in vitro and in vivo. This study demonstrates a new approach for generation of site-specific ADCs, which may lead to the development of a novel class of ADCs with potentially enhanced properties for fighting various human diseases.
MATERIALS AND METHODS
Materials
Synthetic gBlocks DNA fragments and oligonucleotides were purchased from Integrated DNA Technologies (IA, USA). AccuPrime Pfx DNA Polymerase kit (12344024), electroporation cuvettes plus (FB101), Expi293F cells (A14527), Opti-MEM I reduced serum medium (31985070), Ni–nitrilotriacetic acid (NTA) resin (88221), goat anti-human κ light chain secondary antibody with horseradish peroxidase (HRP) (18853), anti-CD38 monoclonal antibody (clone: HIT2) with biotin (13-0389-82), QuantaBlu fluorogenic peroxidase substrate kit (15169), fetal bovine serum (FBS) (26140079), 0.25% trypsin-EDTA with phenol red (25200056), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent (M6494) were purchased from Thermo Fisher Scientific (MA , USA). Anti-human IgG (H+L) antibody (50-668-06) and 4% paraformaldehyde solution in PBS (AAJ19943K2) were purchased from Fisher Scientific (NH, USA). Eco RI DNA restriction enzyme (R3101S), Nhe I DNA restriction enzyme (R3131S), and T4 DNA ligase (M0202S) were purchased from New England Biolabs (MA, USA). Zymoclean gel DNA recovery kit (D4001) was purchased from Zymo Research (CA, USA). Zeocin solution (ant-zn-05) was purchased from InvivoGen (CA, USA). Protein G resin (L00209) and exendin-4 were purchased from GenScript (NJ, USA). Rat liver tritosomes were purchased from XenoTech (KS, USA). PEI MAX transfection grade linear polyethylenimine hydrochloride (24765-1) was purchased from Polysciences Inc. (PA, USA). BalanCD HEK293 medium (91165) was purchased from Irvine Scientific (CA, USA). NGD+ (sc-215563) was purchased from Santa Cruz Biotechnology (TX, USA). Alexa Fluor dye 488 DBCO conjugation reagent (1278-1) was purchased from Click Chemistry Tools (AZ, USA). IRDye 800CW was purchased from LI-COR Biosciences (NE, USA). Tissue culture–treated vented flasks (250 ml) (25-209) were purchased from Genesee Scientific (CA, USA). Multivette 600 LH-Gel (15.1675) for plasma preparation was purchased from SARSTEDT Group (Nümbrecht, Germany). Care touch twist top lancets (30 gauge) for blood sample collection (160104T) were purchased from Care Touch (NY, USA). QuikChange II site-directed mutagenesis kit (200521) was purchased from Agilent Technologies (CA, USA). Black 96-well microplates (655209) and black 96-well microplates with high-binding base (655077) were purchased from Greiner Bio-One (Kremsmünster, Austria). Recombinant human ErbB2/Her2-Fc chimera protein (1129-ER-050) and streptavidin-HRP (DY998) were purchased from R&D Systems (MN, USA). Amicon ultra-15 centrifugal filter units with 10-kDa protein size cutoff (UFC901024) and 30-kDa protein size cutoff (UFC903024) and bovine serum albumin (BSA) (1265925GM) were purchased from MilliporeSigma (MA, USA). Dulbecco’s modified Eagle’s medium (DMEM) (10-017-CV), RPMI 1640 medium (10-040-CV), Dulbecco’s PBS (DPBS) (21-031-CM), and Corning Matrigel matrix (354248) were purchased from Corning Inc. (NY, USA). Erlenmeyer flasks (500 ml) (89095-278), Fluoromount mounting reagent (99990-086), round cover slips (89167-106), U-100 BD micro-fine IV insulin syringes (BD-329424), tissue-culture (TC)–treated 96-well plates (10062-900), and TC-treated 24-well plates (10062-896) were purchased from VWR International (PA, USA).
Cell lines
Breast cancer cell lines HCC1954, MCF7, MDA-MB-231, and MDA-MB-468 were purchased from the American Type Culture Collection (VA, USA).
Chemical synthesis and characterization
The experimental details and results for chemical synthesis of 2′-Cl-araNAD+-N3, alkynyl-MMAF, 2′-Cl-araNAD+–MMAF, 6-MMAF-AMP, 6-MMAF-adenosine, and 6-MMAF-adenine are provided in the Supplementary Materials.
Molecular cloning
Synthetic DNA encoding human CD38 extracellular domain (R45-I300) with four mutated asparagine residues (N100D, N164A, N129D, and N209D) for the removal of N-glycosylation was purchased from Integrated DNA Technologies (IA, USA). The encoded protein sequence is RWRQQWSGPGTTKRFPETVLARCVKYTEIHPEMRHVDCQSVWDAFKGAFISKHPCDITEEDYQPLMKLGTQTVPCNKILLWSRIKDLAHQFTQVQRDMFTLEDTLLGYLADDLTWCGEFATSKINYQSCPDWRKDCSNNPVSVFWKTVSRRFAEAACDVVHVMLDGSRSKIFDKDSTFGSVEVHNLQPEKVQTLEAWVIHGGREDSRDLCQDPTIKELESIISKRNIQFSCKNIYRPDKFLQCVKNPEDSSCTSEI.
The overlap extension polymerase chain reaction (PCR) was adopted to generate DNA fragments encoding (i) the light chain of CD38 N-fusion IgG (the extracellular domain of CD38 fused to the N terminus of Herceptin light chain with a GGS linker between two domains) and (ii) the heavy chain of CD38 C-fusion IgG (the extracellular domain of CD38 fused to the C terminus of Herceptin heavy chain with a GGS linker between two domains). Primers for amplifying DNA fragments and conducting the overlap extension PCRs are listed below.
Primers for amplifying CD38 DNA for constructing CD38 N-fusion IgG light chain were as follows: forward, 5′-TCACGAATTCGAGATGGAGGCAACAATGGTCAGG-3′; reverse, 5′-CGCCACCCCCGATCTCACTAGTACATGAACTATCCTCTGGG-3′. Primers for amplifying Herceptin light chain DNA for constructing CD38 N-fusion IgG light chain were as follows: forward, 5′-TGAGATCGGGGGTGGCGGAAGCGACATCCAGATGACCCAGTCTCC-3′; reverse, 5′-CAGCTAGCACTTATCAACACTCTCCC-3′. Primers for amplifying CD38 DNA for constructing CD38 C-fusion IgG heavy chain were as follows: forward, 5′-GGGGGTGGCGGAAGCAGATGGAGGCAACAATGGTCAGG-3′; reverse, 5′-CCAGCTAGCACTTATCAGATCTCACTAGTACATGAACTATCCTCTGGGTT-3′. Primers for amplifying Herceptin heavy chain DNA for constructing CD38 C-fusion IgG heavy chain were as follows: forward, 5′-CACGAATTCGGAGGTGCAGCTG-3′; reverse, 5′-GCTTCCGCCACCCCCTTTACCCGGAGACAGGGAGAGG-3′.
Bands of the generated DNA fragments for each fusion protein were excised from agarose gels and purified with DNA extraction kits (Zymo Research, CA). Cleaned DNA fragments were processed by DNA restriction enzymes, Nhe I and Eco RI (New England Biolabs, MA), followed by ligation into empty pFUSE vector backbone by DNA T4 ligase (New England Biolabs, MA). Last, ligation products were used to transform DH10B Escherichia coli electrocompetent cells and positive colonies selected from zeocin resistance were picked for DNA sequencing to confirm the designed fusion proteins in the pFUSE expression vectors.
Molecular cloning of CD38 C-fusion IgG E226Q mutant
The generated pFUSE expression vector for CD38 C-fusion IgG heavy chain was used as a template for constructing the pFUSE expression vector for CD38 C-fusion IgG heavy chain E226Q mutant. Site-directed mutagenesis was performed with QuikChange II site-directed mutagenesis kit (Agilent Technologies, CA) per the manufacturer’s instructions using primers listed as follows: forward, 5′-TTCGGAAGTGTTCAGGTACATAACCTCCAACCCGAAAAAGTGC-3′; reverse, 5′-GGAGGTTATGTACCTGAACACTTCCGAAGGTGGAATCTTTATCGA-3′. Upon electroporation with DH10B electrocompetent cells, zeocin-based selection (InvivoGen, CA) was carried out and the identified positive colonies were picked for DNA sequencing to confirm the designed CD38 C-fusion IgG heavy chain E226Q mutant in the pFUSE expression vectors.
Mammalian cell expression and purification of antibodies and antibody fusions
Herceptin IgG, CD38 N-fusion IgG, CD38 C-fusion IgG, and CD38 C-fusion IgG E226Q mutant were expressed and purified by procedures described below. pFUSE expression vectors for antibody light chain (120 μg) and heavy chain (240 μg) in 12 ml of Opti-MEM medium (Thermo Fisher Scientific, MA) were mixed with 960 μl of transfection grade linear polyethylenimine hydrochloride (1 mg/ml) (Polysciences, PA) for the transfection of 240 ml of Expi293 cells (Thermo Fisher Scientific, MA) cultured at the density of 2.5 million ml−1 per the manufacturer’s instructions. After a 6-day incubation on an orbital shaker (125 rpm) in a 37°C incubator with 5% CO2, secreted antibodies or fusion antibodies in the culture media were collected and purified by protein G affinity chromatography (GenScript, NJ) per the manufacturer’s instructions. Eluted proteins by 100 mM glycine (pH 2.7) were dialyzed against PBS buffer (pH 7.4) and concentrated with 30-kDa filters (MilliporeSigma, MA). The purified proteins were examined by SDS-PAGE stained with Coomassie blue. Protein concentrations were determined using a NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific, MA) with their respective calculated molar extinction coefficients.
The pFUSE expression vector for recombinant CD38 extracellular domain with the C-terminal 6-histidine tag was constructed as previously described (30). Briefly, cell culture media with recombinant CD38 was first dialyzed against storage buffer (25 mM Hepes and 250 mM NaCl, pH 7.5) before purification by Ni-NTA resins (Thermo Fisher Scientific, MA). Dialyzed media were gradually passed through the Ni-NTA resins in a gravity flow column twice, followed by washing with 15 column volumes of wash buffer [20 mM tris-HCl (pH 8.0), 200 mM NaCl, and 30 mM imidazole]. Recombinant CD38-His6 were then eluted in 15 column volumes of elution buffer [20 mM tris-HCl (pH 8.0), 200 mM NaCl, and 400 mM imidazole] and were dialyzed into PBS buffer (pH 7.4) for overnight at 4°C and then in the same fresh buffer for another 8 hours under the same conditions. Last, recombinant CD38-His6 was concentrated with 10-kDa cutoff filters (MilliporeSigma, MA). The purified proteins were examined by SDS-PAGE stained with Coomassie blue. Protein concentrations were determined using a NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific, MA) with the calculated molar extinction coefficient.
Binding to recombinant HER2-Fc fusion by ELISA
Recombinant HER2-Fc fusion protein (R&D Systems, MN) (50 μg ml−1) was coated overnight on 96-well ELISA plates at room temperature (Greiner Bio-One, NC) in 80 μl of PBS buffer (pH 7.4). After the coated wells were blocked with 3% BSA (MilliporeSigma, MA) in PBS buffer (pH 7.4) for 2 hours at room temperature followed by three times wash with 200 μl of 0.1% phosphate-buffered saline with Tween-20 (PBST) solution, CD38 N-fusion IgG, CD38 C-fusion IgG, and Herceptin in PBS buffer (pH 7.4) were applied to the wells at various final concentrations (0.0005, 0.005, 0.05, 0.5, 5, 50, 500, and 1000 nM) and incubated at room temperature for 1 hour. After three times wash by PBST, 80 μl of anti-human IgG κ antibody HRP conjugates (Thermo Fisher Scientific, MA) was applied and incubated at room temperature for 1 hour before the three times wash by PBST and subsequent additions of 80 μl of QuantaBlu fluorogenic substrates (Thermo Fisher Scientific, MA). The fluorescence intensities (excitation at 325 nm and emission at 420 nm) were recorded with a Synergy H1 plate reader (BioTek, VT) after a 5-min incubation at room temperature. The sigmoidal dose-response function in GraphPad Prism (GraphPad, CA) was adopted to calculate the EC50 for different constructs against the recombinant HER2 extracellular domain.
Enzymatic activities of CD38 and CD38 IgG fusions
NGD+ (Sigma-Aldrich, MO) was used as a substrate to determine ADP-ribosyl cyclase activity of purified CD38 and CD38 C-fusion IgG. Cyclic guanosine diphosphate–ribose (cGDPR) formed from CD38 cyclase activity is characterized by fluorescence emission at 410 nm (excitation at 300 nm). Reactions were initiated by additions of purified CD38 (20 nM), CD38 C-fusion IgG (10 nM), or Herceptin IgG (10 nM) into assay wells with 100 μM NGD+ in PBS buffer (pH 7.4), followed by monitoring the reactions through fluorescence at 410 nm (excitation at 300 nm) for 5 min using a Synergy H1 plate reader (BioTek, VT).
Inhibition activities of 2′-Cl-araNAD+-N3 for CD38 C-fusion IgG
Various concentrations of 2′-Cl-araNAD+-N3 were incubated with 2 nM CD38 C-fusion IgG and 100 μM NGD+ for 20 min in 50 mM tris buffer (pH 8.5). The formation of cGDPR was monitored with a Synergy H1 plate reader (BioTek, VT) under the kinetic mode with excitation wavelength set at 300 nm and emission wavelength at 410 nm. The initial velocities (vinitial) of cGDPR generation in the presence of different concentrations of 2′-Cl-araNAD+-N3 were used to determine covalent inactivation constant (kinact) by a two-step regression model as follows: [P] = vinitial(1 − exp(−kobst))/kobs and kobs = kinact[I]/([I] + KI), where [P] is the concentration of product formed at given time points, vinitial is the initial reaction rate, kobs is the rate constant, kinact is the inactivation rate constant for the covalent inhibitor, [I] is the concentration of covalent inhibitor, and KI is the concentration of covalent inhibitor giving rise to an inactivation rate equal to half of kinact.
Fluorescent dye conjugation for CD38 C-fusion IgGs and in-gel fluorescence analysis
CD38 C-fusion IgG, CD38 C-fusion IgG E226Q mutant, and Herceptin IgG (2 μM) were first incubated in the absence or presence of 40 μM 2′-Cl-araNAD+-N3 in 50 mM tris buffer (pH 8.5) for 2 hours on ice. The reaction mixtures were then added with Alexa Fluor 488 DBCO (Click Chemistry Tools, AZ) at final concentrations of 200 μM and kept on ice for 0.5 hour before SDS-PAGE analysis. The SDS-PAGE gels were first imaged with ChemiDoc Touch (Bio-Rad, CA) imager for the fluorescence of Alexa Fluor 488 and then stained with Coomassie blue for white-light imaging.
Plasma stability of Alexa Fluor 488–conjugated CD38 C-fusion IgG and fluorescein-labeled exendin-4
To prepare CD38 C-fusion IgG conjugated with Alexa Fluor 488, 5 μM CD38 C-fusion IgG was incubated with 500 μM 2′-Cl-araNAD+-N3 in 50 mM tris buffer (pH 8.5) on ice for 2 hours, followed by buffer exchange with 30-kDa size cutoff filters to remove free 2′-Cl-araNAD+-N3. The resulting CD38 C-fusion IgG labeled with 2′-Cl-arabinose-ADP-N3 was then mixed with Alexa Fluor 488 DBCO (Click Chemistry Tools, AZ) at a molar ratio of 1:50 and incubated on ice for 30 min before buffer exchange for removing free Alexa Fluor 488 DBCO.
To prepare exendin-4 labeled by N-hydroxysuccinimide (NHS)–fluorescein (Thermo Fisher Scientific, MA), exendin-4 (3 mg/ml) was incubated with 1 mM NHS-fluorescein in PBS buffer on ice for 2 hours, followed by passing through Zeba spin desalting columns (7K MWCO; Thermo Fisher Scientific, MA) per the manufacturer’s instruction.
The generated CD38 C-fusion IgG conjugated with 2′-Cl-arabinose–ADP–Alexa 488 and fluorescein-labeled exendin-4 were mixed with fresh CD-1 mouse plasma or PBS at a final concentration of 3 μM for Alexa Fluor 488–conjugated CD38 C-fusion IgG and 0.75 mg/ml for fluorescein-labeled exendin-4 in the presence of penicillin and streptomycin (100 μg/ml) and placed at 37°C in an incubator with 5% CO2 for up to 14 days with fixed amounts of mixtures sampled and frozen at various time points for SDS-PAGE gel analysis at the end of study. SDS-PAGE gels were first imaged using a ChemiDoc Touch (Bio-Rad, CA) imager for the Alexa Fluor 488 or fluorescein signals and then stained with Coomassie blue for white-light imaging. The fluorescence intensities of the protein bands representing intact CD38 C-fusion IgG–2′-Cl-arabinose–ADP–Alexa 488 or fluorescein-labeled exendin-4 were quantified with Image Lab (Bio-Rad, CA).
Cellular uptake assays for Alexa Fluor 488–conjugated CD38 C-fusion IgG
The CD38 C-fusion IgG–2′-Cl-arabinose–ADP–Alexa 488 was prepared as described above in the plasma stability section. HCC1954 cells were cultured in RPMI 1640 medium (Corning, NY) supplemented with 10% FBS, and MDA-MB-468 cells were cultured in DMEM medium (Corning, NY) supplemented with 10% FBS. Both cell lines were kept in a 37°C incubator with 5% CO2. First, 1 × 104 HCC1954 or MDA-MB-468 cells at passage 3 were seeded onto 12-mm round cover slides (VWR, PA) in 24-well plates (VWR, PA) for overnight in the incubator. Culture media were removed next day, followed by three times wash with DPBS (Corning, NY). DPBS (200 μl) containing 200 nM CD38 C-fusion IgG–2′-Cl-arabinose–ADP–Alexa 488 was then added for incubation with cells for 2 hours in a 37°C incubator with 5% CO2 or on ice. As negative controls, both cell lines in DPBS only were also incubated under two conditions mentioned above. Following the incubation, DPBS with CD38 C-fusion IgG–2′-Cl-arabinose–ADP–Alexa 488 was decanted, washed three times with ice-cold DPBS, and fixed by ice-cold 4% paraformaldehyde for 20 min on ice. After the fixation and three times washing with DPBS, the cells were incubated with 0.1% Triton X-100 (MilliporeSigma, MA) for 10 min at room temperature for permeabilization. Last, permeabilized cells washed by DPBS were stained for nuclei with 300 nM 4′,6-diamidino-2-phenylindole (DAPI) water solution (Thermo Fisher Scientific, MA) for 20 min at room temperature and then sealed into slides for confocal imaging using a Leica SP8 confocal laser scanning microscope (Leica, Germany) equipped with a 40×/1.3 oil immersion objective lens (HC PL APO 40×/1.30 Oil CS2). DAPI and Alexa 488 were excited at 405 and 488 nm, respectively. Images were processed with Leica Application Suite X (LAS X) software (Leica, Germany).
Conjugation of 2′-Cl-araNAD+–MMAF to CD38 C-fusion IgG
To determine the conjugation kinetics, 1 mM 2′-Cl-araNAD+–MMAF was incubated with 10 μM CD38 C-fusion IgG in 50 mM tris buffer (pH 8.5) for different amounts of periods (1, 5, 10, 20, 30, 60, 90, and 120 min) on ice. Then, 1 μl of the mixtures was transferred into 100 μl of PBS buffer (pH 7.4) containing 100 μM NGD+ on 96-well plates. The initial rates of formation of cGDPR based on fluorescence intensity measured at 410 nm with a Synergy H1 plate reader (BioTek, VT) under kinetic mode were used to define the residual CD38 catalytic activities. The 100% CD38 enzymatic activities were determined using 1 μl of 10 μM CD38 C-fusion IgG in 50 mM tris buffer (pH 8.5) incubated for 120 min on ice. Data points were fit to the first-order decay function A(t) = A0e(−kobst), where A0 is 100% activity, A(t) is activity measured at a given time t, and kobs is the rate constant.
One-step preparation of ARC-ADC
CD38 C-fusion IgG (5 μM) and 500 μM 2′-Cl-araNAD+–MMAF were incubated in 50 mM tris buffer (pH 8.5) for overnight on ice, followed by buffer exchange using 30-kDa size cutoff filters (MilliporeSigma, MA) to remove free 2′-Cl-araNAD+–MMAF.
Mass spectroscopy study for conjugation of CD38 C-fusion IgG by 2′-Cl-araNAD+–MMAF
All protein samples were diluted to 0.2 mg/ml in 50 mM ammonium bicarbonate (pH 8.0). Glycans were removed using 1:100 (w/w) PNGaseF (New England Biolabs, MA) at 37°C overnight. The intermolecular disulfide bonds were cleaved using 0.1 M dithiothreitol for 5 min at room temperature. Samples (4 μl) were injected onto the Xevo using the method below.
Intact protein samples were analyzed by LC-MS [ACQUITY ultra-performance liquid chromatography (UPLC) H-class system, Xevo G2-XS QTOF, Waters Corporation]. Proteins were separated away from reaction buffer salts using a phenyl guard column (ACQUITY UPLC BEH Phenyl VanGuard Pre-column, 130 Å, 1.7 μm, 2.1 mm × 5 mm, Waters Corporation). The 5-min method used a flow rate (0.2 ml/min) of a gradient of buffer A consisting of 0.1% formic acid in water (water LC-MS #9831-02, JT Baker; formic acid LC-MS #85178, Thermo Fisher Scientific) and buffer B, acetonitrile (acetonitrile UHPLC/MS #A956, Thermo Fisher Scientific). The gradient running program (flow rate set at 0.2 ml/min and curve set as 6) is as follows: maintaining 100% buffer A from 0 to 30 s, adjusting to 10% buffer A and 90% buffer B from 30 to 120 s as gradient, maintaining 10% buffer A and 90% buffer B from 120 to 150 s, reaching to 100% buffer A from 150 to 240 s as a gradient, and maintaining 100% buffer A from 240 to 300 s.
The Xevo Z-spray source was operated in positive MS resolution mode with a capillary voltage of 3000 V and a cone voltage of 40 V (NaCsI calibration, Leu-enkephalin lock-mass). Nitrogen was used as the desolvation gas and a total flow of 800 liter hour−1. Total average mass spectra were reconstructed from the charge state ion series using the MaxEnt1 algorithm from Waters MassLynx software V4.1 SCN949 according to the manufacturer’s instructions. To obtain the ion series described, the major peaks of the chromatogram were selected for integration before further analysis.
Mass spectroscopy study for conjugation site of 2′-Cl-araNAD+–MMAF with CD38 C-fusion IgG
The CD38 C-fusion IgG and anti-HER2 ARC-ADC were subjected to overnight solution digestion with trypsin gold (Promega) at 37°C and pH 8.5. All LC-MS/MS experiments were performed using the same Waters Xevo Q-TOF setup and buffer system as the intact protein analysis described above. Separation of peptides was performed by reversed-phase chromatography using a reversed-phase C4 column (ACQUITY UPLC Protein BEH C4 Column, 300 Å, 1.7 μm, 2.1 mm × 50 mm, Waters Corporation). The gradient running program (flow rate set at 0.3 ml/min and curve set as 6) is as follows: maintaining 97% buffer A and 3% buffer B from 0 to 30 s, reaching to 40% buffer A and 60% buffer B from 30 to 120 s as a gradient, maintaining 40% buffer A and 60% buffer B from 120 to 150 s, reaching to 10% buffer A and 90% buffer B from 150 to 180 s as a gradient, maintaining 10% buffer A and 90% buffer B from 180 to 210 s, reaching to 97% buffer A and 3% buffer B from 210 to 240 s as a gradient, and maintaining 97% buffer A and 3% buffer B from 240 to 300 s.
Peptide data were acquired after a 0.5-min waste diversion to remove buffer salts. The Xevo Z-spray source was operated with a capillary voltage of 3000 V and a cone voltage of 40 V (NaCsI calibration, Leu-enkephalin lock-mass). Nitrogen was used as the desolvation gas and a total flow of 800 liters hour−1. Data were acquired across the 100- to 2000-Da range in MSe continuum mode with alternating 0.5-s scans at alternating collision energy, low energy, 0 V, and a high collision energy ramp (15 to 45 V). The high energy data correspond to the MS2 secondary fragmentation of the low energy scans of a similar time. The data were processed for exact mass peptide matching and b&y ion fragmentation confirmation using BiopharmaLynx software (Version 1.3.5 Waters Corporation). The software generates in silico peptide digestion data including all potential modifications and maps the real data at 30 ppm (parts per million) error to the theoretically calculated masses.
X-ray crystallography of 2′-Cl-araNAD+–human CD38 complex
Recombinant human CD38 catalytic domain was prepared as previously reported (30) and then incubated with 2′-Cl-araNAD+ at a molar ratio of 1:100 on ice for overnight. The covalent protein complex was purified by gel filtration column and kept in 15 mM Hepes (pH 7.5) and 50 mM NaCl at a concentration of 160 μM. Crystallization was performed at 22°C using vapor diffusion method in a hanging drop manner with a 1:1 ratio of 1 μl of the reservoir solution and 1 μl of the protein solution. Initial crystals grew under similar condition as previously published work (30). The optimized condition for the crystal growth resulting in well diffracting crystals was shown to be 100 mM Hepes (pH 7.0) and 100 mM potassium phosphate dibasic and 38 to 42% morpheus precipitant mix 1 (MD2-250-84) from Molecular Dimensions (Newmarket, Suffolk, England), where polyethylene glycol (PEG) 3350 was the essential component. Single crystals appeared after 3 days and grew to their maximum size within 10 to 15 days. Crystals were flash-frozen by liquid nitrogen directly before they were mounted for data collection. The best data were collected at Advance Photon Source, beamline 23-ID-B at Argonne National Laboratory, equipped with an Eiger 16M detector.
The collected data were indexed and integrated with X-ray Detector Software (XDS) and scaled using Scala, a part of the CCP4 suite (37–39). Initial phase information was obtained by molecular replacement using Phaser with the previously solved structure of human CD38 catalytic domain (Protein Data Bank ID: 6EDR) as the search model (40). Waters were added using ArpWarp during the initial round of the refinement, and the ligand was built using Ligand Builder in Coot (41, 42). The structure was improved by iterative rounds of model building and refinement using the programs Coot and Refmac5 (41, 43). The crystals belong to space group P 41 2 2, and it contains two molecules per asymmetric unit with Mathew coefficient of 2.73. Crystallographic details and statistics are listed in table S1.
Flow cytometric analysis
HCC1954 cells were cultured in RPMI 1640 medium (Corning, NY) supplemented with 10% FBS (Thermo Fisher Scientific, MA) in 37°C incubator with 5% CO2. MCF7, MDA-MB-231, and MDA-MB-468 cells were cultured under the same conditions but with DMEM medium (Corning, NY) supplemented with 10% FBS. All cells used for the flow cytometry were at passage 3 with 80% confluency. Cells were detached from 125-ml tissue culture flasks (Genesee Scientific, CA) with 0.5 ml of 0.25% trypsin-EDTA (Thermo Fisher Scientific, MA) for 5 min in a 37°C incubator with 5% CO2, followed with neutralization by the additions of 5 ml of culture media with 10% FBS. After centrifugation at 100g for 5 min and subsequent removals of culture media, cell pellets were washed once by resuspending in 5 ml of DPBS, followed with centrifugation at 100g for 5 min. Cells were then resuspended in 1 ml of ice-cold DPBS containing 200 nM Herceptin IgG and incubated at 4°C for 30 min, followed with three wash cycles with DPBS. Then, cells were resuspended in 1 ml of ice-cold DPBS with 200 nM goat anti-human IgG (H+L) cross-adsorbed secondary antibody with Alexa Fluor 488 (Thermo Fisher Scientific, MA) and incubated at 4°C for 30 min, followed with three wash cycles using 5 ml of DPBS. Last, cells were resuspended in 1 ml of DPBS for flow cytometry analysis with the BD Fortessa X20 Cell Analyzer (BD Biosciences, CA) for cellular Alexa Fluor 488 intensities. Cells treated only with 200 nM goat anti-human IgG (H+L) cross-absorbed secondary antibody with Alexa Fluor 488 were used as controls.
In vitro cytotoxicity assays
All cells used for in vitro cytotoxicity assays were at passage 3. Cells were first seeded into 96-well cell culture plates (VWR, PA) in 90 μl of culture media and incubated at 37°C in an incubator with 5% CO2 for overnight with penicillin-streptomycin (Thermo Fisher Scientific, MA) at following densities: 3000 cells per well for HCC1954, 8000 cells per well for MCF7, 5000 cells per well for MDA-MB-231, and 10,000 cells per well for MDA-MB-468. Then, the anti-HER2 ARC-ADC, CD38 C-fusion IgG, Herceptin IgG, and 2′-Cl-araNAD+–MMAF in culture media at various concentrations were added in triplicates. Plates were placed at 37°C in the incubator with 5% CO2 for 72 hours before adding the MTT reagents (Thermo Fisher Scientific, MA) per the manufacturer’s instructions, followed by cell lysis with 100 μl of lysis buffer (20% SDS in 50% dimethylformamide). After 1-hour incubation at 37°C with 5% CO2, absorbance at 580 nm was measured with a Synergy H1 plate reader (BioTek, VT). Cells treated with culture media and 5 μM paclitaxel (MilliporeSigma, MA) were included as 100 and 0% viability controls, respectively. The data were fitted by the sigmoidal function in GraphPad Prism (GraphPad, CA) to determine EC50 values.
Plasma stability of ARC-ADC based on in vitro cytotoxicity
HCC1954 cells and MDA-MB-468 cells at passage 3 were seeded into 96-well cell culture plates (VWR, PA) with 90 μl of culture media the night before the experiments (3000 cells per well for HCC1954 and 10,000 cells per well for MDA-MB-468). Plasma-incubated ARC-ADC was prepared by mixing ARC-ADC with CD-1 mice plasma at a final concentration of 10 μM and then placed at 37°C in an incubator with 5% CO2 for 72 hours. Freshly prepared and plasma-incubated ARC-ADC were serially diluted into culture media at various concentrations and added into cultured cells on 96-well plates. After 3-day incubation at 37°C with 5% CO2, 10 μl of MTT reagent (Thermo Fisher Scientific, MA) was added, followed by 2-hour incubation at 37°C in an incubator with 5% CO2. Cells were then lysed with the addition of 100 μl of lysis buffer (20% SDS in 50% dimet of the fluorogenic substrates hylformamide) and incubated for 1 hour at 37°C. Absorbance at 580 nm was measured with a plate reader (BioTek, VT). Cells treated with culture media and 5 μM paclitaxel (MilliporeSigma, MA) were included as 100 and 0% viability controls, respectively. The data were fitted by the sigmoidal function in GraphPad Prism (GraphPad, CA) to determine EC50 values.
In vitro drug release of 2′-Cl-araNAD+–MMAF
To examine the degradation of 2′-Cl-araNAD+–MMAF in lysosomes, 2′-Cl-araNAD+–MMAF at a final concentration of 0.5 mM in 50 mM sodium acetate buffer (pH 5) with 10% (v/v) rat liver lysosomal lysates was incubated at 37°C for up to 22 hours with fixed volumes sampled at different time points. Equal volumes of acetonitrile were then added for extraction, followed by HPLC analysis with a C18-A column (Agilent Technologies, CA) through measurements of UV absorbance at 260 nm. The inlet method for the HPLC analysis is as follows: mobile phase A: 0.1% formic acid (aq); mobile B: 0.1% formic acid in acetonitrile; flow rate = 2.0 ml/min; 0 to 2 min: 0 to 4% B; 2 to 4 min: 4 to 10% B; 4 to 6 min: 10 to 20% B; 6 to 12 min: 20 to 50% B; 12 to 17 min: 50 to 100% B; 17 to 20 min: 100 to 0% B. The final degradation product was identified on the basis of retention times of synthesized standards and MS analysis.
To further examine the degradation of rat liver lysosomal lysate–treated 2′-Cl-araNAD+–MMAF in cytosol, 2′-Cl-araNAD+–MMAF at a final concentration of 2 mM in 50 mM sodium acetate buffer (pH 5) with 40% (v/v) rat liver lysosomal lysates was first incubated at 37°C for overnight. Then, the overnight reaction mixtures were diluted by PBS buffer at 1:3 volume ratio containing 10% (v/v) HCC1954 cell lysates and incubated at 37°C for up to 22 hours with fixed volumes sampled at different time points. Equal volumes of acetonitrile were then added for extraction, followed by HPLC analysis with a C18-A column through measurements of UV absorbance at 260 nm. The same HPLC method as mentioned above was used for analysis. The final degradation product was identified on the basis of retention times of synthesized standards and MS analysis.
Pharmacokinetic studies of CD38 C-fusion IgG in mice
Five female CD-1 mice (6 weeks) were given CD38 C-fusion IgG in DPBS by tail vein intravenous injection at 3 mg/kg. Tail venipuncture was then performed for blood collection at different time points (0.5, 5, 9, 24, 48, 72, 120, 168, and 264 hours). Plasma concentrations of CD38 C-fusion IgG were determined using two different sandwich ELISAs. Standards used for the ELISA-based quantification were prepared through serial dilutions of antibody fusion (100 μg ml−1) in mouse plasma.
For the first sandwich ELISA, anti-human IgG (H+L) antibody (8 μg ml−1) (SeraCare, MA) was coated on ELISA plates overnight at room temperature, followed by blocking with 3% BSA in PBS (pH 7.4) for 2 hours at room temperature. After three-cycle wash with 200 μl of 0.1% PBST, 80 μl of 100-fold diluted plasma samples were added in triplicates. Following 2-hour incubation at room temperature and three-cycle wash with 200 μl of 0.05% PBST, 80 μl of 2000-fold diluted anti-human IgG κ antibody HRP conjugate (Thermo Fisher Scientific, MA) was applied and incubated at room temperature for 1 hour. Last, after three times wash with 0.1% PBST, 80 μl of QuantaBlu fluorogenic substrates (Thermo Fisher Scientific, MA) was added and allowed for 5-min incubation before measurements of the fluorescence at 425 nm with a Synergy H1 plate reader (BioTek, VT).
For the second sandwich ELISA, the same capture antibody, anti-human IgG (H+L) antibody, was used to capture the fusion antibody in plasma samples. Following blocking with 3% BSA and incubation with 100-fold diluted plasma samples, biotinylated anti-human CD38 antibody (Thermo Fisher Scientific, MA) was used as the detection antibody and incubated for 1 hour at room temperature. After three times wash with 0.1% PBST, 80 μl of streptavidin-HRP conjugate (R&D Systems, MN) was added and incubated for 1 hour. Following three times wash with 0.1% PBST, 80 μl of the fluorogenic substrate was added and followed by measurements of fluorescence intensities with a plate reader. On the basis of the measured fluorescence intensities, plasma concentrations of the CD38 C-fusion IgG were calculated by fitting into the standard curves generated from the standards prepared on the same plates. Pharmacokinetic parameters including half-life (thalf) were determined by fitting data into a noncompartmental analysis model in SimBiology of MATLAB software package (MathWorks, MA).
Biodistribution of anti-HER2 ARC-ADC in mice
HCC1954 cells (2 million cells per mouse, passage 3) in 50% Matrigel were subcutaneously implanted into the flank of female NSG mice (6 weeks). One week after tumor implantation, mice were administered intravenously though tail vein with IRDye 800CW–labeled anti-HER2 ARC-ADC (5 mg/kg), which was prepared per the manufacturer’s instruction or free IRDye 800CW at the same molar concentration, which was neutralized by diluting in 50 mM tris buffer (pH 7.4). Mice were then imaged at 1, 24, and 48 hours after intravenous injection using IVIS in vivo imaging system (PerkinElmer, MA), followed by euthanasia and imaging of harvested tumors and major organs (heart, lung, liver, spleen, and kidney).
In vivo efficacy studies of anti-HER2 ARC-ADC in mice
HCC1954 cells (1.5 million cells per mouse, at passage 3) in 50% Matrigel (Corning, NY) were subcutaneously implanted into the flank of female NSG mice (6 weeks). When tumor sizes reached 100 mm3, mice (n = 6) were administered intravenously through tail vein with PBS or ARC-ADC (5 mg kg−1) every 3 days for a total of four times. Body weights and tumor sizes were measured every 3 days. The volume of tumor was calculated as mm3 = 0.5 × (length) × (width)2. Mice were euthanized once the tumor sizes exceeded 1000 mm3. Weights of harvested major organs (heart, lung, liver, spleen, and kidney) and body weights were measured. A Kaplan-Meier survival analysis was conducted in GraphPad Prism (GraphPad, CA). All procedures were approved by Institutional Animal Care and Use Committee of the University of Southern California.
In silico prediction for immunogenicity of CD38 C-fusion IgG
The amino acid sequence of CD38 C-fusion IgG heavy chain was uploaded into the NetMHCIIpan 3.2 server for predicting immunogenic peptides, which have high binding affinity to selected representative human MHC class II molecules. Peptide sequences predicted with strong binding potentials to selected MHC class II molecules were recorded.
Among all MHC class II genes in human, the subtypes selected for prediction included all common human leukocyte antigen (HLA)–DR alleles listed in the catalog of common and well-documented HLA alleles (44), five common HLA-DP haplotypes (45), and six common HLA-DQ haplotypes (46). For HLA-DP and HLA-DQ, only haplotypes present in more than 15% of all major populations globally were chosen for prediction. HLA-DPA0103 was the only allele selected to pair with five HLA-DPB alleles, as common HLA-DPA subtypes that account for more than 90% of all populations in total are significantly less polymorphic and their polymorphic regions make minimal contribution to binding of immunogenic peptides (47).
Statistical analysis
Two-tailed unpaired t tests were performed for comparison between two groups. Tumor growth curves of the control and treatment groups were analyzed using two-tailed unpaired t tests. P < 0.05 was defined as statistically significant. Data are shown as means ± SD. Kaplan-Meier method was adopted to compare the survival time between two groups of mice. All statistical analyses were performed using GraphPad Prism (GraphPad, CA).
Supplementary Material
Acknowledgments
We would like to thank the staff at Stanford Synchrotron Radiation Light Source (SSRL) (beamline 12-2) and Advanced Photon Source (APS) beamline 23-ID-B for their excellent support during data collections and also G. W. Han for her advice on the refinement and improving the quality of the final structure. Funding: This work was supported by University of Southern California School of Pharmacy Start-Up Fund for New Faculty, Sharon L. Cockrell Cancer Research Fund, The V Foundation for Cancer Research V Scholar (grant V2016-021) (to Y.Z.), STOP CANCER Research Career Development Award (to Y.Z.), and PhRMA Foundation Research Starter Grant in Translational Medicine and Therapeutics (to Y.Z.). Author contributions: Z.D., X.-N.Z., and Y.Z. designed research. Z.D., X.-N.Z., F.N., Q.C., J.L., and B.B.K. performed research. G.S., H.P., S.G.L., H.-J.L., and R.C.S. provided resources and critical insights. Z.D., X.-N.Z., F.N., and Y.Z. analyzed data. Z.D., X.-N.Z., and Y.Z. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The authors confirm that the data supporting the findings of this study are available from the corresponding author upon request. Coordinates of the solved x-ray structure of human CD38 catalytic domain with covalently attached 2′-Cl-araNAD+ have been deposited in the Protein Data Bank with the PDB ID 6VUA.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/23/eaba6752/DC1
REFERENCES AND NOTES
- 1.Beck A., Goetsch L., Dumontet C., Corvaïa N., Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 16, 315–337 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Jain N., Smith S. W., Ghone S., Tomczuk B., Current ADC linker chemistry. Pharm. Res. 32, 3526–3540 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu R., Wang R. E., Wang F., Antibody-drug conjugates for non-oncological indications. Expert Opin. Biol. Ther. 16, 591–593 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Hamblett K. J., Senter P. D., Chace D. F., Sun M. M., Lenox J., Cerveny C. G., Kissler K. M., Bernhardt S. X., Kopcha A. K., Zabinski R. F., Meyer D. L., Francisco J. A., Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 10, 7063–7070 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Wakankar A. A., Feeney M. B., Rivera J., Chen Y., Kim M., Sharma V. K., Wang Y. J., Physicochemical stability of the antibody-drug conjugate Trastuzumab-DM1: Changes due to modification and conjugation processes. Bioconjug. Chem. 21, 1588–1595 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Junutula J. R., Raab H., Clark S., Bhakta S., Leipold D. D., Weir S., Chen Y., Simpson M., Tsai S. P., Dennis M. S., Lu Y., Meng Y. G., Ng C., Yang J., Lee C. C., Duenas E., Gorrell J., Katta V., Kim A., McDorman K., Flagella K., Venook R., Ross S., Spencer S. D., Lee Wong W., Lowman H. B., Vandlen R., Sliwkowski M. X., Scheller R. H., Polakis P., Mallet W., Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 26, 925–932 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Axup J. Y., Bajjuri K. M., Ritland M., Hutchins B. M., Kim C. H., Kazane S. A., Halder R., Forsyth J. S., Santidrian A. F., Stafin K., Lu Y., Tran H., Seller A. J., Biroc S. L., Szydlik A., Pinkstaff J. K., Tian F., Sinha S. C., Felding-Habermann B., Smider V. V., Schultz P. G., Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. U.S.A. 109, 16101–16106 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruins J. J., Westphal A. H., Albada B., Wagner K., Bartels L., Spits H., van Berkel W. J. H., van Delft F. L., Inducible, site-specific protein labeling by tyrosine oxidation-strain-promoted (4 + 2) cycloaddition. Bioconjug. Chem. 28, 1189–1193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X., Nelson C. G., Nair R. R., Hazlehurst L., Moroni T., Martinez-Acedo P., Nanna A. R., Hymel D., Burke T. R. Jr., Rader C., Stable and potent selenomab-drug conjugates. Cell Chem. Biol. 24, 433, 442.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin S., Yang X., Jia S., Weeks A. M., Hornsby M., Lee P. S., Nichiporuk R. V., Iavarone A. T., Wells J. A., Toste F. D., Chang C. J., Redox-based reagents for chemoselective methionine bioconjugation. Science 355, 597–602 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okeley N. M., Toki B. E., Zhang X., Jeffrey S. C., Burke P. J., Alley S. C., Senter P. D., Metabolic engineering of monoclonal antibody carbohydrates for antibody-drug conjugation. Bioconjug. Chem. 24, 1650–1655 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Tian F., Lu Y., Manibusan A., Sellers A., Tran H., Sun Y., Phuong T., Barnett R., Hehli B., Song F., DeGuzman M. J., Ensari S., Pinkstaff J. K., Sullivan L. M., Biroc S. L., Cho H., Schultz P. G., DiJoseph J., Dougher M., Ma D., Dushin R., Leal M., Tchistiakova L., Feyfant E., Gerber H.-P., Sapra P., A general approach to site-specific antibody drug conjugates. Proc. Natl. Acad. Sci. U.S.A. 111, 1766–1771 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.VanBrunt M. P., Shanebeck K., Caldwell Z., Johnson J., Thompson P., Martin T., Dong H., Li G., Xu H., D'Hooge F., Masterson L., Bariola P., Tiberghien A., Ezeadi E., Williams D. G., Hartley J. A., Howard P. W., Grabstein K. H., Bowen M. A., Marelli M., Genetically encoded azide containing amino acid in mammalian cells enables site-specific antibody–drug conjugates using click cycloaddition chemistry. Bioconjug. Chem. 26, 2249–2260 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Zhou Q., Stefano J. E., Manning C., Kyazike J., Chen B., Gianolio D. A., Park A., Busch M., Bird J., Zheng X., Simonds-Mannes H., Kim J., Gregory R. C., Miller R. J., Brondyk W. H., Dhal P. K., Pan C. Q., Site-specific antibody-drug conjugation through glycoengineering. Bioconjug. Chem. 25, 510–520 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Zhu Z., Ramakrishnan B., Li J., Wang Y., Feng Y., Prabakaran P., Colantonio S., Dyba M. A., Qasba P. K., Dimitrov D. S., Site-specific antibody-drug conjugation through an engineered glycotransferase and a chemically reactive sugar. MAbs 6, 1190–1200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lhospice F., Brégeon D., Belmant C., Dennler P., Chiotellis A., Fischer E., Gauthier L., Boedec A., Rispaud H., Savard-Chambard S., Represa A., Schneider N., Paturel C., Sapet M., Delcambre C., Ingoure S., Viaud N., Bonnafous C., Schibli R., Romagne F., Site-specific conjugation of monomethyl auristatin E to anti-CD30 antibodies improves their pharmacokinetics and therapeutic index in rodent models. Mol. Pharm. 12, 1863–1871 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Strop P., Liu S. H., Dorywalska M., Delaria K., Dushin R. G., Tran T. T., Ho W. H., Farias S., Casas M. G., Abdiche Y., Zhou D., Chandrasekaran R., Samain C., Loo C., Rossi A., Rickert M., Krimm S., Wong T., Chin S. M., Yu J., Dilley J., Chaparro-Riggers J., Filzen G. F., O’Donnell C. J., Wang F., Myers J. S., Pons J., Shelton D. L., Rajpal A., Location matters: Site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 20, 161–167 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Dorywalska M., Strop P., Melton-Witt J. A., Hasa-Moreno A., Farias S. E., Galindo Casas M., Delaria K., Lui V., Poulsen K., Sutton J., Bolton G., Zhou D., Moine L., Dushin R., Tran T. T., Liu S. H., Rickert M., Foletti D., Shelton D. L., Pons J., Rajpal A., Site-dependent degradation of a non-cleavable auristatin-based linker-payload in rodent plasma and its effect on aDC efficacy. PLOS ONE 10, e0132282 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stefan N., Gébleux R., Waldmeier L., Hell T., Escher M., Wolter F. I., Grawunder U., Beerli R. R., Highly potent, anthracycline-based antibody-drug conjugates generated by enzymatic, site-specific conjugation. Mol. Cancer Ther. 16, 879–892 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Beerli R. R., Hell T., Merkel A. S., Grawunder U., Sortase enzyme-mediated generation of site-specifically conjugated antibody drug conjugates with high in vitro and in vivo potency. PLOS ONE 10, e0131177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woitok M., Klose D., Di Fiore S., Richter W., Stein C., Gresch G., Grieger E., Barth S., Fischer R., Kolberg K., Niesen J., Comparison of a mouse and a novel human scFv-SNAP-auristatin F drug conjugate with potent activity against EGFR-overexpressing human solid tumor cells. Onco. Targets Ther. 10, 3313–3327 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nanna A. R., Li X., Walseng E., Pedzisa L., Goydel R. S., Hymel D., Burke T. R. Jr., Roush W. R., Rader C., Harnessing a catalytic lysine residue for the one-step preparation of homogeneous antibody-drug conjugates. Nat. Commun. 8, 1112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mullersteffner H. M., Malver O., Hosie L., Oppenheimer N. J., Schuber F., Slow-binding inhibition of Nad+ glycohydrolase by arabino analogs of β-Nad. J. Biol. Chem. 267, 9606–9611 (1992). [PubMed] [Google Scholar]
- 24.Jiang H., Congleton J., Liu Q., Merchant P., Malavasi F., Lee H. C., Hao Q., Yen A., Lin H., Mechanism-based small molecule probes for labeling CD38 on live cells. J. Am. Chem. Soc. 131, 1658–1659 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shrimp J. H., Hu J., Dong M., Wang B. S., MacDonald R., Jiang H., Hao Q., Yen A., Lin H., Revealing CD38 cellular localization using a cell permeable, mechanism-based fluorescent small-molecule probe. J. Am. Chem. Soc. 136, 5656–5663 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sauve A. A., Deng H. T., Angeletti R. H., Schramm V. L., A covalent intermediate in CD38 is responsible for ADP-ribosylation and cyclization reactions. J. Am. Chem. Soc. 122, 7855–7859 (2000). [Google Scholar]
- 27.Liu Q., Kriksunov I. A., Jiang H., Graeff R., Lin H., Lee H. C., Hao Q., Covalent and noncovalent intermediates of an NAD utilizing enzyme, human CD38. Chem. Biol. 15, 1068–1078 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Q., Graeff R., Kriksunov I. A., Jiang H., Zhang B., Oppenheimer N., Lin H., Potter B. V. L., Lee H. C., Hao Q., Structural basis for enzymatic evolution from a dedicated ADP-ribosyl cyclase to a multifunctional NAD hydrolase. J. Biol. Chem. 284, 27637–27645 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cho H.-S., Mason K., Ramyar K. X., Stanley A. M., Gabelli S. B., Denney D. W. Jr., Leahy D. J., Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756–760 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Dai Z., Zhang X. N., Nasertorabi F., Cheng Q., Pei H., Louie S. G., Stevens R. C., Zhang Y., Facile chemoenzymatic synthesis of a novel stable mimic of NAD+. Chem. Sci. 9, 8337–8342 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sondermann P., Huber R., Oosthuizen V., Jacob U., The 3.2-Å crystal structure of the human IgG1 Fc fragment-FcγRIII complex. Nature 406, 267–273 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Kern J. C., Cancilla M., Dooney D., Kwasnjuk K., Zhang R., Beaumont M., Figueroa I., Hsieh S., Liang L., Tomazela D., Discovery of pyrophosphate diesters as tunable, soluble, and bioorthogonal linkers for site-specific antibody–drug conjugates. J. Am. Chem. Soc. 138, 1430–1445 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Munshi C., Aarhus R., Graeff R., Walseth T. F., Levitt D., Lee H. C., Identification of the enzymatic active site of CD38 by site-directed mutagenesis. J. Biol. Chem. 275, 21566–21571 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Los G. V., Encell L. P., McDougall M. G., Hartzell D. D., Karassina N., Zimprich C., Wood M. G., Learish R., Ohana R. F., Urh M., HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373–382 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Gautier A., Juillerat A., Heinis C., Corrêa I. R. Jr., Kindermann M., Beaufils F., Johnsson K., An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 15, 128–136 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Karosiene E., Rasmussen M., Blicher T., Lund O., Buus S., Nielsen M., NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics 65, 711–724 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kabsch W., Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 26, 795–800 (1993). [Google Scholar]
- 38.Evans P., Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mccoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Langer G., Cohen S. X., Lamzin V. S., Perrakis A., Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murshudov G. N., Vagin A. A., Dodson E. J., Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- 44.Mack S. J., Cano P., Hollenbach J. A., He J., Hurley C. K., Middleton D., Moraes M. E., Pereira S. E., Kempenich J. H., Reed E. F., Common and well-documented HLA alleles: 2012 update to the CWD catalogue. Tissue Antigens 81, 194–203 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sidney J., Steen A., Moore C., Ngo S., Chung J., Peters B., Sette A., Five HLA-DP molecules frequently expressed in the worldwide human population share a common HLA supertypic binding specificity. J. Immunol. 184, 2492–2503 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sidney J., Steen A., Moore C., Ngo S., Chung J., Peters B., Sette A., Divergent motifs but overlapping binding repertoires of six HLA-DQ molecules frequently expressed in the worldwide human population. J. Immunol. 185, 4189–4198 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bugawan T. L., Horn G., Long C., Mickelson E., Hansen J., Ferrara G. B., Angelini G., Erlich H., Analysis of HLA-DP allelic sequence polymorphism using the in vitro enzymatic DNA amplification of DP-alpha and DP-beta loci. J. Immunol. 141, 4024–4030 (1988). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/23/eaba6752/DC1