

CReP recruits translation machinery to the ER and promotes local translation during global protein synthesis repression.
Abstract
Eukaryotic protein synthesis control at multiple levels allows for dynamic, selective responses to diverse conditions, but spatial organization of translation initiation machinery as a regulatory principle has remained largely unexplored. Here we report on a role of constitutive repressor of eIF2α phosphorylation (CReP) in translation of poliovirus and the endoplasmic reticulum (ER)–resident chaperone binding immunoglobulin protein (BiP) at the ER. Functional, proximity-dependent labeling and cell fractionation studies revealed that CReP, through binding eIF2α, anchors translation initiation machinery at the ER and enables local protein synthesis in this compartment. This ER site was protected from the suppression of cytoplasmic protein synthesis by acute stress responses, e.g., phosphorylation of eIF2α(S51) or mTOR blockade. We propose that partitioning of translation initiation machinery at the ER enables cells to maintain active translation during stress conditions associated with global protein synthesis suppression.
INTRODUCTION
Cells respond to environmental stress with coordinated transcriptional, translational, and posttranslational gene expression changes. The pivotal event in the integrated stress response (ISR) is phosphorylation of serine-51 of the α subunit of eukaryotic initiation factor (eIF) 2 (1). The ISR globally curtails translation initiation via the eIF2–guanosine triphosphate (GTP)–initiator methionyl transfer RNA (tRNA) in the ternary complex by inhibiting eIF2B-mediated exchange of guanosine diphosphate for GTP on eIF2 (2). Coincident with this suppression of global protein synthesis is the induction of transcription factor ATF4 and eIF2α–protein phosphatase 1 (PP1) scaffold GADD34, which coordinate downstream ISR programs to restore homeostasis or, with unrelenting stress, activate programmed cell death (3).
Double-stranded RNA (dsRNA) intermediates generated during viral infection induce p-eIF2α(S51) via the dsRNA-activated protein kinase R (PKR), one of four eIF2α(S51)-directed kinases (1). eIF2α(S51) phosphorylation is detrimental to most human viral pathogens, triggering the evolution of elaborate viral countermeasures, e.g., the γ34.5 phosphatase scaffold of type 1 herpes simplex viruses that reverses p-eIF2α(S51) in infected cells (4). Enteroviruses require eIF2 for translation initiation at their (+)strand RNA genomes (5); yet, they maintain viral translation in the face of robust eIF2α(S51) phosphorylation (6, 7). They achieve this without specific mechanisms to counter PKR activation or eIF2α(S51) phosphorylation.
Enteroviruses reorganize the secretory pathway into replication complexes consisting of components derived from endoplasmic reticulum (ER), Golgi, and lysosomes (8), where they concentrate cofactors required by their RNA-dependent RNA polymerases (9). How this locale may be exploited to provide protection from cellular antiviral responses remains largely unexplored.
Our studies of p-eIF2α(S51)’s role in shaping poliovirus:host relations uncovered that CReP, an ER-localized PP1-eIF2α scaffold (10), determines partitioning of translation initiation factors at the ER and defines a spatially and functionally distinct translation initiation compartment at this site. We found that CReP promotes the expression of BiP, an HSP70-type chaperone in the ER and major regulator of the cellular unfolded protein response (11). BiP translation, like poliovirus (PV), is maintained during the ISR (12), and we posit that PV and BiP translation evades inhibitory effects of p-eIF2α(S51) via CReP:eIF2α at the ER. This phenomenon explains earlier seminal findings that BiP translation is maintained during the host protein synthesis shutoff elicited by PV (13). Overall, our investigations demonstrate that CReP engages canonical translation initiation machinery at the ER, allowing for ongoing local protein synthesis in the presence of global translation repression mediated by the ISR.
RESULTS
PV-induced eIF2α(S51) phosphorylation is not inhibitory to viral translation
Infection with wild-type (WT) PV (6), or the highly attenuated derivative PVSRIPO (7), induces prodigious eIF2α(S51) phosphorylation (Fig. 1A). To unravel the source kinase of this response, we depleted either PKR or PKR-like ER kinase (PERK), both of which are eIF2α(S51) kinases plausibly engaged by viral replication at the ER (Fig. 1A) (1). Depletion of PKR, but not PERK, reduced eIF2α(S51) phosphorylation but had no effect on viral translation (expression of viral proteins P2, 2BC, and 2C), the dynamics of host protein synthesis shutdown (assayed by puromycylation), or host cell cytopathogenicity [poly(ADP-ribose) polymerase (PARP) cleavage] upon PVSRIPO infection (Fig. 1A). In addition, ISRIB, a small molecule that abrogates the ISR (14), had minimal effects on viral translation and the host translation shutoff (Fig. 1, B and C). Thus, PV infection-mediated eIF2α(S51) phosphorylation is due to PKR activation, and PV translation must circumvent p-eIF2α(S51)–mediated protein synthesis suppression, as enteroviruses categorically require eIF2 for translation initiation (5).
Fig. 1. PVSRIPO translation persists in the presence of PKR-induced p-eIF2α(S51).
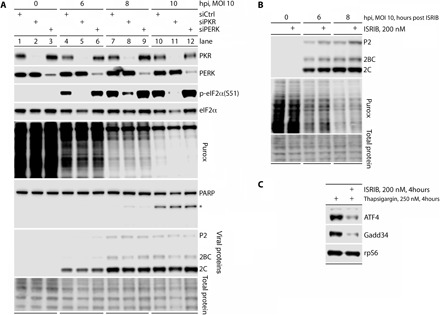
(A) HeLa cells were transfected with control small interfering RNA (siRNA) or siRNAs targeting PKR or PERK (48 hours) and infected with PVSRIPO at a multiplicity of infection (MOI) of 10. At each interval shown, the cells were treated with puromycin (10 μM; 8 min) and harvested for immunoblot analysis of eIF2α status, host translation (puromycylated polypeptides), viral translation (2C, 2BC, P2), or cell death (PARP cleavage; *, cleavage fragment). (n = 3). (B) HeLa cells were infected with PVSRIPO and treated with vehicle or ISRIB (+), puromycylated as described for (A) and lysed at the indicated time points. (n = 3). (C) The biological effect of ISRIB in the assay shown in (B) was validated in HeLa cells treated with thapsigargin as shown.
CReP depletion diminishes PV and BiP translation without inducing p-eIF2α(S51) or the ISR
CReP is a peripheral ER membrane–targeted protein that modulates eIF2α phosphorylation (10, 15). PV’s reliance on eIF2 for viral translation (5), and tethering of PV replication complexes to secretory pathway membranes (9), suggested a role for CReP in viral translation. Preliminary analyses with transient small interfering RNA (siRNA)–mediated CReP depletion supported a role in promoting PVSRIPO translation (fig. S1). To avoid the confounding effects of siRNA transfection on (+)strand RNA virus translation, and to enable CReP reconstitution without the need for multiple transfections, we generated HeLa cell lines expressing a doxycycline (dox)–inducible CReP-targeting short hairpin RNA (shRNA) (Fig. 2).
Fig. 2. CReP depletion inhibits viral and cellular translation without inducing eIF2α(S51) phosphorylation or its downstream response.
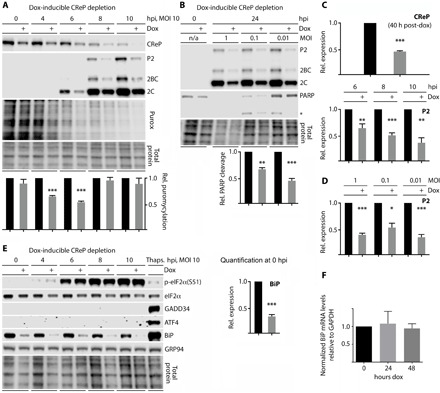
(A and C) HeLa cells expressing a dox-inducible shRNA targeting CReP were mock-treated or treated with dox (4 μg/ml; ~40 hours) and then infected with PVSRIPO. Cells were puromycylated (see Fig. 1) and harvested for immunoblot analysis with the indicated antibodies. (B and D) HeLa cells with dox-inducible CReP depletion were mock- or dox-treated as described in (A), infected with PVSRIPO at MOIs of 1, 0.1, or 0.01 and then lysed (24 hpi) for immunoblot analysis of viral protein and cell death (PARP cleavage). n/a, not applied. (E) The lysates analyzed in (A) were tested by immunoblot alongside lysate from cells treated with thapsigargin (250 nM) and probed for key components of the ISR. (F) CReP dox-inducible knockdown cells were left untreated or treated with dox for 24 or 48 hours, and total RNA was isolated to determine relative BiP/glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels by reverse transcription quantitative polymerase chain reaction (RT-qPCR). These values were then normalized to the no-dox sample and quantified. All quantifications and statistics were performed by Student’s two-tailed t test comparison at the indicated time point, comparing ± dox (n = 3). Bar graphs represent mean and SEM; *P < 0.05; **P < 0.005; ***P < 0.0005.
Dox treatment of HeLa cells with dox-inducible CReP depletion yielded an ~50% decrease in CReP levels and reduced PVSRIPO translation to a similar degree (Fig. 2A). Dox-inducible CReP depletion had a similar effect on the translation of another enterovirus, Coxsackievirus B3 (fig. S2).
Incremental loss of CReP in PVSRIPO-infected cells (Fig. 2A) is due to the inherent instability of CReP [half-time (t1/2) ~ 1.5 hours; fig. S3] and the cells’ inability to replenish CReP levels during virus-induced host protein synthesis shut-off. CReP depletion had a minor effect on global translation at baseline but considerably exacerbated the viral host protein synthesis shut-off at 4 to 6 hours post infection (hpi) (puromycylation assay; Fig. 2A). This effect occurred despite the ~50% reduced rate of viral translation in CReP-depleted cells, indicating that CReP facilitates a portion of global cellular translation that is maintained in the presence of the translation shutdown at 4 to 6 hpi in PVSRIPO-infected HeLa cells. By 8 hpi, due to the severity of the cytopathogenic effect and the extent of the host protein synthesis shut-off, this differential was no longer detected (Fig. 2A).
Tests with PVSRIPO infection over a range of multiplicities of infection (MOIs) confirmed that CReP depletion reduced viral translation and protected cells from virus-induced cell death (PARP cleavage) (Fig. 2B). Protection from viral cytopathogenicity upon CReP depletion demonstrated that the observed effects on PVSRIPO translation are not due to nonspecific toxicity associated with dox-inducible CReP depletion.
CReP has been shown to counteract p-eIF2α(S51) accumulation at basal conditions (10). Thus, our findings could be due to p-eIF2α(S51) build-up upon CReP depletion, thereby preemptively suppressing viral and host protein synthesis. At the ~50% CReP depletion range in our system, this was not the case, as p-eIF2α(S51) levels did not change and the downstream ISR (ATF4/GADD34 induction) failed to materialize (Fig. 2E). Incidentally, we observed that CReP depletion consistently was accompanied by reduced eIF2α levels, suggesting that CReP may play a role in global eIF2α homeostasis (Fig. 2E).
Serendipitously, our studies of the ISR in CReP-depleted cells revealed that levels of BiP, another marker of ER stress (3), were depressed to ~35% of endogenous levels in dox-treated HeLa cells (Fig. 2E). Examining BiP levels in lysates from cells treated with siRNA-mediated CReP depletion confirmed this effect (fig. S1). BiP mRNA levels remained unchanged upon dox treatment, indicating that CReP depletion does not alter BiP transcript levels (Fig. 2F). Seminal earlier work with metabolic labeling in PV-infected HeLa cells demonstrated that BiP biosynthesis persists at high levels during the virus-induced host protein synthesis shut-off (13). Thus, the findings reported by Sarnow (13) and our own observations suggested that CReP may be central to a translation compartment shared by PV and BiP. Like CReP, BiP is a high-turnover protein that is constantly replenished (t1/2 ~ 4.2 hours; fig. S3). These findings suggest that CReP depletion affects translation of select templates, i.e., enteroviral genomic RNA and BiP, without broadly increasing p-eIF2α(S51) levels that would globally repress protein synthesis.
We systematically monitored the effects of CReP depletion on acute ER stress, by testing tunicamycin and thapsigargin-induced ER stress in cells with dox-inducible CReP depletion (fig. S4). CReP depletion did not substantially affect GADD34 or ATF4 induction under these conditions (fig. S4). Moreover, BiP induction remained intact, albeit at reduced levels, likely due to IRE1- or ATF6-mediated transcriptional up-regulation and/or GADD34 compensation (fig. S4) (3). These data suggest that CReP is not involved in orchestrating the acute ER stress response. Last, we investigated a potential role for eIF2A, a 60-kD single-subunit functional homolog of eIF2 previously implicated in BiP translation (16), in the observed effects of CReP knockdown. Depleting eIF2A had no impact on PVSRIPO translation or basal BiP expression (fig. S5), eliminating it as a factor in our studies.
CReP’s influence over PV/BiP translation is due to eIF2α binding
To mechanistically decipher CReP’s role in PV translation, we generated HeLa cell lines with dox-inducible depletion of endogenous CReP with simultaneous reconstitution of WT CReP or CReP(ΔeIF2) (lacking the eIF2α-binding motif; Fig. 3A). Reconstitution with WT CReP rescued the effects of CReP depletion on PVSRIPO and BiP translation (Fig. 3, B and D) and restored total eIF2α levels (Fig. 3B). Meanwhile, CReP(ΔeIF2) reconstitution exacerbated the effects of CReP depletion (compare Figs. 2, A and E and 3, C to E), possibly due to dominant-negative effects. This was particularly obvious in the puromycylation assay, testing global host cell protein synthesis in PVSRIPO-infected cells. CReP(ΔeIF2) reconstitution had a substantial, significant dampening effect on global host protein synthesis (Fig. 3D). As with CReP depletion (Fig. 2A), this effect was greater during infection, coinciding with the induction of PKR-mediated eIF2α phosphorylation (Fig. 3D). In contrast, WT CReP reconstitution eliminated the effect of CReP depletion on host protein synthesis in PVSRIPO-infected cells (Fig. 3D). This evidence corroborates our findings with dox-inducible CReP depletion alone (Fig. 2A) and demonstrates a role for CReP:eIF2α in maintaining protein synthesis during PVSRIPO infection. These reconstitution experiments demonstrate that CReP’s effect on PVSRIPO/BiP translation is dependent on CReP’s capacity to bind to eIF2α.
Fig. 3. The effects of CReP depletion on BiP/PVSRIPO translation are eIF2α dependent.
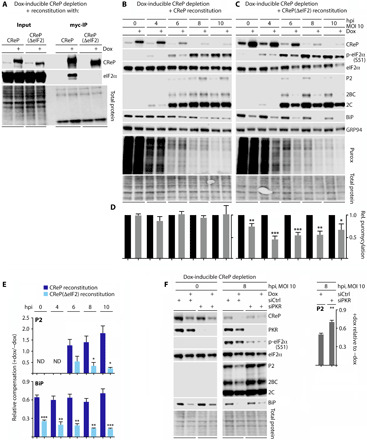
(A) Cells with combined dox-inducible CReP depletion and WT CReP/CReP(ΔeIF2) reconstitution were treated with dox (36 hours) before lysis and IP with anti-Myc beads. Lysates were compared by immunoblot to assess eIF2α binding. (B to E) HeLa cells with endogenous CReP depletion coupled with WT CReP (B) or CReP(ΔeIF2) (C) reconstitution were dox-induced, infected with PVSRIPO, and analyzed by immunoblot and puromycylation assay as described in Fig. 1. ND, not detected. (F) Cells with dox-inducible CReP depletion were mock- or dox-treated (36 hours), transfected with control siRNA or siRNA targeting PKR (36 hours), infected with PVSRIPO, and lysed for immunoblot analysis at the indicated intervals. (D to F) Statistical significance was assessed by Student’s two-tailed t test comparison at each time point between −/+ dox at each time point (D), relative compensation between the two cell lines [WT CReP versus CReP(ΔeIF2)] (E), or −/+ siRNA targeting PKR (F) for the indicated data (bar graphs represent mean and SEM; n = 3); *, **, *** corresponds to P < 0.05, 0.005, and 0.0005, respectively).
CReP protects PV translation from PKR-mediated eIF2α(S51) phosphorylation
One possible explanation for the observed effects of CReP on viral translation could be a role for CReP:eIF2α complexes in maintaining a repository of eIF2, accessible to PVSRIPO at its replication site at the ER, which is protected from PKR-mediated eIF2α(S51) phosphorylation. We tested this possibility by siRNA-mediated knockdown of PKR in cells with dox-inducible CReP depletion. PKR knockdown diminished p-eIF2α(S51) accumulation and neutralized the effect of CReP depletion on viral translation (Fig. 3F). Because PKR depletion had no effect on PVSRIPO translation in cells with WT CReP levels (Fig. 1A), our findings indicate that CReP:eIF2 sustains viral translation in the presence of PKR-induced eIF2α(S51) phosphorylation.
CReP anchors eIF2 to the ER and promotes translation during stress at this site
CReP:eIF2, PV replication complexes, and the site of BiP biosynthesis (17) are localized on the cytoplasmic face of the ER. Thus, CReP:eIF2’s role in promoting translation in the presence of p-eIF2α(S51) accumulation suggests possible compartmentalization of translation machinery. CReP could promote PVSRIPO/BiP translation via local de-phosphorylation of p-eIF2α(S51) or local translation initiation through recruitment of eIF2α/the initiation apparatus to the ER. To investigate this, we carried out assays with dox-inducible CReP depletion followed by sequential detergent fractionation (18). We used eIF4E-binding protein 1 (4EBP1) and glucose-regulated protein 94 (GRP94) as markers for the cytosolic compartment or ER, respectively [Fig. 4, A to C; (19, 20)]. CReP and BiP, heavily enriched in the ER fraction as expected, diminished in abundance with increasing dox exposure (Fig. 4A). This occurred in step with a loss of ER-bound eIF2α, without changes to cytosolic eIF2α levels (Fig. 4A). These events were accompanied by inhibition of global protein synthesis in both compartments, although ER-associated translation was affected more substantially (Fig. 4A).
Fig. 4. CReP anchors eIF2α to the ER and promotes translation at this site.
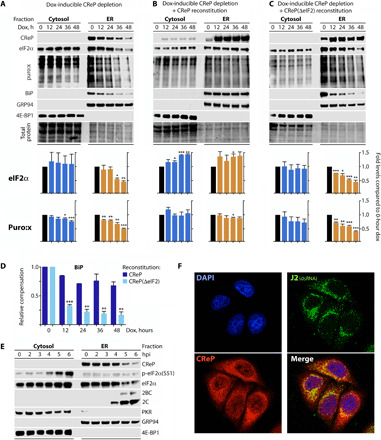
(A to C) Top: HeLa cells with dox-inducible depletion of: endogenous CReP (A); endogenous CReP plus reconstitution with WT CReP (B); or endogenous CReP plus reconstitution with CReP(ΔeIF2) (C) were lysed and processed into cytosolic and ER fractions at various intervals after dox induction. The fractionated lysates were analyzed by immunoblot with the indicated antibodies; ER fraction lysates were loaded at 2× cell equivalency of the cytosolic fraction lysates (see Materials and Methods for further details). GRP94 and eIF4EBP-1 were used as ER and cytosolic markers, respectively. (A to C) Bottom: Quantifications for the partitioning of eIF2α and cytoplasmic/ER-bound protein synthesis (puromycylation assay) with CReP depletion (A), as well as CReP depletion with corresponding WT/ΔeIF2 CReP reconstitution (B and C). Statistical significance was assessed by two-tailed Student’s t test comparison between each time point and time point 0 (graphs represent means ± SEM, n = 3; *, **, *** corresponds to P < 0.05, 0.005, and 0.0005, respectively). (D) Relative BiP expression upon reconstitution with WT CReP or CReP(ΔeIF2) relative to time point 0; statistical significance was assessed as above but comparing the two reconstitutions at each time point. (E) HeLa cells were infected with PVSRIPO (MOI, 10), fractionated, and analyzed by immunoblot with the indicated antibodies (n = 3). (F) WT CReP cells were dox-treated for 24 hours before PVSRIPO infection (MOI, 10; 4.5 hpi); cells were analyzed by confocal microscopy for visualization of the indicated targets. DAPI, 4′,6-diamidino-2-phenylindole.
To test whether the observed effects on compartmentalization were dependent on CReP:eIF2α binding, we fractionated cells with dox-inducible CReP depletion plus WT CReP/CReP(ΔeIF2) reconstitution (Fig. 4, B and C). Reconstitution with WT CReP reversed the loss of BiP expression, ER-bound eIF2α, and ER-associated protein synthesis (Fig. 4, B and D). In contrast, reconstitution with CReP(ΔeIF2) exacerbated the effects of CReP depletion alone (Fig. 4, C and D), as in the reconstitution experiments in PVSRIPO-infected cells (see Fig. 3). These results implicate CReP (i) in controlling the local recruitment of eIF2α to the ER, (ii) in promoting ER-associated protein synthesis in a complex with eIF2α, and (iii) in determining BiP expression (at the ER). Also, these fractionation studies suggested that the depletion of total eIF2α levels observed with CReP depletion/CReP(ΔeIF2) reconstitution are due to a loss of eIF2α from the ER without a concomitant increase in cytosolic eIF2α levels.
ER-bound eIF2α is protected from PKR catalytic activity induced by PVSRIPO infection
These data led us to interrogate the distribution of p-eIF2α(S51) during PVSRIPO infection (Fig. 4E and fig. S7). Viral proteins 2C and 2BC were heavily enriched in the ER fraction, as previously shown (8). Yet, virus-induced p-eIF2α(S51) exclusively occurred in the cytoplasm (Fig. 4E). This effect was evident before eIF2α loss from the ER in PVSRIPO-infected cells (fig. S6). PKR was largely absent from the ER fraction, also in line with a previous report (21). There was substantial overlap between viral dsRNA and CReP staining as visualized by confocal microscopy, suggesting that CReP is present within or adjacent to viral “replication factories” where both viral replication and translation occur (Fig. 4F) (22). Thus, PV may evade PKR-induced p-eIF2α(S51) by accessing a pool of CReP-anchored, ER-localized eIF2α that is protected from S51 phosphorylation and/or subjected to instantaneous, CReP-dependent dephosphorylation.
CReP promotes protein synthesis while mTOR is inhibited
We examined another mode of protein synthesis control potentially affected by subcellular partitioning, based on our observation of complete exclusion of 4EBP1 from the ER (Fig. 4, A to C). Protein synthesis modulation via 4EBP1 involves mTOR-C1 (mammalian target of rapamycin–complex 1), which phosphorylates 4EBP1(S65), thereby promoting eIF4E:eIF4G binding and cap-dependent translation initiation (23). Since 4EBP1 is confined to the cytosol, CReP:eIF2α-controlled translation may be resistant to mTOR-C1 blockade. Treatment of cells with dox-inducible CReP depletion and Torin2, a catalytic inhibitor of mTOR (24), had additive inhibitory effects on global protein synthesis that increased from ~15 to 20% at baseline to ~55% after 3 hours of Torin2 (Fig. 5A; see fig. S7 for technical detail). This suggests that CReP-anchored protein synthesis at the ER remains active in the presence of Torin2, possibly due to exclusion of 4EBPs from the ER. This was evident with the cell cycle regulator p27Kip1 (CDKN1B), a template that is enriched on the ER and induced by amino acid starvation (25, 26). Amino acid starvation–induced p27Kip1 accumulation is unaffected in Torin2-treated cells but diminished in response to CReP depletion (Fig. 5B). Thus, CReP:eIF2α may enable an ER-localized, stress-refractory mode of translation that is protected from mTOR status.
Fig. 5. CReP depletion inhibits basal- and amino acid starvation–induced biosynthesis of p27, which is refractory to mTOR inhibition with Torin2.
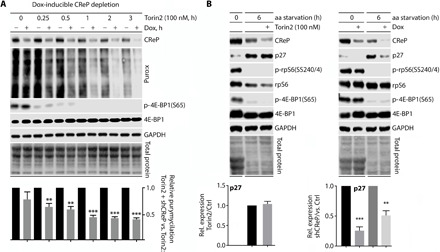
(A) HeLa cells with dox-inducible CReP depletion were mock- or dox-treated (~40 hours), treated with Torin2, puromycylated, and lysed at the indicated time points for immunoblot analysis with the indicated antibodies. Statistical significance was determined by two-tailed Student’s t test comparison between the dox/no-dox at the indicated time point (graphs represent mean and SEM, n = 3; **P < 0.005; ***P < 0.0005). (B) Left: Cells with dox-inducible CReP depletion were placed in aa-deprived media (see Materials and Methods) in the presence or absence of Torin2 and lysed at the indicated time points for immunoblot analysis. (B) Right: Cells with dox-inducible CReP depletion were mock- or dox-treated (~40 hours) before being placed in amino acid–deprived media and lysed at the indicated time points for immunoblot analysis. Bar graphs represent mean comparing dox/no dox at indicated time points and SEM. n = 3; *, **, *** corresponds to P < 0.05, 0.005, 0.0005, respectively.
Proximity-dependent labeling identifies CReP-controlled, ER-localized translation initiation machinery
Collectively, our findings suggest that eIF2α recruited to the ER (via CReP) is part of active initiation complexes, promoting localized translation that is recalcitrant to eIF2α(S51) phosphorylation (and other means of global protein synthesis suppression). To investigate this, we used Bio-ID/proteomics with dox-inducible endogenous CReP depletion with simultaneous overexpression of a BirA-CReP fusion construct, followed by streptavidin pull-down (27) and proteomic analysis (fig. S8). We confirmed that, as with WT CReP reconstitution, the BirA-CReP fusion rescues the effects of CReP depletion (fig. S8). From the list of 265 hits (table S1; see fig. S8 for details of the analytical approach), all eIF2 and PP1 subunits were identified, in addition to valosin-containing protein and β-transducin repeat containing E3 ubiquitin protein ligase, previously implicated in CReP turnover (Fig. 6A) (28, 29). NCK1, another known interactor of CReP (30), which was not identified by proteomics, was readily detected by immunoblot (fig. S9A). The BirA-CReP construct also prominently labeled the viral polyprotein in PVSRIPO-infected cells (table S1).
Fig. 6. CReP recruits the translation initiation apparatus to the ER.
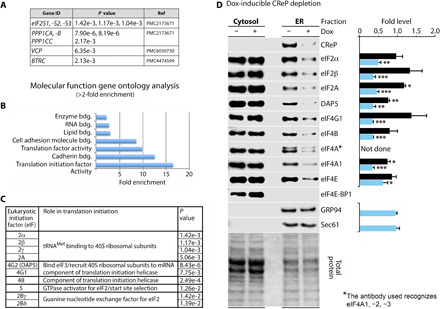
(A) Known CReP interactors identified in the BioID screen. (B) Results from GO molecular function analysis of the top 265 genes that were identified by quantitative liquid chromatography–tandem mass spectrometry (LC-MS/MS) according to P value (n = 3) (fig. S8). (C) Eukaryotic translation initiation factors identified in the screen. (D) Dox-inducible CReP depletion cells were mock- or dox-treated (40 hours) before fractionation as described in Fig. 4. Eukaryotic translation initiation factors in the cytosolic and ER fractions were interrogated by immunoblot and quantified. Statistical significance was determined by two-tailed Student’s t test comparison (black bars, cytosolic ratio + dox/−dox; blue bars, ER ratio + dox/−dox; graphs represent means and SEM; n = 3); *, **, *** corresponds to P < 0.05, 0.005, 0.0005, respectively. Sec61 and GRP94 were used as ER markers. GTPase, guanosine triphosphatase.
PANTHER analysis (31) revealed that the set of 265 hits was heavily enriched in genes encoding for translation and translation initiation factors (Fig. 6, B and C). We validated targets by immunoblot where suitable antibodies were available (fig. S9B). DAP5 (eIF4G2) is homologous with the C-terminal portion of eIF4G1; both assemble with eIF4B in the eIF4G:4A:4B translation initiation helicase (32). eIF2 and eIF5 are components of the 43S preinitiation complex (PIC). Thus, CReP-BirA affinity ligation identified key members of both major assemblies involved in translation initiation, eIF4F and the PIC. We further corroborated these findings by coimmunoprecipitation of CReP with flag-tagged DAP5, which binds directly to the β subunit of eIF2 and was the top hit among initiation factors in our screen (Fig. 6C and fig. S9C).
The CReP-BirA proximity ligation assays suggested CReP-dependent association of translation machinery involved in initiation at the ER. To test this hypothesis, we screened canonical translation initiation factors in fractionated lysates from cells with dox-inducible CReP depletion. CReP depletion dissociated all translation factors identified in the proximity ligation screen from the ER (Fig. 6D). In addition, despite not being identified by the screen, eIF4A1 (a binding partner of eIF4G1/DAP5 and eIF4B in the translation initiation helicase) and eIF4E (the cap-binding protein) lost association with the ER upon CReP depletion (Fig. 6D). Collectively, these data indicate that CReP:eIF2 is associated with active translation initiation complexes, and loss of CReP leads to the elimination of ER-associated translation initiation machinery.
DISCUSSION
Cells readily shut off protein synthesis when encountering stress to reduce energy consumption and readjust cellular activity toward regaining homeostasis. However, even the most severe challenges to cell survival categorically require some ongoing translation. We uncovered selective, protected translation in the presence of severe stress-induced protein synthesis repression, mediated by sequestration of translation initiation machinery via CReP:eIF2 at the ER. ER-localized CReP:eIF2 enabled local translation in the presence of virus-induced eIF2α(S51) phosphorylation and 4EBP-1 activation by mTOR inhibition.
Enteroviruses (e.g., PV/PVSRIPO) engage ribosomes through type 1 internal ribosomal entry sites (IRESs), recruiting eIF4G:eIF4A directly to viral RNA, independent of an m7G-cap and eIF4E (5). A similar scenario has been proposed for BiP (33). However, the small size of the unencumbered BiP 5′ untranslated region (90 nt) and a problematic bicistronic reporter approach put this claim in doubt. We have no evidence that local translation at the ER is restricted to m7G-cap-independent translation initiation; alongside all core translation initiation factors, the m7G-cap-binding protein eIF4E localizes at the ER in a CReP-dependent fashion. Enterovirus (type 1) IRESs are eIF2 dependent (5) and, hence, sensitive to eIF2α(S51) phosphorylation. This indicates that the physiologic significance of ER-resident translation initiation is isolation from p-eIF2α(S51) accumulation during infection rather than providing conditions conducive to cap-independent initiation.
CReP is known as a scaffold that facilitates p-eIF2α(S51) dephosphorylation (10). In this work, we identified a broader role for CReP in recruiting translation initiation machinery to the ER via eIF2 binding. Our proximity-dependent ligation investigations indicate that eIF2A, a single-subunit functional homolog of eIF2 (34), also may bind CReP. Further work must be performed to assess the role of eIF2A in CReP-dependent translation at the ER.
A missense mutation in the CReP eIF2α-binding motif (R658C; fig. S8) is linked to microcephaly, short stature, and premature diabetes (35, 36). The mechanisms defined here, implicating CReP in the cell’s coordination of protein synthesis upon acute stress, could well be at the root of this syndrome, as CReP:eIF2α binding may control rapid responses enabling homeostatic balance in the secretory pathway. In addition to its chaperone activity, BiP is a major regulator of ER calcium channel efflux (37, 38). In cells with high secretory capacity, such as neurons or β-islet cells, CReP:eIF2 may be critical for ER-bound biosynthesis of BiP in response to homeostatic perturbations that require BiP activity.
Our findings resonate with prior reports of protein synthesis in the cytosol and ER compartments being subject to distinct and independent regulation (17), as well as with ER-compartmentalized aminoacyl tRNA charging and hydrolysis (39). Exploitation of this system by viruses to enable viral translation during host protein synthesis suppression further emphasizes the broad cellular significance of CReP-mediated, ER-localized translation.
MATERIALS AND METHODS
Cell lines, siRNA transfections, and CReP expression plasmids
HeLa cells (American Type Culture Collection) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and nonessential amino acids. Transfections were performed 36 hours before infection at ~50% confluency with 50 pM siRNA and 5 μl of Lipofectamine RNAiMax (Invitrogen) per well (six-well plate) in serum-free media. Myc-/flag-tagged CReP was purchased from OriGene. The CReP open reading frame was polymerase chain reaction (PCR)–amplified with corresponding primer pair 5′-cgaaagcttgagcc ggggacaggcggatc-3′/5′-cgactcgagacattgcttgagaacattaagtcc-3′ and cloned into pcDNA5 FRT/TO (Invitrogen) in-between c-myc and flag epitopes similar to what has been described previously with eIF4G (40). The E655Stop mutation eliminating the eIF2 binding domain [CReP(ΔeIF2)] was introduced by a QuikChange Lightning Site-Directed Mutagenesis kit (Agilent) using primer pair 5′-ctgagtattatataagtggtgattaggatcgcaaaggacca-3′/5′-tggtcctttgcgatcctaatcaccacttatataatactcag-3′.
Stable cell lines, puromycylation assays, viral infections, inhibitors, and amino acid deprivation
Stable HeLa cell lines with dox-inducible CReP depletion were established using procedures previously used for dox-inducible eIF4G1 knockdown cell lines (40). Briefly, the miR-4G sequence in pcDNA3.1/TO (40) was replaced with CReP-specific microRNAs designed according to validated shRNA clones: 5′-gcctcgagatctgcgcggcctg tctgtgttaggctaaagtgaagccacagatg-3′/5′-gcctcgaggatccgcatggcctgt ctatgttaggctaaacatctgtggcttcac-3′. CReP knockdown cell lines were selected and maintained in DMEM containing 10% FBS, nonessential amino acids, G418 (800 μg/ml), blasticidin (5 μg/ml), and zeocin (100 μg/ml) (Invitrogen). Reconstitution cell lines were established with WT CReP, CReP(ΔeIF2), and BirA-CReP expression plasmids. The dox-inducible flag-DAP5 cell line has been described previously (41). Reconstitution cell lines were maintained in the same media as above, with hygromycin B (100 μg/ml; Corning) rather than zeocin selection. For puromycylation assays, 10 μM puromycin (Sigma-Aldrich) dissolved in dimethyl sulfoxide (DMSO) was added to the cells 8 min before lysis. PVSRIPO and CBV3 infections were carried out at an MOI of 10, unless indicated otherwise, in DMEM supplemented with 2% FBS and nonessential amino acids. ISRIB, cycloheximide (Sigma-Aldrich), tunicamycin, thapsigargin, and Torin2 (Tocris) were dissolved in DMSO and used at the concentration indicated. Cells were amino acid starved by being placed in DMEM without serum, d-glucose, l-histidine, l-leucine, or sodium pyruvate (Gibco) for the indicated amount of time.
Immunoprecipitation, immunoblotting, antibodies, reverse transcription quantitative PCR analyses
Cell lysate preparation, immunoprecipitation (IP) with anti-c-Myc-agarose beads (Thermo Fisher Scientific), and immunoblotting were performed as described previously (42, 43). For IPs, 70% confluent cultures grown in 150-mm dishes were treated with dox (4 μg/ml; 36 hours) and lysed with radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich) and Halt Protease-Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). After overnight incubation for Myc IP, beads were washed four times in RIPA buffer and processed for immunoblotting. Streptavidin pulldown was performed with the same protocol using anti-streptavidin agarose beads (Thermo Fisher Scientific) and elution with 200 mM NaCl, 100 mM tris (pH 8.0) (Sigma-Aldrich), 2% SDS, and 1 mM Biotin (Sigma-Aldrich) at 60°C (30 min). Lysates from noninduced cells were used as a negative IP control. Before IP, all cell lysates were analyzed by immunoblot to ensure equal loading. Total protein stains were performed with Revert Total Protein Stain (Li-COR) following manufacturer’s protocol. All signal quantifications were normalized to total protein unless indicated otherwise. Antibodies against CReP, GADD34, Sec61B, eIF2A (Proteintech), GRP94 (C. Nicchitta, Duke University), eIF2β (Novus), puromycin (Millipore), PV 2C (42), CVB3 3D (a gift from K. Klingel, University of Tübingen), flag (Sigma-Aldrich), PKR, PERK, p-eIF2α, eIF2α, PARP, ATF4, BiP, p-4EBP1 (s65) 4EBP1, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), rpS6, p27 Kip1, eIF4AI, eIF4A all, eIF4G1, DAP5, eIF4B, p-ERK1/2 (p44/42), ERK1/2, Tubulin, NCK1, eIF4E, and p-rpS6 (S240/4) (all from Cell Signaling Technology) were used in this study. Immunoblots were developed with SuperSignal West Pico (Thermo Fisher Scientific) or WesternBright (BioExpress) chemiluminescence (ECL) kit. For reverse transcription quantitative PCR (RT-qPCR), whole-cell RNA was isolated using TRIzol (Invitrogen) and deoxyribonuclease-treated before cDNA synthesis (Invitrogen SuperScript III) following manufacturer’s protocol. RT-qPCR was performed on a QuantStudio3 machine (Thermo Fisher Scientific) using SYBR green (Thermo Fisher Scientific). BiP Primers: 5′-gaaagaaggttacccatgc agt-3′/5′-caggcca taagcaatagcagc-3′; GAPDH primers: 5′-ggggccatccacagtcttct-3′/5′-atgcctcctgcaccaccaac-3′.
Sequential detergent fractionation, confocal microscopy
Detergent fractionations were performed on ice with cells at 50 to 75% confluence. Cytosol fractions were extracted for 8 min with permeabilization buffer [0.03% digitonin (Sigma-Aldrich)] in 110 mM KCl, 25 mM MgCl2, 1 mM dithiothreitol (DTT), and Halt Protease-Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). After recovery of the cytosolic fraction, the cells were washed in the same buffer supplemented with 0.008% digitonin. The wash fraction was combined with the cytosolic extract. The digitonin-extracted cells were then lysed for 8 min in a buffer containing 2% dodecylmaltoside (Sigma-Aldrich) in 200 mM KCl, 25 mM Hepes (pH 7.4), 10 mM MgCl2, 1 mM DTT, and Halt Protease-Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific) [adapted from (18)]. Subcellular fractions were loaded at equal volumes for immunoblot, resulting in a 2:1 ER:Cytosol cell equivalency ratio. For confocal microscopy, knockdown/WT-CReP reconstitution cells were plated on coverslips, treated with dox for 24 hours before infection (MOI, 10; 4.5 hours). Cells were then washed with phosphate-buffered saline (PBS) and fixed in ice-cold methanol (5 min). After several PBS washes, cells were blocked with 5% goat serum (2 hours, 20°C) and then placed in primary antibody [CReP (Proteintech); J2 (SCICONS)] at a dilution of 1:500 overnight at 4°C. Cells were once again washed and incubated with secondary antibody (1:500; 1 hour, 20°C). In the final 15 min of secondary incubation, 4′,6-diamidino-2-phenylindole (DAPI) stain (Thermo Fisher Scientific; 1:1000) was added. Coverslips were then imaged on a confocal microscope (Leica SP8) at 64× magnification.
Statistical analysis
Quantification of immunoblot signals was performed using the Li-COR Odyssey FC2 imaging system and Image Studio software. All experiments were repeated at least three times, unless indicated otherwise. Quantified immunoblot data were normalized between experiments as described in the figure legends and were represented as averages and SEM. The Student’s t test was used unless indicated otherwise. Significance was defined as a P value of <0.05, and the tests used for each data group are described in the figure legends. GO (gene ontology) analysis with the proteomic dataset was performed with the Panther Classification system “molecular function” annotation dataset.
Proteomics–Sample preparation
The Duke Proteomics Core Facility received nine samples [three of each CReP_BID_N (−dox control sample), CReP_BID_P (+dox sample), and CReP_BID_ Inf (+dox + PVSRIPO sample)]. Samples were reduced with 10 mM DTT (30 min, 80°C) and alkylated with 20 mM iodoacetamide (30 min, 20°C). Next, they were supplemented with 50 μl of 20% SDS in 50 mM triethylammonium bicarbonate (TEAB), a final concentration of 1.2% phosphoric acid, and 1024 μl of S-Trap (ProtiFI) binding buffer (90% MeOH/100 mM TEAB). Proteins were trapped on the S-Trap, digested using sequencing grade trypsin (20 ng/μl; Promega) (1 hour, 47°C), and eluted with 50 mM TEAB, followed by 0.2% FA, and lastly 50% acetonitrile (ACN)/0.2% formic acid. All samples were then lyophilized to dryness and resuspended in 20 μl 1% trifluoroacetic acid/2% ACN containing yeast alcohol dehydrogenase (12.5 fmol/μl; ADH_YEAST). From each sample, 3 μl was removed to create a QC Pool sample, which was run periodically throughout the acquisition period.
Proteomics—Quantitative analysis and methods
Quantitative liquid chromatography–tandem mass spectrometry (LC-MS/MS) was performed on 4 μl of each sample, using a nanoAcquity UPLC system (Waters Corp) coupled to a Thermo Fusion Lumos high-resolution accurate mass tandem mass spectrometer (Thermo) via a nanoelectrospray ionization source. Briefly, the sample was first trapped on a Symmetry C18 20 mm by 180 μm trapping column [5 μl/min at 99.9/0.1 (v/v) water/acetonitrile], after which the analytical separation was performed using a 1.8 μm Acquity HSS T3 C18 75 μm by 250 mm column (Waters Corp.) with a 90-min linear gradient of 5 to 30% acetonitrile with 0.1% formic acid at a flow rate of 400 nl/min with a column temperature of 55°C. Data collection on the Fusion Lumos mass spectrometer was performed in a data-dependent acquisition mode of acquisition with a r = 120,000 [at mass/charge ratio (m/z) 200] full MS scan from m/z 375 to 1500 with a target automatic gain control (AGC) value of 2 × 105 ions followed by 30 MS/MS scans at r = 15,000 (at m/z 200) at a target AGC value of 5 × 104 ions and 45 ms. A 20-s dynamic exclusion was used to increase depth of coverage. The total analysis cycle time for each sample injection was approximately 2 hours. Following 13 total UPLC-MS/MS analyses [excluding conditioning runs but including four replicate quality control (QC) injections; table S1_TAB2], data were imported into Proteome Discoverer 2.2 (Thermo Scientific Inc.), and analyses were aligned based on the accurate mass and retention time of detected ions (“features”) using Minora Feature Detector algorithm in Proteome Discoverer. Relative peptide abundance was calculated on the basis of area under the curve of the selected ion chromatograms of the aligned features across all runs. The MS/MS data were searched against the Swiss-Prot H. sapiens database (downloaded in November 2017) and NCBIprot (“other virus”) with additional proteins, including yeast ADH1, bovine serum albumin, as well as an equal number of reversed-sequence “decoys”) false discovery rate determination. Mascot Distiller and Mascot Server (v 2.5, Matrix Sciences) were used to produce fragment ion spectra and to perform the database searches. Database search parameters included fixed modification on Cys (carbamidomethyl) and variable modifications on Meth (oxidation) and Asn and Gln (deamidation). Peptide Validator and Protein FDR Validator nodes in Proteome Discoverer were used to annotate the data at a maximum 1% protein false discovery rate.
Proteomics—Quantitative LC-MS/MS analysis
Four microliters of peptide digests (~33% of each sample) were analyzed by ultraperformance LC-MS/MS. A QC pool containing an equal mixture of each sample was analyzed at the beginning, middle, and end of the sample set (four times total). Next, data were imported into Proteome Discoverer 2.2 (Thermo Scientific Inc.), and all LC-MS/MS runs were aligned on the basis of the accurate mass and retention time of detected ions (features), which contained MS/MS spectra using Minora Feature Detector algorithm in Proteome Discoverer. Missing values were imputed after sample loading and trim mean (top and bottom 10%) normalization in the following manner. If less than half of the values are missing in a treatment group, then values are imputed with an intensity derived from a normal distribution defined by measured values within the same intensity range (20 bins). If greater than half values are missing for a peptide in a group and a peptide intensity is >5 × 106, then it was concluded that peptide was misaligned and its measured intensity is set to 0. All remaining missing values are imputed with the lowest 5% of all detected values. The following analyses are based on these normalized values. The overall dataset had 327,167 peptide spectral matches. In addition, 644,830 MS/MS spectra were acquired for peptide sequencing by database searching. Following database searching and peptide scoring using Proteome Discoverer validation, the data were annotated at a 1% protein false discovery rate, resulting in identification of 1798 proteins. Raw expression levels of peptides and proteins are presented in table S1_TAB3. Intensity normalized protein level data are shown in table S1_TAB4. Please note that all subsequent analyses were from these normalized protein levels.
Proteomics—Measures of analytical versus biological variability and initial statistical analysis
To assess technical reproducibility, we calculated % coefficient of variation (%CV) for each protein across the four injections of a QC pool that were interspersed throughout the study (table S1_TAB2). To assess biological + technical variability, %CVs were measured for each protein across the individual groups. The mean %CV of the QC pools was 12.7% for all proteins (table S1_TAB4). Variability of the biological samples (analytical variability plus biological variability) was 24.0, 22.7, and 28.7% for the CReP N (−), CReP P (+), and CReP Inf groups, respectively. These data were reflected in the principal components analysis (PCA) of all quantified proteins, where the QC pool samples were tightly clustered (fig. S10). There was a clear differentiation (based on all n = 1798 proteins) between CReP_BID_N and CReP_BID_P samples; there was one outlier according to PCA in the CReP_BID_Inf sample (fig. S10). As an initial statistical analysis, we calculated fold changes between groups based on the median fold change. In addition, we performed a two-tailed heteroscedastic t test on log2-transformed data for each of these comparisons. Briefly, proteins were filtered to include those with a twofold greater expression and a P value of <0.05. This resulted in 355 and 371 proteins in CReP_BID_Inf versus CReP_BID_N (−) (table S1_TAB5) and CReP_BID_P versus CReP_BID_N (table S1_TAB6), respectively. Last, CReP_BID_Inf versus CReP_BID_P (table S1_TAB7) contains proteins that pass the fold change and P value thresholds that are also present in either (table S1_TAB5) or (table S1_TAB6).
Supplementary Material
Acknowledgments
We thank the Duke Proteomics Core Facility: T. Ho (sample preparation, data collection, data analysis, report writing), E. J. Soderblom (study design, data collection, data analysis, report writing), M. A. Moseley (scientific oversight). We thank M. Tremblay (Duke University) for assistance with RT-qPCR. In addition, we thank A. Hoffman (Duke University) for providing the BirA-CReP construct. Funding: This work was supported by NS108773 (M.G.) and a grant from Hope & Gavin Wolfe. Author contributions: J.P.K. and M.G. conceived and designed the study. J.P.K. performed all experiments except for the DAP5 IP’s, which were performed by J.D.B.; E.Y.D. assisted with cloning for the entire project. J.P.K. and M.G. prepared and edited all figures. J.P.K., J.D.B., E.Y.D., and M.G. contributed to the writing and editing of the manuscript and approved the manuscript. Competing interests: M.G. owns intellectual property related to PVSRIPO, which has been licensed to Istari Oncology Inc. M.G. is a co-founder of, advisor to, and equity holder in Istari Oncology Inc. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the corresponding authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/23/eaba0745/DC1
REFERENCES AND NOTES
- 1.Wek R. C., Jiang H. Y., Anthony T. G., Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34, 7–11 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Sudhakar A., Ramachandran A., Ghosh S., Hasnain S. E., Kaufman R. J., Ramaiah K. V., Phosphorylation of serine 51 in initiation factor 2 α (eIF2 α) promotes complex formation between eIF2 α(P) and eIF2B and causes inhibition in the guanine nucleotide exchange activity of eIF2B. Biochemistry 39, 12929–12938 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Walter P., Ron D., The unfolded protein response: From stress pathway to homeostatic regulation. Science 334, 1081–1086 (2011). [DOI] [PubMed] [Google Scholar]
- 4.He B., Gross M., Roizman B., The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the α subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U.S.A. 94, 843–848 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sweeney T. R., Abaeva I. S., Pestova T. V., Hellen C. U., The mechanism of translation initiation on Type 1 picornavirus IRESs. EMBO J. 33, 76–92 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White J. P., Reineke L. C., Lloyd R. E., Poliovirus switches to an eIF2-independent mode of translation during infection. J. Virol. 85, 8884–8893 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown M. C., Holl E. K., Boczkowski D., Dobrikova E., Mosaheb M., Chandramohan V., Bigner D. D., Gromeier M., Nair S. K., Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci. Transl. Med. 9, eaan4220 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlegel A., Giddings T. H. Jr., Ladinsky M. S., Kirkegaard K., Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 70, 6576–6588 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu N. Y., Ilnytska O., Belov G., Santiana M., Chen Y. H., Takvorian P. M., Pau C., van der Schaar H., Kaushik-Basu N., Balla T., Cameron C. E., Ehrenfeld E., van Kuppeveld F. J., Altan-Bonnet N., Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141, 799–811 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jousse C., Oyadomari S., Novoa I., Lu P., Zhang Y., Harding H. P., Ron D., Inhibition of a constitutive translation initiation factor 2α phosphatase, CReP, promotes survival of stressed cells. J. Cell Biol. 163, 767–775 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pobre K. F. R., Poet G. J., Hendershot L. M., The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 294, 2098–2108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novoa I., Zhang Y., Zeng H., Jungreis R., Harding H. P., Ron D., Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 22, 1180–1187 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarnow P., Translation of glucose-regulated protein 78/immunoglobulin heavy-chain binding protein mRNA is increased in poliovirus-infected cells at a time when cap-dependent translation of cellular mRNAs is inhibited. Proc. Natl. Acad. Sci. U.S.A. 86, 5795–5799 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sidrauski C., Acosta-Alvear D., Khoutorsky A., Vedantham P., Hearn B. R., Li H., Gamache K., Gallagher C. M., Ang K. K., Wilson C., Okreglak V., Ashkenazi A., Hann B., Nader K., Arkin M. R., Renslo A. R., Sonenberg N., Walter P., Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife 2, e00498 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kloft N., Neukirch C., von Hoven G., Bobkiewicz W., Weis S., Boller K., Husmann M., A subunit of eukaryotic translation initiation factor 2α-phosphatase (CreP/PPP1R15B) regulates membrane traffic. J. Biol. Chem. 287, 35299–35317 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Starck S. R., Tsai J. C., Chen K., Shodiya M., Wang L., Yahiro K., Martins-Green M., Shastri N., Walter P., Translation from the 5′ untranslated region shapes the integrated stress response. Science 351, aad3867 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stephens S. B., Nicchitta C. V., Divergent regulation of protein synthesis in the cytosol and endoplasmic reticulum compartments of mammalian cells. Mol. Biol. Cell 19, 623–632 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jagannathan S., Nwosu C., Nicchitta C. V., Analyzing mRNA localization to the endoplasmic reticulum via cell fractionation. Methods Mol. Biol. 714, 301–321 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X., Shu L., Hosoi H., Murti K. G., Houghton P. J., Predominant nuclear localization of mammalian target of rapamycin in normal and malignant cells in culture. J. Biol. Chem. 277, 28127–28134 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Kozutsumi Y., Segal M., Normington K., Gething M. J., Sambrook J., The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332, 462–464 (1988). [DOI] [PubMed] [Google Scholar]
- 21.Kim Y., Park J., Kim S., AKim M., Kang M.-G., Kwak C., Kang M., Kim B., Rhee H.-W., Kim V. N., PKR Senses Nuclear and Mitochondrial Signals by Interacting with Endogenous Double-Stranded RNAs. Mol. Cell 71, 1051–1063.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Egger D., Teterina N., Ehrenfeld E., Bienz K., Formation of the poliovirus replication complex requires coupled viral translation, vesicle production, and viral RNA synthesis. J. Virol. 74, 6570–6580 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saxton R. A., Sabatini D. M., mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Q., Xu C., Kirubakaran S., Zhang X., Hur W., Liu Y., Kwiatkowski N. P., Wang J., Westover K. D., Gao P., Ercan D., Niepel M., Thoreen C. C., Kang S. A., Patricelli M. P., Wang Y., Tupper T., Altabef A., Kawamura H., Held K. D., Chou D. M., Elledge S. J., Janne P. A., Wong K. K., Sabatini D. M., Gray N. S., Characterization of Torin2, an ATP-competitive inhibitor of mTOR, ATM, and ATR. Cancer Res. 73, 2574–2586 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reid D. W., Nicchitta C. V., Primary role for endoplasmic reticulum-bound ribosomes in cellular translation identified by ribosome profiling. J. Biol. Chem. 287, 5518–5527 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leung-Pineda V., Pan Y., Chen H., Kilberg M. S., Induction of p21 and p27 expression by amino acid deprivation of HepG2 human hepatoma cells involves mRNA stabilization. Biochem. J. 379, 79–88 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roux K. J., Kim D. I., Burke B., BioID: A screen for protein-protein interactions. Curr. Protoc. Protein Sci. 74, 19.23.1–19.23.14 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Hulsmann J., Kravic B., Weith M., Gstaiger M., Aebersold R., Collins B. C., Meyer H., AP-SWATH Reveals Direct Involvement of VCP/p97 in Integrated Stress Response Signaling Through Facilitating CReP/PPP1R15B Degradation. Mol. Cell. Proteomics 17, 1295–1307 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loveless T. B., Topacio B. R., Vashisht A. A., Galaang S., Ulrich K. M., Young B. D., Wohlschlegel J. A., Toczyski D. P., DNA Damage Regulates Translation through β-TRCP Targeting of CReP. PLOS Genet. 11, e1005292 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Latreille M., Larose L., Nck in a complex containing the catalytic subunit of protein phosphatase 1 regulates eukaryotic initiation factor 2α signaling and cell survival to endoplasmic reticulum stress. J. Biol. Chem. 281, 26633–26644 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Mi H., Dong Q., Muruganujan A., Gaudet P., Lewis S., Thomas P. D., PANTHER version 7: Improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 38, D204–D210 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson R. J., Hellen C. U., Pestova T. V., The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 11, 113–127 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho S., Park S. M., Kim T. D., Kim J. H., Kim K. T., Jang S. K., BiP internal ribosomal entry site activity is controlled by heat-induced interaction of NSAP1. Mol. Cell. Biol. 27, 368–383 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zoll W. L., Horton L. E., Komar A. A., Hensold J. O., Merrick W. C., Characterization of mammalian eIF2A and identification of the yeast homolog. J. Biol. Chem. 277, 37079–37087 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Kernohan K. D., Tétreault M., Liwak-Muir U., Geraghty M. T., Qin W., Venkateswaran S., Davila J.; CareRare Canada Consortium, Holcik M., Majewski J., Richer J., Boycott K. M., Homozygous mutation in the eukaryotic translation initiation factor 2α phosphatase gene, PPP1R15B, is associated with severe microcephaly, short stature and intellectual disability. Hum. Mol. Genet. 24, 6293–6300 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abdulkarim B., Nicolino M., Igoillo-Esteve M., Daures M., Romero S., Philippi A., Senée V., Lopes M., Cunha D. A., Harding H. P., Derbois C., Bendelac N., Hattersley A. T., Eizirik D. L., Ron D., Cnop M., Julier C., A Missense Mutation in PPP1R15B Causes a Syndrome Including Diabetes, Short Stature, and Microcephaly. Diabetes 64, 3951–3962 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schauble N., Lang S., Jung M., Cappel S., Schorr S., Ulucan Ö., Linxweiler J., Dudek J., Blum R., Helms V., Paton A. W., Paton J. C., Cavalié A., Zimmermann R., BiP-mediated closing of the Sec61 channel limits Ca2+ leakage from the ER. EMBO J. 31, 3282–3296 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Higo T., Hamada K., Hisatsune C., Nukina N., Hashikawa T., Hattori M., Nakamura T., Mikoshiba K., Mechanism of ER stress-induced brain damage by IP(3) receptor. Neuron 68, 865–878 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Negrutskii B. S., Deutscher M. P., Channeling of aminoacyl-tRNA for protein synthesis in vivo. Proc. Natl. Acad. Sci. U.S.A. 88, 4991–4995 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobrikov M. I., Shveygert M., Brown M. C., Gromeier M., Mitotic phosphorylation of eukaryotic initiation factor 4G1 (eIF4G1) at Ser1232 by Cdk1:cyclin B inhibits eIF4A helicase complex binding with RNA. Mol. Cell. Biol. 34, 439–451 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bryant J. D., Brown M. C., Dobrikov M. I., Dobrikova E. Y., Gemberling S. L., Zhang Q., Gromeier M., Regulation of HIF-1α during Hypoxia by DAP5-Induced Translation of PHD2. Mol. Cell. Biol. 38, e00647-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobrikov M. I., Dobrikova E. Y., Gromeier M., Ribosomal RACK1:PKCβII modulates intramolecular interactions between unstructured regions of eIF4G that control eIF4E and eIF3 binding. Mol. Cell. Biol. 38, e00306-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dobrikov M. I., Dobrikova E. Y., Gromeier M., Ribosomal RACK1:PKCβII phosphorylates eIF4G1(S1093) to modulate cap-dependent and -independent translation initiation. Mol. Cell. Biol. , (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/23/eaba0745/DC1