

This study suggests that agents able to enhance the activity of β-arrestin-1 in AgRP neurons may be useful as antidiabetic drugs.
Abstract
β-Arrestin-1 and β-arrestin-2 have emerged as important signaling molecules that modulate glucose fluxes in several peripheral tissues. The potential roles of neuronally expressed β-arrestins in regulating glucose homeostasis remain unknown. We here report that mice lacking β-arrestin-1 (barr1) selectively in AgRP neurons displayed impaired glucose tolerance and insulin sensitivity when consuming an obesogenic diet, while mice overexpressing barr1 selectively in AgRP neurons were protected against obesity-associated metabolic impairments. Additional physiological, biochemical, and electrophysiological data indicated that the presence of barr1 is essential for insulin-mediated hyperpolarization of AgRP neurons. As a result, barr1 expressed by AgRP neurons regulates efferent neuronal pathways that suppress hepatic glucose production and promote lipolysis in adipose tissue. Mice lacking β-arrestin-2 (barr2) selectively in AgRP neurons showed no substantial metabolic phenotypes. Our data suggest that agents able to enhance the activity of barr1 in AgRP neurons may prove beneficial as antidiabetic drugs.
INTRODUCTION
The ongoing obesity epidemic represents a major threat to human health worldwide. Obesity is associated with numerous comorbidities, including type 2 diabetes (T2D) and fatty liver disease (1–4). It is well known that distinct areas of the brain play important roles in maintaining euglycemia (5–8). In particular, neuronal subpopulations of the arcuate nucleus of the hypothalamus (ARC) have been studied in great detail because of their known effects on key metabolic functions (6, 9–11). Agouti-related protein (AgRP) neurons of the ARC store and release several orexigenic agents, including AgRP, neuropeptide Y (NPY), and γ-aminobutyric acid, which strongly promote food intake (12–14). Recent studies suggest that distinct AgRP neuronal circuits can also modulate peripheral glucose metabolism and carbohydrate utilization independent of changes in food intake (15, 16).
The function of AgRP neurons and other hypothalamic neurons is regulated by various nutrients, metabolites, and hormones, including insulin and leptin (7, 8, 10, 14, 17). In turn, efferent signals from AgRP neurons and other ARC neurons control hepatic glucose production (HGP), the activity of brown adipose tissue (BAT), and the secretion of hormones from pancreatic islets (5, 16, 18, 19).
Studies with mutant mice selectively lacking the insulin receptor (IR) in AgRP neurons indicated that insulin action on AgRP neurons is essential for insulin-induced suppression of HGP (18). Similar findings were obtained after down-regulation of IR expression in the mediobasal hypothalamus (20). Transsection of the hepatic branch of the vagus nerve abolished the ability of central insulin to suppress HGP (21). These data support the concept that physiological increases in circulating insulin levels activate a brain (hypothalamus)–liver circuit that restrains HGP. Since elevated HGP is a main cause of fasting and postprandial hyperglycemia in T2D (22–24), enhancing hypothalamic insulin signaling may prove clinically beneficial to suppress HGP in T2D.
As is the case with essentially all other cell types, the activity of AgRP neurons is predicted to be regulated by cell surface receptors belonging to the superfamily of G protein–coupled receptors (GPCRs) (25–27). GPCR signaling is modulated by a pair of proteins known as β-arrestin-1 and β-arrestin-2 (barr1 and barr2, respectively), which are well known for their role in mediating GPCR desensitization and internalization (28). However, more recent studies indicate that barr1/2 can also act as signaling proteins in their own right (29–31). Like other key signaling molecules, the two β-arrestins are expressed by virtually every cell type (28, 32).
Studies with whole-body β-arrestin knockout (KO) mice have shown that β-arrestins play important roles in maintaining whole-body glucose and energy homeostasis (33, 34). Several studies have examined β-arrestin–dependent regulation of metabolically relevant, peripheral cell types, including pancreatic β cells (35, 36), hepatocytes (33, 37), and adipocytes (38). These studies demonstrated that β-arrestins expressed in peripheral tissues are required for the maintenance of euglycemia (33, 35, 37, 38).
It should be noted that β-arrestins have been implicated in regulating insulin signaling in hepatocytes and pancreatic β cells (33, 36), raising the possibility that barr1 and/or barr2 may also modulate insulin action in other tissues and cell types including neurons. At present, it remains completely unknown whether neuronal barr1/2 also contributes to the regulation of energy and glucose homeostasis. Thus, we examined whether barr1 and barr2 play a role in modulating the activity of AgRP neurons and, if so, whether this regulation modulates glucose homeostasis including peripheral insulin action. As experimental tools, we generated mice that lacked barr1 or barr2 selectively in AgRP neurons, as well as mice that selectively overexpressed barr1 in AgRP neurons.
We made the unexpected observation that mutant mice lacking barr1 selectively in AgRP neurons (AgRP-barr1-KO mice) showed notable impairments in glucose tolerance and insulin sensitivity when maintained on an obesogenic diet. In agreement with this observation, mice that overexpressed barr1 selectively in AgRP neurons showed pronounced improvements in glucose homeostasis and insulin sensitivity. We also provide additional data that shed light on the mechanisms underlying these unexpected metabolic phenotypes.
To the best of our knowledge, this is the first study demonstrating that a neuronally expressed β-arrestin isoform (barr1) exerts major effects on key metabolic functions including glucose tolerance and insulin sensitivity. Our data suggest that strategies aimed at enhancing the activity of barr1 in AgRP neurons may prove useful for the development of new classes of antidiabetic drugs.
RESULTS
Generation of AgRP-barr1-KO mice
To explore the potential functional roles of barr1 in AgRP neurons, we selectively inactivated barr1 in AgRP neurons. Specifically, we crossed floxed barr1 mice (barr1 f/f mice; genetic background: C57BL/6J) (39) with mice expressing Cre recombinase under the control of AgRP promoter (AgRP-Ires-Cre mice) (40). These matings yielded AgRP-Cre-barr1 f/f mice carrying the AgRP-Cre transgene (AgRP-Cre-barr1 f/f mice) and barr1 f/f control littermates. To confirm that Cre was selectively expressed in AgRP neurons of AgRP-Cre-barr1 f/f mice, we crossed these mutant mice with Z/EG reporter mice that express green fluorescent protein (GFP) in a Cre-dependent manner (41). Figure 1A shows that the resulting AgRP-Cre-Z/EG-barr1 f/f mice selectively expressed GFP in AgRP neurons. Thus, we refer to the AgRP-Cre-barr1 f/f mutant mice simply as AgRP-barr1-KO mice throughout the manuscript. Barr1 f/f mice lacking the AgRP-Cre transgene served as control mice in all experiments where AgRP-barr1-KO mice were studied.
Fig. 1. HFD AgRP-barr1-KO mice show impairments in glucose homeostasis.
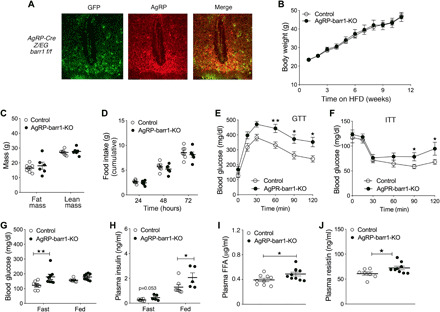
(A) Representative immunofluorescence images showing Cre activity in AgRP neurons of AgRP-Cre-Z/EG-barr1 f/f mice. In the left panel, only Cre-expressing neurons display GFP fluorescence. In the center panel, AgRP neurons were identified with an anti-AgRP antibody. (B) Body weights of AgRP-barr1-KO and control mice maintained on an HFD (HFD feeding was initiated when mice were 6 weeks old). (C) Fat and lean mass of HFD AgRP-barr1-KO and control mice (age, 20 weeks; 14 weeks on HFD). (D) Food intake (cumulative over 3 days) of HFD AgRP-barr1-KO and control mice (age, 20 weeks; 13 weeks on HFD). (E) Glucose tolerance test (GTT; 1 g glucose/kg i.p.) carried out with HFD AgRP-barr1-KO and control mice (age, 14 weeks; 8 weeks of HFD). (F) Insulin tolerance test (ITT; 0.75 U/kg i.p.) performed with HFD AgRP-barr1-KO and control mice (age, 15 weeks; 9 weeks on HFD). (G and H) Fasting and fed blood glucose (G) and plasma insulin (H) levels (age, 14 to 16 weeks; 8 to 10 weeks on HFD). (I and J) Plasma FFA (I) and resistin (J) levels (age, 14 to 16 weeks; 8 to 10 weeks on HFD). Mice had free access to food. Male mice were used for all studies. Data are given as means ± SEM (n = 5 to 9 per group). *P < 0.05; **P < 0.01 [two-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc test (E and F) and two-tailed Student’s t test (G to J)].
AgRP-barr1-KO mice show impaired glucose and insulin tolerance when consuming a calorie-rich diet
When maintained on regular mouse chow, AgRP-barr1-KO mice (males) and their control littermates did not show any significant differences in body weight, glucose tolerance, insulin sensitivity, and blood glucose and plasma insulin levels (fig. S1, A to E). We therefore challenged mice with a high-fat diet (HFD) to induce obesity and obesity-associated metabolic deficits including glucose intolerance and insulin resistance. AgRP-barr1-KO mice and their control littermates consumed the HFD for up to 12 weeks. Figure 1B shows that the lack of barr1 in AgRP neurons had no significant effect on HFD-induced weight gain. Similarly, no significant differences in lean and fat body mass (Fig. 1C) and in food intake (Fig. 1D) were observed between the two groups.
We next subjected the HFD AgRP-barr1-KO and control mice to a series of in vivo metabolic tests. Notably, HFD AgRP-barrr1 KO mice showed significantly impaired glucose tolerance (Fig. 1E) and elevated blood glucose levels 90 and 120 min after injection of insulin [0.75 U/kg intraperitoneally (i.p.); Fig. 1F], as compared to their control littermates. Fasting blood glucose (Fig. 1G), fed plasma insulin (Fig. 1H), and fed plasma free fatty acid (FFA) (Fig. 1I) levels were significantly increased in the barr1 mutant mice. Plasma resistin levels were also markedly elevated in AgRP-barr1-KO mice (Fig. 1J), while the plasma levels of other proinflammatory factors (tumor necrosis factor–α, interleukin-10, and monocyte chemoattractant protein-1) remained unchanged by the lack of barr1 in AgRP neurons (fig. S2, A to C). Glucose-stimulated insulin secretion did not differ significantly between HFD AgRP-barr1-KO and control mice (fig. S2D). These data indicated that HFD AgRP-barr1-KO mice showed impaired glucose homeostasis without concomitant changes in body weight and food intake.
We also demonstrated that the HFD control mice used for these studies (barr1 f/f mice) did not differ significantly from HFD AgRP-Cre and HFD wild-type mice (genetic background of all mice: C57BL/6) in body weight gain, glucose tolerance, and fed and fasting blood glucose and plasma insulin levels (fig. S3; note that mice were maintained on the HFD for up to 17 weeks). This observation indicated that the barr1 f/f mice were well suited to serve as control animals for these metabolic studies.
HFD AgRP-barr1-KO mice display impaired hepatic insulin action
To gain insight into the mechanisms underlying the observed metabolic deficits displayed by the HFD AgRP-barr1-KO mice, we performed euglycemic-hyperinsulinemic clamp studies. The HFD AgRP-barr1-KO mice and their control littermates used for this work had similar body weights, as expected (fig. S4A). During the clamp studies, mice received an insulin infusion at a rate of 3 mU kg−1 min−1. Glucose was infused at a variable rate to maintain euglycemia (Fig. 2A). Plasma glucose specific activity data are shown in fig. S4B to validate steady state. The glucose infusion rate (GIR) required to maintain euglycemia was greatly reduced in the HFD AgRP-barr1-KO mice (Fig. 2B), indicative of systemic insulin resistance. Before the clamp (basal conditions), HGP was significantly increased in the barr1 mutant mice (Fig. 2C). Similarly, the HFD AgRP-barr1-KO mice showed a strong trend toward increased HGP during the hyperinsulinemic clamp (steady-state conditions; 90 to 120 min) (P = 0.053; Fig. 2D), consistent with hepatic insulin resistance. Plasma insulin levels (in nanograms per milliliter) were similar between the two groups of mice during the clamp (KO, 3.41 ± 0.31; control, 2.78 ± 0.98; basal insulin levels before clamping were: KO, 2.85 ± 0.78; control, 0.69 ± 0.13; n = 5 per group). The rate of glucose disposal (Rd) did not differ significantly between barr1 mutant and control mice (fig. S4C).
Fig. 2. HFD AgRP-barr1-KO mice exhibit increased HGP and impaired hepatic insulin signaling.
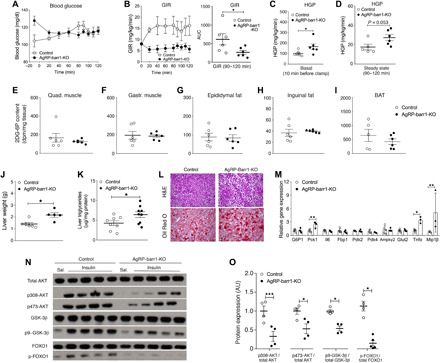
All studies were carried out with male HFD AgRP-barr1-KO and control littermates. (A to D) Euglycemic-hyperinsulinemic clamp studies. (A) Blood glucose levels. (B) GIR. AUC, area under the curve. (C and D) HGP under basal (C) and steady-state conditions (D). (E to I) Tissue glucose uptake by skeletal muscle and adipose tissues after intraperitoneal insulin (0.75 U/kg) injection. (J) Liver weights (HFD for 12 weeks; age, 20 weeks). (K) Liver triglyceride levels (HFD for 12 weeks; age, 20 weeks). (L) Histological staining of liver sections (HFD for 12 weeks; age, 20 weeks). (M) Hepatic expression levels of genes involved in gluconeogenesis and inflammatory processes (HFD for 12 weeks; age, 20 weeks). (N) Hepatic insulin signaling studied by Western blotting analysis after intravenous insulin (5 U per mouse) (HFD for 12 weeks; age, 20 weeks). (O) Quantification of the Western blotting data shown in (N). The data shown in (K) to (O) were obtained with mice that had been fasted for 4 hours. Data are given as means ± SEM (n = 5 to 9 male mice per group). *P < 0.05; **P < 0.01; ***P < 0.001 (Student’s t test). AU, arbitrary units.
In a separate set of experiments, we measured the ability of insulin to stimulate glucose uptake into various insulin-sensitive tissues in vivo. Specifically, we injected AgRP-barr1-KO mice and control littermates maintained on HFD for 14 weeks with insulin (0.75 U/kg i.p.; Humulin) and then measured glucose uptake into different tissues using the 2-deoxy-d-[1-14C] method. As expected, the HFD AgRP-barr1-KO mice showed a severe reduction in insulin sensitivity (fig. S4D). However, insulin-stimulated glucose uptake was similar in barr1 mutant and control mice in skeletal muscle (gastrocnemius and quadriceps muscle) (Fig. 2, E and F), white adipose tissue (WAT) (epididymal and subcutaneous fat) (Fig. 2, G and H), and BAT (Fig. 2I), as well as liver, heart, and brain (fig. S4E). Together, these findings suggest that impaired glucose uptake by peripheral tissues or the brain is not responsible for the metabolic deficits displayed by the HFD AgRP-barr1-KO mice.
Enhanced lipolysis in adipose tissue of AgRP-barr1-KO mice
Since plasma FFA levels were elevated in AgRP-barr1-KO mice (Fig. 1I), we next studied adenosine 3′,5′-monophosphate–dependent protein kinase (PKA) signaling in perigonadal WAT derived from mice at the end of the clamp study (lipolysis in adipocytes is under the control of β-adrenergic receptor–mediated activation of PKA). To assess PKA signaling, we carried out Western blotting studies of WAT protein extracts using an antibody that recognizes phosphorylated PKA target proteins. We found that the amount of phosphorylated PKA target proteins was significantly increased in fat tissue from the barr1 mutant mice (fig. S5, A and B). Among these PKA substrates was phosphorylated perilipin (fig. S5, A and C), the most abundant protein in adipose tissue that is considered a key marker of lipolysis (42). Together, these data are consistent with increased sympathetic outflow to WAT trigging enhanced lipolysis. Phosphorylation of hormone-sensitive lipase (HSL) was also increased in WAT derived from the barr1 mutant mice, although this effect failed to reach statistical significance (fig. S5, D and E).
Lack of barr1 in AgRP neurons increases hepatic lipid content and impairs hepatic insulin signaling
We next examined liver weight, morphology, and other hepatic parameters in HFD AgRP-barr1-KO mice and control littermates that had been maintained on the HFD for 12 weeks. We found that livers from mice lacking barr1 in AgRP neurons weighed significantly more than control livers (Fig. 2J). Moreover, hepatic triglyceride levels were significantly increased in the barr1 mutant mice (Fig. 2K). Microscopic analysis of hematoxylin and eosin (H&E)–stained liver sections revealed increased microvesicular lipid infiltration in the KO mice (Fig. 2L), consistent with Oil Red O staining experiments (Fig. 2L). Liver glycogen levels trended to be decreased in the KO mice (P = 0.14; fig. S4F).
To investigate whether the observed increases in HGP and hepatic lipid content were due to increased gluconeogenesis and lipogenesis, we examined the expression levels of several key enzymes involved in these processes. The hepatic expression of Pck1, which codes for an enzyme that is rate limiting for gluconeogenesis, was significantly increased in the barr1 mutant mice (Fig. 2M). Likewise, the hepatic expression of Tnfa and Mip1b, which code for proinflammatory cytokines, was significantly elevated in HFD AgRP-barr1-KO mice (Fig. 2M). However, we did not observe any significant changes in the hepatic expression levels of genes involved in lipogenesis, including Fas and Srebp1 (fig. S4G), suggesting that lipogenesis is not an important contributor to the increase in hepatic lipid accumulation.
Next, we tested whether the lack of barr1 in AgRP neurons impaired hepatic insulin signaling. We injected HFD AgRP-barr1-KO and control mice with either insulin (5 U per mouse intravenously) or saline and collected liver tissue 5 min later. Western blotting studies showed that the ability of insulin to phosphorylate AKT, glycogen synthase kinase-3β, and FOXO1 was drastically reduced in livers from AgRP-barr1-KO mice, as compared to liver samples from control littermates (Fig. 2, N and O), indicating that the lack of barr1 in AgRP neurons impairs hepatic insulin signaling.
Disruption of vagal input into the liver prevents the metabolic deficits caused by the lack of barr1 in AgRP neurons
Previous studies have shown that insulin and other hormones or nutrients can act on hypothalamic neurons of the ARC to regulate HGP via descending projections that control vagal outflow to the liver. For example, the ability of hypothalamic insulin and FFAs to suppress HGP is blocked by hepatic branch vagotomy but remains unaffected by selective vagal deafferentation (21, 43). On the basis of these observations, we tested the hypothesis that the metabolic deficits caused by the lack of barr1 in AgRP neurons could be prevented by severing the hepatic branch of the vagus.
Specifically, we surgically disrupted the hepatic branch of the vagus nerve in AgRP-barr1-KO and control mice. In the following, we refer to these mice as AgRP-barr1-KO-Vgtm and Control-Vgtm mice, respectively. For control purposes, we also included sham-operated mice in this set of experiments (AgRP-barr1-KO-Sham and Control-Sham mice, respectively). After a 1-week postsurgery recovery period, mice were maintained on an HFD. After 8 weeks of HFD feeding, body weight did not differ significantly between AgRP-barr1-KO mice and their control littermates (Fig. 3, A and B). All mice were then subjected to glucose, insulin, and pyruvate tolerance tests. In agreement with the data shown in Fig. 1 (E and F), the HFD AgRP-barr1-KO-Sham mice showed impaired glucose tolerance and elevated blood glucose levels in an insulin tolerance test (at 60 and 90 min), as compared to HFD Control-Sham mice (Fig. 3, C and E). Notably, these metabolic impairments were no longer observed in mice in which the hepatic branch of the vagus nerve had been severed (Fig. 3, D and F). We also subjected all mice to a pyruvate tolerance test, a test that is commonly used to assess HGP in vivo. Following intraperitoneal injection of pyruvate (2 g/kg), the HFD AgRP-barr1-KO-Sham mice showed significantly greater blood glucose excursions than the HFD Control-Sham mice, indicative of enhanced HGP (Fig. 3G). In contrast, this effect was not observed after dissection of the hepatic branch of the vagus nerve (Fig. 3H). Consistent with the data shown in Fig. 1 (G and H), HFD AgRP-barr1-KO-Sham mice displayed significant increases in blood glucose (fasting conditions) and plasma insulin levels (fed and fasting conditions), as compared to HFD Control-Sham mice (Fig. 3, I and K). In contrast, blood glucose and plasma insulin levels did not differ significantly between HFD AgRP-barr1-KO-Vgtm and HFD Control-Vgtm mice (Fig. 3, J and L). Together, these findings indicate that the metabolic deficits displayed by HFD AgRP-barr1-KO mice require vagal input into the liver.
Fig. 3. Disruption of hepatic vagal innervation prevents the metabolic deficits caused by HFD feeding of AgRP-barr1-KO mice.
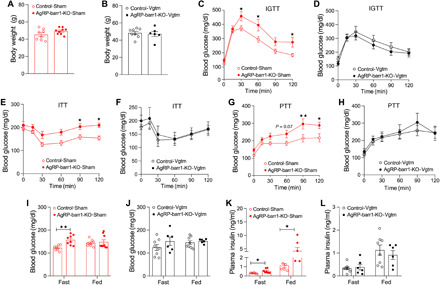
All experiments were carried out with male mice that had been maintained on an HFD for at least 8 weeks. Mice in which the vagal innervation of the liver had been disrupted are referred to as AgRP-barr1-KO-Vgtm and Control-Vgtm mice, respectively. Mice that had been subjected to sham surgery are referred to as AgRP-barr1-KO-Sham and Control-Sham mice, respectively. (A) Body weights of AgRP-barr1-KO-Sham and Control-Sham mice (age, 18 weeks; 8 weeks on HFD). (B) Body weights of AgRP-barr1-KO-Vgtm and Control-Vgtm mice (age, 18 weeks; 8 weeks on HFD). (C and D) Intraperitoneal glucose tolerance test (IGTT; 1 g glucose/kg i.p.) carried out with mice subjected to sham surgery (C) or vagotomy (D) (age, 18 weeks; 8 weeks on HFD). (E and F) Insulin tolerance test (1 U/kg i.p.) performed with mice subjected to sham surgery (E) or vagotomy (F) (age, 19 weeks; 9 weeks on HFD). (G and H) Pyruvate tolerance test (PTT; 2g/kg i.p.) carried out with mice subjected to sham surgery (G) or vagotomy (H) (age, 20 weeks; 10 weeks on HFD). (I and J) Fasting and fed blood glucose levels of sham-operated (I) or vagotomized (J) mice (age, 18 to 19 weeks; 8 to 9 weeks on HFD). (K and L) Fasting and fed plasma insulin levels of sham-operated (K) or vagotomized (L) mice (age, 18 to 19 weeks; 8 to 9 weeks on HFD). Data are given as means ± SEM (n = 6 to 9 male mice per group). *P < 0.05; **P < 0.01 [two-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc test (C, E, and G) and two-tailed Student’s t test (I and K)].
Lack of barr1 in AgRP neurons inhibits insulin-mediated hyperpolarization
Recent work has linked insulin signaling in AgRP neurons to hepatic insulin action (16, 18, 44). This effect requires, at least in part, the hyperpolarization of AgRP neurons by insulin (16, 18, 44). While β-arrestins have been implicated in regulating insulin signaling in peripheral cell types (33, 36), it remains unknown whether barr1 plays a role in modulating insulin action in AgRP neurons. We therefore examined whether barr1 was required for the ability of insulin to hyperpolarize AgRP neurons.
To assess insulin-induced changes in cellular activity, arcuate AgRP neurons were targeted for whole-cell patch-clamp recordings. To be able to identify AgRP neurons for electrophysiological recordings, we generated GFP reporter mice by crossing AgRP-barr1-KO and AgRP-Ires-Cre mice (control mice) with Z/EG reporter mice, which express GFP in a Cre-dependent fashion (Fig. 4, A to D).
Fig. 4. Barr1 deficiency impairs insulin action on AgRP neurons.
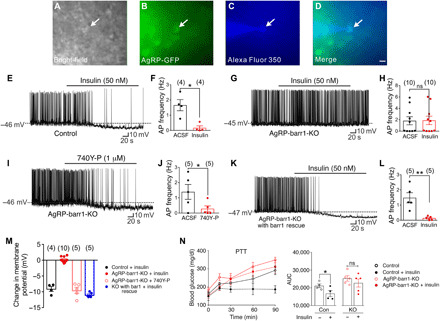
(A to M) Electrophysiological studies with AgRP neurons from 8- to 12-week-old male mice maintained on standard chow. (A to D) Bright-field (A) or fluorescein isothiocyanate (GFP) illumination (B) of an AgRP neuron from AgRP-Z/EG reporter mice. (C) Complete dialysis of Alexa Fluor 350 from the intracellular pipette. (D) Merged image of a targeted AgRP neuron (arrow). Scale bar, 50 μm. (E to H) Current-clamp recording depicting insulin-induced hyperpolarization and reduced action potential (AP) firing frequency of control AgRP neurons (E and F) and lack of these responses in barr1-deficient AgRP neurons (G and H). ACSF, artificial cerebrospinal fluid. (I and J) The PI3 kinase activator 740Y-P elicits insulin-like responses in barr1-deficient AgRP neurons. (K and L) Reexpression of barr1 in AgRP neurons of adult AgRP-barr1-KO mice restores normal insulin responses. (M) Summary of the effects of insulin and 740Y-P on the membrane potential of AgRP neurons. (N) Central administration of insulin (200 μU into the lateral ventricle) fails to inhibit pyruvate (2 g/kg i.p.)–induced hyperglycemia in AgRP-barr1-KO mice (HFD for 12 weeks; 20-week-old males; n = 4 or 5). Data are given as means ± SEM. *P ≤ 0.05; **P ≤ 0.01 [two-tailed Student’s t test (E to L) and one-tailed Student’s t test (N)]. ns, no significant difference.
Consistent with previous reports (18, 44, 45), insulin (50 nM) hyperpolarized arcuate AgRP neurons from control mice (change of resting potential, −9.1 ± 0.8 mV; Fig. 4, E and M) and greatly reduced action potential (AP) firing frequency [artificial cerebrospinal fluid (ACSF): 1.7 ± 0.4 Hz; insulin + ACSF: 0.17 ± 0.10 Hz; *P < 0.05)] (Fig. 4F). In notable contrast, these insulin effects were absent in AgRP neurons lacking barr1 (change of resting potential, 0.2 ± 0.3 mV; AP frequency in ACSF, 1.8 ± 0.7 Hz; insulin + ACSF, 1.9 ± 0.7 Hz) (Fig. 4, G, H, and M).
Insulin is known to hyperpolarize and reduce firing frequency of AgRP neurons by activating KATP channels, involving a phosphoinositide-3 kinase (PI3 kinase)–dependent pathway (18, 46). The cell-permeable PI3 kinase activator, 740Y-P (1 μM), retained the ability to hyperpolarize and reduce the AP firing frequency of AgRP neurons lacking barr1 (change of resting potential, −9.5 ± 1.0 mV; AP frequency in ACSF, 1.4 ± 0.5 Hz; 740Y-P + ACSF, 0.3 ± 0.2 Hz; *P < 0.05) (Fig. 4, I, J, and M), suggesting that that the loss of barr1 in AgRP neurons interferes with insulin signaling upstream of PI3 kinase.
To exclude the possibility that the inability of AgRP neurons to properly respond to insulin in the absence of barr1 was due to deficits caused by the absence of barr1 throughout development, we used a viral approach to reintroduce barr1 into AgRP neurons of adult AgRP-barr1-KO mice. Specifically, we injected the AAV-EF1a-DIO-Barr1-HA-P2A-mCerulean virus or the AAV-hSyn-DIO-GFP control virus bilaterally into the ARC of AgRP-barr1-KO mice.
Reintroduction of barr1 into AgRP neurons of adult AgRP-barr1-KO mice fully restored the ability of insulin (50 nM) to hyperpolarize and drastically decrease the AP firing frequency of AgRP neurons (change of resting potential, −11.0 ± 0.4 mV; AP frequency in ACSF, 1.5 ± 0.3 Hz; insulin + ACSF, 0.1 ± 0.1 Hz; **P < 0.01) (Fig. 4, K to M). These data strongly support the concept that barr1 is necessary for the insulin-induced inhibition of AgRP neurons.
Central administration of insulin does not affect pyruvate-induced hyperglycemia in HFD AgRP-barr1-KO mice
To investigate whether impaired insulin signaling in AgRP neurons was responsible for or contributed to the metabolic deficits displayed by the HFD AgRP-barr1-KO mice, we injected insulin (200 μU) or saline (control) into the lateral ventricle of HFD AgRP-barr1-KO mice and HFD control littermates. Following central saline or insulin treatment, mice were subjected to a pyruvate tolerance test. In this test, the insulin-treated control mice showed significantly reduced blood glucose excursions (Fig. 4N), consistent with previous findings that central (hypothalamic) administration of insulin reduces hepatic glucose output (21, 47, 48). In contrast, central injection of insulin had no significant effect on pyruvate-induced increases in glycemia in the AgRP-barr1-KO mice (Fig. 4N). Although these findings are consistent with the concept that impaired insulin signaling in barr1-deficient AgRP neurons makes an important contribution to the increase in HGP (gluconeogenesis) displayed by the HFD AgRP-barr1-KO mice, they do not provide definite proof. The key caveats are that insulin injected into the mouse lateral ventricle can act on many different neuronal subpopulations and that the outcome of a pyruvate tolerance test does not only depend on changes in HGP (49).
Barr1/IR substrate 1 complex formation and barr1-mediated increase in IR substrate 1 levels in mHypoE-N46 cells
The electrophysiological data shown in Fig. 4 indicated that the loss of barr1 in AgRP neurons impairs insulin signaling upstream of PI3K. Previous work has shown that deletion of barr1 from fibroblasts increases insulin-mediated degradation of IR substrate 1 (IRS-1), while overexpression of barr1 prevents IRS-1 degradation (50). We therefore examined whether a similar mechanism was operative in AgRP neurons.
Specifically, we carried out a series of studies with a clonal hypothalamic mouse cell line (mHypoE-N46 cells) that endogenously expresses AgRP, NPY, and the IR (51). By using a recombinant adenovirus, we overexpressed barr1 in this cell line (barr1-mHypoE-N46 cells). For control purposes, we infected mHypoE-N46 cells with a GFP adenovirus (control mHypoE-N46 cells). We then treated barr1-mHypoE-N46 and control cells with insulin (100 nM) for different periods of time. Barr1 overexpression resulted in significant increases in total IRS-1 expression at the 60-, 120-, and 360-min time points (fig. S6, A and B). In contrast, barr1 overexpression in barr1-mHypoE-N46 cells had no significant effect on IR levels (fig. S6, A and C). These data are consistent with the concept that barr1 regulates IRS-1 expression levels (50).
We next performed coimmunoprecipitation (coIP) assays to examine whether barr1 was able to form a complex with IRS-1 in mHypoE-N46 cells. We first overexpressed a FLAG-tagged version of barr1 in mHypoE-N46 cells by using a recombinant adenovirus. We then treated cells for 1 hour with insulin (100 nM; control cells were left untreated). Subsequently, cell lysates were subjected to immunoprecipitation with either an anti-FLAG antibody or rabbit immunoglobulin G (IgG) (negative control). Immunoprecipitated proteins were probed with an anti–IRS-1 antibody via Western blotting. These studies showed that insulin treatment significantly increased the ability of barr1 to interact with IRS-1 (fig. S6, D and E). As reported previously (50), this interaction is likely to promote insulin signaling by inhibiting IRS-1 degradation. In contrast, IRβ protein could not be detected in the immunoprecipitate, suggesting that barr1 does not form a complex with the IR.
Overexpression of barr1 in AgRP neurons improves whole-body glucose homeostasis
Since barr1 deletion in AgRP neurons led to impaired whole-body glucose homeostasis, increased HGP, and impaired liver insulin signaling, we hypothesized that AgRP neuron–specific overexpression of barr1 might result in enhanced hepatic insulin sensitivity and improved whole-body glucose homeostasis. To test this hypothesis, we overexpressed barr1 in AgRP neurons by injecting the pAAV-EF1a-DIO-Barr1-HA-P2A-mCerulean virus into the ARC of AgRP-Ires-Cre mice. In the same fashion, AgRP-Ires-Cre mice were injected with the AAV-mCherry-DIO control virus. The efficacy and selectivity of the stereotaxic injections were confirmed by immunostaining for mCerulean, which is coexpressed with barr1 from the same virus, and mCherry (control), respectively (Fig. 5A). For simplicity, we refer to the AgRP-Ires-Cre mice injected with the pAAV-EF1a-DIO-Barr1-HA-P2A-mCerulean virus as AgRP-barr1-OE mice.
Fig. 5. Overexpression of barr1 in AgRP neurons improves whole-body glucose tolerance and insulin sensitivity in lean and obese mice.
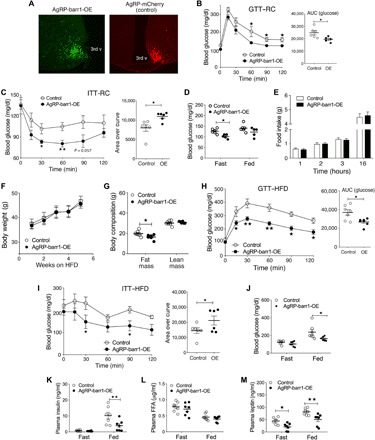
Experiments were carried out with male mice maintained on regular chow (RC) (A to I) or with male mice that had consumed an HFD for at least 7 weeks (F to M). (A) Representative immunofluorescence images showing the expression of mCerulean (left), a marker for barr1 overexpression, and mCherry (control mice; right) in the arcuate nucleus of AgRP-barr1-OE and AgRP-mCherry mice, respectively. 3rd v, third ventricle. (B) Glucose tolerance test (1 g glucose/kg i.p.) carried out with regular chow AgRP-barr1-OE and control mice (age, 12 weeks). (C) Insulin tolerance test (0.75 U/kg i.p.) (11-week-old regular chow mice). (D) Fasting and fed blood glucose levels (regular chow mice, 11 to 12 weeks old). (E) Food intake of regular chow AgRP-barr1-OE and control mice after a 24-hour fast (refeeding; mouse age, 13 weeks). (F) Body weight gain of AgRP-barr1-OE and control mice consuming an HFD (mouse age, 13 to 17 weeks). (G) Fat and lean mass composition of HFD AgRP-barr1-OE and control mice (mouse age, 22 weeks; 9 weeks on HFD). (H) Glucose tolerance test (1 g glucose/kg i.p.) carried out with HFD AgRP-barr1-OE and control mice (mouse age, 21 weeks; 8 weeks on HFD). (I) Insulin tolerance test (0.75 U/kg i.p.) (mouse age, 20 weeks; 7 weeks on HFD). (J to M) Fasting and fed blood glucose (J) and plasma insulin (K), FFA (L), and leptin (M) levels (mouse age, 20 to 22 weeks; 7 to 9 weeks on HFD). Data are given as means ± SEM (n = 6 to 9 mice per group). *P < 0.05; **P < 0.01 [two-way ANOVA followed by Bonferroni’s post hoc test (B, C, H, and I) and two-tailed Student’s t test (D, G, and J to M)].
AgRP-barr1-OE mice and AgRP-mCherry control mice showed similar body weights when maintained on regular chow (fig. S7A). In agreement with our hypothesis, AgRP-barr1-OE mice displayed significantly improved glucose tolerance and insulin sensitivity, as compared to the corresponding control mice (Fig. 5, B and C). AgRP-barr1-OE mice also showed reduced fasting blood glucose levels (Fig. 5D). Fed blood glucose and plasma insulin levels (fasting and fed) did not differ significantly between the two groups (Fig. 5D and fig. S7B). Likewise, fasting and fed plasma leptin (fig. S7C) and FFA levels (fig. S7D) were similar in the two groups of mice. To study the effect of barr1 overexpression on food intake, we measured the consumption of regular chow after a 24-hour fasting period (refeeding). We found that AgRP-barr1-OE mice and control mice consumed similar amounts of food during the 16-hour observation period (Fig. 5E). These data indicate that overexpression of barr1 in AgRP neurons improved glucose homeostasis and insulin tolerance without changes in food intake and body weight.
We next carried out similar metabolic measurements with AgRP-barr1-OE and control mice maintained on an HFD. The two groups of mice showed comparable diet-induced increases in body weight (Fig. 5F). The HFD AgRP-barr1-OE mice displayed a small but statistically significant decrease in fat mass after 12 weeks of HFD feeding (Fig. 5G). Notably, HFD AgRP-barr1-OE mice showed a pronounced improvement in both glucose tolerance and insulin sensitivity, as compared to HFD control mice (Fig. 5, H and I). Moreover, fed blood glucose and plasma insulin levels were significantly reduced in the HFD AgRP-barr1-OE mice (Fig. 5, J and K). Plasma FFA levels were similar in both groups of mice (Fig. 5L), while plasma leptin levels were significantly decreased in the HFD AgRP-barr1-OE mice (Fig. 5M). Collectively, these data indicate that overexpression of barr1 in AgRP neurons protects mice against obesity-associated metabolic deficits.
Overexpression of barr1 in AgRP neurons protects against liver steatosis
Since lack of barr1 in AgRP neurons in HFD AgRP-barr1-KO mice caused increased liver triglyceride content and impaired hepatic insulin signaling, we hypothesized that overexpression of barr1 in AgRP neurons might lead to opposite hepatic phenotypes. To test this hypothesis, we carried out a series of metabolic studies using HFD AgRP-barr1-OE and control mice. Livers from HFD AgRP-barr1-OE mice weighed significantly less than livers from HFD control mice (Fig. 6A). Moreover, hepatic triglyceride levels were significantly reduced in the HFD AgRP-barr1-OE mice (Fig. 6B). In agreement with this observation, H&E and Oil Red O staining experiments also showed reduced hepatic lipid accumulation in the HFD AgRP-barr1-OE mice (Fig. 6C). Moreover, the hepatic transcript levels of several proinflammatory cytokines (Ifng, Mip1a, and Mip1b) were significantly decreased in HFD AgRP-barr1-OE mice, as compared to HFD control mice (Fig. 6D). Liver glycogen content did not differ significantly between the two groups of mice (Fig. 6E).
Fig. 6. Overexpression of barr1 in AgRP neurons protects against diet-induced liver steatosis.
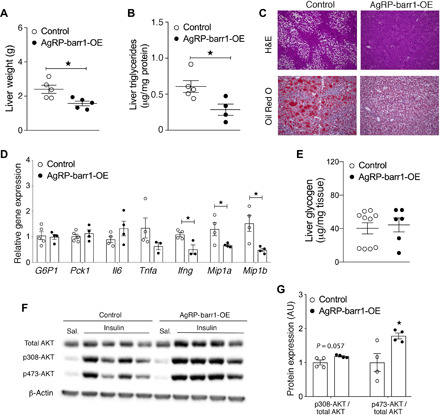
AgRP-barr1-OE and control mice were maintained on an HFD for at least 9 weeks. (A and B) Liver weights (A) and liver triglyceride levels (B) of HFD AgRP-barr1-OE and control mice. (C) Histological staining of liver sections from HFD AgRP-barr1-OE and control mice. (D) Hepatic expression levels of genes involved in gluconeogenesis and proinflammatory genes in HFD AgRP-barr1-OE and control mice. (E) Liver glycogen levels of HFD AgRP-barr1-OE and control mice. (F) Western blotting analysis of hepatic insulin signaling after intravenous insulin treatment (5 U per mouse) of HFD AgRP-barr1-OE and control mice. (G) Quantification of the Western blotting data shown in (F). Data are given as means ± SEM (n = 4 to 6 male mice per group; mouse age, 24 to 25 weeks, 11 to 12 weeks on HFD). *P < 0.05 (two-tailed Student’s t test).
We next studied hepatic insulin signaling in HFD AgRP-barr1-OE and control mice. As expected, insulin-stimulated phosphorylation of AKT was significantly increased in AgRP-barr1-OE mice (Fig. 6, F and G), suggesting that barr1 expressed by AgRP neurons is permissive for hepatic insulin signaling.
Insulin signaling is enhanced in AgRP neurons of AgRP-barr1-OE mice
Since the lack of barr1 impaired insulin signaling in AgRP neurons (Fig. 4), we speculated that overexpression of barr1 in AgRP neurons may have the opposite effect. To test this hypothesis, HFD AgRP-barr1-OE and control mice received a single intravenous injection of insulin (5 U per mouse) or saline. Brains were removed 10 min later, and immunohistochemical studies were performed after preparation of hypothalamic sections. We observed that insulin-stimulated phosphorylation of IRS-1 and AKT was barely detectable in AgRP neurons of control mice but became clearly more pronounced in AgRP neurons of AgRP-barr1-OE mice (fig. S8), indicating that barr1 is an important permissive factor for insulin signaling in AgRP neurons.
Mice lacking barr2 in AgRP neurons show no obvious metabolic phenotype
Since AgRP neurons express both barr1 and barr2, we also generated mice that selectively lacked barr2 in AgRP neurons (26). To obtain AgRP-barr2-KO mice, we crossed floxed barr2 mice with AgRP-Ires-Cre mice (52). As control mice, we used floxed barr2 mice animals that did not harbor the Cre sequence (littermates).
Like the AgRP-barr1-KO mice, the AgRP-barr2-KO mice did not show any obvious metabolic phenotypes when consuming regular mouse chow. We therefore maintained the AgRP-barr2-KO mice on an HFD and subjected these mice to the same metabolic tests as the AgRP-barr1-KO mice. The HFD AgRP-barr2-KO mice and their HFD control littermates showed similar body weight gain and body composition after 8 weeks of HFD feeding (fig. S9, A and B). Both groups of HFD mice also displayed similar impairments in glucose and insulin tolerance (fig. S9, C and D). Blood glucose and plasma insulin levels (fig. S9, E and F), as well as food intake (fig. S9G), did not differ significantly between HFD AgRP-barr2-KO and control mice. Together, these data indicate that the lack of barr2 in AgRP neurons has no significant effect on whole-body glucose homeostasis and insulin sensitivity.
DISCUSSION
Previous work has demonstrated that β-arrestins expressed in peripheral tissues are required for the maintenance of euglycemia (33, 35, 37, 38). However, the potential importance of β-arrestins expressed in the central nervous system in regulating energy and glucose homeostasis has not been studied so far. In the present study, we report the finding that barr1 expressed by arcuate AgRP neurons plays a key role in maintaining whole-body glucose homeostasis and insulin sensitivity.
Specifically, we demonstrated that mutant mice lacking barr1 selectively in AgRP neurons (AgRP-barr1-KO mice) showed pronounced impairments in glucose tolerance and insulin sensitivity when maintained on an obesogenic diet (Fig. 1). Consistent with this observation, mice that overexpressed barr1 selectively in AgRP neurons (AgRP-barr1-OE mice) displayed notable improvements in glucose homeostasis and insulin sensitivity (Figs. 5 and 6).
Several lines of evidence suggest that hepatic insulin resistance plays a role in the metabolic deficits displayed by the AgRP-barr1-KO mice, including the euglycemic-hyperinsulinemic clamp and vagotomy data (pyruvate tolerance test; Fig. 3, G and H). Moreover, the lack of barrr1 in AgRP neurons caused liver steatosis (Fig. 2, J to L), accompanied by impaired hepatic insulin signaling (Fig. 2, N and O).
Peripheral glucose utilization remained unaffected by the barr1 mutation (Fig. 2, E to I, and fig. S4E), suggesting that skeletal muscle glucose metabolism, which plays a central role in determining peripheral glucose utilization, does not contribute to the systemic insulin resistance displayed by the AgRP-barr1-KO mice. HFD AgRP-barrr1 KO mice exhibited significantly increased fed plasma FFA levels (Fig. 1I), without any changes in body weight gain or lean and fat body mass (Fig. 1, B and C). Adipose tissue from the barr1 mutant mice showed enhanced PKA activity (fig. S5), suggesting that the increase in plasma FFA levels displayed by the barr1 mutant mice is caused by increased adipose tissue lipolysis, probably due to enhanced sympathetic outflow to adipose tissue (53). Thus, our data support a model in which barr1 expressed by AgRP neurons participates in the sympathetic control of lipolysis, possibly by promoting insulin signaling in AgRP neurons. In agreement with this concept, insulin infused into the mediobasal hypothalamus suppresses adipose tissue lipolysis in rats (53). Since increased plasma FFA levels are known to impair insulin signaling, increased lipolytic flux from adipose tissue are predicted to contribute to the increase in hepatic lipid content (Fig. 2, K and L), systemic insulin resistance (Fig. 2B), and impaired hepatic insulin signaling (Fig. 2, N and O) displayed by the HFD AgRP-barr1-KO mice.
In contrast to the severe metabolic impairments observed with AgRP-barr1-KO mice maintained on a calorie-rich diet, no substantial metabolic deficits were detectable with AgRP-barr1-KO mice consuming regular mouse chow (fig. S1). This observation indicates that barr1 activity in AgRP neurons is of particular functional relevance in obesity, which is associated with numerous pathophysiological changes. Most likely, lean AgRP-barr1-KO mice do not exhibit any noticeable metabolic impairments because of compensation by other physiological pathways that are able to maintain euglycemia.
Insulin and other hormones or nutrients can act on hypothalamic neurons of the ARC to regulate HGP via descending projections that regulate vagal outflow to the liver (21, 43). Prompted by these previous findings, we examined whether the metabolic impairments caused by the lack of barr1 in AgRP neurons might involve the vagal innervation of the liver. We found that surgical dissection of the hepatic branch of the liver prevented all metabolic deficits displayed by the HFD AgRP-barr1-KO mice (Fig. 3), suggesting that barr1 expressed by AgRP neurons modulates neuronal pathways that promote vagal outflow to the liver.
The metabolic phenotypes displayed by the HFD AgRP-barr1-KO mice were similar to those reported for mutant mice that selectively lacked the IR in AgRP neurons (18). Like the AgRP-barr1-KO mice, the AgRP-IR-KO mice showed normal body weight and food intake but displayed impaired suppression of HGP by insulin. On the basis of these observations, we speculated that the important metabolic role of barr1 expressed by AgRP neurons might depend on the ability of barr1 to regulate insulin signaling in AgRP neurons.
Previous studies have shown that insulin signaling in AgRP neurons contributes to glucose homeostasis via hyperpolarization of AgRP neurons (16, 18, 44). We made the notable observation that AgRP neurons lacking barr1 failed to hyperpolarize in response to insulin, in contrast to AgRP neurons of control mice (Fig. 4, E, F, and I). The inhibitory effect of insulin on the activity of AgRP neurons involves the activation of KATP channels via a PI3 kinase–dependent pathway (18, 46). We found that 740Y-P, a cell-permeable PI3 kinase activator, retained the ability to hyperpolarize AgRP neurons lacking barr1 (Fig. 4, G and I), indicating that barr1 is required for proper insulin signaling in AgRP neurons upstream of PI3 kinase. Consistent with the electrophysiological data, we demonstrated that insulin-mediated phosphorylation of AKT was increased in AgRP neurons of AgRP-barr1-OE mice (fig. S8).
As discussed above, deletion of barr1 in AgRP neurons caused impaired glucose homeostasis only in AgRP-barr1-KO mice maintained on an obesogenic diet but not in lean KO mice consuming standard chow. However, insulin failed to depolarize AgRP neurons isolated from AgRP-barr1-KO mice maintained on standard chow (Fig. 4). One possible explanation for these observations is that insulin-mediated inhibition of AgRP neurons does not lead to noticeable metabolic phenotypes in healthy young AgRP-barr1-KO mice because other central and peripheral signaling pathways involved in maintaining proper glucose homeostasis are fully functional. However, under conditions of impaired glucose tolerance and insulin resistance triggered by the consumption of an obesogenic diet, inhibition of AgRP neurons by insulin appears critical to limit the degree of obesity-induced metabolic deficits.
When the HFD AgRP-barr1-KO mice were subjected to metabolic studies, they were about 1 to 2 months older than the analyzed AgRP-barr1-KO mice maintained on regular chow. We therefore cannot rule out the possibility that increased age may also contribute to the metabolic phenotypes displayed by the HFD AgRP-barr1-KO mice.
It has been shown that deletion of barr1 from fibroblasts increases insulin-mediated degradation of IRS-1, while overexpression of barr1 prevents this process (50). We found that barr1 is able to form a complex with IRS-1, but not with the IR, in mHypoE-N46 cells that endogenously express AgRP, NPY, and the IR (51) (fig. S6). The formation of this complex was promoted by insulin treatment (fig. S6, D and E). Moreover, we showed that barr1 overexpression in mHypoE-N46 cells reduced insulin-induced increases IRS-1 protein degradation (fig. S6, A and B). These findings support a model in which barr1 is required for efficient insulin signaling in AgRP neurons by maintaining proper IRS-1 protein levels. However, it is possible that additional mechanisms contribute to the ability of barr1 to regulate insulin signaling in AgRP neurons. Elucidating the precise molecular mechanisms through which barr1 acts on the insulin signaling cascade in AgRP neurons will be the subject of future investigations.
In contrast to the AgRP-barr1-KO mice, AgRP-barr2-KO mice did not show any noticeable metabolic deficits, independent of the diet that the mice consumed (fig. S9). Since AgRP neurons express both barr1 and barr2, this observation indicates that the two β-arrestins have different functions in AgRP neurons (26). The concept that barr1 and barr2 can mediate divergent functions in the same cell type has been reviewed recently (54). For example, we recently reported that mice lacking barr2 in hepatocytes show pronounced metabolic deficits, while no such changes were observed with hepatocyte-specific barr1-KO mice (37).
The metabolic effects observed after overexpression of barr1 in AgRP neurons were particularly notable. When maintained on an HFD, AgRP-barr1-OE and control mice showed comparable increases in body weight (Fig. 5F). However, despite being obese, AgRP-barr1-OE mice showed pronounced improvements in both glucose tolerance and insulin sensitivity, as compared to obese control mice (Fig. 5, H and I). In addition, blood glucose and plasma insulin levels were also significantly reduced in the barr1 mutant mice (Fig. 5, J and K). Together, these observations indicate that overexpression of barr1 in AgRP neurons protects obese mice against the major metabolic deficits associated with obesity. However, it should be noted that the HFD AgRP-barr1-OE mice displayed a small but statistically significant decrease in fat mass (Fig. 5G), as compared to the corresponding HFD control mice, providing a possible explanation for the observation that plasma leptin levels were reduced in the mutant mice (Fig. 5M). We therefore cannot exclude the possibility that the small decrease in adiposity displayed by the HFD AgRP-barr1-OE mice contributes to the improvements in glucose homeostasis that we found with these mice.
Sexual dimorphism has been reported for the hypothalamic regulation of metabolism including food intake and glucose homeostasis (55, 56). In the present study, we only investigated male mice, primarily because C57BL/6 males develop more severe obesity than females when maintained on an HFD. Further studies will be needed to examine whether barr1 expressed in AgRP neurons of female mice plays a similar functional role as in male mice.
In conclusion, this is the first study demonstrating that a β-arrestin isoform (barr1) expressed in a neuronal subpopulation is essential for maintaining whole-body glucose homeostasis and proper insulin sensitivity. Our findings should be of considerable interest for the development of new classes of antidiabetic drugs.
MATERIALS AND METHODS
Animals
To generate mice lacking barr1 or barr2 in AgRP neurons, (AgRP-barr1-KO and AgRP-barr2-KO mice, respectively), we crossed homozygous floxed barr1 (barr1 f/f mice) (39) or homozygous floxed barr2 mice (barr2 f/f mice) (52) with AgRP-Ires-Cre mice (the Jackson laboratory, stock no. 012899), which express Cre recombinase selectively in AgRP neurons. These matings generated barr1 (or barr2) f/+ mice that carried the AgRP-Cre transgene. These mice were then backcrossed to barr1 (or barr2) f/f mice to generate barr1 (or barr2) f/f AgRP-Cre mice and barr1 (or barr2) f/f littermates that did not carry the Cre transgene. These latter animals served as control mice in all experiments where barr1 (or barr2) f/f AgRP-Cre mice were used. All mice were maintained on a C57BL/6 background, as described previously (35, 39). For electrophysiological recordings, a Cre-dependent GFP reporter was introduced into AgRP-barr1-KO mice by using Z/EG transgenic mice (the Jackson laboratory, stock no. 003920).
Mice overexpressing barr1 selectively in AgRP neurons (AgRP-barr1-OE mice) were generated by stereotactic injections of the AAV-EF1a-DIO-Barr1-HA-P2A-mCerulean virus into the ARC of AgRP-Ires-Cre mice. An AAV-mCherry-DIO virus was injected in the same fashion into the ARC of AgRP-Ires-Cre mice to generate a group of control mice. All animal studies were carried out according to the U.S. National Institutes of Health Guidelines for Animal Research and were approved by the National Institute of Diabetes and Digestive and Kidney Diseases Institutional Animal Care and Use Committee.
Mouse maintenance and diet
Unless stated otherwise, male littermates were used for all experiments. Mice were maintained at room temperature (23°C) on a standard chow (7022 NIH-07 diet, 15% kcal fat, energy density 3.1 kcal/g; Envigo Inc.). Mice were kept on a 12-hour light, 12-hour dark cycle and had free access to water and food. In a subset of experiments, 6- to 8-week-old male mice were switched to an HFD (F3282, 60% kcal fat, energy density 5.5 kcal/g; Bio-Serv). Mice consumed the HFD for at least 8 weeks, unless stated otherwise.
Mouse lean and fat mass composition
The 3-in-1 EchoMRI Analyzer (Echo Medical Systems) was used to measure the lean/fat mass composition of mutant and control mice.
Mouse metabolic tests
All mouse metabolic tests were conducted with adult male mice that were at least 8 weeks old. Intraperitoneal glucose tolerance tests were performed with mice that had been fasted overnight for 12 hours. Mice received glucose (1 or 2 g/kg i.p.), and blood glucose concentrations were measured from the tail vein at specific time points (0, 15, 30, 45, 90, and 120 min after injection) using a portable glucometer (Contour glucometer, Bayer). Insulin tolerance tests were performed after a 4- to 5-hour fast by intraperitoneally injecting mice with human insulin (0.75 or 1 U/kg; Humulin, Eli Lilly). Blood glucose levels were measured at same time points as indicated above. Pyruvate tolerance tests were carried out with mice that had been fasted overnight for 12 hours. Mice received sodium pyruvate (2 g/kg i.p.), and blood glucose levels were measured from the tail vein at defined postinjection time points.
Gene expression analysis
Mice were euthanized, and tissues were quickly frozen on dry ice. The RNeasy mini kit (Qiagen) was used to extract total RNA. Complementary DNA was synthesized using SuperScript III First-Strand Synthesis SuperMix (Invitrogen), and quantitative polymerase chain reaction (PCR) was performed using the SYBR green method (Applied Biosystems). RNA expression data were normalized relative to the expression of 18S ribosomal RNA using the ΔΔCt method. A complete list of primer sequences is provided in table S1. The experimental conditions used for the quantitative reverse transcription PCR studies were similar to those described by Jain et al. (57).
Measurement of plasma insulin and leptin levels
Blood was collected from the tail vein of mice that had free access to food or that had been fasted overnight for 12 to 14 hours. Blood samples were centrifuged at 4°C for 10 min at ~12,000g to obtain plasma. Plasma insulin and leptin levels were measured using enzyme-linked immunosorbent assay kits from Crystal Chem Inc. and R&D Systems, respectively, following the manufacturers’ instructions.
Western blotting studies
Proteins were extracted from various mouse tissues using radioimmunoprecipitation assay (RIPA) buffer supplemented with cOmplete EDTA-free protease inhibitor cocktail (Roche). Protein concentrations in cell lysates were measured using a bicinchoninic acid assay kit (Pierce). Protein samples were denatured at 95°C using NuPAGE lithium dodecyl sulfate (LDS) sample buffer (Thermo Fisher Scientific) and separated using SDS–polyacrylamide gel electrophoresis, followed by transfer to nitrocellulose membranes. Membranes were incubated with primary and secondary antibodies (see table S2 for details), followed by visualization of immunoreactive bands by using SuperSignal West Pico Chemiluminescent Substrate (Pierce).
Plasma adipokine/cytokine measurements
Mice that had been maintained on an HFD for at least 8 weeks were used for the measurement of plasma adipokine/cytokine levels. Blood was collected from the mandibular/jugular vein with K2-EDTA–containing tubes (Microvette, Sarstedt) and then quickly centrifuged at 4°C to collect plasma. Plasma adipokine/cytokine levels were measured using the Bio-Plex Multiplex Immunoassay System (Bio-Rad), following the manufacturer’s instructions.
Intravenous injection of insulin
HFD mice were fasted for 4 hours and then anesthetized with isoflurane. The abdominal cavity was opened, and 5 U of insulin (Humulin, Eli Lilly) dissolved in 100 μl of 0.9% saline was injected into the inferior vena cava. Saline-injected mice served as control animals. Five minutes after insulin or saline injections, liver tissue was collected and frozen for further experiments (58).
Food intake measurements
Initially, mice were housed individually for 8 days. For food intake measurements, mice were provided with a defined amount of regular chow or HFD. Food intake was then measured at defined time points over a period of 3 days.
H&E and Oil Red O staining experiments
Mouse livers were dissected from control and mutant mice and frozen immediately. H&E and Oil Red O staining experiments of liver sections were performed using standard techniques.
Subdiaphragmatic hepatic vagotomy
The hepatic vagal innervation was disrupted in 8-week-old male mice. In brief, following induction of isoflurane anesthesia and aseptic preparation of the surgery site, a ventral midline laparotomy was performed. The hepatic branch of the vagus nerve was selectively exposed and completely sectioned at the proximal end under a dissection microscope (59). Janus Green B dye was used for nerve staining. The surgical site was closed by standard techniques. The procedure was achieved with minimal blood loss and took less than 20 min. Sham-operated mice were subjected to the same conditions, except that the hepatic vagal branch was not severed.
After surgery, the mice were allowed to recover for 1 week. The mice were then kept on an HFD for 8 weeks and subjected to various metabolic tests.
Euglycemic-hyperinsulinemic clamp studies
Male mice maintained on an HFD were used for euglycemic-hyperinsulinemic clamp experiments. Catheters were placed into the internal jugular vein and the carotid artery. After 1 week of recovery, insulin clamps were performed on conscious, freely moving animals. On the day of the experiment, mice were fasted for 4 hours before initiation of the tracer infusion. A primed continuous [3-3H]glucose infusion (5-μCi bolus and 0.05 μCi/min) was given at t = −120 min to measure glucose turnover. The clamp was started at t = 0 min with a continuous insulin infusion (3 mU kg−1 min−1), and the [3-3H]glucose was increased to 0.1 μCi/min to minimize changes in specific activity. Glucose (5 μl) was measured every 10 min, and euglycemia (~120 mg/dl) was maintained using a variable GIR. Samples (10 μl) to determine glucose specific activity were taken at t = −15 and −5 min and every 10 min from t = 80 to 120 min. Rates of basal and insulin-stimulated whole-body glucose turnover were determined as the ratio of the [3H]glucose infusion rate [disintegrations per minute (dpm)] to the specific activity of plasma glucose (dpm per micromole) at the end of the basal period and during the final 30 min of the clamps (90 to 120 min), respectively. HGP (alternative term: endogenous glucose production) during the clamps was determined by subtracting the steady-state GIR from the whole-body glucose turnover rate. Under steady-state conditions for plasma glucose concentration, the rate of glucose disappearance (Rd) equals the rate of glucose appearance (Ra). Ra was determined from the ratio of the infusion rate for [3H]glucose (dpm) and the specific activity of plasma [3H]glucose (dpm) under steady-state conditions. To maintain a stable hematocrit throughout the experiment, donor blood was centrifuged, and erythrocytes were resuspended in saline and infused throughout the clamp.
Western blotting analysis of phospho-PKA substrates in adipose tissue
Perigonadal adipose tissue harvested at the end of the clamp study was homogenized with a bead homogenizer in 20 mM Mops buffer containing 2 mM EGTA, 5 mM EDTA, 30 mM sodium fluoride, 40 mM β-glycerophosphate, 10 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 0.5% NP-40, and complete protease inhibitor cocktail (Roche). Tissue homogenates were then centrifuged at 13,000g for 20 min at −2°C. The supernatant was collected, and protein concentrations were measured using a bicinchoninic acid–based protein quantification kit (Thermo Fisher Scientific). Protein extracts were separated on 4 to 12% NuPAGE gels (Invitrogen) and blotted onto Immobilon FL polyvinylidene difluoride membranes (Millipore). Membranes were blocked at room temperature for 1 hour with Odyssey LI-COR blocking buffer (LI-COR) diluted 1:1 in tris-buffered saline (TBS). Membranes were then incubated with primary antibodies against phosphorylated PKA target proteins, phospho-HSL (S563), and anti–β-actin (see table S2 for details). Membranes were washed consecutively three times for 5 min in TBS buffer containing 0.1% Tween (TBS-T buffer). Blots were then incubated with DyLight 680–conjugated goat anti-rabbit IgG and DyLight 800–conjugated goat anti-mouse IgG (Thermo Fisher Scientific) for 1 hour at room temperature in blocking buffer containing 0.1% TBS-T and 0.1% SDS. Blots were washed three more times in TBS-T, followed by a final wash in TBS, and then scanned with the LI-COR Odyssey Imaging System (LI-COR). Immunoreactive bands were quantified with Odyssey 3.0 software on the basis of a direct fluorescence measurement.
Tissue glucose uptake
Control and mutant mice that had been maintained on an HFD for at least 8 weeks were used for the measurement of tissue glucose uptake. Mice were fasted 4 to 5 hours and then injected with insulin (0.75 U/kg i.p.; Humulin) and 2-deoxy-d-[1-14C] glucose (10 μCi; PerkinElmer). Animals were euthanized 40 min later, followed by the collection of various tissues and glucose uptake measurements (60).
Liver triglyceride and glycogen measurements
Livers were collected from control and mutant mice and snap-frozen. Liver triglycerides were measured using a published procedure (61). Liver glycogen was measured using a commercially available kit (Abnova).
Electrophysiology
Brain slices were prepared from young adult male mice (10 to 18 weeks old), as previously described (44, 62, 63). Briefly, mice were deeply anesthetized with 7% chloral hydrate (i.p.) and transcardially perfused with a modified ice-cold ACSF. The mice were then decapitated, and the entire brain was removed and immediately submerged in ice-cold, carbogen-saturated (95% O2 and 5% CO2) ACSF (126 mM NaCl, 2.8 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 5 mM glucose). Coronal sections (250 μm) were cut with a Leica VT1000S Vibratome and then incubated in oxygenated ACSF at room temperature for at least 1 hour before recording. The slices were bathed in oxygenated ACSF (32° to 34°C) at a flow rate of ∼2 ml/min.
The pipette solution for whole-cell recording was modified to include an intracellular dye (Alexa Fluor 350 hydrazide dye) for whole-cell recording: 120 mM K-gluconate, 10 mM KCl, 10 mM Hepes, 5 mM EGTA, 1 mM CaCl2, 1 mM MgCl2, 2 mM MgATP, and 0.03 mM Alexa Fluor 350 hydrazide dye (pH 7.3). Epifluorescence was briefly used to target fluorescent cells, at which time the light source was switched to infrared differential interference contrast imaging to obtain the whole-cell recording (Zeiss Axioskop FS2 Plus equipped with a fixed stage and a QuantEM:512SC electron-multiplying charge-coupled device camera). Electrophysiological signals were recorded using an Axopatch 700B amplifier (Molecular Devices), low-pass–filtered at 2 to 5 kHz, and analyzed offline on a PC with pCLAMP programs (Molecular Devices). Membrane potential was measured by whole-cell current clamp recordings from AgRP neurons in brain slices. Recording electrodes had resistances of 2.5 to 5 megohms when filled with the K-gluconate internal solution.
Insulin (50 nM) and 740Y-P (1 μM) were added to the ACSF in a subset of experiments. Solutions containing drug were typically perfused for 5 min. A drug effect was required to be associated temporally with the drug’s application, and the response had to be stable within a few minutes.
Stereotaxic injections into the arcuate nucleus
Young adult AgRP-Ires-Cre mice (males) were anesthetized with isoflurane and placed into a stereotaxic apparatus (David Kopf Instruments, model 940A with 923B mouse gas aesthesia head holder). The skull was exposed via a small incision, and a small hole was drilled (0.45-mm drill bit) into the skull. A Hamilton 5-μl syringe with a 30-gauge blunt-end needle was inserted into the brain for the delivery of adeno-associated viruses (AAVs). Bilateral injections of AAVs (200 nl of AAV-EF1a-DIO-Barr1-HA-P2A-mCerulean or AAV-mCherry-DIO) were made into the arcuate nucleus of the hypothalamus. The coordinates were as follows (from bregma): anterior-posterior, 1.46 mm; lateral (from midline), ±0.26 mm; dorsal-ventral, 5.80 mm. Postoperative analgesia was provided [meloxicam SR (sustained release), 1.5 mg/kg subcutaneously; ZooPharm]. Mice were allowed to recover for 10 days before initiating metabolic studies.
Stereotaxic injection of insulin into the lateral ventricle
Mice were prepared for stereotaxic injections as described (27). A Plastics One guide cannula (C315GS-4 W-2) was placed into the mouse lateral ventricle by using a stereotaxic apparatus (David Kopf, model 940). The following coordinates were used: anterior-posterior (from bregma), −0.35 mm; lateral (from midline), −1 mm; dorsal-ventral, −1.8 mm (from surface of the skull). Small screws and dental cement were used to secure the cannula. After a 2-week recovery period, a small injection cannula was placed through the guide cannula at a depth of 2.8 mm to the lateral ventricle. The internal cannula was linked through 22-gauge tubing filled with mineral oil to an SGE 5-μl syringe driven by a KD Scientific infusion pump. The tubing was prefilled with 5 μl of either saline or insulin solution. The infusion rate was 1 μl/min (total infusion volume, 2 μl). Each mouse received a total of 200 μU of insulin (or saline). Forty-five minutes after insulin or saline treatment, mice were subjected to pyruvate tolerance tests (see above).
Preparation of hypothalamic slices and immunohistochemistry
Mice that had received an intravenous injection of insulin (5 U per mouse) or saline were anesthetized and perfused with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer fixative (pH 7.4). The brain was removed and stored for 1 day in 4% PFA. The next day, 40-μm-thick slices of the hypothalamic region containing the arcuate nucleus were prepared by using a vibration microtome. The sections were washed with phosphate-buffered saline (PBS), followed by an overnight incubation with primary antibodies diluted in PBS (1:100; anti–p-AKT and anti–p-IRS-1), supplemented with 2.5% normal goat serum at 4°C. After 72 hours, slices were washed with PBS and incubated with fluorophore-conjugated secondary antibodies (1:400; Alexa Fluor 488 or Alexa Fluor 594) for 2 hours at room temperature. After three washes with PBS, sections were mounted on slides, and antifade mounting medium (Vector Laboratories) was dispensed on the sections to reduce photobleaching and allow long-term storage.
Signaling and coIP studies with mHypoE-N46 cells
We carried out a series of studies with a clonal hypothalamic mouse cell line (mHypoE-N46 cells) that endogenously expresses AgRP, NPY, and the IR (source: Cedarlane, Ontario, Canada) (51). We infected mHypoE-N46 cells with adenoviruses coding for barr1-FLAG or enhanced GFP, respectively. Twenty-four hours later, cells were serum-starved for 16 hours and then treated with 100 nM insulin (Sigma-Aldrich) for 15, 30, 60, 120, and 360 min at 37°C. Subsequently, cells were lysed using RIPA buffer, and cell lysates were subjected to immunoblotting experiments as described under the “Western blotting studies” section.
coIP assays were performed with a Dynabeads coIP kit (Thermo Fisher Scientific) using mHypoE-N46 cells infected with an adenoviruses coding for barr1-FLAG (Vector Biolabs), following the manufacturer’s instructions. Cells were treated with insulin (100 nM) for 1 or 2 hours at 37°C or left untreated. Subsequently, cells were lysed in coIP lysis buffer supplemented with 100 mM NaCl and protease inhibitors (Roche). In brief, anti-FLAG antibody or IgG was covalently coupled to the epoxy beads provided by the kit. Beads were then incubated for 30 min at 4°C with mHypoE-N46 cell lysates. Protein complexes were eluted in elution buffer containing LDS sample buffer (Thermo Fisher Scientific). Samples were analyzed by Western blotting using anti–IRS-1 and anti-barr1 antibodies.
Statistics
Data are expressed as means ± SEM for the indicated number of observations. Before performing specific statistical tests, we performed tests for normality and homogeneity of variance. Data were then tested for statistical significance by two-way analysis of variance (ANOVA), followed by the indicated post hoc tests, or by using a one- or two-tailed unpaired Student’s t test, as appropriate. A P value of less than 0.05 was considered statistically significant. The specific statistical tests that were used are indicated in the figure legends.
Supplementary Material
Acknowledgments
This research was funded by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK, NIH). The floxed barr1 and barr2 mutant mice were kindly provided by Marc G. Caron and Robert J. Lefkowitz (Duke University). We thank Lee Chedester (NIAAA, Rockville, MD) for his help with the vagotomy studies, Jeffrey Reece (NIDDK Advanced Light Microscopy & Image Analysis Core) for his advice regarding the imaging studies, Yinyan Ma for metabolite measurements (NIDDK Mouse Metabolism Core), and Michael Assefa (animal care taker; NIH, Bethesda, MD) for excellent technical assistance. This work was also supported by the Einstein-Mt. Sinai Diabetes Research Center Animal Physiology Core (NIH DK 020541) and NIH grants to C.B. (DK074873, DK083568, DK082724) and K.W.W. (R01 DK100699 and R01 DK119169). Author contributions: S.P.P., Z.C., K.W.W., C.B., and J.W. designed and conceived the experiments. S.P.P., Z.C., Z.H., L.F.B., J.P., D.J.O., and H.E.E. performed and analyzed the experiments. G.J.S. supervised the clamp studies, and C.B., D.J.O., and H.E.E. analyzed tissues and clamp data. Z.H. and K.W.W. carried out and interpreted the electrophysiological experiments. N.U. provided key reagents (DIO AAVs). S.P.P., C.B., K.W.W., and J.W. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/23/eaaz1341/DC1
REFERENCES AND NOTES
- 1.González-Muniesa P., Mártinez-González M.-A., Hu F. B., Després J.-P., Matsuzawa Y., Loos R. J. F., Moreno L. A., Bray G. A., Martinez J. A., Obesity. Nat. Rev. Dis. Primers 3, 17034 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Kusminski C. M., Bickel P. E., Scherer P. E., Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat. Rev. Drug Discov. 15, 639–660 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Saltiel A. R., Olefsky J. M., Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Invest. 127, 1–4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guh D. P., Zhang W., Bansback N., Amarsi Z., Birmingham C. L., Anis A. H., The incidence of co-morbidities related to obesity and overweight: A systematic review and meta-analysis. BMC Public Health 9, 88 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mobbs C. V., Orphaned no more? Glucose-sensing hypothalamic neurons control insulin secretion. Diabetes 65, 2473–2475 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Ruud J., Steculorum S. M., Brüning J. C., Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat. Commun. 8, 15259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arble D. M., Sandoval D. A., CNS control of glucose metabolism: Response to environmental challenges. Front. Neurosci. 7, 20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lam C. K. L., Chari M., Lam T. K. T., CNS regulation of glucose homeostasis. Physiology (Bethesda) 24, 159–170 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Morton G. J., Hypothalamic leptin regulation of energy homeostasis and glucose metabolism. J. Physiol. 583, 437–443 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleinridders A., Könner A. C., Brüning J. C., CNS-targets in control of energy and glucose homeostasis. Curr. Opin. Pharmacol. 9, 794–804 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Loh K., Zhang L., Brandon A., Wang Q., Begg D., Qi Y., Fu M., Kulkarni R., Teo J., Baldock P., Brüning J. C., Cooney G., Neely G. G., Herzog H., Insulin controls food intake and energy balance via NPY neurons. Mol. Metab. 6, 574–584 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krashes M. J., Koda S., Ye C., Rogan S. C., Adams A. C., Cusher D. S., Maratos-Flier E., Roth B. L., Lowell B. B., Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest. 121, 1424–1428 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krashes M. J., Shah B. P., Koda S., Lowell B. B., Rapid versus delayed stimulation of feeding by the endogenously released AgRP neuron mediators GABA, NPY, and AgRP. Cell Metab. 18, 588–595 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morton G. J., Meek T. H., Schwartz M. W., Neurobiology of food intake in health and disease. Nat. Rev. Neurosci. 15, 367–378 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cavalcanti-de-Albuquerque J. P., Bober J., Zimmer M. R., Dietrich M. O., Regulation of substrate utilization and adiposity by Agrp neurons. Nat. Commun. 10, 311 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steculorum S. M., Ruud J., Karakasilioti I., Backes H., Engström Ruud L., Timper K., Hess M. E., Tsaousidou E., Mauer J., Vogt M. C., Paeger L., Bremser S., Klein A. C., Morgan D. A., Frommolt P., Brinkkötter P. T., Hammerschmidt P., Benzing T., Rahmouni K., Wunderlich F. T., Kloppenburg P., Brüning J. C., AgRP neurons control systemic insulin sensitivity via myostatin expression in brown adipose tissue. Cell 165, 125–138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porte D. Jr., Baskin D. G., Schwartz M. W., Leptin and insulin action in the central nervous system. Nutr. Rev. 60, S20–S29 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Könner A. C., Janoschek R., Plum L., Jordan S. D., Rother E., Ma X., Xu C., Enriori P., Hampel B., Barsh G. S., Kahn C. R., Cowley M. A., Ashcroft F. M., Brüning J. C., Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 5, 438–449 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Rosario W., Singh I., Wautlet A., Patterson C., Flak J., Becker T. C., Ali A., Tamarina N., Philipson L. H., Enquist L. W., Myers M. G. Jr., Rhodes C. J., The brain-to-pancreatic islet neuronal map reveals differential glucose regulation from distinct hypothalamic regions. Diabetes 65, 2711–2723 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Obici S., Feng Z., Karkanias G., Baskin D. G., Rossetti L., Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat. Neurosci. 5, 566–572 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Pocai A., Lam T. K. T., Gutierrez-Juarez R., Obici S., Schwartz G. J., Bryan J., Aguilar-Bryan L., Rossetti L., Hypothalamic KATP channels control hepatic glucose production. Nature 434, 1026–1031 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Postic C., Dentin R., Girard J., Role of the liver in the control of carbohydrate and lipid homeostasis. Diabetes Metab. 30, 398–408 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Lin H. V., Accili D., Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 14, 9–19 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unger R. H., Cherrington A. D., Glucagonocentric restructuring of diabetes: A pathophysiologic and therapeutic makeover. J. Clin. Invest. 122, 4–12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cowley M. A., Smith R. G., Diano S., Tschöp M., Pronchuk N., Grove K. L., Strasburger C. J., Bidlingmaier M., Esterman M., Heiman M. L., Garcia-Segura L. M., Nillni E. A., Mendez P., Low M. J., Sotonyi P., Friedman J. M., Liu H., Pinto S., Colmers W. F., Cone R. D., Horvath T. L., The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37, 649–661 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Ren H., Orozco I. J., Su Y., Suyama S., Gutiérrez-Juárez R., Horvath T. L., Wardlaw S. L., Plum L., Arancio O., Accili D., FoxO1 target Gpr17 activates AgRP neurons to regulate food intake. Cell 149, 1314–1326 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakajima K.-I., Cui Z., Li C., Meister J., Cui Y., Fu O., Smith A. S., Jain S., Lowell B. B., Krashes M. J., Wess J., Gs-coupled GPCR signalling in AgRP neurons triggers sustained increase in food intake. Nat. Commun. 7, 10268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pierce K. L., Premont R. T., Lefkowitz R. J., Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Luttrell L. M., Gesty-Palmer D., Beyond desensitization: Physiological relevance of arrestin-dependent signaling. Pharmacol. Rev. 62, 305–330 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rajagopal S., Rajagopal K., Lefkowitz R. J., Teaching old receptors new tricks: Biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 9, 373–386 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gurevich V. V., Gurevich E. V., Overview of different mechanisms of arrestin-mediated signaling. Curr. Protoc. Pharmacol. 67, 2.10.1–2.10.9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gurevich E. V., Gurevich V. V., Arrestins: Ubiquitous regulators of cellular signaling pathways. Genome Biol. 7, 236 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luan B., Zhao J., Wu H., Duan B., Shu G., Wang X., Li D., Jia W., Kang J., Pei G., Deficiency of a β-arrestin-2 signal complex contributes to insulin resistance. Nature 457, 1146–1149 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Zhuang L.-N., Hu W.-X., Zhang M.-L., Xin S.-M., Jia W.-P., Zhao J., Pei G., β-arrestin-1 protein represses diet-induced obesity. J. Biol. Chem. 286, 28396–28402 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu L., Almaça J., Dadi P. K., Hong H., Sakamoto W., Rossi M., Lee R. J., Vierra N. C., Lu H., Cui Y., McMillin S. M., Perry N. A., Gurevich V. V., Lee A., Kuo B., Leapman R. D., Matschinsky F. M., Doliba N. M., Urs N. M., Caron M. G., Jacobson D. A., Caicedo A., Wess J., β-arrestin-2 is an essential regulator of pancreatic β-cell function under physiological and pathophysiological conditions. Nat. Commun. 8, 14295 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ravier M. A., Leduc M., Richard J., Linck N., Varrault A., Pirot N., Roussel M. M., Bockaert J., Dalle S., Bertrand G., β-Arrestin2 plays a key role in the modulation of the pancreatic beta cell mass in mice. Diabetologia 57, 532–541 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Zhu L., Rossi M., Cui Y., Lee R. J., Sakamoto W., Perry N. A., Urs N. M., Caron M. G., Gurevich V. V., Godlewski G., Kunos G., Chen M., Chen W., Wess J., Hepatic β-arrestin 2 is essential for maintaining euglycemia. J. Clin. Invest. 127, 2941–2945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pydi S. P., Jain S., Tung W., Cui Y., Zhu L., Sakamoto W., Jain S., Abel B. S., Skarulis M. C., Liu J., Huynh T., Pacak K., Caron M. G., Gavrilova O., Finkel T., Wess J., Adipocyte β-arrestin-2 is essential for maintaining whole body glucose and energy homeostasis. Nat. Commun. 10, 2936 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim J., Grotegut C. A., Wisler J. W., Li T., Mao L., Chen M., Chen W., Rosenberg P. B., Rockman H. A., Lefkowitz R. J., β-arrestin 1 regulates β2-adrenergic receptor-mediated skeletal muscle hypertrophy and contractility. Skelet. Muscle 8, 39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tong Q., Ye C.-P., Jones J. E., Elmquist J. K., Lowell B. B., Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat. Neurosci. 11, 998–1000 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Novak A., Guo C., Yang W., Nagy A., Lobe C. G., Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis 28, 147–155 (2000). [PubMed] [Google Scholar]
- 42.Granneman J. G., Moore H.-P. H., Location, location: Protein trafficking and lipolysis in adipocytes. Trends Endocrinol. Metab. 19, 3–9 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Pocai A., Obici S., Schwartz G. J., Rossetti L., A brain-liver circuit regulates glucose homeostasis. Cell Metab. 1, 53–61 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Huang Y., He Z., Gao Y., Lieu L., Yao T., Sun J., Liu T., Javadi C., Box M., Afrin S., Guo H., Williams K. W., Phosphoinositide 3-Kinase Is Integral for the Acute Activity of Leptin and Insulin in Male Arcuate NPY/AgRP Neurons. J. Endocr. Soc. 2, 518–532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He Z., Gao Y., Lieu L., Afrin S., Guo H., Williams K. W., Acute effects of zinc and insulin on arcuate anorexigenic proopiomelanocortin neurons. Br. J. Pharmacol. 176, 725–736 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belgardt B. F., Okamura T., Brüning J. C., Hormone and glucose signalling in POMC and AgRP neurons. J. Physiol. 587, 5305–5314 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Obici S., Zhang B. B., Karkanias G., Rossetti L., Hypothalamic insulin signaling is required for inhibition of glucose production. Nat. Med. 8, 1376–1382 (2002). [DOI] [PubMed] [Google Scholar]
- 48.Plum L., Belgardt B. F., Brüning J. C., Central insulin action in energy and glucose homeostasis. J. Clin. Invest. 116, 1761–1766 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hughey C. C., Wasserman D. H., Lee-Young R. S., Lantier L., Approach to assessing determinants of glucose homeostasis in the conscious mouse. Mamm. Genome 25, 522–538 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Usui I., Imamura T., Huang J., Satoh H., Shenoy S. K., Lefkowitz R. J., Hupfeld C. J., Olefsky J. M., β-arrestin-1 competitively inhibits insulin-induced ubiquitination and degradation of insulin receptor substrate 1. Mol. Cell. Biol. 24, 8929–8937 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mayer C. M., Belsham D. D., Insulin directly regulates NPY and AgRP gene expression via the MAPK MEK/ERK signal transduction pathway in mHypoE-46 hypothalamic neurons. Mol. Cell. Endocrinol. 307, 99–108 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Urs N. M., Gee S. M., Pack T. F., McCorvy J. D., Evron T., Snyder J. C., Yang X., Rodriguiz R. M., Borrelli E., Wetsel W. C., Jin J., Roth B. L., O'Donnell P., Caron M. G., Distinct cortical and striatal actions of a β-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc. Natl. Acad. Sci. U.S.A. 113, E8178–E8186 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scherer T., O'Hare J., Diggs-Andrews K., Schweiger M., Cheng B., Lindtner C., Zielinski E., Vempati P., Su K., Dighe S., Milsom T., Puchowicz M., Scheja L., Zechner R., Fisher S. J., Previs S. F., Buettner C., Brain insulin controls adipose tissue lipolysis and lipogenesis. Cell Metab. 13, 183–194 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Srivastava A., Gupta B., Gupta C., Shukla A. K., Emerging Functional Divergence of β-Arrestin Isoforms in GPCR Function. Trends Endocrinol. Metab. 26, 628–642 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Morselli E., Frank A. P., Palmer B. F., Rodriguez-Navas C., Criollo A., Clegg D. J., A sexually dimorphic hypothalamic response to chronic high-fat diet consumption. Int. J. Obes. 40, 206–209 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Mauvais-Jarvis F., Sex differences in metabolic homeostasis, diabetes, and obesity. Biol. Sex Differ. 6, 14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jain S., Ruiz de Azua I., Lu H., White M. F., Guettier J.-M., Wess J., Chronic activation of a designer G(q)-coupled receptor improves β cell function. J. Clin. Invest. 123, 1750–1762 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steculorum S. M., Timper K., Engström Ruud L., Evers N., Paeger L., Bremser S., Kloppenburg P., Brüning J. C., Inhibition of P2Y6 signaling in AgRP neurons reduces food intake and improves systemic insulin sensitivity in obesity. Cell Rep. 18, 1587–1597 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Hiramoto T., Chida Y., Sonoda J., Yoshihara K., Sudo N., Kubo C., The hepatic vagus nerve attenuates Fas-induced apoptosis in the mouse liver via α7 nicotinic acetylcholine receptor. Gastroenterology 134, 2122–2131 (2008). [DOI] [PubMed] [Google Scholar]
- 60.Kim J. K., Gavrilova O., Chen Y., Reitman M. L., Shulman G. I., Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J. Biol. Chem. 275, 8456–8460 (2000). [DOI] [PubMed] [Google Scholar]
- 61.Folch J., Lees M., Sloane Stanley G. H., A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 (1957). [PubMed] [Google Scholar]
- 62.Gao Y., Yao T., Deng Z., Sohn J.-W., Sun J., Huang Y., Kong X., Yu K.-J., Wang R.-T., Chen H., Guo H., Yan J., Cunningham K. A., Chang Y., Liu T., Williams K. W., TrpC5 mediates acute leptin and serotonin effects via pomc neurons. Cell Rep. 18, 583–592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.He Z., Gao Y., Alhadeff A. L., Castorena C. M., Huang Y., Lieu L., Afrin S., Sun J., Betley J. N., Guo H., Williams K. W., Cellular and synaptic reorganization of arcuate NPY/AgRP and POMC neurons after exercise. Mol. Metab. 18, 107–119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/23/eaaz1341/DC1