

STK38, a reader of histone H4 ufmylation, regulates ATM activation.
Abstract
The ATM (ataxia-telangiectasia mutated) kinase is rapidly activated following DNA damage and phosphorylates its downstream targets to launch DDR signaling. Recently, we and others showed that UFM1 signaling promotes ATM activation. We further discovered that monoufmylation of histone H4 at Lys31 by UFM1-specific ligase 1 (UFL1) is an important step in the amplification of ATM activation. However, how monoufmylated H4 enhances ATM activation is still unknown. Here, we report STK38, a kinase in the Hippo pathway, serves as a reader for histone H4 ufmylation to promote ATM activation in a kinase-independent manner. STK38 contains a potential UFM1 binding motif which recognizes ufmylated H4 and recruits the SUV39H1 to the double-strand breaks, resulting in H3K9 trimethylation and Tip60 activation to promote ATM activation. Together, STK38 is a previously unknown player in DNA damage signaling and functions as a reader of monoufmylated H4 at Lys31 to promote ATM activation.
INTRODUCTION
Human genome is constantly exposed to potentially detrimental endogenous and exogenous genotoxic stress. Every cell division cycle has the potential to induce DNA strand breaks. In addition, exogenous genotoxic agents such as cosmic rays, oxidative stress, and chemical mutagens cause different nucleotide modifications and DNA damage (1). Double-strand breaks (DSBs) are one of the most deleterious forms of DNA damage. Ataxia-telangiectasia mutated (ATM) protein kinase is a master regulator of DSB-induced DNA damage response. The ATM gene is located on chromosome 11, and its transcript encodes a 3056–amino acid protein (2–4). ATM protein contains various α-helical HEAT (Huntington-elongation factor 3-protein phosphatase 2A-TOR1) repeats, the conserved FAT (FRAP-ATM-TRRAP) domain, PIKK (Phosphatidylinositol 3-kinase-related kinase) kinase domain, and FAT C-terminal domains (5). Upon DSB induction, ATM is rapidly recruited to DSBs and activated. Activated ATM phosphorylates H2AX at Ser139 (forming γH2AX), and this phosphorylation is recognized by MDC1 (6, 7). MDC1 recruits downstream DNA repair protein and initiates the DNA repair process (8–11). ATM also triggers a second wave of phosphorylation events through its downstream kinase Chk2 and induces cell cycle arrest or cell apoptosis (12). ATM is mutated in patients with ataxia-telangiectasia, whose phenotypes include growth retardation, neurological defects, cancer predisposition, and hypersensitive to radiation (13).
DSB-induced ATM activation is different from oxidative stress–mediated ATM activation, which is due to dimerization of ATM via disulfide bonds (14, 15). Following DSBs, the Mre11-Rad50-Nbs1 (MRN) complex serves as an upstream component of DNA damage signaling and is critical for ATM activation (16–18). Recombinant MRN complex directly binds to double-stranded DNA ends and promotes ATM activation with double-strand DNA in vitro (17). The acetyltransferase Tip60 also contributes to ATM activation (19). Tip60 is activated in a histone H3K9me3-dependent manner and acetylates ATM, thus promoting ATM activation. Recently, we and another group showed that UFM1-specific ligase 1 (UFL1) promotes ATM activation (20, 21). We found that UFL1 ufmylates H4 at Lys31 and enhances the recruitment of SUV39H1 to DSBs, resulting in trimethylation of H3 at Lys9 and activation of Tip60 to enhance ATM activation (21). However, it is still unclear how increased histone H4 ufmylation following DNA damage is connected to H3K9me3. Here, we show that STK38 recognizes monoufmylated histone H4 and recruits SUV39H1 to DSBs to promote ATM activation.
RESULTS
STK38 recognizes monoufmylated H4
To identify potential histone H4 ufmylation readers, we analyzed all the proteins with potential UFM1 binding motif (table S1), which was initially found in UBA5 protein (22). From the potential candidates, we focused on the factors that have been shown to interact with epigenetic regulators. We identified one potential UFM1 binding protein—serine/threonine kinase 38 (STK38) (Fig. 1A). STK38, also named as NDR1, is a member of the AGC kinase family (23), which is implicated in the Hippo pathway. STK38 has been reported to interact with heterochromatin protein 1 (HP1) (24), which forms a complex with KAP-1 and SUV39H1, and helps promote H3K9 trimethylation (25). We carried out immunoprecipitation assay with STK38 antibody and found that STK38 interacted not only with HP1 but also with KAP-1 and SUV39H1 (Fig. 1B). However, we did not observe the interaction between STK38 and the ATM or MRN complex (fig. S1). We therefore hypothesized that STK38 might be a reader for monoufmylated histone H4 and recruit SUV39H1 through the HP1/KAP-1 complex.
Fig. 1. STK38 recognizes monoufmylated H4.
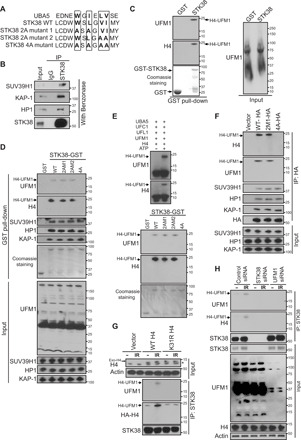
(A) UFM1 binding motif in STK38 protein. (B) STK38 interacts with the SUV39H1–KAP-1–HP1 complex. Rabbit immunoglobulin G (IgG) or STK38 antibody immunoprecipitates from U2OS cells were blotted with indicated antibodies. (C and D) Purified GST or GST-STK38 proteins were incubated with irradiated U2OS cell lysates for 1 hour and then blotted with indicated antibodies. (E) Purified UBA5, UFC1, UFL1, UFM1, and H4 proteins were incubated together in the presence of ATP and MgCl2 at 30°C for 90 min. Reaction products were used for GST pull-down assay with purified wild-type (WT) or mutant GST-STK38 proteins. (F) U2OS cells expressing WT or mutant STK38 were lysed, treated with Benzonase, and then subject to immunoprecipitation with HA agarose beads. Immunoprecipitates were blotted with indicated antibodies. (G and H) The mononucleosomes were purified from WT and K31R H4–expressing U2OS cells with/without 10 Gy IR (G) or U2OS cells transfected with control siRNA, STK38 siRNA, or UFM1 siRNA with/without 10 Gy IR (H) and then incubated with STK38 antibody. The immunoprecipitates were blotted with indicated antibodies. *Maxi PAGE gels that were used to separate the endogenous and exogenous H4. Mini PAGE gels were used for the rest of the blots.
To test this hypothesis, we examined the interaction between STK38 and ufmylated H4. We incubated purified STK38–glutathione S-transferase (GST) fusion protein with irradiated cell lysates and performed pull-down assay. Monoufmylated histone H4 was detected in STK38-GST precipitates, but not in GST precipitates (Fig. 1C). To explore how STK38 binds to ufmylated H4, we mutated critical residues in the potential UFM1 binding domain (LIR/UFIM domain) of STK38 and performed pull-down assay. Mutation of four critical amino acids within the UFM1 binding motif to alanine (4A mutant) abolished the interaction between STK38 and ufmylated H4, without affecting the interaction between STK38 and HP1/KAP-1/SUV39H1 (Fig. 1D). We further performed pull-down assay with purified GST-STK38 and ufmylated H4 generated from in vitro ufmylation assays and found that mutation of the four critical amino acids impaired recognition of ufmylated H4 by STK38 (Fig. 1E). To confirm this result in vivo, we also carried out immunoprecipitation assay. We detected similar impaired interaction between STK38 and ufmylated H4 due to mutation of the four critical amino acids, but no effect on the interaction between STK38 and SUV39H1 complex (Fig. 1F). Previously, we have reported that histone H4 is monoufmylated at Lys31 by the ligase UFL1 (21). To test whether ufmylation of H4 at K31 is important for the interaction between STK38 and H4, we mutated H4 Lys31 into arginine, isolated chromatin fraction, and performed immunoprecipitation assay. We found that mutation of H4K31 abolished the interaction between STK38 and H4 (Fig. 1G). To further confirm this result, we also depleted STK38 and UFM1 separately and found depletion of UFM1, as well as depletion of STK38, attenuated the interaction between STK38 and H4 (Fig. 1H). Our results suggest that STK38 is a H4K31 ufmylation reader and might facilitate the accumulation of SUV39H1 at DSBs.
STK38 is recruited to the DSB
To test further whether STK38 is a reader of monoufmylated H4, we examined whether STK38 was recruited to the DSBs. We found that STK38 proteins accumulated at DNA damage foci following DNA damage (Fig. 2A and fig. S2A). Although STK38 is a serine/threonine kinase, its kinase activity was not essential for its own foci formation, since the K118R kinase dead mutant (26) behaved similar to the wild-type (WT) protein (Fig. 2B and fig. S2B). We have reported that UFL1 is recruited to DSB following DNA damage (21). To test whether the recruitment of STK38 to DSB is mediated by UFL1, we knocked down UFL1 and monitored STK38 foci formation following Ionizing Radiation (IR). We found that depletion of UFL1 attenuated STK38 foci formation (Fig. 2C and fig. S2C), consistent with the role of UFL1 as a histone H4 ufmylation writer. To further confirm that ufmylation of histone H4 protein at Lys31 is critical for STK38 recruitment, we transfected WT and ufmylation-deficient mutant histone H4 K31R plasmids into U2OS cells and monitored STK38 foci formation. We have previously shown that overexpression of H4K31R mutation compromises ATM activation (21), probably by functioning as a dominant-negative mutant. Here, we found that overexpression of H4K31R also suppressed STK38 foci formation (Fig. 2D and fig. S2D), suggesting that ufmylation is important for STK38 recruitment to DNA lesions. To further confirm that ufmylation is important for the recruitment of STK38 to DNA break sites, we knocked down UFM1 and UFSP2 separately and found that loss of UFM1 impaired the STK38 foci formation; in contrast, depletion of UFSP2 enhanced STK38 foci formation (fig. S2, E and F). To further determine that the UFM1 binding motif is critical for the recruitment of STK38 to the DSBs, we transfected WT and 4A mutant into STK38 knockdown cells and found that WT STK38 but not STK38 4A mutant formed foci following IR (Fig. 2E and fig. S2G). Collectively, these results suggest that the recruitment of STK38 to DNA damage site relies on its recognition of monoufmylated H4.
Fig. 2. STK38 is recruited to the DSB.
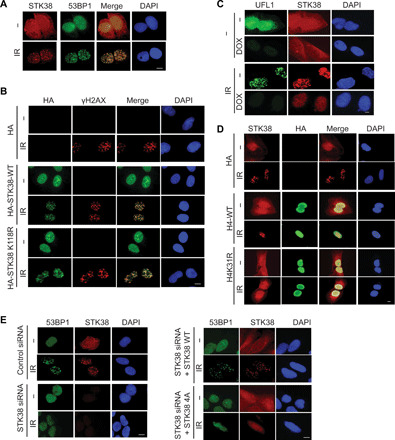
(A) U2OS cells were treated with or without 2 Gy IR. Thirty minutes later, cells were fixed and stained with indicated antibodies. (B) Vector, HA-tagged WT, or kinase-dead mutant (K118R) STK38 plasmids were transfected into U2OS cells. The localization of STK38 was detected by HA antibody. (C) Control and UFL1 knockdown cells were treated with or without 2 Gy IR. Thirty minutes later, cells were fixed and stained with indicated antibodies. (D) Vector, WT H4, or K31R H4–expressing cells were treated with or without 2 Gy IR. Thirty minutes later, cells were fixed and stained with indicated antibodies. (E) STK38 knockdown cells were restored with WT and 4A mutant STK38 and treated with or without 2 Gy IR. Thirty minutes later, cells were fixed and stained with indicated antibodies. Scale bars, 10 μm.
The recruitment of SUV39H1 relies on STK38
We next tested whether the recruitment of SUV39H1 to DSB was dependent on STK38, which recognizes ufmylated histone H4. To monitor the recruitment of SUV39H1 to the defined DSB, we used chromatin immunoprecipitation (ChIP) assay in MDA-MB-231 ROS8 cells (27), since it is difficult to detect SUV39H1 foci. An I-SceI cut recognition site was inserted into a copy of the E-cadherin promoter in MDA-MB-231 ROS8 cells. When cells were treated with doxycycline, I-SceI endonuclease expression was induced and I-SceI created single DSB in the cell. We performed ChIP assay with SUV39H1, H3K9me3, and H3 antibodies and detected the changes in trimethylation of H3K9 and the recruitment of SUV39H1 around the DSB. We found that loss of STK38 attenuated the recruitment of SUV39H1 to DSBs and suppressed H3K9me3 modification at damage sites (Fig. 3A). To further confirm that recognition of ufmylated H4 by STK38 is important for SUV39H1-mediated trimethylation of H3K9 around the DSB, we reintroduced WT and 4A mutant STK38 into STK38 knockdown cells. We found that reconstitution of WT STK38 but not STK38 4A mutant in STK38 knockdown cells partially rescued SUV39H1 recruitment to the DSB and H3K9me3 modification at the DSB (Fig. 3B). These results indicate that recognition of monoufmylated H4 by STK38 is critical for SUV39H1 recruitment to the DSB and subsequent H3K9 trimethylation.
Fig. 3. The recruitment of SUV39H1 relies on STK38.
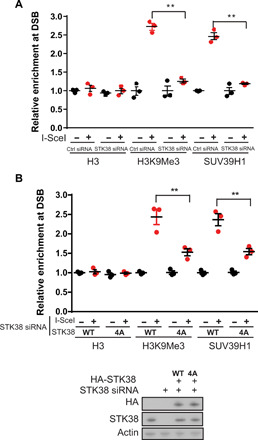
(A) Analysis of H3 and H3K9Me3 status at DSB and recruitment of SUV39H1 in cells that were transfected with control (Ctrl) siRNA or two different STK38 siRNAs by ChIP assay. The y axis represents relative enrichment of target protein binding DNA compared with input. (B) Analysis of H3 and H3K9Me3 status at DSB and recruitment of SUV39H1 in cells that were transfected with STK38 siRNA and reconstituted with WT and 4A mutant STK38 by ChIP assay. Means ± SEM are from three experiments. **P < 0.01. Statistical significance was determined by Student’s t test.
STK38 is important for ATM activation
We had previously shown that UFL1-mediated ufmylation of H4 is important for ATM activation (21). To test whether STK38 is the critical mediator of this process as a histone H4 ufmylation reader, we depleted STK38 in the cells with two different small interfering RNAs (siRNAs) and found that depletion of STK38 greatly impeded ATM activation and its downstream signaling. We also observed the impaired recruitment of Tip60 to chromatin, which is an important step for ATM activation. Accordingly, we observed decreased acetylation of ATM in STK38 knockdown cells (Fig. 4A). To further confirm the role of STK38 in ATM activation, we reintroduced WT ATM, K3016R ATM, or K3016Q ATM mutant into ATM knockout cells. These cells were then transfected with control siRNA or STK38 siRNA. We found that restoration of WT ATM and K3016Q ATM, but not K3016R ATM mutant, enhanced phosphorylation of Chk2, a downstream ATM target. In contrast, knockdown of STK38 suppressed phosphorylation of Chk2 in cells expressing WT ATM but not K3016Q mutant (fig. S3). Since STK38 is a protein kinase, we next examined whether its kinase activity was important for ATM activation. We reintroduced WT or the kinase-dead STK38 into STK38 knockdown cells. Reconstitution of either WT STK38 or the kinase-dead mutant rescued ATM signaling (Fig. 4B), suggesting that STK38 kinase activity is not essential for ATM activation. To confirm the function of STK38 as a monoufmylated H4 reader in ATM activation, we restored STK38 WT or the 4A mutant into knockdown cells and found only WT STK38, but not 4A mutant, rescued ATM activation (Fig. 4C), suggesting that recognition of histone H4 ufmylation by STK38 is important for ATM activation.
Fig. 4. STK38 is important for ATM activation.
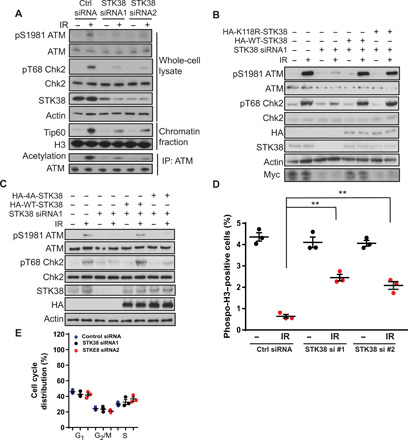
(A) U2OS cells were transfected with control siRNA and two different STK38 siRNAs. Cells were harvested and lysed with whole-cell lysate buffer or underwent chromatin fractionation or NETN buffer for ATM immunoprecipitation. The samples were blotted with indicated antibodies. (B) U2OS cells were transfected with control siRNA or STK38 siRNA with or without reconstitution of either HA-WT-STK38 or HA-K118R-STK38, and cells were then treated with or without 2 Gy IR. Cell lysates were blotted with indicated antibodies. (C) U2OS cells depleted of STK38 were reconstituted with WT and 4A mutant STK38. Cells were lysed and blotted with indicated antibodies. (D) Analysis of phospho-H3 cells in control siRNA and STK38 siRNA–transfected cells. Means ± SEM are from three experiments. **P < 0.01. Statistical significance was determined by Student’s t test. (E) Analysis of cell cycle distribution of cells transfected with control siRNA or two different STK38 siRNAs. The data presented are means ± SEM for three independent experiments. Statistical significance was calculated using two-way analysis of variance (ANOVA).
It has been shown that cells lacking ATM function exhibit a defective G2 checkpoint (28). As a regulator of ATM activation, depletion of STK38 might affect the G2/M checkpoint. We performed G2/M checkpoint assay and found that loss of STK38 compromised the IR-induced G2/M checkpoint (Fig. 4D). To exclude the possibility that the impaired G2/M checkpoint is due to the change in cell cycle, we analyzed the cell cycle profiles of STK38 knockdown cells. We found that loss of STK38 did not notably affect cell cycle distribution (Fig. 4E).
STK38 regulates IR sensitivity
To examine whether the absence of STK38 affects cell radio sensitivity, we performed colony formation assay. Compared with the control group, the survival of STK38 knockdown U2OS cells was significantly reduced following exposure to various doses of radiation (Fig. 5A), suggesting that STK38 is important for cellular radiation response. It is possible that STK38 regulates the DNA damage response through regulation of other cellular processes. To further confirm that the function of STK38 in radiosensitivity is through ATM, we knocked down STK38, ATM, or both in the U2OS cells. We found that loss of STK38 or loss of ATM-sensitized cells to irradiation and depletion of STK38 did not further sensitize cells to IR in ATM knockdown cells (Fig. 5B). When we introduced ATM acetylation mimic mutant K3016Q, we found ATM K3016Q reversed the radiosensitivity caused by the loss of STK38 (fig. S4A). This result suggests that STK38 regulates radiation sensitivity through the regulation of ATM acetylation. To confirm the function of the UFM1 binding motif of STK38 in this response, we restored WT and 4A mutant in STK38 knockdown cells and treated cells with or without ATM inhibitor. We found that reintroduction of WT STK38, but not the STK38 4A mutant, reversed the radiosensitivity caused by knockdown of STK38. ATM inhibitor Ku55933 sensitized cells to IR, and knockdown of STK38 or reintroduction of WT or 4A mutant did not further affect radiosensitivity in the presence of Ku55933 (fig. S4B). Our results suggest that loss of STK38 sensitizes cell to irradiation through ATM.
Fig. 5. STK38 regulates IR sensitivity.
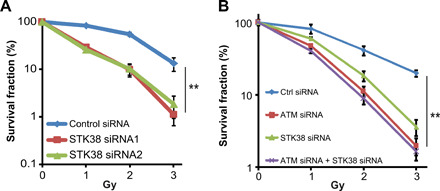
(A) U2OS cells transfected with control siRNA or two different STK38 siRNAs were exposed to the indicated doses of radiation and subjected to colony formation assay. (B) U2OS cells transfected with control siRNA, STK38 siRNA, ATM siRNA, or STK38 siRNA+ ATM siRNA were exposed to the indicated doses of radiation and subjected to colony formation assay. The data are presented as means ± SEM of three independent experiments. **P < 0.01. Statistical significance was calculated by ANOVA with multiple comparisons.
DISCUSSION
There are sets of specialized protein machineries, which add, remove, or recognize modified histones. These proteins are also called histone writers, erasers, and readers (29). Ufmylation, as a previously unidentified histone modification, has its own writer, eraser, and reader. Previous studies suggest that the writer of ufmylated H4 is the E3 ligase UFL1, and the eraser is the deufmylase UFSP2 (21). We have reported that histone H4 is ufmylated at Lys31 by UFL1, and this ufmylation is important for ATM activation (21). Overexpression of UFSP2, on the other hand, decreases ATM activation (21). A study from another group also confirms the critical role of ufmylation for enhancing ATM activation. This study suggests that UFL1 promotes ATM activation through ufmylation of MRE11 and enhances MRE11 loading to the DNA (20). The different substrates identified in both studies might be due to the different experimental models. It is expected that additional substrates of UFL1 will be identified. Further studies are required to reveal the potential redundancy or difference of UFL1 substrates.
So far, the reader for UFM1 was unknown, and it was unclear how histone H4 ufmylation promotes ATM activation. Three different strategies are used for identifying the readers: (i) identification of binding proteins by mass spectrometry, (ii) hypothesis-driven screening between protein domain and ufmylated peptides, and (iii) screening through high-throughput arrays (30). Here, we used the second strategy to identify the UFM1 reader based on the recently defined UFM1 binding domain. We identify an ufmylated H4 reader—STK38, which contains a motif similar to the UFM1 binding domain (22). STK38 is a member of NNDR (nuclear Dbf2-related)/LATS (large tumour suppressor) kinase family and is conserved from yeast to mammals. It is a multifunctional kinase and is involved in many important biological processes. During mitosis, STK38 drives centrosome duplication in a CDK2-dependent manner (31). STK38 is also critical for Fas receptor–induced apoptosis and mediates the RASSF1A/MST1 signaling pathway (32). Under conditions that induce autophagy, STK38 binds to Beclin1 to promote autophagy and serves as a switch between apoptosis and autophagy (33, 34). STK38 also phosphorylates and stabilizes RBM24 and regulates sarcomere assembly and heart contractility (35). Moreover, STK38 is important for immune response. It enhances Mitogen-activated protein/extracellular signal-regulated kinase Kinase Kinase 2 (MEKK2) ubiquitination and suppresses Toll-like receptor 9 (TLR9)–activated inflammatory responses in macrophages (36). STK38 also potentiates nuclear factor κB (NF-κB) activation induced by tumor necrosis factor–α (37).
Recently, a role of STK38 in the DNA damage response has been reported. Following DNA damage, STK38 phosphorylates CDC25A at Ser76, inducing degradation of CDC25A and promoting cell cycle checkpoint activation (38). STK38 also promotes nucleotide excision repair under ultraviolet-induced DNA damage condition (39). Here, we discovered that STK38 has a kinase-independent function in promoting ATM activation. We showed that STK38 contains a UFM1 binding motif. Mutation of conserved amino acids in this motif abolished the interaction between STK38 and ufmylated H4 (Fig. 1, A to C). With the support of in vitro and in vivo data, we propose that STK38 is a reader of ufmylated H4 (Fig. 1, D to H). However, the detailed mechanism of this recognition requires more extensive structural studies in the future. We also further confirmed that STK38 activates ATM signaling, and this process requires the ufmylation binding function of STK38 but not its kinase activity. Consistent with the role of STK38 in ATM activation, Stk38-deficient mice are reported to be prone to develop thymic lymphoma in aged mice, sharing similar tumor spectrum with Atm knockout mice. This observation suggests that both proteins might function to suppress lymphoma in vivo (40–42). Together, we have identified STK38 as a H4 ufmylation reader that facilitates the recruitment of SUV39H1 to DSBs, resulting in the trimethylation of H3K9 and activation of Tip60 to the DSB, thus enhancing ATM activation.
MATERIALS AND METHODS
Cell culture, plasmids, and reagents
U2OS [the American Type Culture Collection (ATCC)] and human embryonic kidney 293T (ATCC) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). MDA-MB-231 ROS8 cells (provided by S. Baylin, Johns Hopkins University) were cultured in DMEM supplemented with 10% FBS and Zeocin and blasticidin treatment. All cell lines were kept in a humidified 37°C 5% CO2 incubator.
STK38-myc was used as the template, and STK38 polymerase chain reaction (PCR) product were inserted into pCMV (cytomegalovirus)–hemagglutinin (HA) vector. STK38 2A mutant 1, 2A mutant 2, and 4A mutant were generated using site-directed mutagenesis kit.
Anti-UFL1 antibody (A303-456A) was purchased from Bethyl Laboratories. Anti-actin (A5316, 1:10,000) and anti-UFL1 (HPA030558 for detecting foci) antibodies were purchased from Sigma-Aldrich. Anti-ATM (2873), anti-pSer1981 ATM (13050), anti-SUV39H1 (8729), anti-SQ/TQ motif (9607, 1:1000 for Western blot), anti-Chk2 (2662), and anti-phosphoChk2 (2197) antibodies were purchased from Cell Signaling Technology. Anti-53BP1 [mab3802 for immunofluorescence (IF)], anti-MDC1 (05-172, 1:1000), anti-SUV39H1 (MABE552, 1:1000 for Western blot), and anti-γH2AX (05-636, 1:1000 for IF) antibodies were purchased from Millipore. Anti-UBA5 (ab177478, 1:1000 for Western blot), anti-UFC1 (ab189252, 1:1000 for Western blot), anti-H3 (ab1791, for Western blot and for ChIP), anti-H4 (ab10158, for Western blot and ChIP), anti-H4K16ac (ab109463, 1:100 for ChIP), and anti-H3K9me3 (ab8898) antibodies were purchased from Abcam. Anti-UFM1 (SC-84652, for IF and Western blot) and anti-Tip60 (SC-25378 for Western blot and ChIP) antibodies were purchased from Santa Cruz Biotechnology. Rabbit 53BP1 antibody (NB100-304) was purchased from Novus Biologicals. For immunoprecipitation assay, anti–immunoglobulin G (IgG) and light chain–specific antibodies were used (Jackson ImmunoResearch). Lipofectamine 2000 Transfection Reagent (Invitrogen) and Mirus TransIT Transfection Reagent (Mirus Bio LLC) were used for carrying out transfections following the manufacturer’s protocols.
RNA interference
UFL1 shRNA sh1 [oligo1 GAAACACTTCTGTGTCAGAAA, targeting 3′ untranslated region (3′UTR)] and UFL1 shRNA sh2 [oligo2 GCTCTGGAACATGGGTTGATA, targeting CoDing Sequence (CDS)] were inserted into Tet-on PLKO.1 vector. Lentiviruses were made according to the manufacturer’s protocol. STK38 siRNA1 (5′-CGUCGGCCAUAAACAGCUdTT-3′, targeting 3′UTR) and STK38 siRNA2 (5′-GCCUGCAACUUAGGCGGAUUGdTT-3′, targeting CDS) were purchased from QIAGEN.
Western blot and immunoprecipitation
Western blot and immunoprecipitation were performed as described previously (43). Cells were lysed with NETN buffer [20 mM tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40] with 50 mM β-glycerophosphate, 10 mM NaF, and 1 mg/ml each of pepstatin A and aprotinin. The cell lysates were incubated with Benzonase to remove DNA and then incubated with antibody against protein of interest and protein A or protein G Sepharose beads (Amersham Biosciences) for 2 hours or overnight at 4°C. The immunoprecipitates were analyzed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gels. Western blots were carried out following standard procedures.
Irradiation
For immunofluorescence studies, the cells were irradiated with 0.5 gray (Gy), and for Western blot/coimmunoprecipitation assays, the cells were irradiated with 2 Gy. Cells were analyzed 30 min following irradiation unless noted otherwise.
Chromatin fractionation assay
The cells were harvested and lysed with low-salt NETN buffer for 20 min. The lysates were centrifuged at 15,000g for 10 min. The supernatant was aspirated off, and the chromatin pellet was washed with phosphate-buffered saline (PBS) and centrifuged at 15,000g for 2 min. This step was repeated three times. The pellet was resuspended in 0.2 M HCl for 30 min on ice. The soluble extraction was neutralized with 1 M NaOH for Western blot.
Colony formation assay
Colony formation assay was carried out as previously described (44). Briefly, 1000 to 3000 U2OS cells were plated in each well of six-well plates and treated as indicated. The next day, cells were exposed to the indicated dose of ionizing radiation and incubated for 10 to 14 days at 37°C to allow colony formation. Colonies were fixed with methanol, stained with methylene blue, and counted. Results were normalized to plating efficiencies.
Immunofluorescence staining
Cells were plated on coverslips and fixed with 3% paraformaldehyde on ice after the indicated treatment. Then, the cells were washed with PBS and permeabilized for 10 min with 0.5% Triton X-100. Cells were blocked and incubated with primary antibodies for 1 hour at room temperature. After washing with PBS, immunofluorescent-labeled secondary antibody (Jackson ImmunoResearch) was added and incubated for 30 min at room temperature. After washes with PBS, cells were incubated with 4′,6-diamidino-2-phenylindole (DAPI). Last, the coverslips were mounted onto glass slides with antifade solution and visualized using a Nikon ECLIPSE E800 fluorescence microscope.
Cell cycle analysis and G2/M checkpoint
U2OS cells were treated as indicated and fixed with 70% precooled ethanol (−20°C) and harvested by centrifugation. Then, the cells were incubated with 69 μM propidium iodide (PI) solution in PBS containing ribonuclease (RNase) A for 30 min at 37°C. Cells were subjected to flow cytometry. The cell cycle distribution was analyzed with FlowJo software. For cell cycle analysis, unstained cells were used as control.
For G2/M checkpoint analysis, cells were harvested and fixed with 70% precooled ethanol (−20°C) and harvested by centrifugation. Then, the cells were incubated with phospho-H3 antibody for 1 hour. After washes with PBS, the cells were incubated with fluorescein isothiocyanate (FITC)–labeled secondary antibody (Jackson ImmunoResearch). Then, cells were stained with 69 μM PI and analyzed by FACS (fluorescence-activated cell sorting). Negative controls were unstained cell, PI-only–, phospho-H3 staining–only–, and control IgG–only–stained cells to set up the gates.
GST fusion protein purification and pull-down assay
STK38, STK38 2A mutant 1 and 2, and STK38 4A mutant were inserted into vector pGEX-4T2. GST fusion proteins were expressed in BL21 Escherichia coli. Bacteria were incubated with 0.25 mM isopropyl β-d-thiogalactoside overnight at 18°C. The bacterial cells were lysed with lysis buffer and sonicated. The lysates were centrifuged, and the supernatant was incubated with glutathione agarose beads at 4°C for 4 hours. After washes with NETN buffer, GST fusion proteins were eluted with glutathione. Then, 1 to 2 μg of GST fusion protein and 40 μl of glutathione agarose beads were added into U2OS cell lysates and incubated at 4°C for 2 hours. After the beads had been washed three times with NETN buffer, the protein was eluted and boiled. The samples were analyzed by SDS-PAGE gel.
ChIP assay
MDA-MB-231 ROS8 cells (5 × 107) were treated as indicated. One day later, the cells were treated with 1% formaldehyde for 10 min at room temperature to cross-link proteins to DNA. Glycine was added to stop the cross-linking. Cells were centrifuged and resuspended in cell lysis buffer [5 mM Pipes (KOH) (pH 8.0), 85 mM KCl, and 0.5% NP-40] with protease inhibitors for 10 min on ice. Nuclei were spun down by centrifugation and resuspended in nuclear lysis buffer [50 mM tris (pH 8.1), 10 mM EDTA, and 1% SDS-containing protease inhibitors]. Sonication was used to shear chromatin to an average size of 0.6 kb. The lysates were precleared with salmon sperm DNA/protein A agarose slurry. Twenty percent of each supernatant was used as input control and processed with the cross-linking reversal step. The rest of the supernatant was incubated with 5 μg the indicated antibody overnight at 4°C with rotation. The immunoprecipitation complexes were washed four times with the following buffers: high-salt buffer [50 mM tris-Cl (pH 8.0), 500 mM NaCl, 0.1% SDS, 0.5% deoxycholate, 1% NP-40, and 1 mM EDTA], LiCl buffer [50 mM tris-Cl (pH 8.0), 250 mM LiCl, 1% NP-40, 0.5% deoxycholate, and 1 mM EDTA], and twice in TE buffer [10 mM tris-Cl (pH 8.0) and 1 mM EDTA (pH 8.0)]. The immunoprecipitation complexes were resuspended in TE containing RNase (50 mg/ml) and incubated for 30 min. Beads washed with elution buffer (1% SDS and 0.1 M NaHCO3) were added for 15 min. Cross-links were reversed by adding RNase (10 μg/ml) and 5 M NaCl to a final concentration of 0.3 M to the eluents and incubated in a 65°C water bath for 4 to 5 hours. Two volumes of 100% ethanol were added to precipitate overnight at −20°C. DNA was pelleted and resuspended in 100 μl of water, 2 μl of 0.5 M EDTA, and 4 μl of 1 M tris (pH 6.5), and 1 μl of proteinase K (20 mg/ml) was added and incubated for 1 to 2 hours at 45°C. DNA was then purified and used in PCRs. The PCR primers for ChIP, close to the I-SceI cut site, were as follows: 5′-CCCTCTCAGTGGCGTCGGAACT-3′ (forward) and 5′-CCCACCCTCTGATGAGTACCT-3′ (reverse). Amplification was performed using the following program: 95°C for 5 min, 1 cycle; 95°C for 45 s, 56°C for 30 s, and 72°C for 30 s, 30 cycles; 72°C for 10 min, 1 cycle. As an internal control for the normalization of the specific fragments amplified, a locus outside the region of the DSB was amplified, in this case FKBP5, using the input control sample as template. The internal control (FKBP5) primers were as follows: 5′-CAGTCAAGCAATGGAAGAAG-3′ (forward) and 5′-CCCGTGCCACCCCTCAGTGA-3′ (reverse). After quantitative PCR (Q-PCR) amplification, the FKBP5 input controls for untransfected (no DSB) and I-SceI transfected (DSB) cells were used to normalize the untransfected and transfected samples, respectively. After normalization, the relative levels of the indicated proteins on a DSB were calculated using the formula: [IP ISce-1/Input ISce-1]/[IP untransfected/Input untransfected]. All Q-PCRs were performed in triplicate, with the SEM values were calculated from three independent experiments.
Statistical analysis
Data in the bar and line graphs are presented as means ± SEM of at least three independent experiments. Comparisons were carried out with a two-tailed unpaired Student’s t test or analysis of variance (ANOVA) with multiple comparison using GraphPad Prism as appropriate (*P < 0.05 and **P < 0.01).
Supplementary Material
Acknowledgment
We thank S. Baylin for providing MDA-MB-231 ROS8 cells. We thank A. Hergovich and A. Califano for STK38 plasmids. We thank W.-G. Zhu for ATM wild-type and mutant plasmids. Funding: This work was supported by NIH grants (CA217183, CA203561, and CA130996). Author contributions: B.Q. and J.Y. designed and conducted the experiments. B.Q., J.Y., S.N., F.Z., L.W., and Z.L. analyzed the data. B.Q., J.Y., and Z.L. wrote the manuscript. All authors read and approved the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/23/eaax8214/DC1
REFERENCES AND NOTES
- 1.Jackson S. P., Bartek J., The DNA-damage response in human biology and disease. Nature 461, 1071–1078 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gatti R. A., Berkel I., Boder E., Braedt G., Charmley P., Concannon P., Ersoy F., Foroud T., Jaspers N. G. J., Lange K., Lathrop G. M., Leppert M., Nakamura Y., O’Connell P., Paterson M., Salser W., Sanal O., Silver J., Sparkes R. S., Susi E., Weeks D. E., Wei S., White R., Yoder F., Localization of an ataxia-telangiectasia gene to chromosome 11q22–23. Nature 336, 577–580 (1988). [DOI] [PubMed] [Google Scholar]
- 3.Savitsky K., Bar-Shira A., Gilad S., Rotman G., Ziv Y., Vanagaite L., Tagle D. A., Smith S., Uziel T., Sfez S., Ashkenazi M., Pecker I., Frydman M., Harnik R., Patanjali S. R., Simmons A., Clines G. A., Sartiel A., Gatti R. A., Chessa L., Sanal O., Lavin M. F., Jaspers N. G., Taylor A. M., Arlett C. F., Miki T., Weissman S. M., Lovett M., Collins F. S., Shiloh Y., A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268, 1749–1753 (1995). [DOI] [PubMed] [Google Scholar]
- 4.Savitsky K., Sfez S., Tagle D. A., Ziv Y., Sartiel A., Collins F. S., Shiloh Y., Rotman G., The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum. Mol. Genet. 4, 2025–2032 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Perry J., Kleckner N., The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell 112, 151–155 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Lou Z., Minter-Dykhouse K., Franco S., Gostissa M., Rivera M. A., Celeste A., Manis J. P., van Deursen J., Nussenzweig A., Paull T. T., Alt F. W., Chen J., MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell 21, 187–200 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Stucki M., Clapperton J. A., Mohammad D., Yaffe M. B., Smerdon S. J., Jackson S. P., MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213–1226 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Burma S., Chen B. P., Murphy M., Kurimasa A., Chen D. J., ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 276, 42462–42467 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Stewart G. S., Wang B., Bignell C. R., Taylor A. M., Elledge S. J., MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421, 961–966 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Lou Z., Minter-Dykhouse K., Wu X., Chen J., MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature 421, 957–961 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Goldberg M., Stucki M., Falck J., D’Amours D., Rahman D., Pappin D., Bartek J., Jackson S. P., MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature 421, 952–956 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Matsuoka S., Huang M., Elledge S. J., Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282, 1893–1897 (1998). [DOI] [PubMed] [Google Scholar]
- 13.Shiloh Y., Ataxia-telangiectasia and the Nijmegen breakage syndrome: Related disorders but genes apart. Annu. Rev. Genet. 31, 635–662 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Barzilai A., Rotman G., Shiloh Y., ATM deficiency and oxidative stress: A new dimension of defective response to DNA damage. DNA Repair 1, 3–25 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Paull T. T., Mechanisms of ATM activation. Annu. Rev. Biochem. 84, 711–738 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Uziel T., Lerenthal Y., Moyal L., Andegeko Y., Mittelman L., Shiloh Y., Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 22, 5612–5621 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee J.-H., Paull T. T., Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304, 93–96 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Lavin M. F., Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 9, 759–769 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Sun Y., Jiang X., Chen S., Fernandes N., Price B. D., A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. U.S.A. 102, 13182–13187 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z., Gong Y., Peng B., Shi R., Fan D., Zhao H., Zhu M., Zhang H., Lou Z., Zhou J., Zhu W.-G., Cong Y.-S., Xu X., MRE11 UFMylation promotes ATM activation. Nucleic Acids Res. 47, 4124–4135 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin B., Yu J., Nowsheen S., Wang M., Tu X., Liu T., Li H., Wang L., Lou Z., UFL1 promotes histone H4 ufmylation and ATM activation. Nat. Commun. 10, 1242 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Habisov S., Huber J., Ichimura Y., Akutsu M., Rogova N., Loehr F., McEwan D. G., Johansen T., Dikic I., Doetsch V., Komatsu M., Rogov V. V., Kirkin V., Structural and functional analysis of a novel interaction motif within UFM1-activating enzyme 5 (UBA5) required for binding to ubiquitin-like proteins and ufmylation. J. Biol. Chem. 291, 9025–9041 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hergovich A., Stegert M. R., Schmitz D., Hemmings B. A., NDR kinases regulate essential cell processes from yeast to humans. Nat. Rev. Mol. Cell Biol. 7, 253–264 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Chakraborty A., Prasanth K. V., Prasanth S. G., Dynamic phosphorylation of HP1α regulates mitotic progression in human cells. Nat. Commun. 5, 3445 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ayrapetov M. K., Gursoy-Yuzugullu O., Xu C., Xu Y., Price B. D., DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. U.S.A. 111, 9169–9174 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bisikirska B. C., Adam S. J., Alvarez M. J., Rajbhandari P., Cox R., Lefebvre C., Wang K., Rieckhof G. E., Felsher D. W., Califano A., STK38 is a critical upstream regulator of MYC’s oncogenic activity in human B-cell lymphoma. Oncogene 32, 5283–5291 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Hagan H. M., Mohammad H. P., Baylin S. B., Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLOS Genet. 4, e1000155 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bao S., Tibbetts R. S., Brumbaugh K. M., Fang Y., Richardson D. A., Ali A., Chen S. M., Abraham R. T., Wang X.-F., ATR/ATM-mediated phosphorylation of human Rad17 is required for genotoxic stress responses. Nature 411, 969–974 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Kinnaird A., Zhao S., Wellen K. E., Michelakis E. D., Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 16, 694–707 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Wilkinson A. W., Gozani O., Histone-binding domains: Strategies for discovery and characterization. Biochim. Biophys. Acta 1839, 669–675 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hergovich A., Lamla S., Nigg E. A., Hemmings B. A., Centrosome-associated NDR kinase regulates centrosome duplication. Mol. Cell 25, 625–634 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Vichalkovski A., Gresko E., Cornils H., Hergovich A., Schmitz D., Hemmings B. A., NDR kinase is activated by RASSF1A/MST1 in response to Fas receptor stimulation and promotes apoptosis. Curr. Biol. 18, 1889–1895 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Joffre C., Dupont N., Hoa L., Gomez V., Pardo R., Gonçalves-Pimentel C., Achard P., Bettoun A., Meunier B., Bauvy C., Cascone I., Codogno P., Fanto M., Hergovich A., Camonis J., The pro-apoptotic STK38 kinase is a new beclin1 partner positively regulating autophagy. Curr. Biol. 25, 2479–2492 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joffre C., Codogno P., Fanto M., Hergovich A., Camonis J., STK38 at the crossroad between autophagy and apoptosis. Autophagy 12, 594–595 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J., Kong X., Lee Y. M., Zhang M. K., Guo L. Y., Lin Y., Lim T. K., Lin Q., Xu X. Q., Stk38 modulates Rbm24 protein stability to regulate sarcomere assembly in cardiomyocytes. Sci. Rep. 7, 44870 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wen M., Ma X., Cheng H., Jiang W., Xu X., Zhang Y., Zhang Y., Guo Z., Yu Y., Xu H., Qian C., Cao X., An H., Stk38 protein kinase preferentially inhibits TLR9-activated inflammatory responses by promoting MEKK2 ubiquitination in macrophages. Nat. Commun. 6, 7167 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Shi D.-D., Shi H., Lu D., Li R., Zhang Y., Zhang J., NDR1/STK38 potentiates NF-κB activation by its kinase activity. Cell Biochem. Funct. 30, 664–670 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Fukasawa T., Enomoto A., Miyagawa K., Serine–Threonine Kinase 38 regulates CDC25A stability and the DNA damage-induced G2/M checkpoint. Cell. Signal. 27, 1569–1575 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Park J. M., Choi J. Y., Yi J. M., Chung J. W., Leem S.-H., Koh S. S., Kang T.-H., NDR1 modulates the UV-induced DNA-damage checkpoint and nucleotide excision repair. Biochem. Biophys. Res. Commun. 461, 543–548 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Cornils H., Stegert M. R., Hergovich A., Hynx D., Schmitz D., Dirnhofer S., Hemmings B. A., Ablation of the kinase NDR1 predisposes mice to the development of T cell lymphoma. Sci. Signal. 3, ra47 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Barlow C., Hirotsune S., Paylor R., Liyanage M., Eckhaus M., Collins F., Shiloh Y., Crawley J. N., Ried T., Tagle D., Wynshaw-Boris A., Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell 86, 159–171 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Petiniot L. K., Weaver Z., Barlow C., Shen R., Eckhaus M., Steinberg S. M., Ried T., Wynshaw-Boris A., Hodes R. J., Recombinase-activating gene (RAG) 2-mediated V(D)J recombination is not essential for tumorigenesis in Atm-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 97, 6664–6669 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qin B., Minter-Dykhouse K., Yu J., Zhang J., Liu T., Zhang H., Lee S. B., Kim J. J., Wang L., Lou Z., DBC1 functions as a tumor suppressor by regulating p53 stability. Cell Rep. 10, 1324–1334 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu J., Qin B., Moyer A. M., Nowsheen S., Liu T., Qin S., Zhuang Y., Liu D., Lu S. W., Kalari K. R., Visscher D. W., Copland J. A., McLaughlin S. A., Moreno-Aspitia A., Northfelt D. W., Gray R. J., Lou Z., Suman V. J., Weinshilboum R., Boughey J. C., Goetz M. P., Wang L., DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J. Clin. Invest. 128, 2376–2388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/23/eaax8214/DC1