

ABSTRACT
Manganese is an essential metal, but elevated brain Mn concentrations produce a parkinsonian-like movement disorder in adults and fine motor, attentional, cognitive, and intellectual deficits in children. Human Mn neurotoxicity occurs owing to elevated exposure from occupational or environmental sources, defective excretion (e.g., due to cirrhosis), or loss-of-function mutations in the Mn transporters solute carrier family 30 member 10 or solute carrier family 39 member 14. Animal models are essential to study Mn neurotoxicity, but in order to be translationally relevant, such models should utilize environmentally relevant Mn exposure regimens that reproduce changes in brain Mn concentrations and neurological function evident in human patients. Here, we provide guidelines for Mn exposure in mice, rats, nematodes, and zebrafish so that brain Mn concentrations and neurobehavioral sequelae remain directly relatable to the human phenotype.
Keywords: manganese, neurotoxicity, animal models, SLC30A10, SLC39A14, ZnT10, ZIP14, Caenorhabditis elegans, zebrafish
Introduction
Manganese is an essential metal required for many biological processes (1). However, modest increases in brain Mn concentrations induce an incurable neurotoxic syndrome (1). A major cause of Mn neurotoxicity in adults is exposure to elevated Mn from occupational sources (e.g., welding, manufacture of dry batteries and steel) (1, 2). Estimates indicate that >100,000 American welders are routinely exposed to elevated Mn (2). The primary manifestation of Mn neurotoxicity in adults is a parkinsonian-like movement disorder, which is induced by the accumulation of Mn in the basal ganglia (1, 3, 4). Moreover, although Mn-induced parkinsonism is pathologically distinct from Parkinson disease (3, 4), elevated Mn concentrations lead to the aggregation of α-synuclein (5) and may enhance the risk of developing Parkinson disease (6). Recent epidemiological studies also indicate that environmental exposure to elevated Mn (e.g., via drinking water, dust, or air) is of substantial concern, especially in children and adolescents who develop fine motor, attentional, emotional, cognitive, and intellectual deficits (7–17). In addition, Mn is predominantly excreted in bile (18–22) and patients with liver dysfunction (e.g., due to cirrhosis) may fail to excrete Mn and may develop Mn neurotoxicity even in the absence of elevated exposure (23–28). Indeed, in 1 clinical study, as many as ∼20% of cirrhotics developed parkinsonism and had elevated blood Mn concentrations along with evidence of Mn deposition in the basal ganglia (29). Finally, over the last few years, 2 inherited forms of Mn neurotoxicity, termed Hypermanganesemia with Dystonia 1 or 2 (HMNDYT1 or 2), have been identified in humans (30–34). HMNDYT1 and HMNDYT2 occur owing to homozygous loss-of-function mutations in the Mn transporters solute carrier family 30 member 10 (SLC30A10) and solute carrier family 39 member 14 (SLC39A14), respectively, that increase blood and brain Mn concentrations (30–37). Overall, Mn neurotoxicity is a major public health problem for which effective treatments are needed.
Animal models are routinely used to investigate the pathobiology of Mn neurotoxicity. But, in order to be translationally relevant, such models should use Mn exposure paradigms that faithfully reproduce the increases in brain Mn concentrations observed in human patients. Normal basal ganglia Mn concentrations in adult humans are ∼1.0–2.5 μg Mn/g tissue (23–25, 28). Reliable information about brain Mn concentrations of humans exposed to elevated Mn via occupational or environmental sources is not available. But, subchronic exposure of rhesus monkeys to amounts of Mn relevant to human occupational exposure by inhalation produced ∼1- to 5-fold increases in basal ganglia Mn concentrations and neurotoxic sequelae (38, 39). Similar results were obtained in rats (40). Thus, Mn neurotoxicity in humans exposed to elevated Mn is expected to be associated with very modest ∼1- to 5-fold increases in basal ganglia Mn. In addition, rigorous data for basal ganglia Mn concentrations of human patients with liver dysfunction are available and show a similar ∼1- to 7-fold elevation (23–25, 27, 28). Below, we provide guidelines for Mn exposure in mice, rats, nematodes, and zebrafish, which are commonly used to study Mn neurotoxicity, so that increases in brain Mn concentrations remain within the range seen in humans. The dosing regimens we describe produce neurobehavioral changes that are analogous to the human phenotype. We also summarize recent progress in understanding the biology of HMNDYT1 or HMNDYT2 using these species [humans with HMNDYT1 or HMNDYT2 exhibit substantially larger (routinely >10-fold) increases in blood and brain Mn concentrations (30–34)].
Rodent Models
Mn dosing regimens to model elevated Mn exposure in rodents
Mice and rats are routinely used in Mn research. In neurobehavioral assays, Mn-treated rodents develop deficits in locomotor, fine motor, and executive functions—behavioral disinhibition, attentional dysfunction, and fine motor defects usually occur in animals exposed to Mn in early life (preweaning or adolescent periods) and may persist into adulthood, whereas exposure in later-life stages has been shown to produce locomotor and fine motor defects (41–52) (Table 1). Thus, there are important similarities in the functional outcomes of Mn toxicity between rodents and humans based on the life-stage at which exposure occurs. Results are broadly comparable between mice and rats; therefore, decisions about the choice of species often depend on the experience of a laboratory, cost (mice are cheaper), requirement for genetic manipulation (genome editing in rats is more challenging than in mice), and the need for sophisticated neurobehavioral analyses (a wider array of motor and cognitive assays and a broader body of evidence are available in rats, which provides a better understanding of functional neurological deficits). Rodents have been exposed to elevated Mn by injection (subcutaneous, intraperitoneal, or directly into the brain), orally (by gavage or in drinking water), and by inhalation. Because humans are primarily exposed to Mn through inhalation under occupational settings and via drinking water or inhalation environmentally, oral delivery and inhalation represent the disease-relevant modes of exposure in rodents. Inhalation exposure requires sophisticated equipment and is experimentally challenging, but Mn delivered by this route has unique pharmacokinetic properties that cannot be recapitulated with other routes of delivery. As examples, compared with the gastrointestinal tract, Mn is more efficiently absorbed into blood from the respiratory system (53), and nasally inspired Mn can be directly transported to the brain by the olfactory nerve (54). Particle size characteristics of inhalation exposures are also important in determining whether particle-associated Mn penetrates into the alveolar lung space for absorption into the blood or particles are deposited higher up in the respiratory tract where they can be coughed up and swallowed. Overall, delivery by inhalation is optimal for studies that seek to model the internal body dose that arises from airborne exposure to Mn in humans (e.g., as seen in occupational settings). We do not discuss inhalation exposure further because these studies are performed in highly specialized laboratories [see (39, 40) for details]. In contrast, oral exposure by drinking water or gavage is more readily accessible. Importantly, in children, elevated Mn concentrations in drinking water, but not food, were associated with increased body Mn concentrations and neurotoxicity (9), suggesting that exposure of rodents to Mn via water is more disease-relevant than other modes of oral exposure (e.g., through food). Therefore, we will focus on Mn delivery via drinking water or oral gavage as translationally relevant models of environmental Mn exposure.
TABLE 1.
Summary of Mn dosing and neurobehavioral outcomes in rodent, nematode, and zebrafish models
| Species | Mn dose | Behavioral test | Outcome | References |
|---|---|---|---|---|
| Rat | 25 or 50 mg Mn · kg–1 · d–1 in preweaning period or lifelong with testing in early life or adulthood | Open field8-arm radial mazeMontoya staircase testFive-choice serial reaction time task | Increased behavioral reactivity (hyperactivity)Spatial learning and memory deficitsDecreased forelimb reaching/grasping skillImpaired focused and selective attention | (41, 42, 46)(42)(43, 44)(45, 49) |
| Mouse | 100 mg Mn · kg–1 · d–1 by gastric gavage in adults | Open field | Hypoactivity | (47) |
| 10 or 30 mg Mn · kg–1 · d–1 by gastric gavage in juveniles | Open field | Increased behavioral reactivity (decreased time spent in margins) | (48) | |
| Caenorhabditis elegans | 10–25 mM Mn for 1 h at L1 | Basal slowing response | Reduction in response | (35) |
| Zebrafish | 25 or 50 μM Mn from 0 to 5 dpf | Spontaneous movement observation | Hypoactivity | (55) |
| 0.5 mM Mn from 0 to 5 dpf | Exploratory behavior | Decreased swimming distance and absolute turn angle | (56) |
Extensive studies in rats have used micropipettes or gavage methods in the preweaning period or drinking water postweaning to deliver daily oral doses of Mn ranging between ∼10 and 125 mg Mn/kg (41–46, 50–52, 57–60). Delivery is in the Mn2+ form, usually as MnCl2, which is the predominant intracellular oxidation state of Mn (61, 62). In 1 particularly well-characterized regimen, animals are provided 25–50 mg Mn · kg–1 · d–1 by micropipette before weaning and in drinking water after weaning (Table 1) (41–46, 57). In this regimen, for dosing before weaning, a stock solution of 225 mg Mn/mL is prepared in water, diluted in a 2.5% (wt:vol) solution of the sweetener stevia dissolved in water (to partly mask the bitter taste of Mn and facilitate intake by pups), and the required dose is administered via a micropipette with a gel loading tip, based on the body weight of the pup measured before dosing (43, 45, 57). The total volume delivered can be as little as ∼10–25 μL/animal. When used in mice, a smaller volume is provided to the animals: ∼5 μL for the first week and ∼10 μL for the remaining 2 wk before weaning. Postweaning exposure is through the animal's drinking water. For this, a 42-mg Mn/mL water stock solution is prepared and diluted to ∼0.2–0.4 mg Mn/mL in the animal's drinking water (43, 45, 57). A similar concentration of drinking water Mn (0.4 mg Mn/mL) has also been used in mice (63). During this period, daily water intake and weights of animals have to be monitored. Water intake is calculated per animal if exposure is per individual, or if to a cage of animals, by dividing the total water intake per cage by the number of animals. Specialized metabolic cages or individual housing of animals are not necessary for oral Mn exposure. The amount of Mn in drinking water can be adjusted on a regular basis (e.g., daily, weekly) to ensure target intake amounts are maintained.
Rats receiving 25 or 50 mg Mn · kg–1 · d–1 via the above regimen from post natal day (PND)1–21 exhibit ∼3-fold increases in brain Mn concentrations in inductively coupled plasma mass spectrometry (ICP-MS) analyses of tissue metal concentrations (43–45). Interestingly, brain Mn concentrations of animals exposed to Mn for longer periods of time (PND1–66 or PND500) are only slightly, although still measurably, higher than in vehicle-treated controls, and substantially lower than in weanling animals exposed over the PND1–21 period (43–45, 57). The lower concentrations of tissue Mn in adult rats who experience the same amount of daily oral Mn exposure as their younger weanling counterparts follow the same age-related decline in tissue Mn concentrations as in unexposed animals (43, 45, 57)—a phenomenon that also occurs in humans (64). This reduction in body Mn concentrations over the lifespan, even in the presence of continuous lifelong Mn exposure, likely reflects reductions in the gastrointestinal uptake of ingested Mn and increased excretion of absorbed Mn in rodents beginning over the second to third week of life and continuing into adulthood (18, 65, 66). Rats exposed to 25 or 50 mg Mn · kg–1 · d–1 by the aforementioned regimen over the preweaning period develop lasting behavioral disinhibition in the open-field test, deficits in fine motor function in the Montoya staircase test, and focused and selective attentional defects in the 5-choice serial reaction time test that persist into adulthood; notably, these behavioral deficits are associated with defects in evoked dopamine and norepinephrine release in the prefrontal cortex and basal ganglia in weanling and adult animals (Table 1) (41–43, 45, 46, 57). It is also noteworthy that, in general, lifelong exposures to the same Mn doses do not measurably worsen these attentional and fine motor deficits caused by early-life exposure, indicating that the early-life preweaning period is particularly susceptible to the lasting neurotoxic effects of elevated Mn exposure (Table 1) (43–45).
Delivery by oral gavage in adolescent and adult animals has also been extensively characterized. In adult mice, exposure to 100 mg Mn · kg body weight–1 · d–1 for 8 wk increases basal ganglia Mn concentrations by ∼2- to 3-fold, produces a hypo-locomotor phenotype in the open-field test, and induces neuronal injury without inducing overt toxicity (Table 1) (47). When mice are exposed as adolescents (PND20–34), a lower concentration of Mn (10–30 mg Mn · kg body weight–1 · d–1) is sufficient to produce a similar ∼2- to 3-fold increase in basal ganglia Mn concentrations and neurobehavioral deficits; in this developmental stage, the primary reported neurobehavioral manifestation is increased behavioral reactivity in the open-field assay (Table 1) (48).
Disease-relevance of the aforementioned dosing regimens based on elevated Mn consumption of rodents
An average adult rodent maintained on regular unpurified diet consumes ∼7%–10% body weight/d or ∼70–100 g unpurified diet · kg body weight–1 · d–1. Unpurified rodent diets contain ∼70–100 μg Mn/g diet. Therefore, rodents obtain ∼5–10 mg Mn · kg body weight–1 · d–1 from unpurified diets. This consumption is several orders of magnitude higher than human dietary Mn intake, which is estimated to be ∼2 mg/d (or 0.02 mg · kg body weight–1 · d–1 for a 70-kg human) (67). Although normal daily dietary requirements for Mn are not known for either infant humans or rodents (68, 69), substantially higher consumption of Mn is also evident in rodents in the preweaning period. Human breast milk contains ∼6 μg Mn/L (70), yielding normal infant intake rates of ∼0.6 μg Mn · kg–1 · d–1, based on infant daily milk consumption rates of ∼0.8 L/d for a 8-kg 6- to 9-mo-old infant. In comparison, rat and mouse milk Mn concentrations are ∼200–300 and ∼50 μg Mn/L, respectively (68, 71), and preweaning rats consume up to ∼260 mL · kg–1 · d–1 over PND1–21 (68, 72), whereas daily milk consumption in mice is ∼200 mL/kg (71, 73). Thus, preweaning rats and mice consume ∼70 and ∼10 μg Mn · kg–1 · d–1, respectively, which is ∼20–100 times higher than normal human infant Mn intake from breast milk. In spite of this higher intake, under physiological conditions, basal ganglia Mn concentrations of rats and mice are similar to that of humans [basal ganglia Mn concentrations in adult rats and mice are ∼1–2 μg Mn/g tissue weight (40, 74), similar to the ∼1.0–2.5 μg Mn/g tissue reported in adult humans (23–25, 28)]. The mechanisms that allow rodents to maintain tissue Mn concentrations at or near concentrations observed in humans in spite of substantially higher intake are unclear, but irrespective of this, a direct implication is that oral Mn dosing regimens in rodents cannot simply recapitulate concentrations of Mn reported to cause disease in environmentally exposed humans [e.g., estimates suggest that drinking water Mn concentrations as low as 133 μg/L are associated with intellectual deficits in children (17)]. Instead, the dosing regimen must 1) reproduce the modest increases in Mn concentrations in the brain and other target organs evident in human patients (i.e., the fold-increase in the tissue Mn concentrations of Mn-treated animals should be in the same range as that reported in humans); and 2) provide a fold-elevation in Mn intake that is comparable with environmental human Mn exposure while accounting for the differences in dietary Mn intake between humans and rodents.
As described in the preceding section, fold-increases in basal ganglia Mn concentrations of rodents orally exposed to 10–100 mg Mn · kg–1 · d–1 are comparable with those in Mn-exposed humans (47, 48, 57). In addition, these doses approximate the fold-increase in exposure that occurs in human infants, children, and adults environmentally exposed to elevated Mn. As an example, the 25 and 50 mg Mn · kg−1 · d−1 exposure amounts in rats over the preweaning period produce relative increases in Mn intake that are ∼350 and ∼700-fold over amounts consumed from lactation alone (∼70 μg Mn · kg−1 · d−1; see above) (41–45, 57). A relevant human comparison is exposure of infants to contaminated water (either directly or indirectly via rehydrated powdered formulas) containing 1.5 mg Mn/L, which is 3 times the US maximum contaminant level guideline and comparable with median well-water concentrations associated with cognitive deficits and other effects in children (16, 45, 69). At 1.5 mg Mn/L water, infants experience daily Mn exposure of ∼200 μg/kg, based on median fluid intake rates of ∼1 L/d for infants <1 y of age, which is ∼300-fold higher than the amount of Mn intake from breast milk (∼0.6 μg Mn · kg−1 · d−1; see above). Further, infants consuming Mn-contaminated well-water containing 1.5 mg Mn/L mixed with a high-Mn soy formula containing ≤1.0 mg Mn/L (total = 2.5 mg Mn/L) (70) would ingest ∼300 μg Mn · kg−1 · d−1, which is ∼500 times higher than the Mn intake from breast milk. In human adults, consumption of water containing 1.5 mg Mn/L provides ∼3 mg Mn/d, based on a daily water consumption of ∼2 L for a 70-kg adult human, which is ∼150% of the estimated daily dietary intake of Mn [∼2 mg/d (67)]. Similarly, a dose of 25–50 mg Mn · kg−1 · d−1 increases the Mn intake of an adult rodent by ∼3- to 6-fold relative to the amount received from unpurified diet (∼5–10 mg Mn · kg body weight −1 · d−1; see above).
Overall, the aforementioned oral Mn dosing regimens provide a means to model human environmental Mn exposure in rodents. We note 2 additional issues. First, purified rodent diets make it feasible to reduce dietary Mn intake of adult (postweaned) rodents (e.g., the well-characterized AIN-93G diet has ∼11 μg Mn/g diet). But, blood, brain, and liver Mn concentrations of mice receiving AIN-93G are comparable with those fed an unpurified diet with ∼84 μg Mn/g diet at 6 wk of age (75), implying that the Mn concentrations in rodent chow, including the reduced Mn in purified diets, do not substantially affect tissue Mn concentrations of wild-type rodents, and by extension, the arguments about the disease-relevance of the oral Mn exposure regimens discussed here. Second, variations in results are, however, possible whenever a new rodent strain or laboratory setting is utilized. Therefore, we suggest that in any new condition, tissue Mn concentrations be validated in a pilot study using standard metal measurement techniques, such as ICP-MS, so that relevance to human Mn neurotoxicity is established before large-scale experiments are initiated.
Injectable modes of Mn delivery
Injectable Mn (e.g., subcutaneous, intraperitoneal) is widely used in rodent studies, usually for ease of experimental manipulation. Some of these exposure regimens produce modest increases in brain Mn concentrations similar to oral exposure. As examples, with intraperitoneal delivery, ∼2- to 3-fold increases in Mn in the striatum are reported in weaned/adult rats exposed to 2.5 mg Mn · kg−1 · d−1 for 8–12 wk (76) or 6 mg Mn/kg 5 d/wk for 4 wk (77); whereas with subcutaneous delivery, analogous increases in brain Mn are observed in adult mice exposed to 7.5 or 15 mg Mn/kg 3 times/wk for 4 wk (78). Despite this, we urge caution in interpreting data from injectable models, especially results of functional neurological assays. Pharmacokinetics of Mn after injection differ from both inhalation and oral delivery in manners that may affect neurotoxic sequelae—as an example, injectable Mn does not undergo gastrointestinal absorption and may exhibit rapid and transient elevations in circulatory concentrations. Moreover, repeated intraperitoneal Mn injections reduce the body weight of rats (76, 77), which may confound neurobehavioral analyses. There is also a risk of infection/inflammation with repeated injections that may influence outcomes—not only because of the tissue-irritant effect of injecting high concentrations of Mn solution, but also because of infection risks associated with repeated injections. In totality, given the ease of oral Mn delivery in rats and mice, a strong rationale for using injectable Mn in rodents is lacking. At the present time, oral (and inhalation) exposure are the translationally relevant modes of Mn exposure for rodent studies.
Slc30a10 knockout mice as a model to study HMNDYT1
In 2012, the first inherited disorder of Mn metabolism, HMNDYT1, was reported to occur owing to homozygous loss-of-function mutations in SLC30A10 (30, 33). Patients accumulate Mn in the liver, brain, and blood with characteristic clinical features including liver cirrhosis, dystonia-parkinsonism, hypermanganesemia, and polycythemia. Parenteral chelation with disodium calcium edetate (Na2CaEDTA) successfully reduces the Mn load with improvement of neurological symptoms and halt of cirrhosis (30). However, this treatment is burdensome due to the need of intravenous administration. Moreover, Na2CaEDTA is unselective and removes other essential metals, causing side effects such as osteopenia and pathological fractures. Animal models (mice, Caenorhabditis elegans, and zebrafish) mimicking the human disorder are playing an important role in elucidating the function of SLC30A10 at the organism level and developing better therapeutics.
Mechanistic assays revealed that SLC30A10 is a cell-surface localized Mn efflux transporter that reduces Mn concentrations and protects against Mn toxicity, whereas disease-causing mutants lack Mn efflux activity (35) [the transport activity of SLC30A10 is specific towards Mn (35, 79, 80)]. At the organism level, SLC30A10 is robustly expressed in the liver and basal ganglia in mice (74, 75, 81) and humans (33, 34), suggesting that the Mn retention and parkinsonism phenotype of HMNDYT1 is likely related to loss-of-function of SLC30A10 in the digestive system as well as the brain. Analyses of full-body and tissue-specific Slc30a10 knockout mice, performed in 2017–2018, provided evidence in support of this hypothesis. Similar to the human condition, blood, brain, and liver Mn concentrations are markedly elevated in full-body Slc30a10 knockout mice fed unpurified rodent diet (∼84 μg Mn/g) by 6 wk of age and lowered when animals are fed the reduced-Mn AIN-93G diet (∼11 μg Mn/g) (75). Surprisingly, however, although biliary excretion is the primary route of Mn elimination (18–20), tissue Mn concentrations are only modestly elevated in liver-specific Slc30a10 knockouts (74), suggesting that activity of SLC30A10 in another part of the gastrointestinal tract likely compensates for loss-of-function in the liver [SLC30A10 localizes to the apical domain of differentiated HepG2 cells that model hepatocytes, implying that its activity should transport Mn from hepatocytes into the bile (81)]. Additional assays focused on the intestines because, although Mn is predominantly excreted in bile, the intestines excrete a smaller amount of Mn directly into feces (19, 21). Further, the intestines express SLC30A10 (74), and SLC30A10 localizes to the apical domain of differentiated CaCO2 cells that model enterocytes (74), suggesting that its transport activity in the intestines should result in the elimination of Mn into feces. Notably, blood, liver, and brain Mn concentrations are robustly elevated, and fecal Mn concentrations lowered, in endoderm-specific Slc30a10 knockout mice that lack SLC30A10 expression in both the liver and intestines (74). In totality, the aforementioned results imply that, under basal physiological conditions, activity of SLC30A10 in the liver and intestines is necessary for Mn excretion, and reduced Mn excretion plays a critical role in increasing tissue Mn concentrations in HMNDYT1 (Figure 1). The role of SLC30A10 in mediating hepatic and intestinal Mn excretion in mice was independently validated in 2019 using radiotracer Mn elimination assays (82).
FIGURE 1.
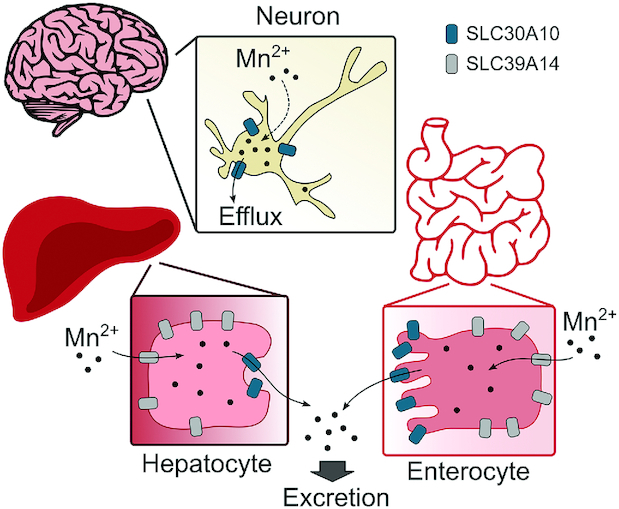
Schematic depicting the mechanisms by which SLC30A10 and SLC39A14 regulate Mn concentrations in the body. SLC30A10, solute carrier family 30 member 10; SLC39A14, solute carrier family 39 member 14.
What then is the function of SLC30A10 in the brain? Current understanding of this important issue comes from analyses of pan-neuronal/glial Slc30a10 knockouts in 2018 (74). Basal ganglia Mn concentrations of pan-neuronal/glial Slc30a10 knockouts are comparable with those of littermate controls under basal physiological conditions (74). However, after exposure to elevated Mn, Mn concentrations in the basal ganglia of the pan-neuronal/glial knockouts are modestly higher than those of littermates (74). Thus, activity of SLC30A10 in the brain may be critical in reducing Mn concentrations in the basal ganglia during elevated exposure as a neuroprotective mechanism (Figure 1).
Three additional points are worth noting about the genetically modified Slc30a10 mice. First, an unexpected finding was that full-body Slc30a10 knockouts develop hypothyroidism owing to the accumulation of Mn in the thyroid gland, which inhibits thyroxine production (75, 81). The role of the thyroid gland in modulating the neurotoxic outcomes of Mn toxicity has not received much attention, but hypothyroidism has now been identified in ≥1 human patient with HMNDYT1 (37). Further work is necessary to determine whether changes in thyroid function are a feature of HMNDYT1 and other forms of Mn neurotoxicity. Second, detailed neurotoxicity analysis of full-body and tissue-specific Slc30a10 knockout mice using neurobehavioral, neurochemical, and microscopy techniques is now ongoing. These studies are expected to provide novel insights into the pathobiology of HMNDYT1 and Mn neurotoxicity. Limited neurobehavioral results available to date (open-field and rotarod assays) suggest that full-body Slc30a10 knockout mice have deficits in generalized locomotion and ambulatory coordination/activity (74), which is broadly consistent with the human phenotype. Finally, the advent of clustered regularly interspaced short palindromic repeats (CRISPR) technology has made genome editing feasible in rats, and a CRISPR-based rat knockout of SLC30A10 is now being characterized. Analyses of the knockout rats should complement work with the knockout mouse models and improve understanding of HMNDYT1 and Mn-induced neurological dysfunction.
Slc39a14 knockout mice as a model to study HMNDYT2
In 2016, the second inherited disorder of Mn metabolism, HMNDYT2, was discovered (31). This disease occurs due to homozygous loss-of-function mutations in SLC39A14 (31). Patients exhibit elevated blood and brain Mn concentrations, but unlike HMNDYT1, the liver is unaffected and polycythemia is absent. Although MRI brain appearances and brain histology are identical to HMNDYT1, chelation treatment with Na2CaEDTA appears to be less effective despite obvious mobilization of Mn from the tissues demonstrated by increased urinary excretion (31). Mouse and zebrafish models have played a major role in better understanding the biology of this disease.
SLC39A14 is a metal importer with capability to transport Mn, Zn, Fe, and Cd from the cell exterior to the cytosol in culture (83). In patients with HMNDYT2, blood concentrations of Fe, Zn, and Cd are normal, suggesting that the primary biological function of SLC39A14 is to transport Mn (31). Further, SLC39A14 is strongly expressed in the liver in humans (31) and rodents (81, 84), and localizes to the basolateral domain of differentiated HepG2 cells (81) or rat hepatocytes (84), suggesting that its activity transports Mn from blood into hepatocytes. Results from full-body Slc39a14 knockout mice in 2017–2018 from 3 separate groups validated this hypothesis (85–87). Similarly to human patients, full-body Slc39a14 knockout mice exhibit increased blood and brain, but not liver, Mn concentrations (85–87). A separate study compared the phenotypes of full-body Slc30a10 or Slc39a14 single knockout mice with a Slc30a10/Slc39a14 double knockout strain, and reported that whereas blood and brain Mn concentrations are elevated in all 3 strains, liver Mn concentrations are only elevated in Slc30a10 single knockouts (and not in Slc39a14 single or Slc30a10/Slc39a14 double knockouts) (81). Overall, these results suggest that SLC39A14 and SLC30A10 act in a cooperative manner to mediate biliary Mn excretion—SLC39A14 transports Mn from blood into hepatocytes, and SLC30A10 then excretes the intrahepatic Mn into bile (Figure 1). Notably, a recent study revealed that activity of SLC39A14 is also necessary to mediate uptake of Mn into intestinal enterocytes, and that blood and brain Mn concentrations are elevated in intestine-specific Slc39a14 knockout mice (88), raising the possibility that SLC39A14 and SLC30A10 also act cooperatively in the intestines to excrete Mn into feces (Figure 1). In sum, a decrease in Mn excretion leads to the increase in tissue Mn concentrations in HMNDYT2.
As with Slc30a10 knockout mice, detailed neurotoxicity analyses of Slc39a14 knockout mice are ongoing. Available neurobehavioral data suggest that full-body Slc39a14 knockouts exhibit motor deficits (85–87), congruent with the human condition. Note, however, that full-body Slc39a14 knockout mice develop torticollis (wry neck) (86, 87, 89), which has not yet been reported in humans and may confound the interpretation of neurobehavioral assays performed in these animals.
C. elegans Model
Over the last few years, the soil-dwelling roundworm C. elegans has provided numerous insights into the underlying mechanisms of Mn-induced neurotoxicity and the control of Mn homeostasis. There are several advantages of the nematode model. C. elegans have a transparent body, allowing observations of intrabody structures. The worms are genetically tractable, making knockdown and over-expression of genes relatively straightforward. Results remain relevant to humans because nematode genes have high homology with human genes (as high as 60%–80%). Finally, Mn-treated worms exhibit deficits in locomotor function (35) (Table 1), providing similarities in the neuromotor consequences of Mn toxicity with humans (behavioral disinhibition and attentional dysfunction induced by early-life Mn exposure have not yet been modeled in nematodes). Below, we provide general guidelines for using C. elegans to study Mn neurotoxicity.
Mn exposure in C. elegans
C. elegans can be exposed to Mn at different larval stages (L1–L4) or as adults. Adapting dose, concentration, and exposure time to the respective larval stage is crucial because bioavailability and toxicity differ greatly over development. Cuticle thickness increases with each larval stage, resulting in reduced bioavailability in adult worms compared with younger stages. The cuticle is 3–6 times thicker in adult worms than in L1 (90). Sensitivity to exogenous agents also varies greatly within the larval stages (91). To study Mn neurotoxicity in C. elegans, both acute and chronic exposure to Mn have been applied. Acute dosing varies between 30 min and 1 h, subacute exposure is carried out ≤4 h, and chronic exposure for days. As the wild-type worm's lifespan is only ∼12 d long (92), incubation times as aforementioned may take up a relevant portion of the nematode's life. However, most studies use the acute exposure regimen with neurotoxicity being examined days after exposure by means of behavioral evaluations, survival/lifespan, Mn determination, and neuronal viability. Acute toxicity is most commonly tested in liquid medium without any food source including S medium (93, 94), or 85 mM sodium chloride + 0.1% Tween (95–97). In contrast, chronic toxicity is assayed on agar plates via the nematodes’ food source, Escherichia coli, but tests may also be conducted in S medium. Here, it is important to note that the E. coli may metabolize the compounds of interest, and prior inactivation of the bacteria may be useful, especially in case the study focuses on species-specific effects.
Unfortunately, the Mn dosing regimens and exposure conditions necessary may vary even within the same larval stage between laboratories because routine conditions, such as room temperature, pH of water, and humidity, may have a strong effect on absorption and metabolism (98). Therefore, a general protocol does not exist for dosing C. elegans with Mn, and summarizing all the applied doses and times in the respective larval stages would go beyond the scope of this review. As a guide, similar to rodents, the Mn2+ form as MnCl2 or MnSO4 is used (99, 100). Owing to the lack of a readily available dosing regimen, we suggest that concentration-effect curves, using survival and lifespan assays, be performed in any C. elegans laboratory to find a suitable Mn exposure concentration for investigations on the desired endpoint. As 1 example, MnCl2 has been tested in the L4 larval stage for both 1 and 4 h, with a calculated median lethal dose of 250 mM and 62 mM, respectively (101). Along with these functional assays, it is critical to directly measure the Mn content in C. elegans after dosing using standard analytical techniques, such as ICP-MS or atomic absorption spectroscopy (98, 102).
Benefit of using C. elegans to study the effects of Mn on the dopaminergic system
In C. elegans, the effects of Mn on the dopaminergic (DAergic) system are studied by assaying for neuron morphology, behavioral changes, neurotransmitter concentrations, and gene expression (103). A benefit of the nematode model is that the morphology of DAergic neurons can be readily evaluated using transgenic worms that express a fluorescent protein in these neurons because the worm has only 8 DAergic neurons: 4 CEP neurons innervate the tip of the nose (2 ventral and 2 dorsal), 2 ADE neurons innervate the head cuticle, and 1 pair of posterior PDE neurons are located in the posterior cuticle. After Mn exposure, morphological endpoints that can be examined as indicative of neurotoxicity include neuronal development, presence of puncta, absence or shrinkage of neurons, neuronal gaps, absence of cell bodies, and reduction in intensity of fluorescence (104). These microscopic parameters can be combined with a functional behavioral assay, the basal slowing response, which measures locomotion and integrity of DAergic neurotransmission in nematodes (35). Mn-treatment affects the performance of worms in the basal slowing response assay (Table 1) (35).
Use of C. elegans to study HMNDYT1
C. elegans do not express a homolog of SLC30A10. Therefore, studies of HMNDYT1 in the nematode model relied on over-expression. Similarly to mammalian systems, wild-type SLC30A10 localizes to the cell surface in body wall muscle cells and DAergic neurons in the nematodes, whereas disease-causing mutants are trapped in the intracellular compartment (35). Further, expression of SLC30A10 wild-type, but not a disease-causing mutant, protects worms against Mn-induced lethality, DAergic neurodegeneration, and locomotor defects (35). Protection is not observed against Zn toxicity (105), consistent with the Mn-specific transport activity of SLC30A10 observed in mammalian cell-lines. Overall, work in the C. elegans system provided results that validated and extended assays in mammalian cell culture and rodents.
Zebrafish Models
In recent years, the zebrafish (Danio rerio) has emerged as an invaluable vertebrate model system for translational research on neurodegenerative human disorders. Their small size, rapid external development, and optical transparency permit in vivo microscopy analysis and real-time imaging of fluorescently labelled proteins throughout development. The central nervous system is organized similarly to that of humans, and many pharmacological targets are shared (106). Zebrafish are easily amenable to genetic manipulation using genome editing technologies such as CRISPR/CRISPR-associated protein 9 and transcription activator-like effector nucleases in order to generate constitutive loss-of-function alleles, transgenic lines, and, more recently, tissue-specific and temporally controlled mutagenesis (107). Their relatively simple brain [∼100,000 neurons at 7 d postfertilization (dpf)] and the wide battery of behavioral tests available allow the analysis of how neural circuits generate behavior and how disease affects these behavioral patterns (108). Due to their large clutch size, zebrafish embryos and larvae are suitable for high-throughput drug screening protocols (109). Chemical compounds can simply be added to the fish medium and their effect on phenotypic readouts such as behavioral patterns or fluorescence intensity of labelled proteins can be determined using automated video tracking and microscopy. Limitations of zebrafish for drug discovery, however, are their ability of neuronal regeneration and the unpredictability of the drug concentrations that have entered the fish (106).
The recent use of zebrafish for the study of Mn neurotoxicity and associated disorders has shown that they present a promising model to dissect the mechanisms of Mn-related disease. Zebrafish have been successfully used as a model of Mn neurotoxicity to study the effects of environmental Mn overexposure through incubation of larvae in medium containing MnCl2 (55, 56, 110). The neuromotor consequences of Mn toxicity in zebrafish are similar to those in humans, rodents, and nematodes because Mn-treated zebrafish also exhibit deficits in locomotor function (55, 56) (Table 1) (but note that, as in nematodes, behavioral hyperreactivity and attentional deficits induced by early-life Mn exposure remain to be modeled in zebrafish). In addition, the use of genome editing tools has allowed the development of disease models for the study of HMNDYT1 and HMNDYT2.
Zebrafish as a model of environmental Mn neurotoxicity
To precipitate a phenotype in wild-type zebrafish larvae due to environmental Mn overexposure, MnCl2 at concentrations ranging from 50 to 100 μM during 0–5 dpf is used in order to observe phenotypic changes as early as 5 dpf (55, 56). The average tissue Mn concentration reached during 50 and 100 μM MnCl2 exposure amounts to 49 and 82 μg Mn/g, respectively (55). Survival is compromised at 10 dpf in larvae exposed to ≥1 mM during 0–5 dpf (56). Locomotor analysis at 5 dpf confirms reduced swimming distance in zebrafish exposed to MnCl2 concentrations as low as 25 μM (Table 1) (55). Concentrations of 0.5 mM during 0–5 dpf lead to diminished swimming distance, absolute turn angle, and mobile time, and increased immobile time (Table 1) (56). MnCl2-exposed larvae display a distinct circular swimming pattern both during spontaneous swimming activity and upon vibrational stimuli which is reversible after the removal of MnCl2. This may be attributed to splayed stereociliary bundles of the haircells within the neuromasts, mechanosensory organs mediating balance in zebrafish (110). Environmental Mn exposure has been demonstrated to result in reduced tyrosine hydroxylase (TH) expression although the number of TH positive cells is unchanged, suggesting that Mn neurotoxicity at least partly involves the DAergic system (56, 110).
slc30a10 loss-of-function in zebrafish as a model of HMNDYT1
The zebrafish slc30a10 loss-of-function mutant has emerged as a suitable model for drug discovery and the study of the pathogenic mechanisms of HMNDYT1 (111). As in humans, slc30a10 expression is largely restricted to the liver and brain in zebrafish larvae. Loss of slc30a10 function in zebrafish causes prominent Mn accumulation from 14 dpf that is accompanied by impaired locomotor behavior with a distinct bradykinesia-type swimming behavior at 4 mo of age. MnCl2 exposure (≥150 μM) for 24 h at 5 dpf leads to impaired locomotion at 6 dpf including tremor, postural instability, disorientation, and impaired balance. In mutant larvae, brain and liver (showing hepatic steatosis) appear discolored upon MnCl2 treatment. Histopathological studies confirm hepatic steatosis and within the brain a reduction of dopamine transporter and TH positive cells as well as increased apoptosis. Na2CaEDTA administration reverses the locomotor phenotype observed, suggesting that the slc30a10 loss-of-function mutant may indeed be an appropriate model for drug screening. Interestingly, during early larval stages slc30a10 mutants appear more resistant to Mn toxicity owing to mechanisms that are yet to be determined, likely involving differential expression of other metal transporters such as SPCA1, a secretory pathway Ca2+-ATPase pump at the Golgi (111).
slc39a14 loss-of-function in zebrafish as a model of HMNDYT2
The slc39a14U801 zebrafish mutant closely resembles the human phenotype with prominent deposition of Mn in the brain and locomotor defects. Under physiological conditions, Mn accumulates in zebrafish larvae as early as 5 dpf. Mutant zebrafish are more susceptible to Mn toxicity, demonstrated by a lower median lethal dose of MnCl2 (376 μM) than in wild-type animals (680 μM). Exposure to 50 μM MnCl2 from 2 to 5 dpf precipitates a locomotor phenotype characterized by reduced swimming activity from 6 dpf that is significantly different from that of wild-type larvae. Na2CaEDTA can effectively reduce Mn accumulation in mutant larvae. Differently than in humans, expression of slc39a14 is restricted to the pronephric duct, suggesting that the kidney plays a major role in the regulation of Mn homeostasis in zebrafish larvae (31).
In summary, zebrafish have proven an invaluable vertebrate organism to model human disorders of Mn dyshomeostasis. Further study of the aforementioned models is likely to unravel novel insights into the regulation of Mn homeostasis in vivo with the view to identify alternative drug targets.
Conclusions
Mn neurotoxicity is an area of intense investigation owing to its human health relevance. Recent discoveries of the genetic disorders of Mn metabolism have only increased interest. Rodents (mice and rats), C. elegans, and zebrafish are routinely used to model Mn neurotoxicity in animals. Major advantages of these species are their ease-of-use and the similarities in the functional neurobehavioral outcomes of Mn toxicity with humans. However, consideration must be given to the Mn dose and the mode of exposure in order to ensure translationally relevant results. We detailed Mn treatment regimens in rodents, nematodes, and zebrafish that recapitulate human Mn exposure. We also provided an update on the current state of knowledge regarding the role of these species in studying genetic disorders of Mn metabolism. Use of the disease-relevant experimental regimens described here will ensure obtained results are directly relevant to Mn-induced neurological dysfunction in humans.
Acknowledgments
We thank Dr. Andrey Selyunin (UT Austin) for generating Figure 1. The authors’ responsibilities were as follows—KT, MA, DRS, and SM: were responsible for the design and final content; and all authors: contributed to the writing of this review and read and approved the final manuscript.
Notes
Supported by National Institute of Environmental Health Sciences grants R01-ES024812 and R01-ES024812S1 (to SM) and R01-ES07331 and R01-ES10563 (to MA); National Institute for Health Research, the Academy of Medical Sciences, Action Medical Research, and Great Ormond Street Hospital Children's Charity UK (to KT); German Research Foundation grant BO 4103/2-1 and DFG Research Unit TraceAge FOR 2558 (to MMN and JB).
Author disclosures: The authors report no conflicts of interest.
CAT and KT contributed equally to this work.
Abbreviations used: CRISPR, clustered regularly interspaced short palindromic repeats; DAergic, dopaminergic; dpf, days postfertilization; HMNDYT, Hypermanganesemia with Dystonia; ICP-MS, inductively coupled plasma mass spectrometry; PND, post natal day; SLC30A10, solute carrier family 30 member 10; SLC39A14, solute carrier family 39 member 14; TH, tyrosine hydroxylase.
References
- 1. Aschner M, Erikson KM, Herrero Hernández E, Tjalkens R. Manganese and its role in Parkinson's disease: from transport to neuropathology. Neuromolecular Med. 2009;11:252–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Racette BA, Criswell SR, Lundin JI, Hobson A, Seixas N, Kotzbauer PT, Evanoff BA, Perlmutter JS, Zhang J, Sheppard L et al.. Increased risk of parkinsonism associated with welding exposure. Neurotoxicology. 2012;33:1356–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olanow CW. Manganese-induced parkinsonism and Parkinson's disease. Ann N Y Acad Sci. 2004;1012:209–23. [DOI] [PubMed] [Google Scholar]
- 4. Perl DP, Olanow CW. The neuropathology of manganese-induced Parkinsonism. J Neuropathol Exp Neurol. 2007;66:675–82. [DOI] [PubMed] [Google Scholar]
- 5. Harischandra DS, Rokad D, Neal ML, Ghaisas S, Manne S, Sarkar S, Panicker N, Zenitsky G, Jin H, Lewis M et al.. Manganese promotes the aggregation and prion-like cell-to-cell exosomal transmission of α-synuclein. Sci Signal. 2019;12:eaau4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson's disease. Neurotoxicology. 1999;20:239–47. [PubMed] [Google Scholar]
- 7. Bhang SY, Cho SC, Kim JW, Hong YC, Shin MS, Yoo HJ, Cho IH, Kim Y, Kim BN. Relationship between blood manganese levels and children's attention, cognition, behavior, and academic performance—a nationwide cross-sectional study. Environ Res. 2013;126:9–16. [DOI] [PubMed] [Google Scholar]
- 8. Bouchard M, Laforest F, Vandelac L, Bellinger D, Mergler D. Hair manganese and hyperactive behaviors: pilot study of school-age children exposed through tap water. Environ Health Perspect. 2007;115:122–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bouchard MF, Sauve S, Barbeau B, Legrand M, Brodeur ME, Bouffard T, Limoges E, Bellinger DC, Mergler D. Intellectual impairment in school-age children exposed to manganese from drinking water. Environ Health Perspect. 2011;119:138–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Claus Henn B, Ettinger AS, Schwartz J, Téllez-Rojo MM, Lamadrid-Figueroa H, Hernández-Avila M, Schnaas L, Amarasiriwardena C, Bellinger DC, Hu H et al.. Early postnatal blood manganese levels and children's neurodevelopment. Epidemiology. 2010;21:433–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khan K, Factor-Litvak P, Wasserman GA, Liu X, Ahmed E, Parvez F, Slavkovich V, Levy D, Mey J, van Geen A et al.. Manganese exposure from drinking water and children's classroom behavior in Bangladesh. Environ Health Perspect. 2011;119:1501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khan K, Wasserman GA, Liu X, Ahmed E, Parvez F, Slavkovich V, Levy D, Mey J, van Geen A, Graziano JH et al.. Manganese exposure from drinking water and children's academic achievement. Neurotoxicology. 2012;33:91–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lucchini RG, Guazzetti S, Zoni S, Donna F, Peter S, Zacco A, Salmistraro M, Bontempi E, Zimmerman NJ, Smith DR. Tremor, olfactory and motor changes in Italian adolescents exposed to historical ferro-manganese emission. Neurotoxicology. 2012;33:687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oulhote Y, Mergler D, Barbeau B, Bellinger DC, Bouffard T, Brodeur ME, Saint-Amour D, Legrand M, Sauve S, Bouchard MF. Neurobehavioral function in school-age children exposed to manganese in drinking water. Environ Health Perspect. 2014;122:1343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Riojas-Rodríguez H, Solís-Vivanco R, Schilmann A, Montes S, Rodríguez S, Ríos C, Rodríguez-Agudelo Y. Intellectual function in Mexican children living in a mining area and environmentally exposed to manganese. Environ Health Perspect. 2010;118:1465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wasserman GA, Liu X, Parvez F, Ahsan H, Levy D, Factor-Litvak P, Kline J, van Geen A, Slavkovich V, LoIacono NJ et al.. Water manganese exposure and children's intellectual function in Araihazar, Bangladesh. Environ Health Perspect. 2006;114:124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kullar SS, Shao K, Surette C, Foucher D, Mergler D, Cormier P, Bellinger DC, Barbeau B, Sauve S, Bouchard MF. A benchmark concentration analysis for manganese in drinking water and IQ deficits in children. Environ Int. 2019;130:104889. [DOI] [PubMed] [Google Scholar]
- 18. Ballatori N, Miles E, Clarkson TW. Homeostatic control of manganese excretion in the neonatal rat. Am J Physiol. 1987;252:R842–7. [DOI] [PubMed] [Google Scholar]
- 19. Bertinchamps AJ, Miller ST, Cotzias GC. Interdependence of routes excreting manganese. Am J Physiol. 1966;211:217–24. [DOI] [PubMed] [Google Scholar]
- 20. Klaassen CD. Biliary excretion of manganese in rats, rabbits, and dogs. Toxicol Appl Pharmacol. 1974;29:458–68. [DOI] [PubMed] [Google Scholar]
- 21. Papavasiliou PS, Miller ST, Cotzias GC. Role of liver in regulating distribution and excretion of manganese. Am J Physiol. 1966;211:211–6. [DOI] [PubMed] [Google Scholar]
- 22. Greenberg DM, Copp DH, Cuthbertson EM. Studies in mineral metabolism with the aid of artificial radioactive isotopes: vii. The distribution and excretion, particularly by way of the bile, of iron, cobalt, and manganese. J Biol Chem. 1943;147:749–56. [Google Scholar]
- 23. Butterworth RF. Parkinsonism in cirrhosis: pathogenesis and current therapeutic options. Metab Brain Dis. 2013;28:261–7. [DOI] [PubMed] [Google Scholar]
- 24. Butterworth RF, Spahr L, Fontaine S, Layrargues GP. Manganese toxicity, dopaminergic dysfunction and hepatic encephalopathy. Metab Brain Dis. 1995;10:259–67. [DOI] [PubMed] [Google Scholar]
- 25. Pomier-Layrargues G, Spahr L, Butterworth RF. Increased manganese concentrations in pallidum of cirrhotic patients. Lancet. 1995;345:735. [DOI] [PubMed] [Google Scholar]
- 26. Spahr L, Butterworth RF, Fontaine S, Bui L, Therrien G, Milette PC, Lebrun LH, Zayed J, Leblanc A, Pomier-Layrargues G. Increased blood manganese in cirrhotic patients: relationship to pallidal magnetic resonance signal hyperintensity and neurological symptoms. Hepatology. 1996;24:1116–20. [DOI] [PubMed] [Google Scholar]
- 27. Layrargues GP, Shapcott D, Spahr L, Butterworth RF. Accumulation of manganese and copper in pallidum of cirrhotic patients: role in the pathogenesis of hepatic encephalopathy?. Metab Brain Dis. 1995;10:353–6. [DOI] [PubMed] [Google Scholar]
- 28. Rose C, Butterworth RF, Zayed J, Normandin L, Todd K, Michalak A, Spahr L, Huet PM, Pomier-Layrargues G. Manganese deposition in basal ganglia structures results from both portal-systemic shunting and liver dysfunction. Gastroenterology. 1999;117:640–4. [DOI] [PubMed] [Google Scholar]
- 29. Burkhard PR, Delavelle J, Du Pasquier R, Spahr L. Chronic parkinsonism associated with cirrhosis: a distinct subset of acquired hepatocerebral degeneration. Arch Neurol. 2003;60:521–8. [DOI] [PubMed] [Google Scholar]
- 30. Tuschl K, Clayton PT, Gospe SM Jr, Gulab S, Ibrahim S, Singhi P, Aulakh R, Ribeiro RT, Barsottini OG, Zaki MS et al.. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet. 2012;90:457–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tuschl K, Meyer E, Valdivia LE, Zhao N, Dadswell C, Abdul-Sada A, Hung CY, Simpson MA, Chong WK, Jacques TS et al.. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Commun. 2016;7:11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tuschl K, Mills PB, Parsons H, Malone M, Fowler D, Bitner-Glindzicz M, Clayton PT. Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia—a new metabolic disorder. J Inherit Metab Dis. 2008;31:151–63. [DOI] [PubMed] [Google Scholar]
- 33. Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, Severijnen LA, Di Toro Mammarella L, Mignarri A, Monti L et al.. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet. 2012;90:467–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lechpammer M, Clegg MS, Muzar Z, Huebner PA, Jin LW, Gospe SM Jr. Pathology of inherited manganese transporter deficiency. Ann Neurol. 2014;75:608–12. [DOI] [PubMed] [Google Scholar]
- 35. Leyva-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, Morrisett RA, Bowman AB, Aschner M, Mukhopadhyay S. SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J Neurosci. 2014;34:14079–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mukhopadhyay S. Familial manganese-induced neurotoxicity due to mutations in SLC30A10 or SLC39A14. Neurotoxicology. 2018;64:278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anagianni S, Tuschl K. Genetic disorders of manganese metabolism. Curr Neurol Neurosci Rep. 2019;19:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dorman DC, Struve MF, Marshall MW, Parkinson CU, James RA, Wong BA. Tissue manganese concentrations in young male rhesus monkeys following subchronic manganese sulfate inhalation. Toxicol Sci. 2006;92:201–10. [DOI] [PubMed] [Google Scholar]
- 39. Erikson KM, Dorman DC, Lash LH, Aschner M. Manganese inhalation by rhesus monkeys is associated with brain regional changes in biomarkers of neurotoxicity. Toxicol Sci. 2007;97:459–66. [DOI] [PubMed] [Google Scholar]
- 40. Dorman DC, McManus BE, Parkinson CU, Manuel CA, McElveen AM, Everitt JI. Nasal toxicity of manganese sulfate and manganese phosphate in young male rats following subchronic (13-week) inhalation exposure. Inhal Toxicol. 2004;16:481–8. [DOI] [PubMed] [Google Scholar]
- 41. Kern CH, Smith DR. Preweaning Mn exposure leads to prolonged astrocyte activation and lasting effects on the dopaminergic system in adult male rats. Synapse. 2011;65:532–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kern CH, Stanwood GD, Smith DR. Preweaning manganese exposure causes hyperactivity, disinhibition, and spatial learning and memory deficits associated with altered dopamine receptor and transporter levels. Synapse. 2010;64:363–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beaudin SA, Nisam S, Smith DR. Early life versus lifelong oral manganese exposure differently impairs skilled forelimb performance in adult rats. Neurotoxicol Teratol. 2013;38:36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beaudin SA, Strupp BJ, Lasley SM, Fornal CA, Mandal S, Smith DR. Oral methylphenidate alleviates the fine motor dysfunction caused by chronic postnatal manganese exposure in adult rats. Toxicol Sci. 2015;144:318–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Beaudin SA, Strupp BJ, Strawderman M, Smith DR. Early postnatal manganese exposure causes lasting impairment of selective and focused attention and arousal regulation in adult rats. Environ Health Perspect. 2017;125:230–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Conley TE, Beaudin SA, Lasley SM, Fornal CA, Hartman J, Uribe W, Khan T, Strupp BJ, Smith DR. Early postnatal manganese exposure causes arousal dysregulation and lasting hypofunctioning of the prefrontal cortex catecholaminergic systems. J Neurochem. 2019:e14934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu X, Sullivan KA, Madl JE, Legare M, Tjalkens RB. Manganese-induced neurotoxicity: the role of astroglial-derived nitric oxide in striatal interneuron degeneration. Toxicol Sci. 2006;91:521–31. [DOI] [PubMed] [Google Scholar]
- 48. Moreno JA, Yeomans EC, Streifel KM, Brattin BL, Taylor RJ, Tjalkens RB. Age-dependent susceptibility to manganese-induced neurological dysfunction. Toxicol Sci. 2009;112:394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Beaudin SA, Strupp BJ, Uribe W, Ysais L, Strawderman M, Smith DR. Methylphenidate alleviates manganese-induced impulsivity but not distractibility. Neurotoxicol Teratol. 2017;61:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Amos-Kroohs RM, Bloor CP, Qureshi MA, Vorhees CV, Williams MT. Effects of developmental exposure to manganese and/or low iron diet: changes to metal transporters, sucrose preference, elevated zero-maze, open-field, and locomotion in response to fenfluramine, amphetamine, and MK-801. Toxicol Rep. 2015;2:1046–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Amos-Kroohs RM, Davenport LL, Atanasova N, Abdulla ZI, Skelton MR, Vorhees CV, Williams MT. Developmental manganese neurotoxicity in rats: cognitive deficits in allocentric and egocentric learning and memory. Neurotoxicol Teratol. 2017;59:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sprowles JLN, Amos-Kroohs RM, Braun AA, Sugimoto C, Vorhees CV, Williams MT. Developmental manganese, lead, and barren cage exposure have adverse long-term neurocognitive, behavioral and monoamine effects in Sprague-Dawley rats. Neurotoxicol Teratol. 2018;67:50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Roels H, Meiers G, Delos M, Ortega I, Lauwerys R, Buchet JP, Lison D. Influence of the route of administration and the chemical form (MnCl2, MnO2) on the absorption and cerebral distribution of manganese in rats. Arch Toxicol. 1997;71:223–30. [DOI] [PubMed] [Google Scholar]
- 54. Dorman DC, Brenneman KA, McElveen AM, Lynch SE, Roberts KC, Wong BA. Olfactory transport: a direct route of delivery of inhaled manganese phosphate to the rat brain. J Toxicol Environ Health A. 2002;65:1493–511. [DOI] [PubMed] [Google Scholar]
- 55. Tu H, Fan C, Chen X, Liu J, Wang B, Huang Z, Zhang Y, Meng X, Zou F. Effects of cadmium, manganese, and lead on locomotor activity and neurexin 2a expression in zebrafish. Environ Toxicol Chem. 2017;36:2147–54. [DOI] [PubMed] [Google Scholar]
- 56. Altenhofen S, Wiprich MT, Nery LR, Leite CE, Vianna MRMR, Bonan CD. Manganese(II) chloride alters behavioral and neurochemical parameters in larvae and adult zebrafish. Aquat Toxicol. 2017;182:172–83. [DOI] [PubMed] [Google Scholar]
- 57. Lasley SM, Fornal CA, Mandal S, Strupp BJ, Beaudin SA, Smith DR. Early postnatal manganese exposure reduces rat cortical and striatal biogenic amine activity in adulthood. Toxicol Sci. 2019;173(1):144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McDougall SA, Reichel CM, Farley CM, Flesher MM, Der-Ghazarian T, Cortez AM, Wacan JJ, Martinez CE, Varela FA, Butt AE et al.. Postnatal manganese exposure alters dopamine transporter function in adult rats: potential impact on nonassociative and associative processes. Neuroscience. 2008;154:848–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Reichel CM, Wacan JJ, Farley CM, Stanley BJ, Crawford CA, McDougall SA. Postnatal manganese exposure attenuates cocaine-induced locomotor activity and reduces dopamine transporters in adult male rats. Neurotoxicol Teratol. 2006;28:323–32. [DOI] [PubMed] [Google Scholar]
- 60. Vorhees CV, Graham DL, Amos-Kroohs RM, Braun AA, Grace CE, Schaefer TL, Skelton MR, Erikson KM, Aschner M, Williams MT. Effects of developmental manganese, stress, and the combination of both on monoamines, growth, and corticosterone. Toxicol Rep. 2014;1:1046–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gunter KK, Aschner M, Miller LM, Eliseev R, Salter J, Anderson K, Hammond S, Gunter TE. Determining the oxidation states of manganese in PC12 and nerve growth factor-induced PC12 cells. Free Radic Biol Med. 2005;39:164–81. [DOI] [PubMed] [Google Scholar]
- 62. Gunter TE, Miller LM, Gavin CE, Eliseev R, Salter J, Buntinas L, Alexandrov A, Hammond S, Gunter KK. Determination of the oxidation states of manganese in brain, liver, and heart mitochondria. J Neurochem. 2004;88:266–80. [DOI] [PubMed] [Google Scholar]
- 63. Krishna S, Dodd CA, Hekmatyar SK, Filipov NM. Brain deposition and neurotoxicity of manganese in adult mice exposed via the drinking water. Arch Toxicol. 2014;88:47–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Oulhote Y, Mergler D, Bouchard MF. Sex- and age-differences in blood manganese levels in the U.S. general population: national health and nutrition examination survey 2011–2012. Environ Health. 2014;13:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Miller ST, Cotzias GC, Evert HA. Control of tissue manganese: initial absence and sudden emergence of excretion in the neonatal mouse. Am J Physiol. 1975;229:1080–4. [DOI] [PubMed] [Google Scholar]
- 66. Keen CL, Bell JG, Lonnerdal B. The effect of age on manganese uptake and retention from milk and infant formulas in rats. J Nutr. 1986;116:395–402. [DOI] [PubMed] [Google Scholar]
- 67. Freeland-Graves JH, Mousa TY, Kim S. International variability in diet and requirements of manganese: causes and consequences. J Trace Elem Med Biol. 2016;38:24–32. [DOI] [PubMed] [Google Scholar]
- 68. Keen CL, Lönnerdal B, Clegg M, Hurley LS. Developmental changes in composition of rat milk: trace elements, minerals, protein, carbohydrate and fat. J Nutr. 1981;111:226–36. [DOI] [PubMed] [Google Scholar]
- 69. Ljung K, Vahter M. Time to re-evaluate the guideline value for manganese in drinking water?. Environ Health Perspect. 2007;115:1533–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Frisbie SH, Mitchell EJ, Roudeau S, Domart F, Carmona A, Ortega R. Manganese levels in infant formula and young child nutritional beverages in the United States and France: comparison to breast milk and regulations. PLoS One. 2019;14:e0223636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Barnett SA, Dickson RG. Milk production and consumption and growth of young of wild mice after ten generations in a cold environment. J Physiol. 1984;346:409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cramer CP, Blass EM. Mechanisms of control of milk intake in suckling rats. Am J Physiol. 1983;245:R154–9. [DOI] [PubMed] [Google Scholar]
- 73. Rath EA, Thenen SW. Use of tritiated water for measurement of 24-hour milk intake in suckling lean and genetically obese (ob/ob) mice. J Nutr. 1979;109:840–7. [DOI] [PubMed] [Google Scholar]
- 74. Taylor CA, Hutchens S, Liu C, Jursa T, Shawlot W, Aschner M, Smith DR, Mukhopadhyay S. SLC30A10 transporter in the digestive system regulates brain manganese under basal conditions while brain SLC30A10 protects against neurotoxicity. J Biol Chem. 2019;294:1860–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hutchens S, Liu C, Jursa T, Shawlot W, Chaffee BK, Yin W, Gore AC, Aschner M, Smith DR, Mukhopadhyay S. Deficiency in the manganese efflux transporter SLC30A10 induces severe hypothyroidism in mice. J Biol Chem. 2017;292:9760–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nielsen BS, Larsen EH, Ladefoged O, Lam HR. Subchronic, low-level intraperitoneal injections of manganese (IV) oxide and manganese (II) chloride affect rat brain neurochemistry. Int J Toxicol. 2017;36:239–51. [DOI] [PubMed] [Google Scholar]
- 77. O'Neal SL, Lee JW, Zheng W, Cannon JR. Subacute manganese exposure in rats is a neurochemical model of early manganese toxicity. Neurotoxicology. 2014;44:303–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jursa T, Smith DR. Ceruloplasmin alters the tissue disposition and neurotoxicity of manganese, but not its loading onto transferrin. Toxicol Sci. 2009;107:182–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zogzas CE, Aschner M, Mukhopadhyay S. Structural elements in the transmembrane and cytoplasmic domains of the metal transporter SLC30A10 are required for its manganese efflux activity. J Biol Chem. 2016;291:15940–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zogzas CE, Mukhopadhyay S. Putative metal binding site in the transmembrane domain of the manganese transporter SLC30A10 is different from that of related zinc transporters. Metallomics. 2018;10:1053–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liu C, Hutchens S, Jursa T, Shawlot W, Polishchuk EV, Polishchuk RS, Dray BK, Gore AC, Aschner M, Smith DR et al.. Hypothyroidism induced by loss of the manganese efflux transporter SLC30A10 may be explained by reduced thyroxine production. J Biol Chem. 2017;292:16605–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mercadante CJ, Prajapati M, Conboy HL, Dash ME, Herrera C, Pettiglio MA, Cintron-Rivera L, Salesky MA, Rao DB, Bartnikas TB. Manganese transporter Slc30a10 controls physiological manganese excretion and toxicity. J Clin Invest. 2019;129(12):5442–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jeong J, Eide DJ. The SLC39 family of zinc transporters. Mol Aspects Med. 2013;34:612–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nam H, Wang CY, Zhang L, Zhang W, Hojyo S, Fukada T, Knutson MD. ZIP14 and DMT1 in the liver, pancreas, and heart are differentially regulated by iron deficiency and overload: implications for tissue iron uptake in iron-related disorders. Haematologica. 2013;98:1049–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Aydemir TB, Kim M-H, Kim J, Colon-Perez LM, Banan G, Mareci TH, Febo M, Cousins RJ. Metal transporter zip14 (Slc39a14) deletion in mice increases manganese deposition and produces neurotoxic signatures and diminished motor activity. J Neurosci. 2017;37:5996–6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jenkitkasemwong S, Akinyode A, Paulus E, Weiskirchen R, Hojyo S, Fukada T, Giraldo G, Schrier J, Garcia A, Janus C et al.. SLC39A14 deficiency alters manganese homeostasis and excretion resulting in brain manganese accumulation and motor deficits in mice. Proc Natl Acad Sci U S A. 2018;115:E1769–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yongjuan X, Gao H, Wang J, Qiang Y, Imam MU, Li Y, Wang J, Zhang R, Zhang H, Yu Y et al.. Manganese transporter Slc39a14 deficiency revealed its key role in maintaining manganese homeostasis in mice. Cell Discovery. 2017;3:17025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Scheiber IF, Wu Y, Morgan SE, Zhao N. The intestinal metal transporter ZIP14 maintains systemic manganese homeostasis. J Biol Chem. 2019;294:9147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hojyo S, Fukada T, Shimoda S, Ohashi W, Bin BH, Koseki H, Hirano T. The zinc transporter SLC39A14/ZIP14 controls G-protein coupled receptor-mediated signaling required for systemic growth. PLoS One. 2011;6:e18059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cox GN, Staprans S, Edgar RS. The cuticle of Caenorhabditis elegans: II. Stage-specific changes in ultrastructure and protein composition during postembryonic development. Dev Biol. 1981;86:456–70. [DOI] [PubMed] [Google Scholar]
- 91. Jones CA, Hartman PS. Replication in UV-irradiated Caenorhabditis elegans embryos. Photochem Photobiol. 1996;63:187–92. [DOI] [PubMed] [Google Scholar]
- 92. Riddle DL, Blumenthal T, Meyer BJ, Priess JR, C. elegans II. 2nd ed Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1997. [PubMed] [Google Scholar]
- 93. Lewis JA, Fleming JT. Basic culture methods. Methods Cell Biol. 1995;48:3–29. [PubMed] [Google Scholar]
- 94. Sulston J, Hodgkin J. Methods. In: Wood WB, editor The nematode Caenorhabditis elegans. 1st ed Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1988. pp. 587–606. [Google Scholar]
- 95. Henze A, Homann T, Rohn I, Aschner M, Link CD, Kleuser B, Schweigert FJ, Schwerdtle T, Bornhorst J. Caenorhabditis elegans as a model system to study post-translational modifications of human transthyretin. Sci Rep. 2016;6:37346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Melov S, Ravenscroft J, Malik S, Gill MS, Walker DW, Clayton PE, Wallace DC, Malfroy B, Doctrow SR, Lithgow GJ. Extension of life-span with superoxide dismutase/catalase mimetics. Science. 2000;289:1567–9. [DOI] [PubMed] [Google Scholar]
- 97. Petrascheck M, Ye X, Buck LB. An antidepressant that extends lifespan in adult Caenorhabditis elegans. Nature. 2007;450:553–6. [DOI] [PubMed] [Google Scholar]
- 98. Bornhorst J, Chakraborty S, Meyer S, Lohren H, Grosse Brinkhaus S, Knight AL, Caldwell KA, Caldwell GA, Karst U, Schwerdtle T et al.. The effects of pdr1, djr1.1 and pink1 loss in manganese-induced toxicity and the role of α-synuclein in C. elegans. Metallomics. 2014;6:476–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lin YT, Hoang H, Hsieh SI, Rangel N, Foster AL, Sampayo JN, Lithgow GJ, Srinivasan C. Manganous ion supplementation accelerates wild type development, enhances stress resistance, and rescues the life span of a short-lived Caenorhabditis elegans mutant. Free Radic Biol Med. 2006;40:1185–93. [DOI] [PubMed] [Google Scholar]
- 100. Avila DS, Benedetto A, Au C, Bornhorst J, Aschner M. Involvement of heat shock proteins on Mn-induced toxicity in Caenorhabditis elegans. BMC Pharmacol Toxicol. 2016;17:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Schetinger MRC, Peres TV, Arantes LP, Carvalho F, Dressler V, Heidrich G, Bowman AB, Aschner M. Combined exposure to methylmercury and manganese during L1 larval stage causes motor dysfunction, cholinergic and monoaminergic up-regulation and oxidative stress in L4 Caenorhabditis elegans. Toxicology. 2019;411:154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Schunk PF, Kalil IC, Pimentel-Schmitt EF, Lenz D, de Andrade TU, Ribeiro JS, Endringer DC. ICP-OES and micronucleus test to evaluate heavy metal contamination in commercially available Brazilian herbal teas. Biol Trace Elem Res. 2016;172:258–65. [DOI] [PubMed] [Google Scholar]
- 103. Martinez-Finley EJ, Gavin CE, Aschner M, Gunter TE. Manganese neurotoxicity and the role of reactive oxygen species. Free Radic Biol Med. 2013;62:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Benedetto A, Au C, Avila DS, Milatovic D, Aschner M. Extracellular dopamine potentiates Mn-induced oxidative stress, lifespan reduction, and dopaminergic neurodegeneration in a BLI-3-dependent manner in Caenorhabditis elegans. PLoS Genet. 2010;6:e1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chen P, Bowman AB, Mukhopadhyay S, Aschner M. SLC30A10: a novel manganese transporter. Worm. 2015;4:e1042648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Saleem S, Kannan RR. Zebrafish: an emerging real-time model system to study Alzheimer's disease and neurospecific drug discovery. Cell Death Discov. 2018;4:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Albadri S, De Santis F, Di Donato V, Del Bene F. CRISPR/Cas9-mediated knockin and knockout in zebrafish. In: Jaenisch R, Zhang F, Gage F, Genome editing in neurosciences. Cham (Switzerland): Springer; 2017. pp. 41–9. [PubMed] [Google Scholar]
- 108. Sumbre G, de Polavieja GG. The world according to zebrafish: how neural circuits generate behavior. Front Neural Circuits. 2014;8:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Gehrig J, Pandey G, Westhoff JH. Zebrafish as a model for drug screening in genetic kidney diseases. Front Pediatr. 2018;6:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Bakthavatsalam S, Das Sharma S, Sonawane M, Thirumalai V, Datta A. A zebrafish model of manganism reveals reversible and treatable symptoms that are independent of neurotoxicity. Dis Model Mech. 2014;7:1239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Xia Z, Wei J, Li Y, Wang J, Li W, Wang K, Hong X, Zhao L, Chen C, Min J et al.. Zebrafish slc30a10 deficiency revealed a novel compensatory mechanism of Atp2c1 in maintaining manganese homeostasis. PLos Genet. 2017;13:e1006892. [DOI] [PMC free article] [PubMed] [Google Scholar]