

Abstract
Oxidized LDL (oxLDL) is a known risk factor for atherogenesis. This study aimed to reveal structural features of oxLDL present in human circulation related to atherosclerosis. When LDL was fractionated on an anion-exchange column, in vivo-oxLDL, detected by the anti-oxidized PC (oxPC) mAb, was recovered in flow-through and electronegative LDL [LDL(−)] fractions. The amount of the electronegative in vivo-oxLDL, namely oxLDL in the LDL(−) fraction, present in patients with acute MI was 3-fold higher than that observed in healthy subjects. Surprisingly, the LDL(−) fraction contained apoA1 in addition to apoB, and HDL-sized particles were observed with transmission electron microscopy. In LDL(−) fractions, acrolein adducts were identified at all lysine residues in apoA1, with only a small number of acrolein-modified residues identified in apoB. The amount of oxPC adducts of apoB was higher in the LDL(−) than in the L1 fraction, as determined using Western blotting. The electronegative in vivo-oxLDL was immunologically purified from the LDL(−) fraction with an anti-oxPC mAb. The majority of PC species were not oxidized, whereas oxPC and lysoPC did not accumulate. Here, we propose that there are two types of in vivo-oxLDL in human circulating plasma and the electronegative in vivo-oxLDL accompanies oxidized HDL.
Keywords: atherosclerosis, oxidized low density lipoprotein, oxidized high density lipoprotein, apolipoproteins, proteomics, lipidomics, high density lipoprotein
Accumulating evidence shows that oxidative modification of lipoproteins plays important roles in the initiation and progression of atherosclerosis (1–3). Oxidized LDL (oxLDL) is generated through enzymatic and nonenzymatic oxidation reactions on lipid molecules and apoB together with subsequent protein modifications with oxidized PC (oxPC) and reactive lipid aldehydes, including malondialdehyde (MDA) and acrolein (1). Generally, oxidative modification of LDL increases the net negative charge of LDL particles, allowing them to gain high affinity for scavenger receptors while losing their affinity for the LDL receptor (4, 5). Uptake of oxLDL via scavenger receptors leads to atherosclerotic lesion development through foam cell formation. The presence of oxLDL in atherosclerotic legions in Watanabe heritable hyperlipidemic rabbits and apoE-knockout mice, but not in the normal aorta, has been immunohistochemically demonstrated (6–8). In human coronary arteries and carotid arteries, the accumulation of oxLDL in foam cells has been verified in previous studies (9, 10). These results provide convincing evidence that oxLDL is present in atherosclerotic lesions.
A highly sensitive quantification procedure for oxLDL was developed using the anti-oxPC mAb, DLH3, recognizing oxPC containing a truncated chain (11, 12). Additionally, alternative sandwich ELISA systems using other mAbs have also been developed to quantify circulating oxLDL. The mAb clones include E06 specific for the phosphorylcholine head group of oxidized phospholipids (PLs) but not for native PLs (13, 14), 4E6 specific for copper-induced oxLDL (15), and MDA2 specific for MDA-modified LDL (16). A sandwich assay using a recombinant form of LOX-1, a scavenger receptor, was developed to detect circulating oxLDL-like ligands (17). Studies using these mAbs have demonstrated associations between elevated oxLDL levels in human circulation and cardiovascular events (18). These observations raise the possibility that oxLDL could be a useful biomarker of the development and progression of CVDs. Notably, in vivo-oxLDL levels increased transiently in patients with acute MI (AMI) (19–21). Hence, in AMI patients, in vivo-oxLDL is considered to be released from the ruptured plaques (22).
The structure of in vivo-oxLDL, on the whole, is yet to be clarified. Several in vitro-oxLDL preparations have been developed and employed to assess the roles of oxLDL in pro-inflammatory and pro-atherogenic events. Copper-induced oxLDL (Cu-oxLDL) is the most widely used in vitro-oxLDL. Previously, our laboratory identified oxidative modifications of apoB amino acid residues (23) and determined PC and oxPC molecular species (24) in Cu-oxLDL by LC-MS/MS analyses. Another type of in vitro-oxLDL preparation, minimally modified LDL (MM-LDL), was produced by treatment with a low concentration of ferrous ions or by coculture with endothelial cells (25). Protein modification of MM-LDL is moderate, allowing MM-LDL recognition by the LDL receptor but not by the scavenger receptor. It contains substantial amounts of oxPC, enabling the binding of anti-oxPC mAb to MM-LDL (26). In addition, myeloperoxidase (MPO)-mediated oxidation reactions have been proposed as a potential mechanism of in vivo-oxLDL generation under inflammatory conditions (27). Numerous PC oxidation products containing truncated acyl chains are formed in MPO-induced oxLDL (28). Although Cu-oxLDL has been frequently utilized for decades, it remains unknown whether such properties of in vitro-oxLDL are consistent with those of in vivo-oxLDL.
The presence of subfractions of LDL in human circulation has been widely accepted. Electronegative LDL [referred to as LDL(−) or L5 in the literature] can be separated by anion-exchange chromatography (29) and has been correlated with CAD (30) and CVD (31). It is proposed that LDL(−) contains increased amounts of free fatty acids and lysoPC, which may contribute to its electronegativity and pathological effects (32, 33). However, whether LDL(−) corresponds to in vivo-oxLDL recognized by anti-oxPC antibodies has not been investigated.
HDL is known as an anti-atherogenic lipoprotein owing to its role in reverse cholesterol transport, anti-inflammatory effects, and its capacity for protecting LDL from oxidative damage (34). The capacity of HDL to inhibit LDL oxidation is due to apoA1 and several enzymes in HDL particles, including paraoxonase 1 (PON1), LCAT, and lipoprotein-associated phospholipase A2 (Lp-PLA2) (35, 36). Oxidation of HDL decreases the capability to inhibit LDL oxidation, and its anti-atherogenic properties change drastically to pro-atherogenic properties. Huang et al. (37) have revealed that oxidized apoA1-containing modifications generated by MPO-dependent reactions accumulate in human atherosclerotic plaques. Therefore, it should be noted that oxidized HDL (oxHDL) could be an important atherosclerotic risk factor, as well as oxLDL.
The purpose of this study is to clarify the structural features of in vivo-oxLDL. We focused on in vivo-oxLDL of patients with AMI, comparing it with that of healthy subjects. The criterion of in vivo-oxLDL in this study is modified with oxPC based on its antigenicity against anti-oxPC mAb. Our data demonstrated that there are two types of in vivo-oxLDL in human plasma, which were separated on anion-exchange chromatography. In vivo oxLDL in the LDL(−) fraction was more abundant in patients with AMI than in healthy subjects. In addition, the LDL(−) fraction contained heavily modified apoA1 and oxPC-modified apoB. The LDL(−) fraction was shown to be a mixture of LDL- and HDL-sized particles by electron microscopy. These features are distinct from Cu-oxLDL prepared in vitro.
MATERIALS AND METHODS
Materials
The anti-oxPC mAb, DLH3, was prepared as described previously (11); the mAb was in part provided as a gift from Kyowa Medex Co. Ltd. (Tokyo, Japan) (38). Anti-apoB-100 mAb (#H45640M) for sandwich ELISA was from Meridian Life Science (Memphis, TN). Monoclonal IgM κ-isotype control (#010-001-339) was from Rockland (Philadelphia, PA). Anti-human apoB-100 polyclonal antibody (pAb) (sheep IgG) (#PC086) and alkaline phosphatase (ALP)-conjugated anti-sheep IgG pAb (donkey IgG) (#AP184A) for sandwich ELISA were from Binding Site (Birmingham, UK) and Chemicon (Temecula, CA), respectively. Anti-apoB-100 pAb (#R1032P) and anti-apoA-1 pAb (#GTX112692) for Western blotting were from Acris Antibodies (San Diego, CA) and GeneTex (Irvine, CA), respectively. 1,2-Didecanoyl-sn-glycero-3-phosphocholine and 1-pentadecanoyl-2-hydroxy-sn-glycero-3-phosphocholine were purchased from Avanti Polar Lipids (Alabaster, AL). Acetonitrile, 2-propanol, ultrapure water (LC-MS grade), and ammonium formate were purchased from Fujifilm Wako Pure Chemical Co. (Osaka, Japan).
Isolation of human lipoproteins from patients with AMI and healthy subjects
This study was conducted in accordance with the principles of the Declaration of Helsinki and was approved by the Ethical Committee of Showa University (No. 214), with informed consent obtained from all healthy subjects and patients before the study. Plasma samples were separated from the blood of seven healthy volunteers (age 22–56 years), after a 12 h overnight fast, by centrifugation at 700 g for 15 min, after which EDTA was added to the plasma (final concentration 0.25 mM) and immediately used. Twenty-five patients with AMI (age 25–85 years; 23 patients presented ST-segment elevation MI and two patients had non-ST-segment elevation MI) received cardiac catheter treatment at Showa University Hospital, and then, aliquots of blood samples suctioned from the site of infarction were used in this study. Sera obtained from the blood samples were collected after centrifugation and stored at −80°C until analysis.
Total LDL and HDL were prepared as described previously (12). Briefly, LDL (1.019 < d < 1.063 g/ml) and HDL (1.063 < d <1.210 g/ml) fractions were separated from the healthy plasma using ultracentrifugation (Himac CS150GX, HITACHI), with the addition of KBr to adjust the density, and then dialyzed three times against PBS containing 0.25 mM EDTA to remove the KBr. LDL and HDL fractions of AMI patients were isolated from pooled sera, where each batch of the patients’ sera was prepared from three to six individuals. The protein concentrations of LDL and HDL were determined by the BCA method using BSA as standard. All lipoprotein samples were maintained under Ar gas at 4°C until analysis to avoid spontaneous oxidation. To minimize possible damage during storage, analyses of lipids or proteins of all lipoprotein samples were performed within 1 week after isolation.
In vitro modification of LDL
In vitro Cu-oxLDL was prepared by oxidation of LDL from healthy subjects (0.2 mg protein per milliliter), with 5 μM copper sulfate for 3 h at 37°C, according to the method previously described (11) with few modifications.
Separation of LDL subfractions on anion-exchange chromatography
LDL collected from plasma or serum was fractionated into subfractions using anion-exchange chromatography as reported previously (29). Tandem Q-Sepharose Fast Flow 7/25 mm columns (GE Healthcare, Chicago, IL) were connected to a peristaltic pump. The counter-ion was introduced by washing the columns with 10 ml of elution buffer B [10 mmol/l Tris-HCl, 1 mol/l NaCl, 1 mmol/l EDTA (pH7.4)], and then equilibrated with 20 ml of buffer A [10 mmol/l Tris-HCl, 1 mmol/l EDTA (pH 7.4)]. The LDL was replaced in buffer A using a PD-10 desalting column (Bio-Rad, Hercules, CA), and then 3–4 mg protein of LDL was injected into Q-Fast Flow columns at a flow rate of 1 ml/min. The ratio of buffer B to buffer A was increased in a stepwise manner, and eluates collected as 1 ml aliquots were kept on ice. The flow-through fraction was named L1, and the eluted fraction (using 0.5 mol/l NaCl) was termed LDL(−), referring to electronegative LDL. The elution buffers were degassed before the column elution; samples and recovered fractions were maintained under Ar gas. The absorbance of each fraction was measured at 280 nm using NanoDrop 2000c (Thermo Fisher Scientific, Waltham, MA).
Determination of oxLDL and apoB levels by sandwich ELISA
The oxLDL and apoB contents in the fractions eluted from Q-Fast Flow columns were determined using a sandwich ELISA method previously described (11). Briefly, microtiter wells (Costor®3369, Corning) were precoated with 1.0 μg/100 μl of DLH3 or 0.5 μg/100 μl of anti-apoB-100 mAb for more than 2 h at room temperature. The plates were blocked with 1% BSA-TBS, and then 100 μl of diluted or undiluted column elution fractions were added overnight at 4°C. After extensive washing, the oxLDL or LDL captured in the wells was detected with anti-human apoB-100 pAb (sheep IgG) and alkaline phosphatase-conjugated anti-sheep IgG pAb (donkey IgG). In each microtiter plate, either diluted Cu-oxLDL (0.1–3 ng/well) or diluted LDL (0.5–10 ng/well) were run simultaneously to determine the standard curves.
Gel electrophoresis and immunoblotting analysis
The samples for electrophoresis were concentrated to the required amount using ultrafiltration membranes (Amicon® Ultra 10K device; Merck, Darmstadt, Germany) when necessary. Native PAGE was performed on 3–20% Tris-glycine gels loaded with 2 μg of protein per lane and run at 20 mA for 1.5 h under nondenaturing conditions with Tris-glycine buffer. For the agarose gel electromobility assay, 0.6% agarose gels were loaded with 12 μg of protein per lane and run at 50 V for 2 h on ice using barbital buffer at pH 8.6. The agarose gels were stained with Coomassie Brilliant Blue stain. Proteins were separated either on native PAGE gels or on agarose gels, and then they were transferred to PVDF membranes and probed with antibodies against oxPC, apoB, or apoA1. The blots were detected by enhanced chemiluminescence (ECL Prime; GE Healthcare) or ImmunoStar LD (Fujifilm Wako).
Electron microscopy
Lipoprotein particles present in the L1 and LDL(−) fractions were observed microscopically using a Hitachi H7600 transmission electron microscope (HITACHI, Tokyo, Japan). After anion exchange chromatography, lipoprotein samples were diluted to <0.01 μg/μl. Samples were absorbed on carbon-coated grids and processed for negative staining with 2% uranyl acetate solution, pH 7.0.
The particle size of lipoproteins was measured using ImageJ software. The ranges of LDL and HDL were defined as 16.0–31.3 nm and 7.6–16.0 nm based on the literature regarding the classification of lipoprotein subclasses (39).
Affinity extraction of in vivo-oxLDL by an anti-oxPC mAb
The microtiter wells (Costor®3369) were precoated with 1.0 μg/100 μl of DLH3 or monoclonal IgM κ isotype control at room temperature for 2 h. The plate was blocked with Pierce™ blocking buffer (#37572; Thermo Fisher) for 1 h, and then incubated with the L1 or LDL(−) fractions and Cu-oxLDL solution overnight at 4°C. After washing with cold-PBS three times, the lipids in the lipoproteins bound to the antibodies were extracted with 100% ethanol containing internal standard lipids (1,2-didecanoyl-sn-glycero-3-phosphocholine and 1-pentadecanoyl-2-hydroxy-sn-glycero-3-phosphocholine). Similarly, proteins bound to the antibody-coated microtiter wells were extracted with 50 mM Tris-HCl, pH 8.0, containing 0.1% SDS.
Detection of oxidatively modified amino acid residues by LC-MS/MS
Analysis of the oxidative modification of apolipoproteins using LC-MS/MS was carried out according to the method previously reported (23). After agarose gel electrophoresis, blotted proteins on the PVDF membrane were stained with Ponceau-S solution, and the protein bands were dissected. In vivo-oxLDL separated from the L1 and LDL(−) fractions using anti-oxPC mAb was spotted on PVDF membranes (5 × 5 mm). All PVDF membrane pieces were washed with chloroform/methanol (1/1, v/v) to deplete lipids. Then, the lipid-depleted proteins were reduced with 10 mM dithiothreitol/80 mM ammonium hydrogen carbonate/20% acetonitrile for 1 h at 60°C and alkylated with 55 mM iodoacetamide/80 mM ammonium hydrogen carbonate/20% acetonitrile for 45 min at room temperature in the dark. After washing with Milli-Q water followed by 2% acetonitrile/Milli-Q water, the proteins were digested with 1 μg trypsin/70% acetonitrile/30 mM ammonium hydrogen carbonate overnight at 37°C. The tryptic digests were extracted twice with 1% formic acid/70% acetonitrile and dried in a SpeedVac evaporator. The extracted peptides were dissolved in 0.1% formic acid and analyzed using a DiNa Nano-LC system (KYA Tech Co., Tokyo, Japan) coupled online with Triple TOF 5600 (Sciex, Framingham, MA), as described previously (40). The oxidized peptides were identified using ProteinPilot™ version 4.5 software (Sciex). The search engine and sequence database used were Paragon™ Algorithm 4.5.0.0, 1654, and UniProt, respectively. The number of entries in the database was 15,908. The protease used was trypsin that cleaves the C-terminal end of lysine and arginine residues. At most, five missed cleavages were permitted. Mass tolerance for precursor ion and that for fragment ion were both 0.05. Threshold score/expected value was set at 1.30/95% (Unused Prot Score). All fixed and variable modifications attached are listed in supplemental Table S1. The original proteome datasets were deposited to JPOST project number JPST000758 (https://globe.jpostdb.org/entry/JPST000758).
Analysis of PC molecular species by LC-MS/MS
Lipids were quantified using a LC-MS/MS system (QTRAP5500, Sciex; coupled to a Shimadzu Prominence HPLC system). Normal-phase HPLC, for the analysis of PCs, was performed on an Inertsil SIL-100A column with the elution solvent acetonitrile/methanol/3% formic acid (18:11:1, v/v/v) as described previously (24). The samples were dried under N2 gas, dissolved in 100 μl of mobile phase solvent, and 10 μl of the sample were injected. The PCs were detected by multiple reaction monitoring in which 42 elected ions that released fragment ions of m/z = 184.1 were detected simultaneously (supplemental Table S2). The relative amount of PC species in in vivo-oxLDL was calculated semi-quantitatively, and the amounts of PCs that bound to monoclonal IgM κ isotype control as nonspecific reaction were subtracted.
Statistical analysis
Statistical analysis was performed using JMP Pro13 software. Statistical differences between groups were tested by the nonparametric Mann-Whitney U test. For comparisons across multiple groups, the data were analyzed using the parametric Tukey-Kramer test and nonparametric Dunn’s test. Data compared with control was analyzed using the parametric Dunnett’s test. A value of P < 0.05 was considered statistically significant.
RESULTS
Elution profiles of oxLDL on an anion-exchange column
LDL fractions from AMI patients and healthy subjects were fractionated on the anion-exchange column (Fig. 1). The absorbance of eluates from the column at 280 nm showed four peaks corresponding to the stepwise separation. The first peak at fraction numbers 3–5 is referred to as the L1 fraction, which is the flow-through fraction, and the fourth peak at fraction numbers 25–27 is referred to as the LDL(−), which was eluted from the column using 0.5 M NaCl. The absorbance for the LDL(−) fraction in AMI patients was 2- to 3-fold higher than that in healthy subjects. ApoB was detected in all four peaks and oxLDL was detected mostly in the L1 and LDL(−) fractions when the elution profiles of apoB and oxLDL were examined by sandwich ELISA (Fig. 1A, B). The concentration of oxLDL in the LDL(−) fraction was approximately 3-fold higher in AMI patients than in healthy subjects. The oxLDL/apoB ratios of the LDL(−) fraction in AMI patients and healthy controls were 3- and 6-fold higher than those observed in the L1 fraction, respectively. The use of plasma or serum made a minimal difference because the elution profiles of LDL separated from the plasma and serum from healthy subjects were well-matched with each other (Fig. 1A, C).
Fig. 1.

The elution profiles of in vivo-oxLDL using anion-exchange chromatography. A, B: The LDL fraction from healthy controls (A) and patients with AMI (B) were separated on Q-Fast Flow columns. The concentrations of oxLDL (red triangles) and apoB (blue circles) were measured by sandwich ELISA. Note that the units for oxLDL (left vertical axis) is nanograms per microliter while that for apoB (right vertical axis) is micrograms per microliter. The protein elution profile measured by absorbance at 280 nm (black solid line) and NaCl concentration of the elution buffer (dashed line) are indicated. The horizontal axis represents the fraction number of eluates. Each point represents the mean (SD) of three sets of independent experiments. C: Elution profile of the LDL fraction from the serum of a healthy subject. D: Elution profiles of Cu-oxLDL and native HDL fractions on the anion-exchange chromatography were measured under the same conditions.
ApoB protein in the LDL(−) fraction shows increased oxPC modifications
Oxidative modifications of apoB protein in the L1 and LDL(−) fractions were detected by native PAGE followed by immunoblotting. Modification of apoB with oxPC was more abundant in the LDL(−) fractions than in the L1 fractions in both healthy and AMI samples (Fig. 2).
Fig. 2.
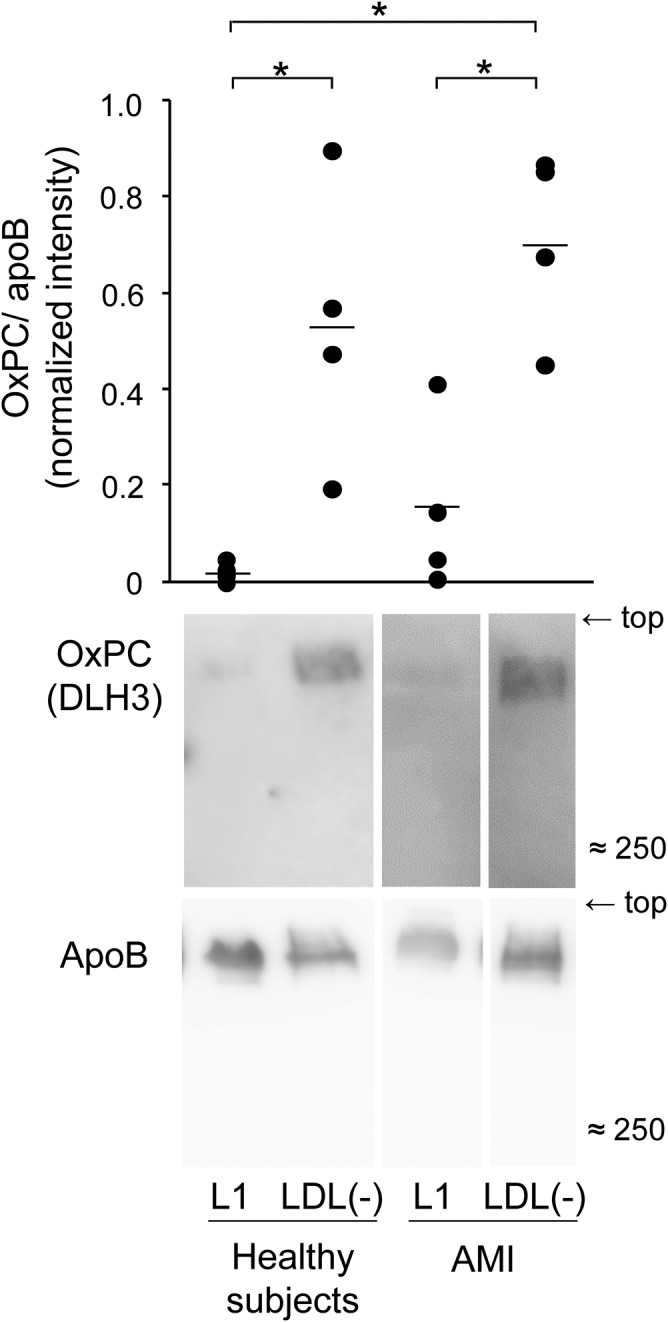
Modification of apoB by oxPC was detected by Western blotting. The L1 and LDL(−) fractions (2 μg protein per lane) were separated on native PAGE and immunoblot analyses were carried out to detect oxPC and apoB. The band images of oxPC-modified apoB in the L1 and LDL(−) fractions were quantified using ImageJ software. The graphs show results of the band intensities after normalization with apoB in each sample. Data represent the mean and each data point for four independent experiments. *P < 0.05.
Electromobility of apoB protein in the LDL(−) fraction of AMI patients
Electrophoretic mobility in agarose gels of the L1 and LDL(−) fractions and Cu-oxLDL were compared to evaluate the extent of apoB protein denaturation (Fig. 3A–C). Previous experiments showed that electromobility of LDL markedly shifted toward the anode in agarose gel electrophoresis by copper-induced oxidation (24). Interestingly, the LDL(−) fraction contained two bands. ApoB appeared to be the lower band in the Western blot (Fig. 3A, C). In AMI patients, there was a statistically significant difference in the electromobilities between the LDL(−) and L1 fractions (Fig. 3B). ApoB in the LDL(−) fractions tended to shift farther than in the L1 fractions. ApoB in the LDL(−) fractions had lower electromobility than did Cu-oxLDL, not only in healthy subjects but also in AMI patients. However, Cu-oxLDL eluted at the same fractions as LDL(−) from the anion-exchange column (Fig. 1D). The electronegative property of LDL(−) in this fraction may not be solely attributed to apoB modification.
Fig. 3.

Comparison of electromobility of the L1 and LDL(−) fractions on agarose gel electrophoresis. A: L1 and LDL(−) fractions separated from human LDL of AMI patients or healthy subjects and Cu-oxLDL were analyzed by 0.6% agarose gel electrophoresis (12 μg of protein per lane). Protein bands were stained with Coomassie Brilliant Blue stain. B: Relative electromobility of apoB protein in the L1 and LDL(−) fractions from AMI patients or healthy subjects and Cu-oxLDL were quantitated. Data represent the mean and each data point for six independent experiments. C, D: Detection of the apolipoproteins in the LDL(−) fractions from healthy subjects and AMI patients. Proteins separated on 0.6% agarose gel were transferred to PVDF membrane and probed with antibodies against apoB (C) or apoA1 (D). These are representative data of three independent experiments.
Presence of HDL in the LDL(−) fraction
To identify the proteins in upper bands detected only in LDL(−) fractions (Fig. 3A), both upper and lower bands were analyzed by LC-MS/MS with ProteinPilot software. The numbers of identified protein species, except for trypsin, are shown in Table 1. Within these identified proteins, only apoA1, apoE, and apoB peptides had greater confidence than trypsin peptides (Table 1). ApoE is known to associate with LDL and large HDL particles. Accordingly, the presence of apoA1 and apoE suggested that HDL particles were recovered in the LDL(−) fractions of both healthy subjects and AMI patients, although the LDL fraction was separated from HDL by ultracentrifugation before the anion-exchange column. In addition, the HDL fraction, separated by ultracentrifugation, eluted mostly at fraction number 15 when subjected to the anion-exchange column (Fig. 1D). The presence of apoA1 in the upper band of the LDL(−) fractions was verified by Western blotting (Fig. 3D).
TABLE 1.
Identification of proteins contained in the L1 and LDL(−) fractions of healthy subjects and AMI patients
| Name | %Cov | N | Number of Identified Protein Species | |
| Healthy subject | ||||
| LDL(−)_upper | ApoA1 | 82.4 | 1 | 4 |
| ApoE | 46.4 | 2 | ||
| LDL(−)_lower | ApoB-100 | 85.3 | 1 | 10 |
| ApoA1 | 91.0 | 2 | ||
| ApoE | 80.8 | 3 | ||
| L1 | ApoB-100 | 95.1 | 1 | 39 |
| ApoE | 67.2 | 3 | ||
| AMI patient | ||||
| LDL(−)_upper | ApoA1 | 100.0 | 1 | 23 |
| ApoE | 71.9 | 2 | ||
| LDL(−)_lower | ApoB-100 | 69.2 | 1 | 35 |
| ApoE | 65.6 | 2 | ||
| L1 | ApoB-100 | 94.7 | 1 | 26 |
| ApoE | 82.3 | 2 |
Proteins contained in the L1 and LDL(−) fractions of healthy subjects and AMI patients were separated using 0.6% agarose gel electromobility (Fig. 3A) and transferred to PVDF membrane. The protein bands on the PVDF membrane detected with Ponceau-S staining were dissected and digested with trypsin. Peptides extracted from the PVDF membrane were dissolved in 0.1% formic acid and analyzed using LC-MS/MS. N indicates the rank of the specified protein within the list of all the detected proteins in each band. %Cov indicates the percentage of matching amino acids from identified peptide signals within the total number of amino acids in the sequence. The data are representative results of each of the multiple replicates (n = 3).
To examine the morphological properties of lipoprotein particles detected in the LDL(−) fractions, direct observation of lipoproteins in the L1 and LDL(−) fractions using transmission electron microscopy (TEM) was performed (Fig. 4). In the L1 fractions, most of the particles were LDL, circular with a diameter over 20 nm, and similar results were observed in both AMI patients and healthy subjects. On the contrary, many smaller particles, as well as LDL particles, were observed in the LDL(−) fraction. The diameter of the particles observed in the L1 and LDL(−) fractions from healthy and AMI samples together with the HDL fraction from healthy subjects were measured (Fig. 4C). Compared with LDL particles in the LDL(−) fractions, the particle size of LDL in the L1 fractions was significantly larger. The LDL(−) from healthy subjects contained intermediate-sized particles (15–19 nm), and the mean diameter of HDL particles was significantly different from that of from AMI patients. Although a few HDL particles were in close contact with LDL particles (Fig. 4A, black arrows), most of particles were separately present.
Fig. 4.

Transmission electron micrograph images showing lipoprotein particles in the L1 and LDL(−) fractions of healthy subjects and AMI patients. A: L1 and LDL(−) fractions were diluted to <0.01 μg/μl, and then 5 μl of samples were dropped onto carbon-coated grids and stained with 2% uranyl acetate solution, pH 7.0. Scale bars indicate 20 nm. B: Native LDL and HDL fractions separated from healthy plasma were observed under the same conditions. Scale bars indicate 20 nm. C: The diameters of each particle observed by TEM were measured. According to previous literature on lipoprotein classification (39), particles with diameters of 16–31.3 nm were considered as LDL; 7.6–16 nm were considered as HDL. The mean diameter (SD) and number of LDL particles of healthy L1, AMI L1, healthy LDL(−), AMI LDL(−), and native LDL are 28.4 nm (2.16) (n = 72), 27.1 nm (2.45) (n = 56), 23.4 nm (4.01) (n = 96), 25.1 nm (2.68) (n = 19), and 24.6 nm (2.70) (n = 211), and those of HDL particles of healthy LDL(−), AMI LDL(−), and native HDL are 12.9 nm (1.78) (n = 43), 11.2 nm (1.72) (n = 23), 10.7 nm (1.55) (n = 152), respectively. *P < 0.05, **P < 0.01, ***P < 0.001.
ApoA1 in the LDL(−) fraction is oxidatively modified
The apoA1 in the LDL(−) fractions shifted higher in the agarose gel than that in native HDL (Fig. 3D), suggesting the presence of oxidatively modified apoA1 in the LDL(−) fraction, which may contribute to the electronegative properties of this fraction.
Oxidatively modified amino acid residues of apoA1 were analyzed using ProteinPilot software following the identification of the proteins, and those of apoA1in the LDL(−) fraction and the control HDL fraction were compared. In the apoA1 present in the LDL(−) fraction, a wide variety of oxidized amino acid residues of apoA1 were identified, with the total number of sites of oxidized amino acid residues significantly higher when compared with native HDL (Table 2). The %Cov of each analysis of apoA1, which is the percentage of matching amino acids from identified peptide signals within the total number of amino acids in the sequence, was >80%. ApoA1 contains 21 lysine residues among 243 amino acids, and acrolein or MDA adducts were detected at nearly all the lysine residues in apoA1 in the LDL(−) fractions of healthy subjects as well as AMI patients (Table 2). According to the review by Shao (41), the levels of modified apoA1 peptides, including chloro-tyrosine and nitro-tyrosine, are elevated in circulating HDL from CAD patients. These halogenated amino acids were found in a few peptides in apoA1 in the LDL(−) fraction (Table 2). Figure 5 shows the MS/MS spectra identifying peptides modified with nitrotyrosine, chlorotyrosine, and the hydroxynonenal-adduct of lysine residues. Although quantitative evaluation could not be applied to this analysis, it is likely that adducts of reactive lipid aldehydes to lysine residues increased in apoA1 in the LDL(−) fractions when compared with native HDL (supplemental Tables S3–S7).
TABLE 2.
Number of modified amino acid residues in apoA1 of HDL from LDL(−) fractions and native HDL
| Number of Various Amino Acid Residues | |||||
| HDL in LDL(−) Fraction | Native HDL | ||||
| Modification Types | AMI Patient | Healthy Subject | (A) | (B) | (C) |
| Adducts of reactive lipid aldehydes | |||||
| Acrolein addition +38 (K) | 13 | 14 | 10 | 0 | 5 |
| Acrolein addition +56 (K) | 19 | 16 | 9 | 0 | 11 |
| Acrolein addition +76 (K) | 19 | 13 | 8 | 2 | 6 |
| Acrolein addition +94 (K) | 16 | 14 | 7 | 0 | 4 |
| Acrolein addition +112 (K) | 19 | 18 | 8 | 1 | 8 |
| Reduced acrolein addition +58 (K) | 20 | 21 | 13 | 4 | 8 |
| Reduced acrolein addition +96 (K) | 13 | 11 | 6 | 1 | 7 |
| Reduced HNE (H) | 4 | 4 | 3 | 1 | 5 |
| ONE addition +154 (K) | 12 | 16 | 11 | 1 | 10 |
| Hexanoyl addition +98 (K) | 14 | 18 | 7 | 0 | 5 |
| HPNE addition +172 (K) | 12 | 17 | 13 | 0 | 8 |
| MDA adduct +54 (K) | 17 | 18 | 4 | 1 | 4 |
| MDA adduct +62 (K) | 19 | 15 | 11 | 0 | 4 |
| Halogenation/nitration | |||||
| Chloro (Y) | 0 | 1 | 0 | 0 | 0 |
| Nitro (W) | 0 | 1 | 0 | 0 | 0 |
| Nitro (Y) | 4 | 2 | 1 | 2 | 0 |
| Oxidized amino acids | |||||
| Dioxidation (K) | 1 | 1 | 0 | 1 | 0 |
| Dioxidation (P) | 2 | 0 | 1 | 0 | 1 |
| Dioxidation [R] | 4 | 1 | 4 | 0 | 0 |
| Dioxidation (W) | 4 | 4 | 4 | 2 | 3 |
| Dioxidation (Y) | 2 | 1 | 3 | 0 | 1 |
| Oxidation (D) | 7 | 2 | 1 | 1 | 1 |
| Oxidation (F) | 2 | 0 | 1 | 0 | 1 |
| Oxidation (H) | 4 | 2 | 3 | 2 | 1 |
| Oxidation (K) | 8 | 0 | 0 | 0 | 1 |
| Oxidation (N) | 3 | 1 | 0 | 0 | 0 |
| Oxidation (P) | 10 | 5 | 5 | 0 | 5 |
| Oxidation (R) | 4 | 4 | 3 | 0 | 2 |
| Oxidation (W) | 4 | 4 | 4 | 0 | 2 |
| Oxidation (Y) | 5 | 2 | 2 | 2 | 0 |
| Trioxidation (W) | 3 | 4 | 1 | 0 | 0 |
| Trioxidation (Y) | 1 | 0 | 2 | 0 | 1 |
| Others | |||||
| Trp→hydroxykynurenine (W) | 4 | 4 | 2 | 0 | 0 |
| Trp→kynurenine (W) | 4 | 4 | 4 | 0 | 1 |
| Total | 273 | 238 | 151 | 21 | 105 |
LDL(−) upper bands and native HDL bands from three healthy subjects in agarose gel electrophoresis were analyzed by LC-MS/MS to detect modified peptide fragments of apoA1. In total, 34 modification types were identified in the apoA1 sequence. Total sites of the oxidized amino acid modifications were counted for each sample. The name of the original amino acid residue was written in one-letter code; (D), aspartic acid; (F), phenylalanine; (H), histidine; (K), lysine; (N), asparagine; (P), proline; (R), arginine; (W), tryptophan; (Y), tyrosine. The representative data from HDL in LDL(–) fraction from an AMI patient and a healthy subject, and native HDL fraction from three healthy subjects (A, B, C) are indicated. See supplemental Tables S3–S7 for detailed results.
Fig. 5.

Identification of modified peptides. The MS/MS spectra annotated for modified peptide fragments from apoA1 containing 3-nitrotyrosine (A) or 3-chlorotyrosine (B) and 4-hydroperoxynonal-adduct of lysine are shown.
PC profile of in vivo-oxLDL
In vivo-oxLDL bound to anti-oxPC mAb was collected from the L1 or LDL(−) fractions, and the profiles of PC molecular species were determined by LC-MS/MS. Similarly, in vitro-oxLDL exhibiting antigenicity to anti-oxPC mAb was extracted from Cu-oxLDL and PC molecular species were analyzed. Figure 6 shows the relative amount of each PC species in oxLDL in L1 and LDL(−) from AMI patients and healthy subjects and in in vitro-oxLDL. The PC species are classified into five categories. Among the nonoxidized PC in in vivo-oxLDL, PC34:2 (m/z = 758.6) was the highest, followed by PC34:1 (m/z = 760.6) and PC36:2 (m/z = 786.6) (Fig. 6A, C). The long-chain oxPC species derived from PC34:2 and PC36:4 were marginally increased in oxLDL in the LDL(−) of AMI patients (Fig. 6D). Among cleaved oxPC species, oxPC650 [representative species is 1-palmitoyl-2-(9-oxo-nonanoly) PC; m/z = 650.6] was the most detected in the in vivo-oxLDL of healthy subjects but not AMI patients (Fig. 6E). The major lysoPC species, 16:0-LPC and 18:0-LPC, were detected in the in vivo-oxLDL obtained from AMI patients as well as in vitro oxLDL; however, their contents in the in vivo-oxLDL from healthy subjects were low (Fig. 6B). LysoPC demonstrated a good contrast with oxPC650, which was high in the oxLDL from healthy subject but low in AMI patients. Overall, the PC species was most detected in oxLDL in the LDL(−) of AMI patients, implying that the amount of oxLDL bound to the anti-oxPC mAb was the highest in the LDL(−) of AMI patients. However, most of the PC species in in vivo-oxLDL were nonoxidized PCs, and lysoPC and cleaved oxPC did not accumulate. On the contrary, lysoPC accounted for a considerably higher percentage in in vitro-oxLDL separated from Cu-oxLDL, where the profile of PC species was in good agreement with our previous results (24).
Fig. 6.

Box-plot diagrams showing the relative amount of 42 PC species in in vivo-oxLDL in L1 and in vivo-oxLDL in LDL(−) separated from healthy subjects (n = 7) and AMI patients (n = 7) together with in vitro oxLDL (Cu-oxLDL) (n = 7). The PC molecular species include saturated/monounsaturated fatty acid-containing PC (A), PUFA-containing PC (B), long chain oxPC (C), cleaved oxPC (D), and lysoPC (E). ¶P < 0.05 [compared with oxLDL in LDL(−) (healthy) or oxLDL in LDL(−) (AMI)], *P < 0.05 [oxLDL in L1 (healthy) vs. oxLDL in LDL(−) (AMI)], #P < 0.05 [oxLDL in L1 (healthy) vs. in vitro oxLDL], ‡P < 0.05 [oxLDL in LDL(−) (healthy) vs. in vitro oxLDL], & P < 0.05 [in vitro oxLDL vs. oxLDL in L1 (AMI)], and $P < 0.05 [in vitro oxLDL vs. oxLDL in LDL(−) (AMI)].
Identification of oxidatively modified apoB amino acid residues in in vivo-oxLDL
The in vivo-oxLDL separated from the LDL(−) and L1 fractions was recovered by SDS solution, and apoB peptides were analyzed by LC-MS/MS. Table 3 shows the list of oxidized amino acid residues in in vivo-oxLDL. The sequence coverage of apoB using this procedure was only 30–40% for oxLDL in the L1 of AMI patients and Cu-oxLDL, and more than 50% for others. Acrolein and MDA-modified lysine residues were identified from samples captured by anti-oxPC mAb, and many of the modified lysine residues were found in certain domains in apoB (approximately residues 1–1,000, 2,100–2,570, and 4,050–4,500). ApoB in the in vivo-oxLDL particles contained modified amino acid residues comparable to some extent to those observed in oxPC-positive Cu-oxLDL (supplemental Tables S8–S17).
TABLE 3.
Modified amino acid residues detected in apoB in oxLDL bound to anti-oxPC mAb
| Oxidation Types | Modified Residues | |
| Healthy subject | ||
| In vivo-oxLDL in L1 | Acrolein addition +38 (K) | K2238, K2773 |
| Acrolein addition +94 (K) | K1751 | |
| Dioxidation (R) | R540 | |
| Dioxidation (W) | W694*, W936*, W1114*, W1210*, W1954, W2104*, W2221*, W2546*, W3126, W4087* | |
| Hexanoyl addition +98 (K) | K2769 | |
| Reduced acrolein addition +96 (K) | K1754 | |
| MDA adduct +62 (K) | K2243 | |
| Trp→Kynurenin (W) | W4087* | |
| In vivo-oxLDL in LDL(−) | Acrolein addition +38 (K) | K4372 |
| Acrolein addition +56 (K) | K4436 | |
| Acrolein addition +76 (K) | K894, K4511 | |
| MDA adduct +62 (K) | K896, K903, K4434 | |
| ONE addition +154 (K) | K2291 | |
| AMI patient | ||
| In vivo-oxLDL in L1 | Dioxidation (W) | W2104*, W4087* |
| In vivo-oxLDL in LDL(−) | Acrolein addition +38 (K) | K627 |
| Dioxidation (W) | W694, W1114, W1210, W1434, W2104, W2221, W2546, W2894, W3126, W4087 | |
| MDA adduct +54 (K) | K614 | |
| MDA adduct +62 (K) | K642 | |
| Oxidation (P) | P1436 | |
| Trp→Kynurenin (W) | W4087 | |
| In vitro Cu-oxLDL | Acrolein addition +56 (K) | K1697, K3136 |
| Acrolein addition +112 (K) | K130 | |
| Dioxidation (W) | W2104, W4087 | |
| Hexanoyl addition +98 (K) | K1710, K2313 | |
| MDA adduct +62 (K) | K1529, K2306 | |
| Reduced acrolein addition +96 (K) | K4044 | |
| Trp→Kynurenin (W) | W4087 |
Oxidatively modified apoB peptides in oxLDL bound to anti-oxPC mAb were identified by LC-MS/MS. L1, LDL(−) from healthy subjects and AMI patients, and Cu-oxLDL were added to microtiter wells precoated with anti-oxPC mAb, and the antigens bound to the mAb were subjected to LC-MS/MS analysis. Proteins bound to IgM κ isotype control were recovered simultaneously with the nonspecific reaction. oxLDL possessing antigenicity against the anti-oxPC mAb in in vitro Cu-oxLDL was also extracted under the same conditions. The proteins spotted onto PVDF membranes were digested by trypsin followed by reductive alkylation, and extracted peptides were analyzed by LC-MS/MS. ApoB peptides with modifications were identified using ProteinPilot software. The table shows oxidatively modified apoB-100 amino acid residues that were identified in two different experiments. The amino acid residues with an asterisk (*) were also detected in the control wells that were precoated with monoclonal IgM κ isotype control. The name of the original amino acid residue was written in one-letter code; (K), lysine; (P), proline; (R), arginine; (W), tryptophan. See supplemental Tables S8–S17 for detailed results.
DISCUSSION
A large volume of evidence to show the involvement of oxLDL in the initiation and progression of atherosclerosis has accumulated over the past quarter century. However, a few critical questions remain unanswered, including establishing the actual form of in vivo-oxLDL. In this study, we succeeded in separating in vivo-oxLDL from the blood of healthy subjects and patients with AMI and investigated its characteristics. The in vivo-oxLDL recognized by the anti-oxPC mAb was divided into two types, oxLDL in L1 and oxLDL in LDL(−). To the best of our knowledge, this is the first study to directly indicate that in vivo-oxLDL consisted of at least two unique fractions, distinct from in vitro Cu-oxLDL. In addition, our data indicated the presence of heavily modified HDL in the LDL(−) fraction.
Several research groups have investigated in vivo-modified LDLs, like electronegative LDL and lipoprotein (a) [Lp(a)], that fit well with the oxLDL hypothesis. Avogaro, Bon, and Cazzolato (42) first demonstrated that LDL(−) can be separated using anion-exchange chromatography, and the reported LDL(−) particles may be heterogeneously aggregated in a way independent of S-S bonds. LDL(−) has been characterized by increased electronegativity, phospholipolysis, and the presence of many apolipoproteins, in addition to apoB (43–45). Additionally, it has an ability to stimulate endothelial cells through the LOX-1 receptor (46, 47). In our study, it was demonstrated that LDL(−) contained one type of oxPC-positive in vivo-oxLDL. One of the major findings of this study was that oxHDL accompanied in vivo-oxLDL in the LDL(−) fraction. This was supported by the following reasons: First, while the LDL fraction (d = 1.019–1.063) was separated from other lipoproteins by ultracentrifugation, oxHDL was recovered in the LDL(−) fraction. Second, the control HDL fraction was mostly eluted at 0.22 mol/l NaCl, whereas the LDL(−) fraction was eluted at 0.5 mol/l NaCl. Remarkably, the apoA1 in the LDL(−) fraction was heavily modified. Gao and Podrez (48) have reported that apoA1 is also susceptible to oxidative modifications based on a precise analysis of modified apoA1 peptides. Therefore, it is likely that the electronegative property of oxLDL in LDL(−) was due to, at least in part, its association with oxHDL. LDL- and HDL-sized particles were mostly separated under TEM, and they were separately electrophoresed on agarose gel. The association between them was possibly electrostatic interaction, but not covalent binding.
HDL is considered to be anti-atherosclerotic and anti-oxidative. However, recent advances in HDL investigations have revealed more complex and pivotal roles of HDL in atherogenesis (34). The presence of chlorinated and nitrated oxHDL, which are MPO-dependent modifications, in human atherosclerotic lesions as well as plasma has been reported (37, 49, 50). An ELISA system using the recombinant LOX-1 receptor showed the presence of oxidatively modified HDL in human plasma (51). Reportedly, a significant association exists between plasma oxHDL and inflammatory states in patients with psoriasis (52). Notably, HDL turns to atherogenic once it is oxidatively modified. Our current observations included a characteristic of oxHDL in vivo; oxHDL could interact with oxLDL and work together as electronegative lipoproteins.
Our current data showed that, in patients with AMI, the oxLDL concentration in the LDL(−) fraction was about 3-fold higher than in healthy subjects, as determined by sandwich ELISA (Fig. 1). The blood samples of AMI patients were collected from the infarct regions during catheter treatments. It is possible that oxLDL in LDL(−), at least in part, is derived from the ruptured plaque at the acute phase, corresponding with the current hypothesis regarding the origin of circulating oxLDL based on the transient increase of plasma oxLDL levels in patients with AMI (19, 22). Hazen and colleagues have reported the presence of oxHDL in the plasma and in atherosclerotic plaques, while modified apoA1 in atherosclerotic lesions was lipid-poor and aggregated (37, 50). These distinct features of modified apoA1 in the plaque failed to correspond with oxHDL in the LDL(−) fraction. The oxHDL that associates with oxLDL in the LDL(−) fraction might be generated in circulation presumably through the interaction of HDL with oxLDL.
The electronegative in vivo-oxLDL, namely oxLDL in LDL(−), contains oxLDL and heavily modified oxHDL. Interestingly, the apoB in this fraction is immune-positive to anti-oxPC mAb but has fewer oxidatively modified amino acid residues than apoA1. In our previous study, the apoB fragments modified with oxPC became resistant to lysosomal hydrolysis, staying longer in macrophage-derived foam cells (53). Thus, increased oxPC-apoB adducts in the LDL(−) fraction could be derived from accumulated oxLDL in foam cells. Another type of oxLDL existed in the L1 fractions of healthy subjects as well as AMI patients. The oxLDL in the L1 fraction demonstrated a marginal increase in electromobility and minimal apoB modification with oxPC but contained comparable amounts of extractable oxPC. These characteristics correspond to MM-LDL (25, 26, 54). While heavily oxidized LDL is quickly cleared from the circulation by the liver (55), MM-LDL binds to the LDL receptor but not to scavenger receptors, remaining in circulation longer than the heavily oxidized LDL. In addition to the acute diffusion of oxidized lipoproteins from the ruptured plaque and quick clearance by the liver, oxidized lipoproteins might spontaneously transfer between arterial tissue and circulation (22).
When LDL is treated with copper sulfate, both lipids and proteins are extensively modified. The accumulation of oxidative modifications of apoB amino acids, oxidized lipids, and lysoPC, as well as a significant increase in the negative charge in Cu-oxLDL particles, were observed (23, 24). Interestingly, apoB in LDL(−) is covalently modified with oxPC, whereas the site of modified amino acid residues in apoB is minimal, and the electronegative property of the LDL is moderate. In addition, the amounts of oxPC and lysoPC in in vivo-oxLDL were substantially lower compared with in vitro-oxLDL. The preparation of Cu-oxLDL in a small test tube in which only LDL is present in a limited volume of buffer could be the reason for the differences. During the oxidation process in vitro, any intermediate products, including reactive aldehydes and oxPC, remain in a closed system and easily react with proteins; whereas, under in vivo conditions, oxidative intermediates formed in the LDL particles diffuse into the circulation or transfer to other lipoproteins. Because HDL has protective effects toward LDL oxidation (56), HDL is likely to interact with LDL particles to protect them from further oxidative modification by transferring oxPCs from LDL to HDL and by scavenging radicals with apoA1 molecules. This antioxidative action of HDL against oxLDL may lead to oxHDL generation (57).
We observed that apoA1 in LDL(−) was heavily modified with acrolein. This was consistent with a previous report showing preferable modification of apoA1 with acrolein in plasma HDL (48). Acrolein is a lipid peroxidation product; however, it is also produced through the enzymatic action of MPO (58). Because inflammatory responses occur in atheromatous plaques, MPO derived from macrophages and neutrophils could participate in the process of acrolein modification of lipoproteins.
Because the LDL(−) fraction could contain heterogeneous forms of lipoproteins, we separated oxPC-bound oxLDL particles in the LDL(−) and L1 fractions to analyze their molecular features. Notably, modifications of apoB amino acid residues were limited in both fractions. The profiles of extracted PC molecular species demonstrated little difference among oxLDL in L1 or LDL(−) from AMI patients or healthy controls, except for POVPC (m/z = 650) and lysoPC (Fig. 6). The lysoPC in in vivo-oxLDL could be produced by hydrolysis of cleaved oxPC by Lp-PLA2. A meta-analysis on Lp-PLA2 levels revealed that the enzyme activity and mass were associated with long-term risks of CHD events (59). In this study, Lp-PLA2 activity in the LDL fraction was higher in AMI patients than in healthy subjects (AMI patients: 16.8 U/l vs. healthy subjects: 11.6 U/l), and the cleaved oxPC could be hydrolyzed enzymatically. Compared with Cu-oxLDL, the contents of oxPCs and lysoPC were much smaller, suggesting that the oxidative change in the in vivo-oxLDL was limited. Furthermore, it has been proposed that the smaller HDL subfraction, HDL3, mediates reduction of PCOOH to PCOH of LDL utilizing methionine residues of apoA1, so that oxidation of LDL lipids may be prevented by HDL3 (35). In addition, it is possible that oxPC or lysoPC generated in oxLDL can transfer between two lipoproteins, e.g., oxLDL and HDL particles, and this functional interaction can yield oxHDL (34, 57). Currently, we are attempting to elucidate the transfer action of oxPC and lysoPC between lipoproteins using stable isotope-labeled probes.
Lp(a) is more electronegative than LDL and is a known risk factor for atherosclerosis (60–62). The density of Lp(a) ranges between 1.050 and 1.082 g/ml, distributed in both LDL and HDL fractions by density gradient ultracentrifugation. One previous report has proposed a model in which LDL(−) (referred to as L5) contains several apolipoproteins, including apoA1 and apo(a) in addition to apoB in its particle (63). Our LC-MS/MS analysis of the LDL(−) lower bands, identified apoE and apoA1 in addition to apoB; however, apo(a) was identified as one of the minor proteins, which is consistent with previous reports (29, 30). In our LDL(−) samples, any lipoproteins with particle size 32.5–40 nm corresponding to Lp(a) were not found by TEM. Additionally, it is known that the serum concentration of Lp(a) has great variety among different ethnic groups; the Lp(a) concentration in the Asian population is lower than other ethnic groups (62). Although we cannot rule out the presence of Lp(a) in the LDL(−) fraction, we consider its contribution would be limited.
In addition to apoA1, HDL contains some other apolipoproteins like apoA2. Recently, it was reported that HDL particles containing apoA2 have different properties from those without apoA2 (64). Although apoA2 was detected in the LDL(−) fraction as a minor constituent in our experiments, it is possible that a subclass of HDL may have a selective contribution to the HDL-LDL complex.
Several studies have shown that small dense LDL (sdLDL) is an atherogenic subfraction of LDL (65, 66). It is hypothesized that sdLDL is susceptible to oxidative modification; however, direct proof remains unavailable. From the TEM images of the LDL(−) fraction, we observed that it consists of mainly HDL and normal-sized LDL; however, LDL(−) from healthy subjects may in part contain smaller-sized LDL that corresponds to sdLDL. More work is needed to clarify the relationship between sdLDL and in vivo-oxLDL.
Our current study succeeded in separating in vivo-oxLDL from human plasma. We observed that there are two forms of in vivo-oxLDL, one of them, oxLDL in L1, contains oxPC but is not electronegative and the other, oxLDL in LDL(−), is modified covalently with oxPC and is electronegative, presumably due to association with heavily oxidized HDL. The latter oxLDL increased in the acute phase of AMI patients. Both of these oxLDLs demonstrated different features from Cu-oxLDL. There might be many different forms of in vivo-oxLDL; however, our observations clearly provide new insights into the structural features of in vivo-oxLDL and help in understanding the mechanisms of atherogenesis.
Data availability
The original proteome datasets together with the list of identified peptides were deposited to JPOST (https://repository.jpostdb.org/entry/JPST000758). All other data is contained in the article.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the work of past and present members of the Division of Biological Chemistry who contributed to this study. Without their contribution, this work would not have been possible. The authors would like to thank Editage (www.editage.com) for English language editing.
Footnotes
Abbreviations:
- AMI
- acute MI
- Cu-oxLDL
- copper-induced oxidized LDL
- L1
- flow-through fraction of LDL from anion-exchange column
- LDL(−)
- electronegative LDL fraction eluted from anion-exchange column
- Lp(a)
- lipoprotein (a)
- Lp-PLA2
- lipoprotein-associated phospholipase A2
- MDA
- malondialdehyde
- MM-LDL
- minimally modified LDL
- MPO
- myeloperoxidase
- oxHDL
- oxidized HDL
- oxLDL
- oxidized LDL
- oxPC
- oxidized PC
- pAb
- polyclonal antibody
- PL
- phospholipid
- sdLDL
- small dense LDL
- TEM
- transmission electron microscopy
This research was supported in part by Japan Society for the Promotion of Science KAKENHI Grants JP15K07944, JP21790092, JP24590094, and JP24790087; the Private University Research Branding Project; Strategic Research Foundation at Private Universities Grant S1001011; Showa University Research Grants for Young Researchers (2017 for N.S., 2007 for T.O.); and Japan Science Society Sasakawa Scientific Research Grant 22-418. The authors declare that they have no conflicts of interest with the contents of this article.
The online version of this article (available at https://www.jlr.org) contains a supplement.
REFERENCES
- 1.Itabe H. 2003. Oxidized low-density lipoproteins: what is understood and what remains to be clarified. Biol. Pharm. Bull. 26: 1–9. [DOI] [PubMed] [Google Scholar]
- 2.Maiolino G., Rossitto G., Caielli P., Bisogni V., Rossi G. P., and Calo L. A.. 2013. The role of oxidized low-density lipoproteins in atherosclerosis: the myths and the facts. Mediators Inflamm. 2013: 714653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartley A., Haskard D., and Khamis R.. 2019. Oxidized LDL and anti-oxidized LDL antibodies in atherosclerosis - novel insights and future directions in diagnosis and therapy. Trends Cardiovasc. Med. 29: 22–26. [DOI] [PubMed] [Google Scholar]
- 4.Gillotte K. L., Hörkko S., Witztum J. L., and Steinberg D.. 2000. Oxidized phospholipids, linked to apolipoprotein B of oxidized LDL, are ligands for macrophage scavenger receptors. J. Lipid Res. 41: 824–833. [PubMed] [Google Scholar]
- 5.Steinbrecher U. P., Witztum J. L., Parthasarathy S., and Steinberg D.. 1987. Decrease in reactive amino groups during oxidation or endothelial cell modification of LDL. Correlation with changes in receptor-mediated catabolism. Arteriosclerosis. 7: 135–143. [DOI] [PubMed] [Google Scholar]
- 6.Rosenfeld M. E., Palinski W., Ylä-Herttuala S., Butler S., and Witztum J. L.. 1990. Distribution of oxidation specific lipid-protein adducts and apolipoprotein B in atherosclerotic lesions of varying severity from WHHL rabbits. Arteriosclerosis. 10: 336–349. [DOI] [PubMed] [Google Scholar]
- 7.Itabe H., Takeshima E., Iwasaki H., Kimura J., Yoshida Y., Imanaka T., and Takano T.. 1994. A monoclonal antibody against oxidized lipoprotein recognizes foam cells in atherosclerotic lesions. Complex formation of oxidized phosphatidylcholines and polypeptides. J. Biol. Chem. 269: 15274–15279. [PubMed] [Google Scholar]
- 8.Palinski W., Ord V. A., Plump A. S., Breslow J. L., Steinberg D., and Witztum J. L.. 1994. ApoE-deficient mice are a model of lipoprotein oxidation in atherogenesis. Demonstration of oxidation-specific epitopes in lesions and high titers of autoantibodies to malondialdehyde-lysine in serum. Arterioscler. Thromb. 14: 605–616. [DOI] [PubMed] [Google Scholar]
- 9.Ehara S., Ueda M., Naruko T., Haze K., Itoh A., Otsuka M., Komatsu R., Matsuo T., Itabe H., Takano T., et al. 2001. Elevated levels of oxidized low density lipoprotein show a positive relationship with the severity of acute coronary syndromes. Circulation. 103: 1955–1960. [DOI] [PubMed] [Google Scholar]
- 10.Nishi K., Itabe H., Uno M., Kitazato K. T., Horiguchi H., Shinno K., and Nagahiro S.. 2002. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arterioscler. Thromb. Vasc. Biol. 22: 1649–1654. [DOI] [PubMed] [Google Scholar]
- 11.Itabe H., Yamamoto H., Imanaka T., Shimamura K., Uchiyama H., Kimura J., Sanaka T., Hata Y., and Takano T.. 1996. Sensitive detection of oxidatively modified low density lipoprotein using a monoclonal antibody. J. Lipid Res. 37: 45–53. [PubMed] [Google Scholar]
- 12.Itabe H., Yamamoto H., Suzuki M., Kawai Y., Nakagawa Y., Suzuki A., Imanaka T., and Takano T.. 1996. Oxidized phosphatidylcholines that modify proteins. Analysis by monoclonal antibody against oxidized low density lipoprotein. J. Biol. Chem. 271: 33208–33217. [DOI] [PubMed] [Google Scholar]
- 13.Hörkkö S., Bird D. A., Miller E., Itabe H., Leitinger N., Subbanagounder G., Berliner J. A., Friedman P., Dennis E. A., Curtiss L. K., et al. 1999. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J. Clin. Invest. 103: 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsimikas S., Kiechl S., Willeit J., Mayr M., Miller E. R., Kronenberg F., Xu Q., Bergmark C., Weger S., Oberhollenzer F., et al. 2006. Oxidized phospholipids predict the presence and progression of carotid and femoral atherosclerosis and symptomatic cardiovascular disease: five-year prospective results from the Bruneck study. J. Am. Coll. Cardiol. 47: 2219–2228. [DOI] [PubMed] [Google Scholar]
- 15.Holvoet P., Donck J., Landeloos M., Brouwers E., Luijtens K., Arnout J., Lesaffre E., Vanrenterghem Y., and Collen D.. 1996. Correlation between oxidized low density lipoproteins and von Willebrand factor in chronic renal failure. Thromb. Haemost. 76: 663–669. [PubMed] [Google Scholar]
- 16.Kotani K., Maekawa M., Kanno T., Kondo A., Toda N., and Manabe M.. 1994. Distribution of immunoreactive malondialdehyde-modified low-density lipoprotein in human serum. Biochim. Biophys. Acta. 1215: 121–125. [DOI] [PubMed] [Google Scholar]
- 17.Inoue N., Lkamura T., Kokubo Y., Fujita Y., Sato Y., Nakanishi M., Yanagida K., Kakino A., Iwamoto S., Watanabe M., et al. 2010. LOX index, a novel predictive biochemical marker for coronary heart disease and stroke. Clin. Chem. 56: 550–558. [DOI] [PubMed] [Google Scholar]
- 18.Itabe H., and Ueda M.. 2007. Measurement of plasma oxidized low-density lipoprotein and its clinical implications. J. Atheroscler. Thromb. 14: 1–11. [DOI] [PubMed] [Google Scholar]
- 19.Naruko T., Ueda M., Ehara S., Itoh A., Haze K., Shirai N., Ikura Y., Ohsawa M., Itabe H., Kobayashi Y., et al. 2006. Persistent high levels of plasma oxidized low-density lipoprotein after acute myocardial infarction predict stent restenosis. Arterioscler. Thromb. Vasc. Biol. 26: 877–883. [DOI] [PubMed] [Google Scholar]
- 20.Tsimikas S., Willeit P., Willeit J., Santer P., Mayr M., Xu Q., Mayr A., Witztum J. L., and Kiechl S.. 2012. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J. Am. Coll. Cardiol. 60: 2218–2229. [DOI] [PubMed] [Google Scholar]
- 21.Holvoet P., Vanhaecke J., Janssens S., Van de Werf F., and Collen D.. 1998. Oxidized LDL and malondialdehyde-modified LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation. 98: 1487–1494. [DOI] [PubMed] [Google Scholar]
- 22.Itabe H., Kato R., Sawada N., Obama T., and Yamamoto M.. 2019. The significance of oxidized low-density lipoprotein in body fluids as a marker related to diseased conditions. Curr. Med. Chem. 26: 1576–1593. [DOI] [PubMed] [Google Scholar]
- 23.Obama T., Kato R., Masuda Y., Takahashi K., Aiuchi T., and Itabe H.. 2007. Analysis of modified apolipoprotein B-100 structures formed in oxidized low-density lipoprotein using LC-MS/MS. Proteomics. 7: 2132–2141. [DOI] [PubMed] [Google Scholar]
- 24.Sasabe N., Keyamura Y., Obama T., Inoue N., Masuko Y., Igarashi Y., Aiuchi T., Kato R., Yamaguchi T., Kuwata H., et al. 2014. Time course-changes in phosphatidylcholine profile during oxidative modification of low-density lipoprotein. Lipids Health Dis. 13: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berliner J. A., Territo M. C., Sevanian A., Ramin S., Kim J. A., Bamshad B., Esterson M., and Fogelman A. M.. 1990. Minimally modified low density lipoprotein stimulates monocyte endothelial interactions. J. Clin. Invest. 85: 1260–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Itabe H., Mori M., Fujimoto Y., Higashi Y., and Takano T.. 2003. Minimally modified LDL is an oxidized LDL enriched with oxidized phosphatidylcholines. J. Biochem. 134: 459–465. [DOI] [PubMed] [Google Scholar]
- 27.Podrez E.A., Febbraio M., Sheibani N., Schmitt D., Silverstein R. L., Hajjar D. P., Cohen P. A., Frazier W. A., Hoff H. F., and Hazen S. L.. 2000. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J. Clin. Invest. 105: 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Podrez E. A., Poliakov E., Shen Z., Zhang R., Deng Y., Sun M., Finton P. J., Shan L., Gugiu B., Fox P. L., et al. 2002. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J. Biol. Chem. 277: 38503–38516. [DOI] [PubMed] [Google Scholar]
- 29.De Castellarnau C., Sánchez-Quesada J. L., Benítez S., Rosa R., Caveda L., Vila L., and Ordóñez-Llanos J.. 2000. Electronegative LDL from normolipemic subjects induces IL-8 and monocyte chemotactic protein secretion by human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 20: 2281–2287. [DOI] [PubMed] [Google Scholar]
- 30.Chu C. S., Chan H. C., Tsai M. H., Stancel N., Lee H. C., Cheng K. H., Tung Y. C., Chan H. C., Wang C. Y., Shin S. J., et al. 2018. Range of L5 LDL levels in healthy adults and L5′s predictive power in patients with hyperlipidemia or coronary artery disease. Sci. Rep. 8: 11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu J. F., Chou T. C., Lu J., Chen S. H., Chen F. Y., Chen C. C., Chen J. L., Elayda M., Ballantyne C. M., Shayani S., et al. 2014. Low-density lipoprotein electronegativity is a novel cardiometabolic risk factor. PLoS One. 9: e107340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benítez S., Camacho M., Arcelus R., Vila L., Bancells C., Ordóñez-Llanos J., and Sánchez-Quesada J. L.. 2004. Increased lysophosphatidylcholine and non-esterified fatty acid content in LDL induces chemokine release in endothelial cells. Relationship with electronegative LDL. Atherosclerosis. 177: 299–305. [DOI] [PubMed] [Google Scholar]
- 33.Benítez S., Camacho M., Bancells C., Vila L., Sánchez-Quesada J. L., and Ordóñez-Llanos J.. 2006. Wide proinflammatory effect of electronegative low-density lipoprotein on human endothelial cells assayed by a protein array. Biochim. Biophys. Acta. 1761: 1014–1021. [DOI] [PubMed] [Google Scholar]
- 34.Lüscher T. F., Landmesser U., von Eckardstein A., and Fogelman A. M.. 2014. High-density lipoprotein: vascular protective effects, dysfunction, and potential as therapeutic target. Circ. Res. 114: 171–182. [DOI] [PubMed] [Google Scholar]
- 35.Zerrad-Saadi A., Therond P., Chantepie S., Couturier M., Rye K. A., Chapman M. J., and Kontush A.. 2009. HDL3-mediated inactivation of LDL-associated phospholipid hydroperoxides is determined by the redox status of apolipoprotein A-I and HDL particle surface lipid rigidity: relevance to inflammation and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 29: 2169–2175. [DOI] [PubMed] [Google Scholar]
- 36.Soran H., Schofield J. D., and Durrington P. N.. 2015. Antioxidant properties of HDL. Front. Pharmacol. 6: 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang Y., DiDonato J. A., Levison B. S., Schmitt D., Li L., Wu Y., Buffa J., Kim T., Gerstenecker G. S., Gu X., et al. 2014. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 20: 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohno H., Sueshige N., Oguri K., Izumidate H., Masunari T., Kawamura M., Itabe H., Takano T., Hasegawa A., and Nagai R.. 2000. Simple and practical sandwich-type enzyme immunoassay for human oxidatively modified low density lipoprotein using antioxidized phosphatidylcholine monoclonal antibody and antihuman apolipoprotein-B antibody. Clin. Biochem. 33: 243–253. [DOI] [PubMed] [Google Scholar]
- 39.Okazaki M., and Yamashita S.. 2016. Recent advances in analytical methods on lipoprotein subclasses: Calculation of particle numbers from lipid levels by gel permeation HPLC using “spherical particle model”. J. Oleo Sci. 65: 265–282. [DOI] [PubMed] [Google Scholar]
- 40.Moriya Y., Obama T., Aiuchi T., Sugiyama T., Endo Y., Koide Y., Noguchi E., Ishizuka M., Inoue M., Itabe H., et al. 2017. Quantitative proteomic analysis of gingival crevicular fluids from deciduous and permanent teeth. J. Clin. Periodontol. 44: 353–362. [DOI] [PubMed] [Google Scholar]
- 41.Shao B. 2012. Site-specific oxidation of apolipoprotein A-I impairs cholesterol export by ABCA1, a key cardioprotective function of HDL. Biochim. Biophys. Acta. 1821: 490–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Avogaro P., Bon G. B., and Cazzolato G.. 1988. Presence of a modified low density lipoprotein in humans. Arteriosclerosis. 8: 79–87. [PubMed] [Google Scholar]
- 43.Hodis H. N., Kramsch D. M., Avogaro P., Bittolo-Bon G., Cazzolato G., Hwang J., Peterson H., and Sevanian A.. 1994. Biochemical and cytotoxic characteristics of an in vivo circulating oxidized low density lipoprotein (LDL-). J. Lipid Res. 35: 669–677. [PubMed] [Google Scholar]
- 44.Bancells C., Sánchez-Quesada J. L., Birkelund R., Ordóñez-Llanos J., and Benítez S.. 2010. HDL and electronegative LDL exchange anti- and pro-inflammatory properties. J. Lipid Res. 51: 2947–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bancells C., Canals F., Benítez S., Colomé N., Julve J., Ordóñez-Llanos J., and Sánchez-Quesada J. L.. 2010. Proteomic analysis of electronegative low-density lipoprotein. J. Lipid Res. 51: 3508–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu J., Yang J. H., Burns A. R., Chen H. H., Tang D., Walterscheid J. P., Suzuki S., Yang C. Y. C., Sawamura T., and Chen C. H.. 2009. Mediation of electronegative low-density lipoprotein signaling by LOX-1: a possible mechanism of endothelial apoptosis. Circ. Res. 104: 619–627. [DOI] [PubMed] [Google Scholar]
- 47.Chan H. C., Ke L. Y., Chu C. S., Lee A. S., Shen M. Y., Cruz M. A., Hsu J. F., Cheng K. H., Chan H. C. B., Lu J., et al. 2013. High electronegative LDL from patients with ST-elevation myocardial infarction triggers platelet activation and aggregation. Blood. 122: 3632–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao D., and Podrez E. A.. 2018. Characterization of covalent modifications of HDL apoproteins by endogenous oxidized phospholipids. Free Radic. Biol. Med. 115: 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pennathur S., Berg C., Shao B., Byun J., Kassim S. Y., Singh P., Green P. S., McDonald T. O., Brunzell J., Chait A., et al. 2004. Human atherosclerotic intima and blood of patients with estblished coronary artery disease contain high density lipoprotein damaged by reactive nitrogen species. J. Biol. Chem. 279: 42977–42983. [DOI] [PubMed] [Google Scholar]
- 50.DiDonato J. A., Huang Y., Aulak K. S., Even-Or O., Gerstenecker G., Gogonea V., Wu Y., Fox P. L., Tang W. H. W., Plow W. F., et al. 2013. Function and Distribution of apolipoprotein A1 in the artery wall are markedly distinct from those in plasma. Circulation. 128: 1644–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kakino A., Usami Y., Horiuchi S., Fujita Y., Kotani K., Chen C. H., Okamura T., and Sawamura T.. 2019. A novel cell-free, non-fluorescent method to measure LOX-1-binding activity corresponding to the functional activity of HDL. J. Atheroscler. Thromb. 26: 947–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sorokin A. V., Kotani K., Elnabawi Y. A., Dey A. K., Sajja A. P., Yamada S., Ueda M., Harrington C. L., Baumer Y., Rodante J. A., et al. 2018. Association between oxidation-modified lipoproteins and coronary plaque in psoriasis. Circ. Res. 123: 1244–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Itabe H., Suzuki K., Tsukamoto Y., Komatsu R., Ueda M., Mori M., Higashi Y., and Takano T.. 2000. Lysosomal accumulation of oxidized phosphatidylcholine-apolipoprotein B complex in macrophages: intracellular fate of oxidized low density lipoprotein. Biochim. Biophys. Acta. 1487: 233–245. [DOI] [PubMed] [Google Scholar]
- 54.Miller Y. I., Viriyakosol S., Worrall D. S., Boullier A., Butler S., and Witztum J. L.. 2005. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler. Thromb. Vasc. Biol. 25: 1213–1219. [DOI] [PubMed] [Google Scholar]
- 55.Van Berkel T. J., De Rijke Y. B., and Kruijt J. K.. 1991. Different fate in vivo of oxidatively modified low density lipoprotein and acetylated low density lipoprotein in rats. Recognition by various scavenger receptors on Kupffer and endothelial liver cells. J. Biol. Chem. 266: 2282–2289. [PubMed] [Google Scholar]
- 56.Emert B., Hasin-Brumshtein Y., Springstead J. R., Vakili L., Berliner J. A., and Lusis A. J.. 2014. HDL inhibits the effects of oxidized phospholipids on endothelial cell gene expression via multiple mechanisms. J. Lipid Res. 55: 1678–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rasmiena A. A., Barlow C. K., Ng T. W., Tull D., and Meikle P. J.. 2016. High density lipoprotein efficiently accepts surface but not internal oxidised lipids from oxidised low density lipoprotein. Biochim. Biophys. Acta. 1861: 69–77. [DOI] [PubMed] [Google Scholar]
- 58.Anderson M. M., Hazen S. L., Hsu F. F., and Heinecke J. W.. 1997. Human neutrophils employ the myeloperoxidase-hydrogen peroxide-chloride system to convert hydroxy-amino acids into glycolaldehyde, 2-hydroxypropanol, and acrolein. J. Clin. Invest. 99: 424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li D., Zhao L., Yu J., Zhang W., Du R., Liu X., Liu Y., Chen Y., Zeng R., Cao Y., et al. 2017. Lipoprotein-associated phospholipase A2 in coronary heart disease: review and meta-analysis. Clin. Chim. Acta, 465: 22–29. [DOI] [PubMed] [Google Scholar]
- 60.Nestel P. J., Barnes E. H., Tonkin A. M., Simes J., Fournier M., White H. D., Colquhoun D. M., Blankenberg S., and Sullivan D. R.. 2013. Plasma lipoprotein (a) concentration predicts future coronary and cardiovascular events in patients with stable coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 33: 2902–2908. [DOI] [PubMed] [Google Scholar]
- 61.Tsimikas S., Lau H. K., Han K. R., Shortal B., Miller E., Segev A., Curtiss L. K., Witztum J. L., and Strauss B. H.. 2004. Percutaneous coronary intervention results in acute increases in oxidized phospholipids and lipoprotein (a): short-term and long-term immunologic responses to oxidized low-density lipoprotein. Circulation. 109: 3164–3170. [DOI] [PubMed] [Google Scholar]
- 62.Schmidt K., Noureen A., Kronenberg F., and Utermann G.. 2016. Structure, function, and geneteics of lipoporotein (a). J. Lipid Res. 57: 1339–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Akyol S., Lu J., Akyol O., Akcay F., Armutcu F., Ke L. Y., and Chen C. H.. 2017. The role of electronegative low-density lipoprotein in cardiovascular diseases and its therapeutic implications. Trends Cardiovasc. Med. 27: 239–246. [DOI] [PubMed] [Google Scholar]
- 64.Kido T., Kurata H., Kondo K., Itakura H., Okazaki M., Urata T., and Yokoyama S.. 2016. Bioinformatic analysis of plasma apolipoproteins A-I and A-II revealed unique features of A-I/A-II HDL particles in human plasma. Sci. Rep. 6: 31532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hirano T., Ito Y., Koba S., Toyoda M., Ikejiri A., Saegusa H., Yamazaki J., and Yoshino G.. 2004. Clinical significance of small dense low-density lipoprotein cholesterol levels determined by the simple precipitation method. Arterioscler. Thromb. Vasc. Biol. 24: 558–563. [DOI] [PubMed] [Google Scholar]
- 66.Nishikura T., Koba S., Yokota Y., Hirano T., Tsunoda F., Shoji M., Hamazaki Y., Suzuki H., Itoh Y., Katagiri T., et al. 2014. Elevated small dense low-density lipoprotein cholesterol as a predictor for future cardiovascular events in patients with stable coronary artery disease. J. Atheroscler. Thromb. 21: 755–767. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original proteome datasets together with the list of identified peptides were deposited to JPOST (https://repository.jpostdb.org/entry/JPST000758). All other data is contained in the article.