

Abstract
Survival for patients with newly diagnosed cervical cancer has not significantly improved over the past several decades. We sought to identify a clinically relevant set of prognostic genes for squamous cell carcinoma of the cervix (SCCC), the most common cervical cancer subtype. Using RNA-sequencing data and survival data from 203 patients in The Cancer Genome Atlas (TCGA), we conducted a series of analyses using different decile cutoffs for gene expression to identify genes that could indicate large and consistent survival differences across different decile cutoffs of gene expression. Those analyses identified 42 high-risk genes. A patient’s survivability could be estimated by simply counting the number of high-risk genes with extremely high expression (above the 90th percentile) or estimating a transcriptomic risk score (TRS) using a machine learning algorithm with 9 of the 42 genes. On multivariate analysis, the significant predictors of mortality included high TRS (HR = 44.8), stage IV (HR = 28.1), intermediate TRS (HR = 4.75), and positive lymph node status (HR = 2.92). Approximately 18% of earlier-stage patients were identified as a poor-prognosis subgroup with high TRS. In patients with SCCC, transcriptomic risk appears to better predict survival than clinical prognostic factors, including stage.
Keywords: Cervical cancer, squamous cell carcinoma, gene expression, RNA, TCGA, transcriptomic risk, prognostic biomarker
Introduction
Worldwide, cervical cancer is the most common and deadliest gynecologic malignancy, accounting for an estimated 570,000 new cases and 311,000 deaths each year [1]. Despite efforts in screening and human papillomavirus (HPV) vaccine adoption, cervical cancer remains a persistent health challenge for women in the United States, with 13,170 new cases and 4,250 deaths estimated for 2019 [2]. Survival for women with cervical cancer has not significantly improved since the mid-1970s, in contrast to the majority of other common cancers in the United States [3]. While early-stage cervical cancer can be successfully treated, with 5-year overall survival (OS) rates as high as 97%, metastatic cervical cancer is virtually incurable, with 5-year OS rates below 10% [4]. For patients with recurrent cervical cancer, their prognosis remains poor. The mortality risk for metastatic or recurrent cervical cancer is high, with median OS remaining limited to less than 1.5 years, even with the 3.5 month gain in median OS shown in GOG 240 by adding bevacizumab to first-line systemic platinum-based combination chemotherapy [5,6]. Therefore, new approaches are needed to better identify and treat patients with cervical cancer at high risk of recurrence and death.
A major focus in improving systemic treatment of cervical cancer involves developing a better understanding of the genomic, transcriptomic, and proteomic underpinnings and heterogeneity of the disease. The central tenet in the pathogenesis of cervical cancer is the involvement of HPV, which can be found in up to 99.7% of cervical cancers [7]. Despite the near-universal contribution of HPV to cervical carcinogenesis, there is wide variance in the risk of cancer associated with the different types of carcinogenic HPV, as well as the association of types of carcinogenic HPV with the different histologic subtypes (squamous cell carcinoma and adenocarcinoma) of cervical cancer [8].
To further advance the molecular understanding of cervical cancer, The Cancer Genome Atlas (TCGA) project recently published their analysis of 228 primary cervical cancers [9]. While the results from that study noted a number of novel molecular features, the integrated clustering, which identified 3 main subgroups (keratin-low squamous, keratin-high squamous, adenocarcinoma), was not based on patient outcomes such as survival. A proteomic grouping was associated with differences in survival, but that grouping was (a) not primarily based on patient outcomes and (b) used as a small component of the integrative clustering that resulted in the featured novel subgroups (of note, the prognostic value of the proteomic grouping was recently validated by a separate group and dataset [10]). Further, no data was reported by TCGA to show that differences in the main novel cervical cancer subgroups were associated with differences in clinically relevant outcomes. Several other studies have investigated the genomic contributions to differences in clinical outcomes in cervical cancer, but outcomes were typically not a starting point in those studies, and their sample sizes were much smaller than TCGA [11-14]. Other groups have evaluated the potential of micro-RNA signatures for use as prognostic biomarkers, but results have been mixed and the most promising of those signatures did not validate [15-17]. Further, it is unclear whether the findings in above studies were confounded by fundamental differences between the 2 major histologic subtypes of cervical cancer (squamous cell vs. adenocarcinoma), which arise from separate sites of the cervix and have different molecular profiles [14].
The objective of this study was to conduct an outcome-based transcriptomic analysis of squamous cell carcinoma of the cervix (SCCC), the most common histologic subtype of cervical cancer, using TCGA cervical SCC dataset. Our goal was to identify sets of genes that can identify subgroups with large and clinically meaningful survival differences.
Materials and methods
TCGA cervical squamous cell carcinoma (SCC) dataset
The RNAseq data (IlluminaHiSeq: log2-normalized_count+1) for SCCC from TCGA was downloaded from UCSC Xena [18]. The details regarding the clinical characteristics of this dataset are available in a recent publication from TCGA [9]. We used TCGA dataset for this study because it has the largest number of patients and the highest quality gene expression data of any publicly available dataset of patients with cervical cancer. Given the inherent molecular differences between the 2 histologic subtypes of cervical cancer, we chose to focus this analysis on SCC. Our rationale was that SCC is the most common cervical cancer subtype and there were far more patient-derived samples for SCC than for adenocarcinoma in TCGA cervical cancer dataset. RNA-seq data for a total of 20,530 genes was available for each patient sample analyzed in this study. Samples were included in this study if they were SCCC and had both RNAseq and OS data available. Accordingly, samples were excluded from the study if they (a) did not contain SCC, (b) contained SCC but were mixed with another histologic subtype (e.g., a mixed SCC and adenocarcinoma tumor), (c) did not contain RNA-seq data, or (d) did not contain OS data.
A total of 203 patients with SCCC met inclusion criteria for this analysis. Median age of the sampled population was 47 years. Median follow-up was 27.3 months. Stage distribution was as follows: I (102; 50.2%), II (50; 24.6%), III (32; 15.8%), IV (14; 6.9%), unknown (5; 2.5%). As of last follow-up, 143 (70.4%) of patients were alive, and 60 (29.6%) had died.
Statistical analyses
All statistical analyses were performed using the R language and environment for statistical computing (R version 3.2.2; R Foundation for Statistical Computing; www.r-project.org). We used Cox proportional hazards models to evaluate the impact of gene expression levels on overall survival. Overall survival data (diagnosis to date of death) were downloaded from TCGA patient phenotype files. Patients who were alive were censored at the date of last follow-up visit. Kaplan-Meier survival analysis and log-rank test were used to compare differences in overall survival between groups classified using different cut-offs of expression level.
Identification of survival-associated genes
We initially examined survival differences associated with each gene using 10 different cutoffs corresponding to each decile. For example for the 90% cutoff, the top 10% of patients with the highest expression levels for a given gene were assigned to a “high expression” group and the bottom 90% of patients are assigned to a “low expression” group and the two groups of patients were analyzed using a univariate Cox regression analysis. Similarly, the top 80% of patients with the highest expression could also be compared to the remaining 20% of patients. For individual genes, the difference in survival for above and below the cut-off was assessed using hazard ratio (HR) and log-rank test, with a significance level of P < 0.01. This process was repeated for each gene and at each cutoff.
We employed a 2-step selection procedure to identify genes with large survival differences that may be generalizable to other datasets. First, we identified genes with large survival differences at every decile cutoff. In the second step, we eliminated genes that were not consistently associated with survival at other cutoffs. This procedure allowed identification of genes that had large survival differences and could consistently predict survival at different cutoffs.
To accomplish these goals, survival analysis was systematically conducted for every gene and at every decile cutoff. Examination of the results suggested that larger survival differences were usually observed at the 90% and 10% cutoffs, although survival differences were also seen at the middle cutoffs for many genes. We therefore focused our subsequent attention on genes identified using the 90th and 10th percentile cutoffs. At the 90th percentile cutoff, the 10% of patients with the highest gene expression levels for each gene were compared to the bottom 90% of the patients with lower expression. At the 10th percentile cutoff, for each gene, the 90% of patients with higher expression levels were compared to the 10% of patients with the lowest expression levels.
Building the SCCC gene signature and TRS stratifier
The individual genes with high survival differences were used to construct a survival prediction model using a machine learning method. The least absolute shrinkage selection operator (LASSO) algorithm was used to select and fit the regression coefficients for each gene in a penalized Cox proportional hazard model [19,20]. This process allowed us to select a subset of the genes, with weighted expression values, to use in calculating a survival risk score for each patient. The risk scores were then used to stratify all patients into 3 transcriptomic risk score (TRS) groups. The stratification was optimized using the log-rank test. For the univariate analysis, major clinical characteristics with prognostic relevance were fitted to a Cox regression model after removal of patients with unknown clinical information. All clinical variables that were significant on univariate analysis (stage and lymph node status) were combined with TRS for the multivariate Cox model. Although LASSO is capable of selecting genes, it is not possible to apply LASSO to the entire genomic dataset with over 20,000 genes and come up with the best model. Therefore, our approach of pre-selecting genes using unigene survival analyses and then fitting a LASSO model represents a practical and efficient way of developing multivariate models.
Results
Identification of genes associated with poor survival
Using selection criteria of HR > 3.5 and p-values < 0.01, 41 genes had good survival prediction potential as shown by the Kaplan-Meier survival curves for representative genes (Figure 1). The HR for these 41 selected genes ranged from 3.5 to 5.2, while the p-values were all less than 10-5 except for 1 gene which had a value of 1.6×10-4, suggesting highly significant associations between extremely high expression of the 41 genes and poor survival of SCCC patients.
Figure 1.
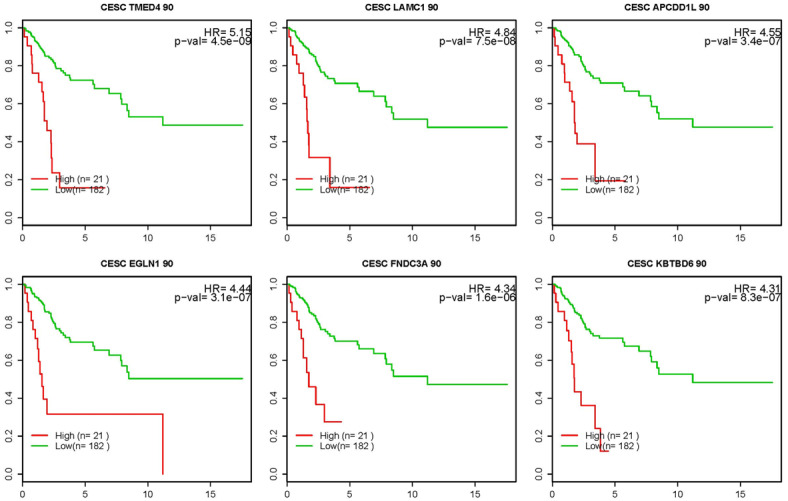
Representative Kaplan-Meier survival curves (6 shown) for the top 41 high-risk genes for which patients above the 90th percentile (or in the top 10%) for expression level had a HR > 3.5 for death and p-value of < 0.01. X-axis: time (years); Y-axis: survival probability.
When the 41 genes were assessed for survival prediction at every decile cutoff for patients with high vs. low expression, the choice of the 90th percentile as the cutoff point was confirmed. As shown by the Kaplan-Meier survival curves in Figure 2A, the survival prognosis worsened with increasing expression levels, with the best survival usually seen at the first decile and worst survival seen at the tenth decile. These observations provided further confidence that the 41 genes selected using the 90th percentile cutoff may be excellent survival prediction genes.
Figure 2.

A. Representative survival curves (6 shown) across different expression-level deciles for the 41 high-risk genes. For each gene, patients were divided into deciles based on expression level. Patients in the middle deciles were grouped together as they showed similar survival. X-axis: time (years); Y-axis: survival probability. G1 = first decile, G2 = second decile, G3-5 = 3rd-5th decile groups combined. G6-8 = 6th-8th deciles combined. G9 = 9th decile, G10 = 10th decile. B. Survival curves for patients based on decile level of expression for SDF4.
There were some notable findings from examining, for each patient, the number of the 41 high-risk genes for which they were above the 90th percentile cutoff for expression level. Interestingly, some patients had many of the high-risk genes above the cutoff, while some patients had no high-risk genes above the cutoff (Figure 3A). The patients who died mostly had large numbers of high-risk genes, while patients who survived in the follow-up period had lower numbers of genes above the cutoff. Further, none of the patients with zero highrisk genes above cutoff had died (Figure 3A). Survival was then compared between 3 groups of patients, with 0-1 gene (group 1), 2-4 genes (group 2) and ≥ 5 genes (group 3) above the 90th percentile cutoff, respectively (Figure 3B). As expected, group 3 has the worst survival (HR = 17.2, median survival = 1.7 years) and group 2 has intermediate survival (HR = 3.5, median survival = 7.9 years) compared to group 1 as reference (median survival not yet reached). These data suggest that the survival outcome is in part determined by the load of high-risk gene expression in a patient with SCCC.
Figure 3.

Additive model for transcriptomic risk. A. Heatmap showing, for each patient, the number the 41 high-risk/high-expression genes for which they were in the top 10% for expression level. Each gene with top 10% expression level is marked by a blue bar. Top bar of x-axis: blue = alive; red = deceased. Each patient identifier is listed along the bottom of the x-axis. B. Survival curves based on the number of the 41 high-risk genes for which a patient was above the 90th percentile cutoff for level of expression. Group 1 (G1) = 0-1; G2 = 2-4; G3 = 5+genes.
The functions of 41 high-risk genes were evaluated by pathway analysis supplemented by manual curation. Fifteen of the 41 genes (ANGPTL4, FNDC3A, GALNT2, GALNT3, GLG1, KBTBD6, LAMC1, LIF, MMS19, MTDH, NRP1, PFKP, PLOD1, QSOX1, ZNF281) are implicated in metastasis, migration and/or invasion [21-40]; 11 genes (ANGPTL4, APCDD1L, COPA, FNDC3A, GALNT3, KBTBD6, LIF, MTDH, NRP1, PLAGL1, RPS6KA2) in cell proliferation [22,25,27,32,34,35,41-45]; 4 genes (CD46, CD59, KBTBD2, NRP1) in immune suppression [46-50]; and 3 genes (GRB10, NRP1, PEAR1) in angiogenesis [43,51,52]. The functions of the genes are consistent with their association with poor survival as observed in this study.
Similar analyses were carried out using the 10th percentile cutoff for each gene. There were 319 genes with HR > 3.5 and P < 0.05. Of those 319 genes, 9 appeared to be very interesting in that they were associated with very large HR values because no patient with expression below the 10th percentile was dead. Lowering the cutoff to the 8th percentile identified 25 additional genes with similar pattern. To further assess the potential utility of these 34 genes, of which 2 were also identified by the 90th percentile, we examined the survival in each of the 10 deciles. The survival differences were not consistent across different decile cutoffs for all genes, except for SDF4. Similar to the patterns observed for the 41 genes identified from the 90th percentile cutoff, survival worsened as SDF4 expression level increased (Figure 2B). SDF4 encodes a calcium-binding protein and is mutated in prostate cancer and overexpressed in pancreatic cancer [53,54]. For these reasons, of the low-percentile cutoff genes, only SDF4 was of interest to further pursue.
Transcriptomic risk score (TRS) using machine learning
Using the 41 genes identified through the 90th percentile analysis plus SDF4, a LASSO algorithm was used to identify the gene signature optimized for predicting survival prognosis among those 42 genes. The best model uses 9 genes (TMED4, EGLN1, PLOD1, PEAR1, DKFZP434L187, TM2D1, SLN, CD46, and SDF4), of which 8 are from the 90th percentile and 1 (SDF4) is from the 8th percentile. These 9 genes can be used to calculate a transcriptomic risk score (TRS) for mortality. When the TRS was applied to TCGA SCCC patient population, patients were stratified into 3 TRS groups (Figure 4A): low (n = 41), intermediate (n = 121), and high (n = 41). In the follow-up period, patients who died included 2 (4.9%) in the lowrisk TRS group, 26 (21.5%) in the intermediate-risk TRS group, and 32 (78.0%) in the high-risk TRS group.
Figure 4.

A. Survival curves stratified by TRS group based on the weighted expression of 9 genes identified by the LASSO machine learning algorithm. The genes are: TMED4, EGLN1, PLOD1, PEAR1, DKFZP434L187, TM2D1, SLN, CD46, and SDF4. The TRS groups are: low (n = 41), intermediate (n = 121), and high (n = 41). The cutoffs of 20th and 80th percentile for the three risk groups were arbitrary but determined before the statistical test. These cutoffs were selected to identify subjects with most discrepant survival. B. Survival curves for the TRS groups when patients with stage IV disease were excluded. C. Survival curves for the major clinical oncologic variables for SCCC. X-axes = time (years).
Univariate and multivariate analyses with TRS and clinical parameters
Clinical characteristics of the TRS groups identified within TCGA SCCC population can be found in Table 1. Across the TRS groups, median age was similar, the most patients were stage I, the majority of patients with known lymph node status had negative lymph nodes, and grades 2-3 were similarly represented. Median overall survival was 1.56 years for the high-risk TRS group, 8.48 years for the intermediate-risk TRS group, and not yet reached for the low-risk TRS group.
Table 1.
Clinical characteristics of patients in the low, intermediate, and high transcriptomic risk groups
| Characteristic | Transcriptomic risk group | ||
|---|---|---|---|
|
| |||
| Low (n = 41) | Intermediate (n = 121) | High (n = 41) | |
| Age at diagnosis, years | |||
| Median | 47 | 45 | 48 |
| Range | 28-80 | 21-79 | 20-79 |
| Stage, n (%) | |||
| I | 26 (25.5) | 58 (56.9) | 18 (17.6) |
| II | 6 (12.0) | 35 (70.0) | 9 (18.0) |
| III | 7 (21.9) | 17 (53.1) | 8 (25.0) |
| IV | 0 (0) | 8 (57.1) | 6 (42.9) |
| Unknown | 2 (40.0) | 3 (60.0) | 0 (0) |
| LN status, n (%) | |||
| Negative | 18 (23.4) | 47 (61.0) | 12 (15.6) |
| Positive | 12 (29.3) | 24 (58.5) | 5 (12.2) |
| Unknown | 11 (12.9) | 50 (58.8) | 24 (28.2) |
| OS, years | |||
| Median | NYR | 8.48 | 1.56 |
| Grade, n (%) | |||
| 1 | 4 (40.0) | 5 (50.0) | 1 (10.0) |
| 2 | 22 (22.9) | 56 (58.3) | 18 (18.8) |
| 3 | 13 (17.1) | 48 (63.2) | 15 (19.7) |
| 4 | 0 (0) | 1 (100) | 0 (0) |
| Unknown | 2 (10.0) | 11 (55.0) | 7 (35.0) |
NYR, not yet reached.
Stage-by-stage distribution of the TRS groups can also be found in Table 1. Two observations from this part of the analysis are worth highlighting. First, approximately 18% (17.6% for stage I and 18.0% for stage II) of earlier-stage patients were a poor-survival subgroup with high TRS. Second, 21.9% of stage III patients had low TRS, belonging to the good-survival TRS group.
Univariate analysis of major clinical variables for SCCC found that stage and lymph node status were each significantly associated with survival, but grade was not (Figure 4C and Table 2). Stage IV patients had very poor survival, while survival was not significantly different between stage I, II and III patients. On univariate analysis, the high-risk TRS group was 43.7 times more likely to die compared to the low-risk TRS group (HR = 43.7, P < 0.001). The intermediate-risk TRS group had a HR of 4.94 compared to the low risk group (P = 0.03).
Table 2.
Hazard ratios for TRS and major clinical factors
| Characteristic | Univariate analysis | Multivariate analysis | ||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| HR | 95% CI | P | HR | 95% CI | P | |
| Transcriptomic risk | ||||||
| Low | Ref | Ref | Ref | Ref | Ref | Ref |
| Intermediate | 4.94 | 1.16 to 21.1 | 0.03 | 4.75 | 1.07 to 21.1 | 0.04 |
| High | 43.7 | 10.2 to 186 | < 0.001 | 44.8 | 8.62 to 233 | < 0.001 |
| Stage | ||||||
| I | Ref | Ref | Ref | Ref | Ref | Ref |
| II | 0.66 | 0.28 to 1.52 | 0.33 | 0.36 | 0.0841 to 1.56 | 0.17 |
| III | 1.23 | 0.58 to 2.63 | 0.59 | 0.42 | 0.0547 to 3.27 | 0.41 |
| IV | 6.22 | 2.89 to 13.4 | < 0.001 | 28.1 | 3.73 to 212 | 0.001 |
| Grade | ||||||
| 1 | Ref | Ref | Ref | |||
| 2 | 1.06 | 0.25 to 4.47 | 0.94 | Not included | ||
| 3 | 1.01 | 0.23 to 4.34 | 0.99 | |||
| Lymph Node Status | ||||||
| Negative | Ref | Ref | Ref | Ref | Ref | Ref |
| Positive | 2.16 | 1.03 to 4.55 | 0.043 | 2.92 | 1.32 to 6.44 | 0.008 |
Given that stage was the most significant clinical factor associated with survival and the high TRS and stage IV survival curves appeared similar, survival analysis was further carried out on stage I-III patients stratified by TRS (Figure 4B). The TRS-stratified survival pattern for stage I-III patients was almost identical to that observed with stage IV patients, confirming that TRS-based survival differences were not confounded by stage. In addition, multivariate analysis using TRS as the dependent variable and clinical variables that were significant on univariate analysis as co-variables revealed high TRS as the most important survival predictor (HR 44.8; 95% CI, 8.62 to 233; P < 0.001). The next most important survival predictor was stage IV (HR 28.1; 95% CI, 3.73 to 212; P < 0.001), followed by intermediate TRS (HR 4.75; 95% CI, 1.07 to 21.1; P = 0.04), and positive lymph node status (HR 2.92; 95% CI, 1.32 to 6.44; P = 0.008).
Discussion
This study identified 42 genes that individually predict SCCC patient survival. The majority of identified genes have been associated with key cancer hallmarks such as cellular proliferation, migration/invasion, and/or metastasis. The survival prognosis appears to be influenced not only by the expression level of each high-risk gene but also the number of the genes with the highest expression levels. Survival gradually worsened as expression level of the 42 genes increased. Poorest survival was observed in patients with highest expression for 5 or more genes; best survival was observed in patients with 0-1 genes with highest expression. These results suggest that the risk for dying of SCCC is determined by the patient’s transcriptomic risk burden.
Machine learning identified a 9-gene signature that appears sufficiently accurate to predict survival. A transcriptomic risk score for mortality can be computed with the 9 genes utilized to stratify patients into low, intermediate, and high TRS groups. Based on our analyses of TCGA SCCC patient population, while stage IV was a very good predictor for poor survival, TRS was not entirely confounded by stage or any other clinical variables. Indeed, multivariate analyses using TRS and the prognostically significant clinical parameters for SCCC demonstrated that TRS was by far a better survival predictor than stage. And even in a patient population that did not have a significant survival difference among stages I-III, TRS could identify patients at high, intermediate, and low risk of mortality.
In current clinical practice, there is no prognostic biomarker for cervical cancer. Factors that inform adjuvant treatment for early cervical cancer include: a risk stratification based on stromal invasion, lymphovascular space invasion, and tumor diameter [55] (intermediate-risk disease: give pelvic radiotherapy); criteria for high-risk of recurrence and death (positive margins, positive lymph nodes, parametrial involvement) that merit chemoradiation [56]. For locally advanced cervical cancer, chemoradiation is standard of care; the benefit of additional chemotherapy given after chemoradiation is currently under investigation (NCT01414608). Stage and lymph node status can influence treatment planning for cervical cancer, but those factors may miss some patients at high risk for mortality.
Data from this study raises the concern that early stage may underestimate mortality in some patients, as approximately 18% of stage I and II patients in the studied TCGA SCCC population were high TRS and poor survivors. Given our study’s finding that TRS appears to outperform stage and lymph node status as a prognostic variable, it warrants further investigation as a biomarker for SCCC. Such would be especially important to a poor-prognosis subgroup of earlier-stage SCCC patients with high TRS, who might be under-treated relative to their prognosis based on clinical factors alone.
Another important observation was that 21.9% of stage III SCCC had low TRS associated with good survival, which would suggest that a subset of stage III SCCC patients may have an overestimation of mortality risk with clinical factors alone. Further investigation in more patients would be needed to confirm the presence and degree of prognosis-modifying impact of low TRS in patients with stage III SCCC. However, the finding of 2 within-stage TRS subgroups prognostically different than expected based on stage alone strongly suggests that TRS is not completely confounded by stage.
This work also provides a novel perspective on gene expression in SCCC with respect to survival. Our approach is quite different from prior studies in several respects: clinical outcomes were not a starting point in those studies, sample sizes were much smaller than TCGA, analysis was limited to specific gene types (e.g., micro-RNAs), and/or the inclusion of both major histologic subtypes may have confounded the genomic analyses [9,11-17]. In contrast, our study leveraged the relatively high number of SCCC patients with both gene expression and survival data and avoided the pitfalls of grouping multiple histologic subtypes into a single -omic analysis. Further, we conducted an analysis through the lens of clinical relevance (i.e., who survived and who died?). While our finding of a transcriptomic risk gene signature for SCCC has not yet been validated with a separate data set, a strength of this study is its focus on genes showing large and consistent survival differences at multiple cutoffs. Such genes are more likely to be validated in other datasets and be clinically relevant.
In conclusion, this study identified 42 genes highly associated with survival in SCCC. Among TCGA SCCC patients analyzed, survival prognosis worsened with (a) increasing expression level for each individual high-risk gene or (b) a greater number of those genes with high expression level in a patient’s tumor. These findings suggest the importance of the transcriptomic risk load on survival. Furthermore, a patient’s survivability can be estimated by simply counting the number of genes with extremely high expression or with a machine learning-based 9-gene signature. Both methods appear to have better prognostic ability than any reported prognostic marker for SCCC, including stage and lymph node status. Although the clinical application of these discoveries will require validation in other datasets, our study provides a roadmap towards a clinically meaningful prognostic biomarker for SCCC.
Acknowledgements
JJW was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development under award number K12HD085817. JXS was partly supported by the Georgia Research Alliance Academy as an eminent scholar. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure of conflict of interest
None.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Ward EM, Johnson CJ, Cronin KA, Ma J, Ryerson B, Mariotto A, Lake AJ, Wilson R, Sherman RL, Anderson RN, Henley SJ, Kohler BA, Penberthy L, Feuer EJ, Weir HK. Annual report to the nation on the status of cancer, 1975-2014, featuring survival. J Natl Cancer Inst. 2017;109 doi: 10.1093/jnci/djx030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quinn MA, Benedet JL, Odicino F, Maisonneuve P, Beller U, Creasman WT, Heintz AP, Ngan HY, Pecorelli S. Carcinoma of the cervix uteri. FIGO 26th annual report on the results of treatment in gynecological cancer. Int J Gynaecol Obstet. 2006;95(Suppl 1):S43–103. doi: 10.1016/S0020-7292(06)60030-1. [DOI] [PubMed] [Google Scholar]
- 5.Tewari KS, Sill MW, Long HJ 3rd, Penson RT, Huang H, Ramondetta LM, Landrum LM, Oaknin A, Reid TJ, Leitao MM, Michael HE, Monk BJ. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370:734–743. doi: 10.1056/NEJMoa1309748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tewari KS, Sill MW, Penson RT, Huang H, Ramondetta LM, Landrum LM, Oaknin A, Reid TJ, Leitao MM, Michael HE, DiSaia PJ, Copeland LJ, Creasman WT, Stehman FB, Brady MF, Burger RA, Thigpen JT, Birrer MJ, Waggoner SE, Moore DH, Look KY, Koh WJ, Monk BJ. Bevacizumab for advanced cervical cancer: final overall survival and adverse event analysis of a randomised, controlled, open-label, phase 3 trial (gynecologic oncology group 240) Lancet. 2017;390:1654–1663. doi: 10.1016/S0140-6736(17)31607-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 8.Li N, Franceschi S, Howell-Jones R, Snijders PJ, Clifford GM. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: variation by geographical region, histological type and year of publication. Int J Cancer. 2011;128:927–935. doi: 10.1002/ijc.25396. [DOI] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543:378–384. doi: 10.1038/nature21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rader JS, Pan A, Corbin B, Iden M, Lu Y, Vellano CP, Akbani R, Mills GB, Simpson P. Identification and validation of a prognostic proteomic signature for cervical cancer. Gynecol Oncol. 2019;155:324–330. doi: 10.1016/j.ygyno.2019.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barron EV, Roman-Bassaure E, Sanchez-Sandoval AL, Espinosa AM, Guardado-Estrada M, Medina I, Juarez E, Alfaro A, Bermudez M, Zamora R, Garcia-Ruiz C, Gomora JC, Kofman S, Perez-Armendariz EM, Berumen J. CDKN3 mRNA as a biomarker for survival and therapeutic target in cervical cancer. PLoS One. 2015;10:e0137397. doi: 10.1371/journal.pone.0137397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Espinosa AM, Alfaro A, Roman-Basaure E, Guardado-Estrada M, Palma I, Serralde C, Medina I, Juarez E, Bermudez M, Marquez E, Borges-Ibanez M, Munoz-Cortez S, Alcantara-Vazquez A, Alonso P, Curiel-Valdez J, Kofman S, Villegas N, Berumen J. Mitosis is a source of potential markers for screening and survival and therapeutic targets in cervical cancer. PLoS One. 2013;8:e55975. doi: 10.1371/journal.pone.0055975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Medina-Martinez I, Barron V, Roman-Bassaure E, Juarez-Torres E, Guardado-Estrada M, Espinosa AM, Bermudez M, Fernandez F, Venegas-Vega C, Orozco L, Zenteno E, Kofman S, Berumen J. Impact of gene dosage on gene expression, biological processes and survival in cervical cancer: a genome-wide follow-up study. PLoS One. 2014;9:e97842. doi: 10.1371/journal.pone.0097842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wright AA, Howitt BE, Myers AP, Dahlberg SE, Palescandolo E, Van Hummelen P, MacConaill LE, Shoni M, Wagle N, Jones RT, Quick CM, Laury A, Katz IT, Hahn WC, Matulonis UA, Hirsch MS. Oncogenic mutations in cervical cancer: genomic differences between adenocarcinomas and squamous cell carcinomas of the cervix. Cancer. 2013;119:3776–3783. doi: 10.1002/cncr.28288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.How C, Pintilie M, Bruce JP, Hui AB, Clarke BA, Wong P, Yin S, Yan R, Waggott D, Boutros PC, Fyles A, Hedley DW, Hill RP, Milosevic M, Liu FF. Developing a prognostic micro-RNA signature for human cervical carcinoma. PLoS One. 2015;10:e0123946. doi: 10.1371/journal.pone.0123946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu B, Ding JF, Luo J, Lu L, Yang F, Tan XD. Seven protective miRNA signatures for prognosis of cervical cancer. Oncotarget. 2016;7:56690–56698. doi: 10.18632/oncotarget.10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeng Y, Wang KX, Xu H, Hong Y. Integrative miRNA analysis identifies hsa-miR-3154, hsa-miR-7-3, and hsa-miR-600 as potential prognostic biomarker for cervical cancer. J Cell Biochem. 2018;119:1558–1566. doi: 10.1002/jcb.26315. [DOI] [PubMed] [Google Scholar]
- 18.Goldman M, Craft B, Hastie M, Repečka K, Kamath A, McDade F, Rogers D, Brooks AN, Zhu J, Haussler D. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv. 2019:326470. [Google Scholar]
- 19.Simon N, Friedman J, Hastie T, Tibshirani R. Regularization paths for Cox’s proportional hazards model via coordinate descent. J Stat Softw. 2011;39:1–13. doi: 10.18637/jss.v039.i05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 21.Liao YH, Chiang KH, Shieh JM, Huang CR, Shen CJ, Huang WC, Chen BK. Epidermal growth factor-induced ANGPTL4 enhances anoikis resistance and tumour metastasis in head and neck squamous cell carcinoma. Oncogene. 2017;36:2228–2242. doi: 10.1038/onc.2016.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan MJ, Teo Z, Sng MK, Zhu P, Tan NS. Emerging roles of angiopoietin-like 4 in human cancer. Mol Cancer Res. 2012;10:677–688. doi: 10.1158/1541-7786.MCR-11-0519. [DOI] [PubMed] [Google Scholar]
- 23.Shan SW, Lee DY, Deng Z, Shatseva T, Jeyapalan Z, Du WW, Zhang Y, Xuan JW, Yee SP, Siragam V, Yang BB. MicroRNA MiR-17 retards tissue growth and represses fibronectin expression. Nat Cell Biol. 2009;11:1031–1038. doi: 10.1038/ncb1917. [DOI] [PubMed] [Google Scholar]
- 24.Lin MC, Huang MJ, Liu CH, Yang TL, Huang MC. GALNT2 enhances migration and invasion of oral squamous cell carcinoma by regulating EGFR glycosylation and activity. Oral Oncol. 2014;50:478–484. doi: 10.1016/j.oraloncology.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Wang ZQ, Bachvarova M, Morin C, Plante M, Gregoire J, Renaud MC, Sebastianelli A, Bachvarov D. Role of the polypeptide N-acetylgalactosaminyltransferase 3 in ovarian cancer progression: possible implications in abnormal mucin O-glycosylation. Oncotarget. 2014;5:544–560. doi: 10.18632/oncotarget.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yasmin-Karim S, King MR, Messing EM, Lee YF. E-selectin ligand-1 controls circulating prostate cancer cell rolling/adhesion and metastasis. Oncotarget. 2014;5:12097–12110. doi: 10.18632/oncotarget.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Genau HM, Huber J, Baschieri F, Akutsu M, Dotsch V, Farhan H, Rogov V, Behrends C. CUL3-KBTBD6/KBTBD7 ubiquitin ligase cooperates with GABARAP proteins to spatially restrict TIAM1-RAC1 signaling. Mol Cell. 2015;57:995–1010. doi: 10.1016/j.molcel.2014.12.040. [DOI] [PubMed] [Google Scholar]
- 28.Lietard J, Musso O, Theret N, L’Helgoualc’h A, Campion JP, Yamada Y, Clement B. Sp1-mediated transactivation of LamC1 promoter and coordinated expression of laminin-gamma1 and Sp1 in human hepatocellular carcinomas. Am J Pathol. 1997;151:1663–1672. [PMC free article] [PubMed] [Google Scholar]
- 29.Nishikawa R, Goto Y, Kojima S, Enokida H, Chiyomaru T, Kinoshita T, Sakamoto S, Fuse M, Nakagawa M, Naya Y, Ichikawa T, Seki N. Tumor-suppressive microRNA-29s inhibit cancer cell migration and invasion via targeting LAMC1 in prostate cancer. Int J Oncol. 2014;45:401–410. doi: 10.3892/ijo.2014.2437. [DOI] [PubMed] [Google Scholar]
- 30.Pasqualini L, Bu H, Puhr M, Narisu N, Rainer J, Schlick B, Schafer G, Angelova M, Trajanoski Z, Borno ST, Schweiger MR, Fuchsberger C, Klocker H. miR-22 and miR-29a Are members of the androgen receptor cistrome modulating LAMC1 and Mcl-1 in prostate cancer. Mol Endocrinol. 2015;29:1037–1054. doi: 10.1210/me.2014-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zu C, Liu T, Zhang G. MicroRNA-506 inhibits malignancy of colorectal carcinoma cells by targeting LAMC1. Ann Clin Lab Sci. 2016;46:666–674. [PubMed] [Google Scholar]
- 32.Li X, Yang Q, Yu H, Wu L, Zhao Y, Zhang C, Yue X, Liu Z, Wu H, Haffty BG, Feng Z, Hu W. LIF promotes tumorigenesis and metastasis of breast cancer through the AKT-mTOR pathway. Oncotarget. 2014;5:788–801. doi: 10.18632/oncotarget.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang JL, Wang HY, Yang Q, Lin SY, Luo GY, Zhang R, Xu GL. Methyl-methanesulfonate sensitivity 19 expression is associated with metastasis and chemoradiotherapy response in esophageal cancer. World J Gastroenterol. 2015;21:4240–4247. doi: 10.3748/wjg.v21.i14.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi X, Wang X. The role of MTDH/AEG-1 in the progression of cancer. Int J Clin Exp Med. 2015;8:4795–4807. [PMC free article] [PubMed] [Google Scholar]
- 35.Yao Y, Gu X, Liu H, Wu G, Yuan D, Yang X, Song Y. Metadherin regulates proliferation and metastasis via actin cytoskeletal remodelling in non-small cell lung cancer. Br J Cancer. 2014;111:355–364. doi: 10.1038/bjc.2014.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang YJ, Liu XC, Du J. MiR-152 regulates metastases of non-small cell lung cancer cells by targeting neuropilin-1. Int J Clin Exp Pathol. 2015;8:14235–14240. [PMC free article] [PubMed] [Google Scholar]
- 37.Moon JS, Kim HE, Koh E, Park SH, Jin WJ, Park BW, Park SW, Kim KS. Kruppel-like factor 4 (KLF4) activates the transcription of the gene for the platelet isoform of phosphofructokinase (PFKP) in breast cancer. J Biol Chem. 2011;286:23808–23816. doi: 10.1074/jbc.M111.236737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gilkes DM, Bajpai S, Wong CC, Chaturvedi P, Hubbi ME, Wirtz D, Semenza GL. Procollagen lysyl hydroxylase 2 is essential for hypoxia-induced breast cancer metastasis. Mol Cancer Res. 2013;11:456–466. doi: 10.1158/1541-7786.MCR-12-0629. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Lake DF, Faigel DO. The emerging role of QSOX1 in cancer. Antioxid Redox Signal. 2014;21:485–496. doi: 10.1089/ars.2013.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hahn S, Hermeking H. ZNF281/ZBP-99: a new player in epithelial-mesenchymal transition, stemness, and cancer. J Mol Med (Berl) 2014;92:571–581. doi: 10.1007/s00109-014-1160-3. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi M, Fujita M, Furukawa Y, Hamamoto R, Shimokawa T, Miwa N, Ogawa M, Nakamura Y. Isolation of a novel human gene, APCDD1, as a direct target of the beta-Catenin/T-cell factor 4 complex with probable involvement in colorectal carcinogenesis. Cancer Res. 2002;62:5651–5656. [PubMed] [Google Scholar]
- 42.Sudo H, Tsuji AB, Sugyo A, Kohda M, Sogawa C, Yoshida C, Harada YN, Hino O, Saga T. Knockdown of COPA, identified by loss-of-function screen, induces apoptosis and suppresses tumor growth in mesothelioma mouse model. Genomics. 2010;95:210–216. doi: 10.1016/j.ygeno.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 43.Jubb AM, Strickland LA, Liu SD, Mak J, Schmidt M, Koeppen H. Neuropilin-1 expression in cancer and development. J Pathol. 2012;226:50–60. doi: 10.1002/path.2989. [DOI] [PubMed] [Google Scholar]
- 44.Juma AR, Damdimopoulou PE, Grommen SV, Van de Ven WJ, De Groef B. Emerging role of PLAG1 as a regulator of growth and reproduction. J Endocrinol. 2016;228:R45–56. doi: 10.1530/JOE-15-0449. [DOI] [PubMed] [Google Scholar]
- 45.Milosevic N, Kuhnemuth B, Muhlberg L, Ripka S, Griesmann H, Lolkes C, Buchholz M, Aust D, Pilarsky C, Krug S, Gress T, Michl P. Synthetic lethality screen identifies RPS6KA2 as modifier of epidermal growth factor receptor activity in pancreatic cancer. Neoplasia. 2013;15:1354–1362. doi: 10.1593/neo.131660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cui W, Zhao Y, Shan C, Kong G, Hu N, Zhang Y, Zhang S, Zhang W, Zhang Y, Zhang X, Ye L. HBXIP upregulates CD46, CD55 and CD59 through ERK1/2/NF-kappaB signaling to protect breast cancer cells from complement attack. FEBS Lett. 2012;586:766–771. doi: 10.1016/j.febslet.2012.01.039. [DOI] [PubMed] [Google Scholar]
- 47.Kesselring R, Thiel A, Pries R, Fichtner-Feigl S, Brunner S, Seidel P, Bruchhage KL, Wollenberg B. The complement receptors CD46, CD55 and CD59 are regulated by the tumour microenvironment of head and neck cancer to facilitate escape of complement attack. Eur J Cancer. 2014;50:2152–2161. doi: 10.1016/j.ejca.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 48.Chaudhary B, Khaled YS, Ammori BJ, Elkord E. Neuropilin 1: function and therapeutic potential in cancer. Cancer Immunol Immunother. 2014;63:81–99. doi: 10.1007/s00262-013-1500-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li B, Lin H, Fan J, Lan J, Zhong Y, Yang Y, Li H, Wang Z. CD59 is overexpressed in human lung cancer and regulates apoptosis of human lung cancer cells. Int J Oncol. 2013;43:850–858. doi: 10.3892/ijo.2013.2007. [DOI] [PubMed] [Google Scholar]
- 50.Dai K, Huang Y, Chen Z, Sun X, Yang L, Jiang Y. Kbtbd2 inhibits the cytotoxic activity of immortalized NK cells through down-regulating mTOR signaling in a mouse hepatocellular carcinoma model. Eur J Immunol. 2018;48:683–695. doi: 10.1002/eji.201747281. [DOI] [PubMed] [Google Scholar]
- 51.Giorgetti-Peraldi S, Murdaca J, Mas JC, Van Obberghen E. The adapter protein, Grb10, is a positive regulator of vascular endothelial growth factor signaling. Oncogene. 2001;20:3959–3968. doi: 10.1038/sj.onc.1204520. [DOI] [PubMed] [Google Scholar]
- 52.Vandenbriele C, Kauskot A, Vandersmissen I, Criel M, Geenens R, Craps S, Luttun A, Janssens S, Hoylaerts MF, Verhamme P. Platelet endothelial aggregation receptor-1: a novel modifier of neoangiogenesis. Cardiovasc Res. 2015;108:124–138. doi: 10.1093/cvr/cvv193. [DOI] [PubMed] [Google Scholar]
- 53.Kumar A, White TA, MacKenzie AP, Clegg N, Lee C, Dumpit RF, Coleman I, Ng SB, Salipante SJ, Rieder MJ, Nickerson DA, Corey E, Lange PH, Morrissey C, Vessella RL, Nelson PS, Shendure J. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc Natl Acad Sci U S A. 2011;108:17087–17092. doi: 10.1073/pnas.1108745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gronborg M, Kristiansen TZ, Iwahori A, Chang R, Reddy R, Sato N, Molina H, Jensen ON, Hruban RH, Goggins MG, Maitra A, Pandey A. Biomarker discovery from pancreatic cancer secretome using a differential proteomic approach. Mol Cell Proteomics. 2006;5:157–171. doi: 10.1074/mcp.M500178-MCP200. [DOI] [PubMed] [Google Scholar]
- 55.Sedlis A, Bundy BN, Rotman MZ, Lentz SS, Muderspach LI, Zaino RJ. A randomized trial of pelvic radiation therapy versus no further therapy in selected patients with stage IB carcinoma of the cervix after radical hysterectomy and pelvic lymphadenectomy: a gynecologic oncology group study. Gynecol Oncol. 1999;73:177–183. doi: 10.1006/gyno.1999.5387. [DOI] [PubMed] [Google Scholar]
- 56.Peters WA 3rd, Liu PY, Barrett RJ 2nd, Stock RJ, Monk BJ, Berek JS, Souhami L, Grigsby P, Gordon W Jr, Alberts DS. Concurrent chemotherapy and pelvic radiation therapy compared with pelvic radiation therapy alone as adjuvant therapy after radical surgery in high-risk early-stage cancer of the cervix. J. Clin. Oncol. 2000;18:1606–1613. doi: 10.1200/JCO.2000.18.8.1606. [DOI] [PubMed] [Google Scholar]