

Abstract
Purpose:
The potential biological determinants of aggressive prostate cancer (PCa) in African American (AA) men are unknown. Here we characterize PCa genomic alterations in the largest cohort to date of AA men with clinical follow-up for metastasis, with the aim to elucidate the key molecular drivers associated with poor prognosis in this population.
Experimental Design:
Targeted sequencing was retrospectively performed on 205 prostate tumors from AA men treated with radical prostatectomy (RP) to examine somatic genomic alterations and percent genome alteration (PGA). Cox proportional hazards analyses assessed the association of genomic alterations with risk of metastasis.
Results:
At RP, 71% (145/205) of patients had grade group ≥3 disease, and 49% (99/202) were non-organ-confined. The median PGA was 3.7% (IQR 0.9–9.4%) and differed by pathologic grade (p<0.001) and stage (p=0.02). Median follow-up was 5 years. AA men with the highest quartile of PGA had increased risks of metastasis (multivariable: HR 13.45, 95% CI 2.55–70.86, p=0.002). The most common somatic mutations were SPOP (11.2%), FOXA1 (8.3%), and TP53 (3.9%). The most common loci altered at the copy number level were CDKN1B (6.3%), CHD1 (4.4%), and PTEN (3.4%). TP53 mutations and deep deletions in CDKN1B were associated with increased risks of metastasis on multivariable analyses (TP53: HR 9.5, 95% CI 2.2–40.6, p=0.002; CDKN1B: HR 6.7, 95% CI 1.3–35.2, p=0.026).
Conclusions:
Overall, PGA, somatic TP53 mutations, and a novel finding of deep deletions in CDKN1B were associated with poor prognosis in AA men. These findings require confirmation in additional AA cohorts.
Introduction:
African American (AA) men have a 1.8-fold higher incidence of prostate cancer (PCa) than European American (EA) men and are twice as likely to die from their disease.1 Numerous studies show that AA men still exhibit worse oncologic and mortality outcomes even after controlling for socioeconomic contributors.2–3 The biological determinants of aggressive PCa in AA men are still unclear and remain an unmet need in cancer health disparities research. There is growing evidence that molecular subtypes of PCa occur at different frequencies in AA and EA men;4–9 yet these differences do not explain the adverse cancer-specific outcomes in AA men. There have been several large-scale genome sequencing studies that have identified unique molecular drivers in PCa,10–12 however these have been largely confined to men of European ancestry. The Cancer Genome Atlas (TCGA) sequencing effort contains only 14% (n=43) AA men, of whom only 17 have clinically significant cancer (grade group [GG] ≥3). Though there have been subsequent sequencing studies of AA patients,13–17 many of these have been limited in sample size, number of men with clinically significant cancer, and clinical outcome data necessary to draw prognostic associations. Thus, there remain significant gaps in our understanding of PCa genomics in men of African ancestry. Without representative genomic studies in AA men, we risk worsening the current health care disparities in PCa.18–19 Using a comprehensive panel designed to target cancer-associated genes, we performed somatic genomic sequencing of the largest cohort of AA men with radical prostatectomy (RP)-treated clinically localized PCa and long-term follow-up for biochemical recurrence (BCR) and metastasis. Our aim is to elucidate the key molecular drivers associated with poor prognosis in this population.
Materials and Methods:
Cohort description:
Institutional Review Board approval was obtained from participating institutions (Johns Hopkins Medical Institutions [JHMI] and University of Michigan) in accordance with the U.S. Common Rule and the study was conducted under a waiver of consent. The AA cohort was selected in two groups, as previously reported.4,20 First, we identified all consecutive AA men treated by RP at JHMI from 1995–2005 with available clinical follow-up. For RP GG1 (n=304 consecutive cases) and RP GG2 (n=144 consecutive cases), 50 AA men were randomly selected from each category but only those with available tissue were included in the study (n=28 GG1, n=36 GG2). All other RP GG categories (GG3–5) had less than 50 AA men treated from 1995–2005, thus all consecutive AA men in these GG categories with available tissue were included (n=48 GG3, n=26 GG4, n=14 GG5). For the second group, to increase the cohort size and also enrich for higher GG, all AA RP cases from 2006–2010 at JHMI with GG≥3 were additionally included. The final cohort included 220 self-identified AA men: 28 GG1, 36 GG2, 96 GG3, 34 GG4, and 26 GG5. All cases were reviewed by pathologists on this study during cohort selection.
DNA isolation:
For all cases, formalin-fixed paraffin embedded (FFPE) RP specimens were obtained, and 5 x 0.6 mm punches from the dominant tumor nodule (highest GG) were procured for somatic DNA extraction. DNA was extracted from FFPE material using the Qiagen AllPrep FFPE DNA/RNA Kit (Qiagen, Valencia, CA). DNA concentrations were quantified with the Qubit fluorometer, using a Quant-iT dsDNA High Sensitivity Assay Kit (Invitrogen, Carlsbad, CA). Germline DNA from normal tissue was not obtained.
Next generation sequencing and bioinformatic analysis:
We performed targeted, multiplexed PCR-based DNA next generation sequencing (NGS) as described21–22 using a custom designed expanded pan-genitourinary and other cancer (Pan-GU) panel comprised of 3,127 amplicons targeting 135 genes on all autologs and the X chromosome to facilitate genome-wide copy number profiling.22 The genes included: ABL1, ALK, APC, AR, ATM, B2M, BAP1, BCL2L1, BCL2L11, BRAF, BRCA1, BRCA2, BRWD1, CCND1, CCNE1, CD274, CDH1, CDH19, CDK12, CDK4, CDK6, CDKN1A, CDKN1B, CDKN2A, CHD1, CTNNA1, DEK, E2F3, EGFR, ERBB2, ERBB3, ERCC2, ERF, ESR1, ETS2, ETV1, FBXW7, FGFR1, FGFR2, FGFR3, FGFR4, FH, FLCN, FOXA1, FOXP1, GNAS, JAK1, JAK2, KEAP1, KIF5C, KIT, KLF3, KRAS, LSAMP, MDM2, MDM4, MECOM, MED12, MED12L, MET, MITF, MSH2, MSH6, MTOR, MYC, MYCL, MYCN, NCOR1, NF2, NKX3–1, PBRM1, PDGFRA, PIK3CA, PIK3R1, PPARG, PRKCQ, PTEN, PTPN14, PTPRM, RB1, RET, RYBP, SAV1, SCUBE1, SDHA, SDHB, SDHC, SDHD, SETD2, SMARCB1, SPOPL, STAG2, TCEB1, TFE3, TFEB, TP53, TSC1, TSC2, UBE2G2, WT1, YWHAZ, ZBTB16, ZC3H13, ZFHX3, ZMYM3, ZNF292. We used 20–24 ng of DNA per sample for library construction using the Ion Ampliseq library kit 2.0 (Life Technologies, Carlsbad, CA) with barcode incorporation and sequencing on the Ion Torrent Proton sequencer as described.21–22 Data analysis was performed as described to identify high-confidence, prioritized somatic mutations and copy number alterations (CNA) using validated pipelines based on Torrent Suite 5.0.4.0.21–24 Samples meeting the following requirements were considered for mutation and CNA calling: ≥1.5M reads, ≥70% reads on target, ≥80% uniformity, and ≥500x mean coverage depth. Samples meeting the following requirements were considered for CNA calling only: ≥1.5M reads, ≥50% reads on target, ≥80% uniformity, and ≥400x mean coverage depth. High-confidence somatic variants occurring at hotspots (>3 observations impacting that residue in COSMIC or cBioPortal) in oncogenes, inframe indels in oncogenes or tumor suppressor genes, or hotspot or deleterious (nonsense/frameshift/splice site altering) variants of tumor suppressor genes were considered driving variants.21,24 Log2 copy number ratio cutoffs to define deep deletions, shallow deletions, gains and amplifications, respectively were <−0.807, 0.415 to −0.807, 0.322 to 0.807, and >0.807. Percent of the genome with copy number alterations (PGA) was defined as the number of genes with any CNA (as just described) / total number of genes assessed for CNA (n=106). PGA was examined as a continuous variable and by quartiles, similar to previous studies of EA men25: ≤0.93% (≤25th percentile), 0.94–3.74% (25th-50th percentile), 3.75–9.35% (50th-75th percentile), and >9.35% (>75th percentile).
Of the 220 cases sequenced, a total of 205 (93%) resulted in interpretable data for CNA/PGA calls; 184 of those 205 cases (90%) had interpretable sequencing data for mutations. All 205 sequenced cases had clinical, pathologic, and long-term follow-up information, including biochemical recurrence (BCR) and metastasis. At our institution, men were followed up with PSA assays and digital rectal examinations every 3 months after surgery for the first year, semiannually for the second year, and annually thereafter. A detectable serum PSA level of at least 0.2 ng/mL was evidence of biochemical recurrence. Time to metastasis was defined as the time from RP to the first detection of distant metastasis on imaging. Distant metastases were diagnosed by radionuclide bone scan, chest radiograph, or other body imaging, which was performed at the time of biochemical recurrence and annually thereafter.
Immunohistochemistry for p27:
p27 immunohistochemistry was performed manually on standard histologic sections of formalin fixed paraffin embedded prostate tumors using a mouse monoclonal antibody for p27 (SX53G8.5, Cell Signaling Technologies) and an anti-mouse secondary detection kit (Leica). Immunostaining was scored by two pathologists (TLL and DCS), where tumors with any loss (focal or homogeneous) of p27 nuclear staining were considered to have p27 loss, using the prostatic stroma and benign glands as well as inflammatory cells as internal positive controls. Because p27 immunostaining is sensitive to fixation as previously reported,26 cases without internal control staining were not scored for p27 loss and were considered inevaluable.
Statistical analysis:
All statistical analyses were performed using Stata 15.0 (StataCorp, College Station, TX). Continuous variables were compared using the rank-sum calculation or Kruskwal-Wallis test when appropriate, and proportions were compared with the chi-squared test. BCR-free survival (BFS) and metastasis-free survival (MFS) were analyzed using the Kaplan-Meier method and log-rank statistic. Univariable and multivariable Cox proportional hazards models evaluated the association of genomic alterations with BCR and metastasis; multivariable models were adjusted for age, PSA level, pathologic GG, and pathologic stage. Patients were censored at last follow-up if they did not develop either BCR or metastasis.
Results:
Targeted NGS of known cancer-related loci was interpretable for CNA in 205 of 220 AA patients (93%). Table 1 describes their clinico-pathologic characteristics. Median PSA level was 7.3 ng/ml (IQR 5.24–13.0 ng/ml), and median age was 58 years (IQR 53–63 years). Seventy percent of patients had GG3–5 disease (145/205), and 49% of tumors were non-organ-confined. Median duration of follow-up was 5 years (IQR 2–10 years): 39% of patients developed BCR, and 11% developed metastasis. The 10-year BFS and MFS survival rates were, respectively, 46.8% (95% CI 36.7–56.2%) and 81.6% (95% CI 71.1–88.6%).
Table 1.
Clinico-pathologic characteristics and percent genome alteration (PGA) for African-American cohort (Copy number alteration data available for n=205 of 220 patients).
| Variable | AA (n=205) |
|---|---|
| Median PSA, ng/ml (IQR) | 7.30 (5.24–13.00) |
| Median age, yr (IQR) | 58 (53–63) |
| Clinical T stage, n (%) | |
| T1c | 154 (75.1) |
| T2a | 34 (16.6) |
| T2b/c | 17 (8.3) |
| Biopsy grade group, n (%) | |
| 1 (GS 6) | 72 (35.1) |
| 2 (GS 3+4) | 49 (23.9) |
| 3 (GS 4+3) | 48 (23.4) |
| 4 (GS 8) | 26 (12.7) |
| 5 (GS 9–10) | 10 (4.9) |
| RP grade group, n (%) | |
| 1 (GS 6) | 28 (13.7) |
| 2 (GS 3+4) | 32 (15.6) |
| 3 (GS 4+3) | 91 (44.4) |
| 4 (GS 8) | 33 (16.1) |
| 5 (GS 9–10) | 21 (10.2) |
| Pathologic T stage, n (%) | |
| T2N0 | 87 (43.1) |
| T3aN0 | 71 (35.2) |
| T3bN0 | 28 (13.9) |
| N1 | 16 (7.9) |
| Median length of follow-up, yr (IQR) | 5 (2–10) |
| Biochemical recurrence, n (%) | 74 (39.2) |
| Median time to biochemical recurrence, yr (IQR) | 2 (1–7) |
| 5-yr BCR-free survival (95% CI) | 0.603 (0.518, 0.678) |
| 10-yr BCR-free survival (95% CI) | 0.468 (0.367, 0.562) |
| Metastasis, n (%) | 20 (10.6) |
| Median time to metastasis, yr (IQR) | 5 (2–9) |
| 5-yr metastasis-free survival (95% CI) | 0.918 (0.852, 0.956) |
| 10-yr metastasis-free survival (95% CI) | 0.816 (0.711, 0.886) |
| Median PGA (IQR) | 3.74 (0.93–9.35) |
| Mean PGA | 5.85 |
| Median PGA, high CNV only (IQR) | 0 (0,0) |
| Mean PGA, high CNV only | 0.45 |
Median PGA of JHMI AA men was 3.7% (IQR 0.9–9.4%). Overall, PGA increased with pathologic grade (p<0.001) (Table 2) and T-stage (p=0.019). When examined as a continuous variable, PGA was significantly associated with metastasis (Table 3; multivariable HR 1.15 per 1% change in PGA, 95% CI 1.07–1.23, p<0.001) but not with BCR (Supplementary Table S1). Additionally, we found that the highest quartile of PGA was significantly associated with metastasis on multivariable analysis (Figure 1A and Table 3; HR 13.45, 95% CI 2.55–70.86, p=0.002) and with BCR on univariable analysis (HR 1.91, 95% CI 1.06–3.45, p=0.032) (Supplementary Table S1).
Table 2.
Percent genome alteration in JHMI AA cohort (n=205) and TCGA cohort (n=313) by pathologic grade.
| JHMI AA (n=205) | TCGA AA (n=43) | TCGA EA (n=270) | ||||
|---|---|---|---|---|---|---|
| PGA, median (IQR) | p-value | PGA, median (IQR) | p-value | PGA, median (IQR) | p-value | |
| Total | 3.74 (0.93–9.35) | 6.48 (0.45–12.34) | 0.877^ | 6.08 (1.74–11.71) | ||
| RP grade group | <0.001* | <0.001* | <0.001* | |||
| 1 | 0.94 (0–4.21) | 0.61 (0.004–6.39) | 0.486^ | 2.11 (0.05–4.99) | ||
| 2 | 1.87 (0–3.27) | 3.89 (0.09–7.53) | 0.480^ | 4.68 (1.36–7.94) | ||
| 3 | 3.74 (0.94–10.28) | 9.57 (6.34–16.73) | 0.154^ | 6.94 (2.68–11.38) | ||
| 4 | 8.41 (2.80–11.22) | 12.03 (8.86–15.51) | 0.780^ | 13.74 (7.40–22.20) | ||
| 5 | 4.67 (0.94–9.35) | 26.2 (15.2–34.9) | 0.032^ | 8.97 (3.53–17.90) | ||
P-value signifies Kruskal Wallis calculation to detect differences in PGA between grade groups within each cohort.
P-value signifies rank sum calculation to detect differences in PGA between TCGA AA and TCGA EA cohorts within each grade group.
Table 3.
Univariable and multivariable Cox proportional hazards models assessing association of PGA with metastasis
| Variable | Univariable | Multivariable* | ||
|---|---|---|---|---|
| HR (95% CI) | p-value | HR (95% CI) | p-value | |
| PGA (continuous) ‡ | 1.11 (1.06, 1.17) | <0.001 | 1.15 (1.07, 1.23) | <0.001 |
| PGA (quartile)^ | ||||
| ≤25th percentile | 1.00 (reference) | -- | 1.00 (reference) | -- |
| 25–50th percentile | 0.80 (0.07, 8.88) | 0.859 | 0.58 (0.05, 6.43) | 0.655 |
| 50–75th percentile | 5.28 (1.02, 27.41) | 0.048 | 3.31 (0.60, 18.25) | 0.170 |
| >75th percentile | 14.43 (3.13, 66.65) | 0.001 | 13.45 (2.55, 70.86) | 0.002 |
Multivariable models were adjusted for age at RP, PSA, RP grade group <4 or ≥4, and pathologic stage. RP grade group was analyzed as dichotomous variable due to low number of metastatic events per grade group.
Per 1% change in PGA
≤0.93% (≤25th percentile), 0.94–3.74% (25th-50th percentile), 3.75–9.35% (50th-75th percentile), and >9.35% (>75th percentile)
Figure 1. Correlation of clinical outcomes and genomic alterations in African American cohort.

(A) Kaplan-Meier plot of metastasis-free survival by quartile of percent genome alteration. (B) Oncoprint showing distribution of somatic mutations and copy number alterations for AA cohort. (C) Kaplan-Meier plot of metastasis-free survival by presence of TP53 mutation compared to wild type. (D) Kaplan-Meier plot of metastasis-free survival by presence of CDKN1B deep deletions compared to wild type.
Though it is not possible to compare PGA between different cohorts due to different sequencing methodology, for comparisons of PGA between EA and AA men within a single cohort (Table 2), we examined the TCGA dataset which includes 43 self-identified AA men (with race confirmed in 98% [42/43] by evidence of germline African ancestry27) (Supplementary Table S2) with available PGA data. The median PGA for TCGA AA men was 6.5% (IQR 0.5–12.3%) and increased with GG (p<0.001). Similarly, median PGA for TCGA EA men was 6.1% (IQR 1.7–11.7%) and also increased with GG (p<0.001). There did not appear to be any differences in PGA by ancestry except among GG5 disease (AA 26.2% vs EA 9.0%, p=0.032), though sample numbers were very small for AA men with GG5 cancer (n=4).
Of the 205 JHMI AA men, 184 had both somatic mutation and CNA data available, and this subset of men was analyzed to identify any associations between genomic alterations (somatic mutations or CNA) and oncologic outcomes in our cohort. Supplementary Table S3 describes the clinico-pathologic characteristics of this subset. Overall, 35% (65/184) of this subset had pathogenic somatic mutations detected using this targeted sequencing panel, consistent with the relatively low mutation burden reported in primary prostate cancer (~1 mutation/Mb) .11 The most common somatic mutations and CNA are listed in Figure 1B and Supplementary Table S4–S5. The three most commonly mutated genes were SPOP (11.2%), FOXA1 (8.3%), and TP53 (3.9%). The three loci most commonly altered at the copy number level were CDKN1B (6.3%), CHD1 (4.4%), and PTEN (3.4%).
Because most alterations were present at a low prevalence in this cohort, we selected the six alterations with >3% prevalence (FOXA1 mutation, SPOP mutation, TP53 mutation, CDKN1B deep deletion, CHD1 deep deletion, and PTEN deep deletion) and assessed associations with oncologic outcome. Among the mutated genes, TP53 mutation was the only one significantly associated with metastasis on univariable analysis and remained significant on multivariable analysis (Figure 1C and Table 4; HR 9.45, 95% CI 2.20–40.55, p=0.002). TP53 mutation did not meet the threshold for statistical significance for association with BCR on univariable analysis (Supplementary Table S1; HR 2.36, 95% CI 0.95–5.69, p=0.065) but did reach significance on Kaplan-Meier analysis (p=0.038). Among the CNA, deep deletions in CDKN1B were associated with increased risk of metastasis (Figure 1D and Table 4; multivariable HR 6.66, 95% CI 1.26–35.22, p=0.026) but were not significantly associated with BCR (Supplementary Table S1). Deep deletions in PTEN did not meet the threshold for statistical significance for association with metastasis on multivariable analysis (Table 4; HR 5.16, 95% CI 0.97–27.48, p=0.055) but were significantly associated with BCR on multivariable analysis (Supplementary Table S1; HR 2.94, 95% CI 1.08–8.00, p=0.035). No metastatic events were observed in patients with deep deletions in CHD1. Though NKX3–1 CNA had a prevalence of 3.4% (Supplementary Table S4), these were evenly split between amplifications and deletions, thus association of NKX3–1 with outcomes was not performed.
Table 4.
Univariable and multivariable Cox proportional hazards models assessing association of genetic alterations with metastasis.
| Variable | Univariable | Multivariable* | ||
|---|---|---|---|---|
| HR (95% CI) | p-value | HR (95% CI) | p-value | |
| CDKN1B deep deletion | 5.71 (1.23, 26.61) | 0.026 | 6.66 (1.26, 35.22) | 0.026 |
| CHD1 deep deletion† | -- | -- | -- | -- |
| PTEN deep deletion | 4.19 (0.94, 18.62) | 0.060 | 5.16 (0.97, 27.48) | 0.055 |
| FOXA1 mutation | 0.57 (0.08, 4.29) | 0.586 | 0.34 (0.04, 2.76) | 0.310 |
| SPOP mutation | 1.05 (0.30, 3.62) | 0.942 | 1.34 (0.34, 5.32) | 0.678 |
| TP53 mutation | 7.01 (2.01, 24.41) | 0.002 | 9.45 (2.20, 40.55) | 0.002 |
Multivariable models were adjusted for age at RP, PSA, RP grade group <4 or ≥4, and pathologic stage. RP grade group was analyzed as dichotomous variable due to low number of metastatic events per grade group.
No metastatic events were observed for CHD1 genetic alterations, hence HR and p-values were not calculated.
As deep deletions in CDKN1B have not previously been described as a recurrent genomic alteration in AA men associated with prognosis, we aimed to further characterize the patients harboring this genomic lesion. Median PSA for these men was 11.9 ng/ml (IQR 6.3–21.0 ng/ml), and 60% (6/10) of the lesions occurred in men who were younger than the cohort’s median age (58 years). Of the ten tumors with deep deletions in CDKN1B, 50% (5/10) were GG4–5, even though GG4–5 disease only represented about 25% of the entire cohort. Additionally, 90% (9/10) of the tumors were non-organ confined at RP. To confirm the presence of CDKN1B deep deletions in this subgroup, we performed immunohistochemistry for p27, the protein encoded by CDKN1B. As previously reported, p27 immunostaining is highly sensitive to tissue fixation conditions,26 thus of the 10 cases with CDKN1B deep deletions, only 5 had adequate internal control staining in benign glands and stroma to allow evaluation of p27 expression levels in tumor cells. Of those with adequate staining, 60% (3/5) had focal loss of p27 protein in some, but not all tumor cells while 40% had homogeneous loss of p27 (Figure 2), confirming the copy number calls.
Figure 2. p27 immunostaining in African-American prostate tumors with CDKN1B deep deletions.
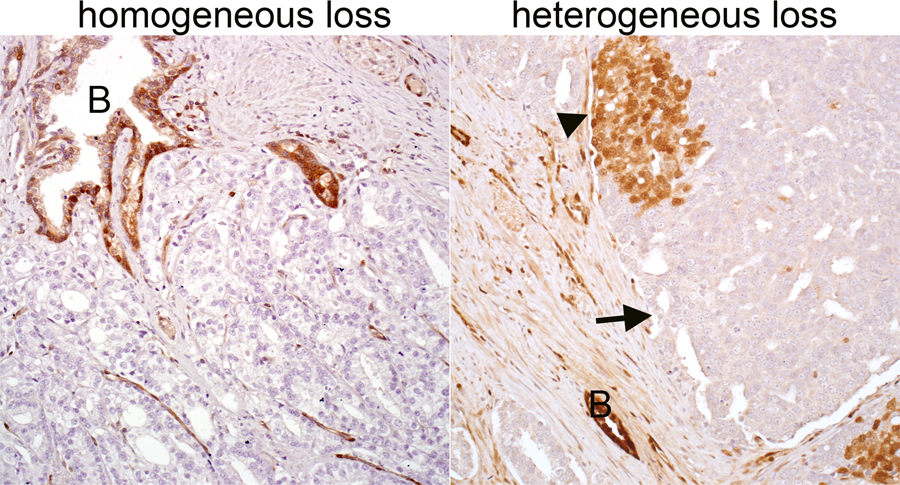
Both cases showed deep deletions of CDKN1B by sequencing, however the tumor on the left demonstrates homogeneous loss of p27 protein, suggestive of a clonal alteration, while the tumor on the right shows heterogeneous loss of p27 in some (arrow) but not all (arrowhead) tumor cells, suggestive of a subclonal alteration in CDKN1B. In both cases, benign glands (B) provide an internal positive control for p27 staining. All images reduced from 200x magnification.
Discussion:
There is limited understanding of the underlying genetic mechanisms of aggressive disease in AA men with clinically localized prostate tumors. Here we performed targeted NGS in the largest cohort of AA men to date, enriched with intermediate and high grade disease. We demonstrated that somatic TP53 mutations, deletions in CDKN1B, and extent of PGA were associated with risk of metastasis in AA men in multivariable models controlling for clinical-pathologic parameters, including Gleason grade group. This study is the first to correlate somatic genomic sequencing data with risk of metastasis in AA PCa. Notably, the poor prognosis of patients with deep deletions in CDKN1B is a novel finding that has important clinical implications for AA men. These deep deletions were more common in younger men with higher grade and stage disease and correlated with p27 protein loss.
Several large-scale sequencing efforts have greatly contributed to our understanding of the genomic landscape of PCa.11–12 TCGA identified seven recurrent molecular subtypes of PCa, including four gene fusion events (ERG, ETV1/4, and FLI1) and three mutational events (SPOP, FOXA1, and IDH1). However, their cohort of 333 prostate tumors included 43 from AA patients and only 17 AA patients with GG≥3 disease, limiting the generalizability and clinical relevance of their results. Additionally, current genomic risk classifiers used in risk stratification of PCa are derived from primarily EA cohorts, and their use in underrepresented racial groups may exacerbate health disparities.18–19 Our work in this current study contributes to the much needed effort to understand the genomics of AA PCa and reduce the pervasive racial gap in cancer health care.
Other groups have included exclusively men of African ancestry in their sequencing studies,13–16 and several unique genomic drivers of carcinogenesis have been identified. However, with the exception of Huang et al,13 these studies have generally had small numbers of AA men with clinically significant disease. Importantly, none have included long-term clinical outcome data for metastasis. In their cohort of 192 AA tumors, Huang et al13 identified recurrent mutations in SPOP (8%), FOXA1 (6%), and TP53 (7%) along with ERF (5%), a novel ETS transcription factor somatic alteration not previously recognized as a recurrent mutation in primary PCa. In our cohort, we found 10 cases with CNA in ERF (1 deep deletion, 6 shallow deletions, and 3 low level amplifications) but no mutations. Petrovics et al14 sequenced 7 AA prostate tumors and 7 EA tumors and discovered novel deletions in the LSAMP region enriched in AA patients and associated with BCR. LSAMP deletions were seen in 3 cases of our AA cohort (1 deep deletion, 2 shallow deletions).
In the current study we identified two genomic alterations that were associated with increased risk of metastasis among AA men: TP53 mutations and deletions in CDKN1B. The TP53 tumor suppressor gene is arguably the most frequently mutated gene in human cancer, present in more than 40% of tumors overall.28 However, in PCa surgical cohorts, TP53 mutations are relatively infrequent: 8% in the TCGA cohort (EA men) and 4% in the AA men of the current study. This prevalence increases up to 50% in metastatic castration-resistant PCa, though again, predominantly EA men were studied.29 Accordingly, the presence of TP53 mutation is associated with an aggressive PCa phenotype and PCa-specific mortality.30–31 Recent genome analyses by racial ancestry in pan-cancer cohorts reveal that TP53 mutations are significantly more common among AA cancer patients than EA patients,27 a finding that does not seem to be replicated in PCa based on the TCGA data described above and additional studies by our group.32 Importantly, while the overall prevalence of TP53 mutations is generally low in AA men with clinically localized PCa, it remains associated with outcomes as seen in EA cohorts, reminiscent of findings with PTEN loss.4
To our knowledge, no prior studies have identified deletions in CDKN1B as a recurrent somatic genomic lesion in AA PCa that is associated with metastasis, though there is some indication of a similar association in previous EA cohorts. CDKN1B encodes for p27, a cyclin-dependent kinase inhibitor that negatively regulates cell proliferation and whose loss is associated with poor prognosis in several cancer types. In older studies of predominantly EA cohorts,33–36 prostate tumors that had decreased expression of p27 protein were found to have an increased risk of BCR after surgery. However other studies have shown that the absence or low expression of p27 has no impact on PCa prognosis,36–38 and it often occurs in association with other known PCa molecular subtypes (eg, PTEN loss or ERG-negative tumors).35,37 Many of these studies are limited in their sample size and also lack a uniform cut-off between high and low p27 protein expression, typically measured by immunohistochemistry. In addition, prior studies have established that p27 immunostaining is highly sensitive to formalin fixation time, which introduces a significant pre-analytic variable into evaluation of p27 immunostaining, and makes many cases inevaluable.26 In some linkage studies, germline polymorphisms in CDKN1B have been associated with hereditary PCa.39–41 Additionally, loss of heterozygosity at the CDKN1B locus (12p13) has been identified as a recurrent alteration in metastatic PCa, and it is associated with higher GG disease, lymph node metastases, and advanced tumor stage among predominantly EA cohorts.42–43 In our current study, deep deletions in CDKN1B occurred in 5–6% of AA men and were independently associated with risk of metastasis, findings which have not been previously described in this population. We were able to confirm subclonal (heterogeneous) or clonal (homogeneous) p27 protein loss by immunohistochemistry in a subset of evaluable samples with CDKN1B deep deletions in our cohort, confirming the copy number calls in this subset of cases and providing additional information on likely clonality. However, we acknowledge that p27 immunostaining is likely not adequate for screening large cohorts due to variability in pre-analytic conditions. Additionally, we found CDKN1B deletions were more common in younger men who presented with higher stage and grade disease, suggesting this cell cycle pathway may be important in the more aggressive disease state found in AA PCa.
Consistent with an association of CDKN1B loss with aggressive PCa, previous sequencing studies have found the prevalence of CDKN1B deep deletions is enriched in metastatic compared to primary disease (Supplementary Figure S1). In most primary tumor cohorts, such as the TCGA, CDKN1B deep deletions are reported in only 1–2% of cases overall, while the prevalence is increased to 2–6% of metastatic cohorts. Unfortunately, the low prevalence in the TCGA cohort makes it impossible to determine whether these deletions are more common among AA patients. In the Huang et al13,44 study of AA tumors, deep deletions of CDKN1B occurred in 2.2% of primary tumors, but there was no matched EA cohort included in this study. As mentioned above, our AA cases were significantly higher grade and stage compared to those included in TCGA and Huang et al, potentially explaining why the rate of CDKN1B loss in the current study is somewhat higher.
To better compare deep deletion frequency between AA and EA patients, we interrogated data from the AACR Project GENIE cohort45 (Supplementary Figure S1) and found that CDKN1B deep deletions occurred in 1.2% (14/1206) of primary PCa among EA patients and 1.8% (2/109) of primary PCa among AA patients. Consistent with our finding that CDKN1B deletion is associated with risk of metastasis, the rate of deletion was higher in both races in metastatic tumors (4.5% or 35/764 in EA patients and 5.7% or 4/70 in AA patients), though this difference between primary and metastatic disease only reached statistical significance in the EA samples (p<0.0001). However, the EA and AA cohorts were not matched in the GENIE samples and may be confounded by selection for specific clinical-pathologic variables45. Thus, it remains uncertain whether the relatively high rate of CDKN1B deletions in our AA primary tumor cohort (6%) is due to the higher grade and stage of these tumors relative to other cohorts, or whether this high rate is a cohort-specific or race-specific finding. Additional AA cohorts will be required to resolve these questions. In addition, future research is warranted to determine if CDKN1B deep deletions play a contributory role in outcome disparities between races.
PGA in tumors from men of African ancestry has only been described in a few sequencing studies.13,15–16 The extent of CNA, or PGA, was an important predictor of adverse prognosis in the AA men in our cohort. Patients with the highest quartile of PGA had increased risks of metastasis and BCR. Previous work by Hieronymus et al25 demonstrated in predominantly EA cohorts that CNA burden is a prognostic factor in PCa relapse and metastasis and can also be assessed in biopsy specimens, allowing for improved risk stratification and treatment guidance in the diagnostic setting. Our study further corroborates the association of PGA with high risk pathology and increased metastasis among AA men with PCa. Similar to our findings, PGA in the TCGA cohort increased with pathologic grade in both AA and EA men. While the only difference in PGA between the AA and EA men of TCGA was among GG5 disease, this comparison is severely limited due to the small number of men with GG5 tumors (AA n=4, EA n=38). Continued sequencing efforts are necessary to robustly compare the PGA profile between AA and EA men and determine its precise role as a biomarker.
The strengths of our study include the large cohort of intermediate and high grade AA prostate tumors (n=145 GG≥3) and the long-term clinical outcome data, which allowed for the first correlation of somatic genomic sequencing with risk of metastasis in AA men to our knowledge. The inclusion of men of African ancestry in PCa genomic studies is crucial to understanding the role of race in health disparities. AA race was determined by self-identification in our study, but we did not confirm this with racial ancestry informative genetic markers. A major limitation of our study is the lack of a grade-matched EA cohort that would have allowed comparisons between races. However, it is important to note that the value of studying the AA population is not principally in its comparison to a reference EA cohort; rather, it is critical to fill in the gaps in our knowledge about AA PCa genomics so that we can better tailor risk stratification and precision therapies in the AA population that is disproportionately affected by PCa mortality. Importantly, while TP53 mutations and deletions in CDKN1B were shown to be associated with an increased risk of metastasis in AA men after surgery, overall these genomic alterations remain relatively infrequent in AA men with clinically localized PCa, and the multivariable models are knowingly over-fitted with estimates that could deviate from truth. However, they are presented here for comparison to univariable hazard ratios. Additional AA cohorts are needed to corroborate our findings. Finally, it is important to note that because this study was designed to look specifically at outcomes in AA patients relative to the most common genomic alterations, we used targeted sequencing and did not perform whole genome or whole exome sequencing to look for novel low prevalence alterations in AA tumors.
In conclusion, targeted somatic sequencing of AA prostate tumors reveals significant associations of genomic alterations with metastasis in AA men. Notably, our study is the first to describe deletions in CDKN1B as a recurrent genomic lesion in AA men that is associated with poor prognosis. Furthermore, the observation that these deletions may occur in younger men with high grade tumors suggests that this pathway may contribute to a more aggressive disease course. This work begins to fill in the significant gaps in our understanding of the biology of aggressive PCa in AA men. Continued research in AA PCa genomics will guide the advancement of precision medicine in underrepresented populations and help alleviate cancer health disparities.
Supplementary Material
Translational Relevance:
In this retrospective study, we performed targeted genomic sequencing of 205 prostate tumors from African American men treated with radical prostatectomy, with the aim to elucidate the key molecular drivers associated with poor prognosis in this population. We found that percent of the genome with copy number alterations (PGA), TP53 mutations, and CDKN1B deep deletions were associated with increased risks of metastasis in multivariable models, though these findings must be confirmed in additional African American cohorts. This is the first study to correlate somatic genomic alterations with long-term outcomes after radical prostatectomy in African American men and to identify deletions in CDKN1B as associated with metastasis in this population. Though confirmatory studies are required, these findings begin to fill in the significant gaps in our understanding of the biology of aggressive prostate cancer in African American men and will guide the advancement of precision medicine in underrepresented populations.
Acknowledgements:
This work was supported by a Health Disparity Research Awards from the CDMRP-PCRP (W81XWH-15–1-0661 to S. Tomlins, E. Schaeffer and T. Lotan); U01CA196390 (E. Schaeffer), R01 CA183857 to S. Tomlins, the NIH/NCI Prostate SPOREs P50 CA58236, P50 CA186786–05 and P50 CA180995; and the NCI Cancer Center Support Grant 5P30CA006973–52. E. Schaeffer is also supported by Prostate Cancer Foundation and Polsky Urologic Cancer Institute. The authors would like to acknowledge the American Association for Cancer Research and its financial and material support in the development of the AACR Project GENIE registry, as well as members of the consortium for their commitment to data sharing. Interpretations are the responsibility of study authors.
Conflict of Interest Statement: TLL has had sponsored research agreements for other projects from Roche/Ventana Medical Systems and GenomeDx Biosciences. TLL has served as a consultant for Janssen. EMS has served as a consultant for AbbVie. SAT has received travel support from and had a sponsored research agreement with Compendia Bioscience/Life Technologies/Thermo Fisher Scientific. The University of Michigan has been issued patents on ETS gene fusions in prostate cancer, on which SAT is a coinventor. The diagnostic field of use was licensed to Hologic/Gen-Probe Inc., which has sublicensed rights to Roche/Ventana Medical Systems. SAT has served as a consultant for and received honoraria from Janssen, AbbVie, Sanofi, Almac Diagnostics, and Astellas/Medivation. SAT has sponsored research agreements with Astellas/Medivation and GenomeDX. SAT is an equity holder in, prior consultant for, and current employee of Strata Oncology.
References:
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019; 69: 7–34. [DOI] [PubMed] [Google Scholar]
- 2.Dash A, Lee P, Zhou Q, et al. The impact of socioeconomic factors on prostate cancer outcomes in black patients treated with surgery. Urology 2008; 72: 641–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahal BA, Berman RA, Taplin ME, et al. Prostate cancer-specific mortality across Gleason scores in black vs nonblack men. JAMA 2018; 320: 2479–81. [DOI] [PubMed] [Google Scholar]
- 4.Tosoian JJ, Almutairi F, Morais CL, et al. Prevalence and prognostic significance of PTEN loss in African American and European American men undergoing radical prostatectomy. Eur Urol. 2017; 71: 697–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamoah K, Johnson MH, Choeurng V, et al. Novel biomarker signature that may predict aggressive disease in African American men with prostate cancer. J Clin Oncol. 2015; 33: 2789–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faisal FA, Sundi D, Tosoian JJ, et al. Racial variations in prostate cancer molecular sybtupes and androgen receptor signaling reflect anatomic tumor location. Eur Urol. 2016; 70: 14–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farrell J, Young D, Chen Y, et al. Predominance of ERG-negative high grade prostate cancers in African American men. Mol Clin Oncol. 2014; 2: 982–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Powell IJ, Dyson G, Land S, et al. Genes associated with prostate cancer are differentially expressed in African American and European American men. Cancer Epidemiol Biomarkers Prev. 2013; 22: 891–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khani F, Mosquera JM, Park K, et al. Evidence for molecular differences in prostate cancer between African American and Caucasian men. Clin Cancer Res. 2014; 20: 4925–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barbieri CE, Baca SC, Lawrence MS, et al. Exome sequencing identifies recurrent SPOP, FOXA1, and MED12 mutations in prostate cancer. Nat Genet. 2012; 44: 685–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research Network. The molecular taxonomy primary prostate cancer. Cell 2015; 163: 1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraser M, Sabelnykova VY, Yamaguchi TN, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017; 541: 359–64. [DOI] [PubMed] [Google Scholar]
- 13.Huang FW, Mosquera JM, Garofalo A, et al. Exome sequencing of African American prostate cancer reveals loss of function ERF mutations. Cancer Discov. 2017; 7: 973–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrovics G, Li H, Stumpel T, et al. A novel genomic alteration of LSAMP associates with aggressive prostate cancer in African American men. E Bio Medicine. 2015; 2: 1957–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindquist KJ, Paris PL, Hoffman TJ, et al. Mutational landscape of aggressive prostate tumors in African American men. Cancer Res. 2016; 76: 1860–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaratlerdsiri W, Chan E, Gong T, et al. Whole-genome sequencing reveals elevated tumor mutational burden and initiating driver mutations in African men with treatment-naïve high risk prostate cancer. Cancer Res. 2018; 78: 6736–46. [DOI] [PubMed] [Google Scholar]
- 17.Mahal BA, Alshalalfa M, Spratt DE, et al. Prostate cancer genomic-risk differences between African-American and white men across Gleason scores. Eur Urol. 2019;1038–40. [DOI] [PubMed]
- 18.Martin AR, Kanai M, Kamatani Y, et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51:584–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spratt DE, Chan T, Waldron L, et al. Racial/ethnic disparities in genomic sequencing. JAMA Oncol. 2016;2:1070–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaur HB, Guedes LB, Lu J, Maldonado L, Reitz L, Barber JR, et al. Association of tumor-infiltrating T-cell density with molecule subtype, racial ancestry and clinical outcomes in prostate cancer. Mod Pathol. 2018; e-pub ahead of print 3 May 2018; doi: 10.1038/s41379-018-0083-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hovelson DH, McDaniel AS, Cani AK, et al. Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumors. Neoplasia 2015; 17: 385–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lazo de la Vega L, Samaha MC, Hu K, et al. Multiclonality and marked branched evolution of low-grade endometrioid endometrial carcinoma. Mol Cancer Res. 2019; 17: 731–40. [DOI] [PubMed] [Google Scholar]
- 23.Grasso C, Butler T, Rhodes K, et al. Assessing copy number alterations in targeted, amplicon-based next-generation sequencing data. J Mol Diagn. 2015; 17 :53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDaniel AS, Stall JN, Hovelson DH, et al. Next-generation sequencing of tubal intraepithelial carcinomas. JAMA Oncol. 2015; 1: 1128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hieronymus H, Schultz N, Gopalan A, et al. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci. 2014; 111: 11139–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Marzo AM, Fedor HH, Gage WR, et al. Inadequate formalin fixation decreases reliability of p27 immunohistochemical staining: probing optimal fixation time using high-density tissue microarrays. Hum Pathol. 2002; 33: 756–60. [DOI] [PubMed] [Google Scholar]
- 27.Yuan J, Hu Z, Mahal BA, et al. Integrated analysis of genetic ancestry and genomic alterations across cancers. Cancer Cell 2018; 34: 549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013; 502: 333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015; 161: 1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Che M, DeSilvio M, Pollack A, et al. Prognostic value of abnormal p53 expression in locally advanced prostate cancer treated with androgen deprivation therapy and radiotherapy: a study based on RTOG 9202. Radiat Oncol Biol Phys. 2007; 15: 1117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guedes LB, Almutairi F, Haffner MC, et al. Analytic, pre-analytic, and clinical validation of p53 immunohistochemistry for detection of TP53 missense mutation in prostate cancer. Clin Cancer Res. 2017; 23: 4693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaur HB, Lu J, Guedes LB, et al. TP53 missense mutation is associated with increased tumor-infiltrating T-cells in primary prostate cancer. Hum Pathol. 2019; e-pub ahead of print 6 Mar. 2019; doi: 10.1016/j.humpath.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsihlias J, Kapusta LR, DeBoer G, et al. Loss of cyclin-dependent kinase inhibitor p27Kip1 is a novel prognostic factor in localized human prostate adenocarcinoma. Cancer Res. 1998; 58: 542–8. [PubMed] [Google Scholar]
- 34.Yang RM, Naitoh J, Murphy M, et al. Low p27 expression predicts poor disease-free survival in patients with prostate cancer. J Urol. 1998; 159: 941–5. [PubMed] [Google Scholar]
- 35.Halvorsen OJ, Haukaas SA, and Akslen LA. Combined loss of PTEN and p27 expression is associated with tumor cell proliferation by Ki-67 and increased risk of recurrent disease in localized prostate cancer. Clin Cancer Res. 2003; 9: 1474–9. [PubMed] [Google Scholar]
- 36.Swanson GP and Quinn D. Using molecular markers to help predict who will fail after radical prostatectomy. Prostate Cancer 2011; doi: 10.1155/2011/290160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sirma H, Broemel M, Stumm L, et al. Loss of CDKN1B/p27Kip1 expression is associated with ERG fusion-negative prostate cancer, but is unrelated to patient prognosis. Oncol Lett. 2013; 6: 1245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Erdamar S, Yang G, Harper JW, et al. Levels of expression of p27Kip1 protein in human prostate and prostate cancer: an immunohistochemical analysis. Mod Pathol. 1999; 12: 751–5. [PubMed] [Google Scholar]
- 39.Chang B, Zheng SL, Isaacs SD, et al. A polymorphism in CDKN1B gene is associated with increased risk of hereditary prostate cancer. Cancer Res. 2004; 64: 1997–9. [DOI] [PubMed] [Google Scholar]
- 40.Suarez BK, Lin J, Burmester JK, et al. A genome screen of multiplex sibships with prostate cancer. Am J Hum Genet. 2000; 66: 933–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsieh CL, Oakley-Girvan I, Balise RR, et al. A genome screen of families with multiple cases of prostate cancer: evidence of genetic heterogeneity. Am J Hum Genet. 2001; 69: 148–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kibel AS, Faith DA, Bova GS, et al. Loss of heterozygosity at 12P12–13 in primary and metastatic prostate adenocarcinoma. J Urol. 2000; 164: 192–6. [PubMed] [Google Scholar]
- 43.Kluth M, Ahrary R, Hube-Magg C, et al. Genomic deletion of chromosome 12p is an independent prognostic marker in prostate cancer. Oncotarget 2015; 6: 27966–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Armenia J, Wankowicz SAM, Liu D, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet. 2018; 50: 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.AACR Project GENIE Consortium. AACR Project GENIE: Powering precision medicine through an international consortium. Cancer Discov. 2017; 7: 818–31. Version 6.1-public. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.