

Abstract
In autosomal dominant polycystic kidney disease (ADPKD), the inexorable growth of numerous fluid-filled cysts leads to massively enlarged kidneys, renal interstitial damage, inflammation, and fibrosis, and progressive decline in kidney function. It has long been recognized that interstitial fibrosis is the most important manifestation associated with end-stage renal disease; however, the role of abnormal extracellular matrix (ECM) production on ADPKD pathogenesis is not fully understood. Early evidence showed that cysts in end-stage human ADPKD kidneys had thickened and extensively laminated cellular basement membranes, and abnormal regulation of gene expression of several basement membrane components, including collagens, laminins, and proteoglycans by cyst epithelial cells. These basement membrane changes were also observed in dilated tubules and small cysts of early ADPKD kidneys, indicating that ECM alterations were early features of cyst development. Renal cystic cells were also found to overexpress several integrins and their ligands, including ECM structural components and soluble matricellular proteins. ECM ligands binding to integrins stimulate focal adhesion formation and can promote cell attachment and migration. Abnormal expression of laminin-332 (laminin-5) and its receptor α6β4 stimulated cyst epithelial cell proliferation; and mice that lacked laminin α5, a component of laminin-511 normally expressed by renal tubules, had an overexpression of laminin-332 that was associated with renal cyst formation. Periostin, a matricellular protein that binds αVβ3- and αVβ5-integrins, was found to be highly overexpressed in the kidneys of ADPKD and autosomal recessive PKD patients, and several rodent models of PKD. αVβ3-integrin is also overexpressed by cystic epithelial cells, and the binding of periostin to αVβ3-integrin activates the integrin-linked kinase and downstream signal transduction pathways involved in tissue repair promoting cyst growth, ECM synthesis, and tissue fibrosis. This chapter reviews the roles of the ECM, integrins, and focal adhesion signaling in cyst growth and fibrosis in PKD.
Keywords: Autosomal dominant polycystic kidney disease, cyst growth, basement membrane, ECM, integrin-linked kinase, focal adhesion kinase
1. Introduction
Polycystic kidney disease (PKD) is a family of monogenic disorders characterized by the development of numerous fluid-filled cysts in the kidneys. Autosomal dominant polycystic kidney disease (ADPKD) is the most common form of the disease and is one of the most frequently inherited disorders with a gene frequency of 1 in ~800 births affecting nearly 12 million people worldwide [1]. Approximately one-half of ADPKD patients progress to renal failure by age 60 and require dialysis or renal replacement therapy, making it the third leading cause of end-stage renal disease (ESRD) after diabetes mellitus and hypertension. ADPKD is caused by mutations in PKD1 and PKD2, which encode transmembrane proteins polycystin-1 (PC-1) and polycystin-2 (PC-2), respectively [2–4]. In ADPKD, cysts develop in all nephron segments; however, the majority of visible cysts in an end-stage kidney appears to be derived from the collecting ducts. Autosomal recessive polycystic kidney disease (ARPKD), a less common juvenile form of PKD, has an estimated prevalence of 1 in ~12,000 live births and is associated with neonatal mortality and childhood morbidity. ARPKD is caused by mutations in PKHD1, which encodes fibrocystin (also called polyductin). In ARPKD kidneys, there is a uniform fusiform dilation of the collecting ducts leading to massive kidney enlargement and renal functional decline in the neonates. Despite identification of the causative genes for ADPKD and ARPKD more than 20 years ago, the functions of the PKD proteins and the mechanisms responsible for initial cyst formation remain poorly understood.
PC-1 is a receptor-like protein with a large extracellular N-terminal region and PC-2 is a transient receptor potential channel (also called TRPP2). These proteins co-localize to multiple cellular domains including primary cilia and basolateral membranes and interact to form a regulated non-selective cation channel permeable to Ca2+ [5]. The polycystins are thought to function in intracellular Ca2+ signaling in response to ligand binding or mechanosensation and mutations in the polycystin genes disrupt intracellular Ca2+ homeostasis [6–8]. Mutations in PKD1 or PKD2 cause a similar disease phenotype; however, patients with PKD1 mutations have an early onset of ESRD (average of 54 years) compared to those patients harboring a PKD2 mutation (average age 74 years) [9]. This difference in severity is thought to be caused by the development of more cysts in PKD1 mutant kidneys rather than a difference in the rate of cyst growth [10]. Moreover, PKD1 patients have highly variable age of onset for ESRD, which is determined by the type of mutation (truncating vs. non-truncating), the number of somatic mutations in genes of disease modifiers [11], and other confounding factors.
In ADPKD, renal cyst enlargement involves aberrant epithelial cell proliferation, accumulation of fluid in the cyst cavity, and remodeling of the extracellular matrix (ECM) [12]. Several signaling pathways have been implicated in PKD pathogenesis [13]; however, the 3′, 5′-cyclic adenosine monophosphate (cAMP) pathway appears to be central for cyst growth by stimulating both cell proliferation and transepithelial fluid secretion [12–15]. Although renal cysts are benign neoplasms, progressive enlargement of the cysts leads to renal insufficiency through damage to adjacent parenchyma, vascular remodeling, extensive nephron loss and development of interstitial fibrosis. Recent evidence indicates that excessive production and aberrant composition of the ECM induce specific responses to cyst-lining cells that contribute to cyst growth and fibrosis [16–21]. While renal fibrosis has been well-described in human ADPKD kidneys and animal models of PKD [19, 22, 23], the role of abnormal ECM in PKD pathogenesis has not been rigorously studied.
2. Aberrant expression of extracellular matrix in PKD
2.1. Early alterations in the basement membrane of cysts
The ECM plays an essential role in tissue development and homeostasis by providing structural and mechanical integrity to the surrounding cells and contributing to the regulation of the microenvironment [24]. The cellular basement membrane is a highly specialized ECM component that anchors the epithelium to the surrounding connective tissue [24, 25]. ECM remodeling and basement membrane abnormalities are common features of cysts in human ADPKD and ARPKD and rodent models of PKD [20, 23, 26]. Early studies by Grantham and associates showed that nearly every cyst in pathological specimens of human ADPKD kidneys had a thickened or extensively laminated basement membrane, but these structures were highly variable in appearance [23]. Similarly, multilayering of the tubule basement membrane was observed in renal biopsy specimens of young individuals with early-stage ADPKD [27], suggesting that ECM changes occur early in cyst development. Subsequent evidence showed abnormal regulation of gene expression for several basement membrane components. Expression of laminin β (formally B1) and γ (B2) chains and collagen IV were found to be abnormal in cystic basement membranes of a murine model of PKD [28]. There also were defects in the synthesis and intracellular transport of sulfated proteoglycans by human ADPKD cyst epithelial cells compared to normal kidney cells [29]. As reviewed by Calvet in 1993 [30], an early hypothesis for the underlying cause of PKD pathogenesis was that a defect in the expression of a basement membrane or ECM component decreases tensile strength or increases elasticity of the tubule basement membrane, leading to cystic dilations and subsequent cyst formation [31]. However, a subsequent study found that normal and cystic basement membranes were equally compliant [32], confirming that altered deformability of the tubule basement membrane does not account for initial cystogenesis in ADPKD. A review of the data suggested that changes in ECM gene expression may be a compensatory response in an attempt to repair the abnormal tubule morphology during cystogenesis [30, 33]
2.2. The role of abnormal ECM in PKD
Human ADPKD cells in culture produce thicker basement membranes than normal human kidney cells, and their basement membranes contain spherical aggregates composed, in part, of sulfated proteoglycans and bundles of banded collagen similar to interstitial type I collagen [26]. These observations indicate that primary cultures of ADPKD cells retain an abnormal cellular phenotype and that aberrant ECM production is independent of factors present in situ. ADPKD cells plated on various matrix components grew at different rates with the greatest proliferative response on type I collagen > type IV collagen > fibronectin > laminin, indicating that abnormal basement membrane and ECM production may contribute to excessive growth of cyst-lining epithelial cells [20].
In the Han:SPRD (Cy) rat model of ADPKD, certain cystic portions of the tubule were found to be associated with dramatic thickening of the underlying basement membrane [34]. In the portion of the renal tubule undergoing cystic transformation (Figure 1), the hyperplastic cystic region contained cells expressing elevated levels of phosphorylated ERK (p-ERK), a key component of the mitogenic pathway involved in cyst epithelial cell proliferation [8, 35]. There was a sharp boundary in the thickness of the basement membrane between p-ERK expressing cells and adjacent normal cells within the same developing cyst. However, in other studies it was shown that while cystic cells have abnormal ECM production [17, 20, 28, 36, 37], there was not a strict association between basement membrane thickening and cyst formation. Moreover, thickened basement membranes were often observed associated with normal-appearing tubules in cystic kidneys and in several non-cystic renal pathological conditions. These observations make it difficult to assign a causative relationship between changes in the ECM and cyst formation.
Figure 1. Early changes in the basement membrane of dilated tubules undergoing cystic transformation in a Han:SPRD Cy/+ rat kidney.
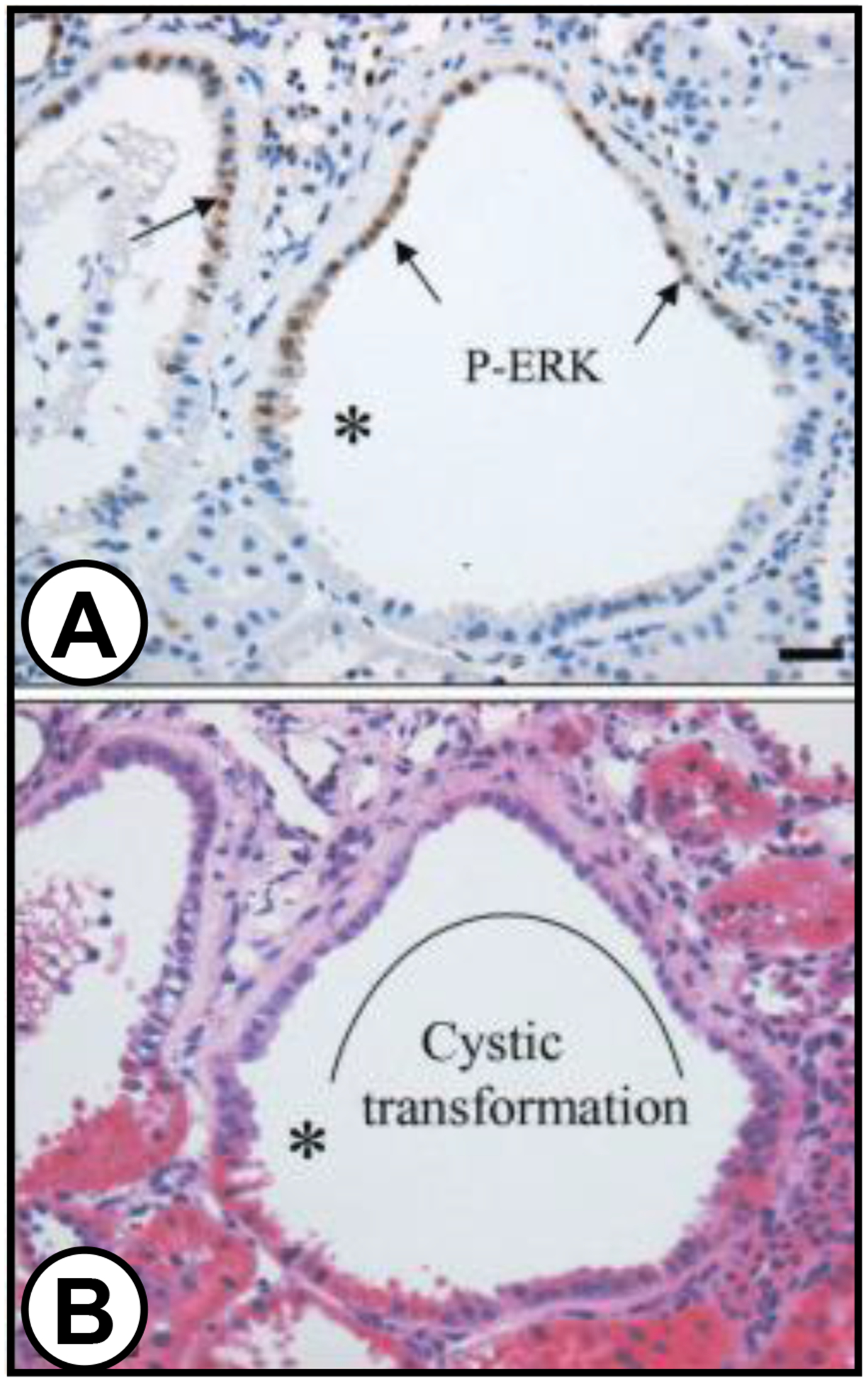
(A) P-ERK expression in patches of epithelial cells lining cysts (arrows) and the transition (*) between abnormal p-ERK expressing cells and adjacent cells with a normal phenotype. Tubule basement membrane is thickened beneath p-ERK expressing cells and the subjacent interstitium is widened and infiltrated with mononuclear cells. (B) Hematoxylin and eosin section of same field. The cytoplasm of the p-ERK expressing cells in the field stains faint pink, whereas other mural cells stain dark pink. Curved line marks segment of renal tubule that has undergone cystic transformation. Republished with permission from Nagao et al. Kidney Int. 63: F427–437, 2003.
Although ECM abnormalities may not be the primary cause for cyst formation, altered composition of the cystic basement membrane likely affects cell-matrix interactions, contributing to epithelial-to-mesenchymal transition (EMT), incomplete cellular differentiation, aberrant proliferation, and changes in tubule morphogenesis [38, 39]. Mangos et al. showed that knockout of Pkd1 or Pkd2 orthologs in zebrafish resulted in ectopic and persistent expression of multiple collagen mRNAs and proteins, including Col2a1, in the notochord and caused dorsal axis curvature, a PKD phenotype in zebrafish [17]. Gene knockdown of Col2a1 or inhibition of collagen crosslinking restored a normal phenotype, suggesting that abnormalities in ECM composition or amount are developmental defects that are directly linked to polycystin function and not a consequence of tissue damage or inflammation [40]. This evidence supports the hypothesis that cystic transformation and aberrant ECM production may be caused by arrested development of the cystic epithelial cells [41]. However, the cellular response to tissue injury and incomplete cellular differentiation, due to the loss of the polycystins, would be difficult to distinguish.
2.3. Aberrant expression of ECM components and matricellular proteins in PKD
Differential microarray analysis was used to compare autonomous gene expression in cultured ADPKD and normal human kidney (NHK) cells to identify potential factors that are aberrantly expressed by the cystic cells and may contribute to changes in the microenvironment (Table 1) [42]. Consistent with earlier studies [20, 36, 43–45], mRNAs of several genes involved in ECM production and tissue remodeling were elevated in ADPKD cystic cells compared with normal kidney cells. Table 1 shows the relative expression of selected ECM components in ADPKD versus normal human kidney (NHK) cells determined by microarray analysis using the Affymetrix GeneChip® Human genome U133 Plus 2.0 (Santa Clara, CA). Many collagen molecules were highly overexpressed in ADPKD cells compared to normal cells. Other structural proteins including lumican, laminin, heparan sulfate proteoglycan, and chondroitin sulfate proteoglycan 2 were also overexpressed by the cystic cells. In addition, there was overexpression of soluble ECM molecules including matrix metalloproteinases and ADAMTS, as well as matricellular proteins (periostin, SPARC, and thrombspondin) and TGF-β, the master regulator of ECM production. Surprisingly, periostin, a 90-kDa secreted ECM molecule originally thought to be a bone factor (previously named osteoblast specific factor 2 or OSF-2), was one of the most highly overexpressed genes in our dataset [42]. Periostin is the newest member of the family of matricellular proteins that includes thrombospondins, osteopontin, SPARC, tenascin-C, connective tissue growth factors and transforming growth factor-β (TGF-β)-induced protein (βig-H3) [46–49]. Matricellular proteins are rapidly turned over and have regulatory roles, including the activation of cellular pathways involved in tissue injury repair [46, 50–55]. The expression of many of these factors were elevated in the ADPKD cells (Table 1 and unpublished data).
Table 1. Microarray analysis of selected ECM components in ADPKD vs. normal human kidney (NHK) cells.
ADPKD and NHK cells (N = 2 kidney preparations each) were grown to 70–80% confluency in 0.002% fetal bovine serum and total RNA was isolated for whole genome expression using the Affymetrix GeneChip® Human Genome U133 Plus 2.0 Array. SPARC = secreted protein, acidic, cysteine-rich (osteonectin) and βig-H3 = transforming growth factor, beta-induced protein. Republished from Wallace et al. Am J Physiol Renal Physiol 295: F1463-F1471, 2008.
| Gene Name | GenBankID | Ave. Fold Increase |
|---|---|---|
| Collagen I | K01228 | 15.7 |
| Periostin (probe 1) | D13665 | 15.3 |
| Periostin (probe 2) | AY140646 | 14.3 |
| Collagen III | AU144167 | 12.4 |
| Lumican | NM_002345 | 4.6 |
| Laminin | NM_018897 | 3.2 |
| Matrix metalloproteinase-2 | NM_004530 | 2.7 |
| TGF-β | AU145950 | 2.6 |
| βig-H3 | NM_000358 | 2.4 |
| Heparan sulfate proteoglycan 2 | AI991033 | 2.2 |
| SPARC | NM_003118 | 2.0 |
| Thrombospondin | AI812030 | 2.0 |
3. The role of laminin in renal cystic disease
Laminins are heterotrimeric glycoproteins containing an α-chain, a β-chain, and a γ-chain, and are major components of the basement membrane. Currently, there are 15 reported heterotrimers that are named according to their chain composition. Laminin-511 (α5, β1, γ1; formerly termed laminin-10) is a major component of the tubule basement membrane; whereas, laminin-332 (α3, β3, γ2; formerly termed laminin-5) is not typically observed. Laminin-332 and integrin β4 subunit, a component of α6β4, the membrane receptor for laminin-332, are highly overexpressed in ADPKD cells [43]. Joly et al. showed that ADPKD cells, grown as in vitro cysts within semi-solid Matrigel 3-D culture, secreted laminin-332 into the matrix. The addition of laminin-332 increased ADPKD in vitro cyst formation [16], whereas, a function blocking antibody for the laminin reduced cyst formation. This effect of laminin-332 on cyst growth appeared to be mediated by the activation of ERK-dependent cell proliferation. In cancer, laminin-332 and α6β4 are important for cell adhesion, proliferation, migration, and invasion of cancer cells, and are potential targets for treatment [56]. In support of the hypothesis that abnormalities in basement membrane laminin is associated with PKD pathogenesis, Miner and associates showed that a hypomorphic mutation in mouse laminin α5 was sufficient to cause PKD. The basement membranes of these cysts were thickened containing an aberrant accumulation of laminin-332. These mice developed proteinuria and subsequent death due to renal failure [57].
In PCK rats, an orthologous model of ARPKD with focal corticomedullary cysts similar to human ADPKD, there was aberrant expression of laminin-γ2 as early as postnatal day 2 and elevated laminin-332 protein by day 30, coinciding with cystogenesis [58]. The cystic basement membranes appeared to be focally evaginated into the interstitial compartment (Figure 2). Laminin-511, which has been implicated in terminal differentiation, was absent in the basement membranes of the PCK cysts. Laminin-332 mRNA was elevated in human ARPKD (2-fold for α3; 4-fold for β3; and 10-fold for γ2). Immunohistochemistry analysis, using an antibody that recognizes trimeric laminin-332, revealed abundant staining from laminin-332 in a human ARPKD kidney, but no staining in normal human kidneys [58]. In this study, cystic cells derived from the Oak Ridge Polycystic Kidney (ORPK) mice also had elevated expression of laminin-332 and higher proliferation rates compared to laminin-332 rescued cells. Treatment with a function-blocking antibody to laminin-332 inhibited ORPK cell proliferation, further supporting a role for this laminin in cystogenesis.
Figure 2. Basement membrane abnormalities begin as early as postnatal day (PN) 21 in PCK rat kidneys.
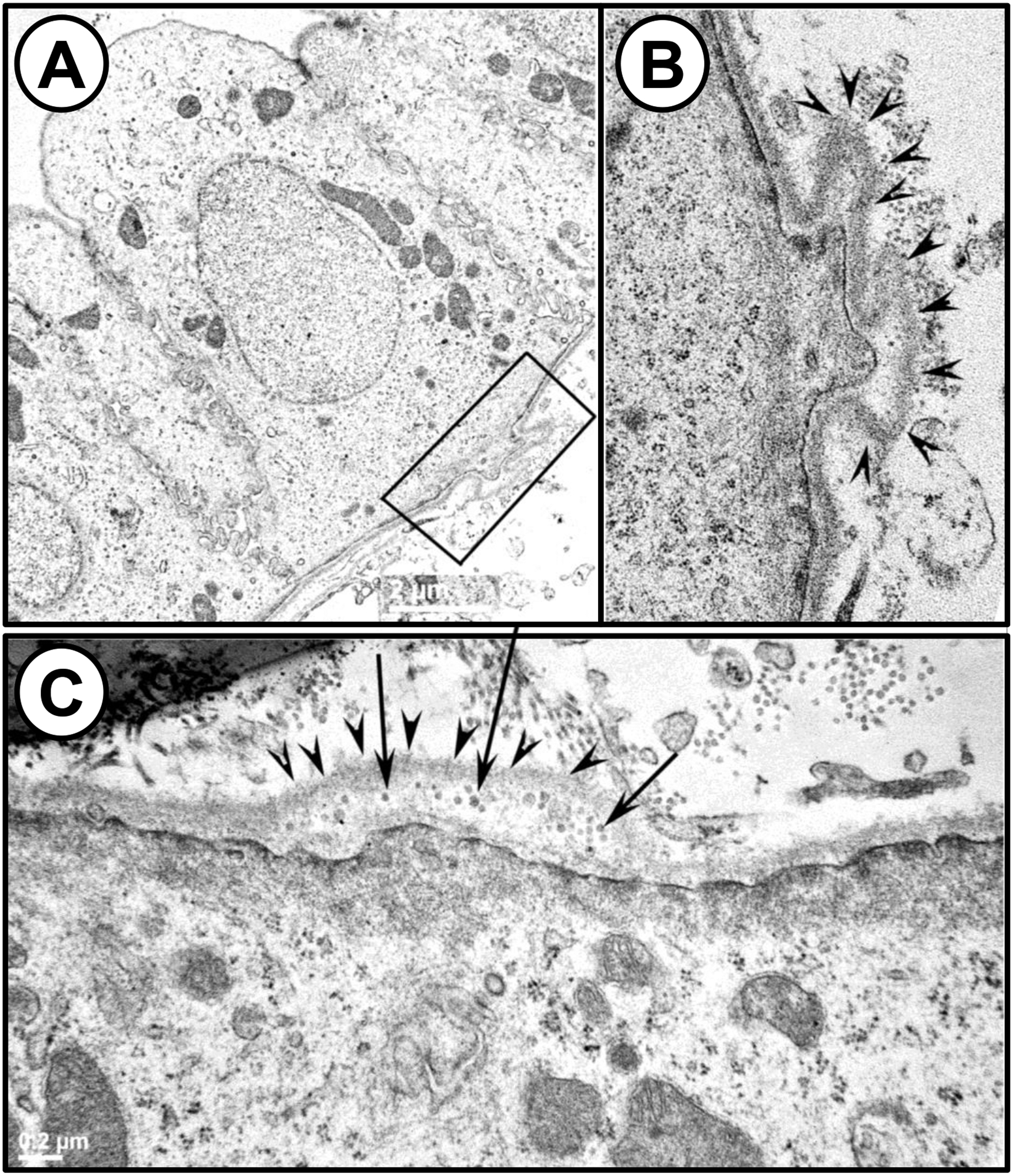
(A) TEM image of pericystic epithelia from a PN 21 PCK rat kidney. The region highlighted by the rectangular box shows evaginations of the basement membrane into the interstitial compartment. (B) Magnified view of the region highlighted by the rectangular box in A. The evaginations of the basement membrane are marked with arrowheads in this image. (C) A different image of a PN21 PCK pericystic kidney region, revealing the presence of vesicle-like structures (arrows) in the evaginations of the basement membrane (arrowheads) on the basal surface of the pericystic epithelia. Republished from Vijayakumar et al. Am J Physiol Renal Physiol 306: F640-F654, 2014.
4. Integrin signaling in PKD
4.1. Integrins mediate ECM-cell communication
Integrins are the main receptor proteins that bind the ECM allowing cells to respond to changes in ECM composition. Integrins are composed of two noncovalently associated transmembrane glycoproteins - the α and β subunits. Currently, there are 18 α subunits and 8 β subunits that make up 24 distinct integrins (αβ heterodimers) [59]. Integrins functionally link the cell cytoskeleton to the ECM by connecting to bundles of actin filaments and anchoring the cell to its surrounding environment. Integrins mediate cell signal transduction by two general mechanisms - “inside-out” signaling and “outside-in” signaling [60]. The outside-in signaling involves integrin binding to its ligand leading to activation of cellular pathways involved in cell attachment, migration, survival, differentiation, and proliferation. After an integrin binds to its ligand, the β subunit binds intracellular anchor proteins, such as talin, α-actinin and filamin. The anchor proteins either directly bind actin or other anchor proteins, such as vinculin, linking the integrin to the cellular actin cytoskeleton. This leads to integrin clustering and formation of focal adhesions between the cell and the ECM [60]. It appears that this interaction provides a stronger bond by locking the integrin in a conformation that allows the integrin to bind to the ECM ligand more tightly. The formation of focal adhesions is accompanied by the recruitment of focal adhesion kinase (FAK) and Src, and several adaptor proteins such as integrin-linked kinase (ILK) and paxillin. This, in turn, leads to the activation of intracellular signaling pathways involved in cell migration, survival, and proliferation, and alterations in gene expression [60]. Cellular changes, in turn, affect the composition and organization of the ECM components. This inside-out signaling can also involve cellular regulation of the affinity of the integrin to the ECM ligand. This is thought to involve a conformational change from the cytoplasmic domain of the integrin that affects extracellular binding, which in turn affects cell adhesion and the response to ligand binding. This bidirectional communication between the cell and its ECM is referred to as “dynamic reciprocity” and is important for proper tissue development and homeostasis [61].
4.2. Upregulation of integrins alters adhesion and motility in PKD
Dysregulation of integrin expression is well-established in ADPKD and is a key feature of tissue remodeling during cyst formation. The β1-integrin subunit is the most prevalent β-subunit in the kidney, and it combines with several different α-integrin subunits. In normal fetal kidneys, α2- and α3- subunits are expressed in distal tubules and collecting ducts, whereas the α6-subunit is expressed in all nephron segments [62–64]. There is a marked increase in the α1-subunit in precystic and cystic collecting duct cells in fetal ADPKD and ARPKD kidneys, indicating that increased expression of the α1 subunit may be an early event in cyst formation. There were also irregular staining patterns for α2-, α3-, and α6-integrin subunits; whereas, expression of the α5-subunit appeared to be unchanged [64]. Human ADPKD cells have elevated expression of α2β1-integrin and increased adhesion to type 1 collagen [65], and incubation with a function blocking monoclonal antibody to the α2-subunit inhibited this attachment. The α8-subunit, which heterodimerizes exclusively with β1 and is typically expressed in vasculature and glomerular mesangium, was shown to have de novo expression in cystic epithelial cells [66]. This may be particularly important since, tenascin and osteopontin, ligands for α8β1 integrin, are also overexpressed by cyst lining cells [67–69].
α6β4-integrin and its ligand laminin-332 (laminin 5) were shown to be overexpressed and that their interaction enhanced ADPKD cell adhesion and migration [43]. More recently, αVβ3 integrin, a receptor for vitronectin and periostin, was found to be overexpressed 9-fold in human ADPKD cells [42]. A growing body of evidence supports the hypothesis that changes in integrin expression influence the development of cysts, possibly through alterations in the cell-matrix interactions [20, 70] that stimulate pathways found to be important in cyst growth. Concomitant increases in integrins and their ligands may trigger an activation loop that supports the survival and proliferation of cystic cells.
4.3. Role of integrins in cystogenesis
The β-integrin subunits physically interact with the cytoskeleton at focal contacts and associate with the actin-filament binding molecules talin and α-actinin, providing the structural link between the ECM and the cytoskeleton [71, 72]. Early studies showed that the β1-subunit is required for kidney collecting duct morphogenesis and proper renal function. Embryonic collecting duct-specific deletion of the β1-integrin subunit using the Itgb1f/f mouse crossed with the HoxB7-Cre recombinase mouse led to renal dysfunction, loss of medullary collecting duct (CD) expression of aquaporin-2 and arginine vasopressin receptor 2 (AVP V2R), and hypoplastic medullary CDs [73]. There was a range of defects from small kidneys with cysts and dilated tubules to renal agenesis. It is likely that the cysts were a consequence of tubule malformation, cell shedding due to apoptosis leading to tubule obstruction and elevated intratubular fluid, rather than a direct mitogenic effect on the cells. Lee et al. used a conditional double knockout mouse in which both Pkd1 and Itgb1 were inactivated specifically in the CD using an AQP2-Cre recombinase to investigate the role of β1-integrin subunits on PKD progression [74]. The use of a mouse in which Cre was under the control of the AQP2 promoter allowed Itgb1 and Pkd1 inactivation at a later developmental stage, circumventing the lethality associated with early ablation of either Pkd1 or Itgb1. Single knockout of Pkd1 using the AQP2-Cre resulted in a significant increase in kidney weight (KW), represented as a percentage of body weight (BW) in the mice by 4 weeks of age due to extensive cyst growth, leading to fibrosis and elevated blood urea nitrogen (BUN). The loss of collecting duct expression of the β1-integrin subunit in the double knockout mice almost eliminated cyst formation in the kidneys. There was no change in KW/BW, fibrosis, or BUN compared to the wildtype mice, and 100% of the PKD mice with CD-specific knockout of the β1-integrin subunit survived beyond 15 months [74].
Kreidberg et al. showed that α3 integrin, which is expressed during renal development, plays a key role in renal development [75]. A targeted mutation in Itga3 resulted in fewer and shorter CDs, indicating a disruption in branching morphogenesis and/or tubule extension. Interestingly, this group also found small cysts in the α3 integrin mutant kidneys, suggesting that dysregulation of the α3β1 integrin may be related to cystogenesis. This group later showed that in Pkd1−/− cells, α3β1 integrin was mostly sequestered in the Golgi, along with the E3 ubiquitin ligase c-Cbl and there was subsequent overexpression of c-MET, a receptor tyrosine kinase target of c-Cbl [76]. Increased c-MET resulted in elevated mTOR activation, a key signaling pathway in cyst growth. Additional studies will be needed to delineate the specific roles of the β1-integrins in PKD progression.
4.4. Periostin, a ligand for αVβ3 integrin, promotes cyst growth and fibrosis in PKD
Our laboratory was the first to demonstrate that periostin, a ligand for αVβ3-integrins, plays an important role in the progression of PKD [42]. In normal adults, periostin expression is restricted to collagen-rich tissues under mechanical stress such as the periosteum, periodontal ligaments, and heart values and ventricles [46, 48]. Periostin is thought to maintain the integrity of the ECM and regulate the biomechanical property of connective tissue by promoting collagen cross-linking [47, 77]. Periostin is unique in that it contains four FAS1 domains, which have homology to fasciclin-1, a neural adhesion protein in insects. Periostin is secreted into the ECM and can bind αVβ3- and αVβ5-integrins through its FAS1 domains to regulate several cell functions, including cell survival, migration, and proliferation. Evidence indicates that periostin stimulates signaling pathways involved in tissue repair, including epithelial-mesenchymal transition (EMT), cell adhesion, proliferation, survival, and matrix production [47, 78–84]. Periostin binding to αVβ3-integrin is also thought to promote the release of TGF-β from latent TGF-β-binding protein. Increased levels of active TGF-β, in turn, stimulate ECM production and periostin secretion through the SMAD signaling pathway [85, 86], potentially contributing to a positive feedback mechanism leading to periostin/αVβ3-integrin/TGF-β-induced fibrosis.
Periostin is expressed within the nephrogenic zone during renal development; however, it is not expressed in healthy adult kidney, indicating that the protein does not play a role in normal renal function [55, 87]. We discovered that periostin is highly overexpressed by cyst-lining epithelial cells and accumulates within the matrix adjacent to renal cysts of human ADPKD and ARPKD patients, and several rodent models of PKD [42]. ADPKD cyst epithelial cells also overexpress the αV-integrin subunit, and binding of periostin to αVβ3-integrin activates AKT/mTOR signaling and the proliferation of ADPKD cells. Global knockout of periostin in pcy/pcy mice, a slowly progressive PKD model, significantly reduced renal mTOR signaling, cell proliferation, cyst number, cystic index, and KW/BW. Periostin ablation also reduced renal fibrosis, preserved renal function, and significantly extended the lifespan of the mice [88]. In a follow-up study, a conditional periostin (Postn) transgenic mouse was used to selectively overexpress periostin in cells of the renal collecting duct (CD), a predominant site for cyst formation [19]. Interestingly, CD-specific overexpression of periostin was not sufficient to induce cyst formation in normal mice. On the other hand, CD overexpression of periostin in pcy/pcy mice increased renal AKT/mTOR signaling and cell proliferation, leading to accelerated cyst growth, severe renal fibrosis, and a more rapid decline in kidney function. These studies provided strong evidence that renal expression of periostin and αVβ3-integrin signaling contribute importantly to cyst growth and fibrosis in PKD downstream of cyst initiation.
4.5. The role of integrin-linked kinase (ILK) on cyst epithelial cell proliferation
The binding of integrin to ECM ligands leads to integrin clustering, recruitment of multiple structural and signaling molecules, and the formation of focal adhesions. Focal adhesions are the principal cellular structure for cell-matrix interactions and are composed of a large multi-protein complex that connects the actin cytoskeleton to the ECM [89]. ILK, a major component of the focal adhesion, interacts directly with the cytoplasmic domain of the β-integrin subunit and forms a signaling complex with paxillin, α-parvin, actin, and several adaptor proteins, thereby providing a physical link between the ECM/integrin and actin cytoskeleton. ILK is also an essential intermediate for communication between the ECM and cellular signaling pathways, including those involved in EMT and cell proliferation [90, 91]. ILK was originally characterized as a protein kinase [92–94]; however, several studies have shed doubt as to ILK being a bona fide kinase [95–97]. It is now generally agreed that ILK functions as a scaffolding protein with adaptor proteins PINCH and α-parvin. Activation of the ILK-PINCH-Parvin (IPP) signaling complex leads to the phosphorylation of downstream molecules such as the AKT/mTOR and GSK-3β pathways.
We demonstrated that basal ILK activity was elevated in human ADPKD cells compared to NHK cells and that periostin increased ILK activity, the phosphorylation of AKT and S6 kinase, and stimulated ADPKD cell proliferation [98]. CDP-22, a small-molecule ILK inhibitor, completely blocked periostin-induced phosphorylation of AKT and S6 kinase, suggesting that ILK may be a potential therapeutic target to slow renal cyst growth. To examine renal ILK in vivo, Ilkfl/fl mice were crossed with Pkhd1-Cre mice to knock out ILK in CD cells. Mice with CD-specific knockout of ILK (ILK−/− CD mice) had lower BW and KW/BW and developed a urinary concentrating defect [98, 99]. These mice had declining renal function and typically died by 20 weeks of age. ILK−/− CD mice had apoptotic cells within the lumens of the CDs, consistent with caspase-3-mediated anoikis [100]. By contrast, the loss of only one ILK allele in Ilkfl/+: Pkhd1-Cre mice was well-tolerated [98]. These mice had normal body weight, KW/BW, renal morphology and function, and lived beyond one year.
The effect of ILK knockdown was tested in two PKD mouse models - Pkd1fl/fl: Pkhd1-Cre mice, a rapidly progressive orthologous ADPKD model, and the NPHP3 pcy/pcy mice [98]. Decreased CD expression of ILK in Pkd1fl/fl: Ilk fl/+: Pkhd1-Cre mice reduced the number of renal cysts, cystic area, mTOR activity and cell proliferation. These mice had decreased KW/BW and BUN and had a 27% increase in lifespan. These results were confirmed by knocking down ILK in CDs of pcy/pcy mice. These pcy/pcy: Ilk fl/+: Pkhd1-Cre mice had decreased in renal cystic disease and fibrosis, improved renal function, and a 20% increase in lifespan compared to pcy/pcy mice with normal ILK expression. These studies demonstrated that ILK is a central intermediate for periostin-induced activation of AKT/mTOR signaling and proliferation of cystic epithelial cells, and that persistent ILK activation contributes importantly to PKD progression and the decline in renal function.
5. The polycystins associate with focal adhesion components and ECM proteins
5.1. The role of the polycystins in the focal adhesion.
Polcystin-1 (PC-1) localizes at cell-cell and cell-matrix contacts, as well as primary cilia [5]. PC-1 was shown to co-localize and co-immunoprecipitate with α2β1-integrin, focal adhesion proteins (FAK and paxillin), actin-binding proteins (vinculin, talin, and α-actinin), and cell adherens junction proteins (E-cadherin and β-catenin) in normal human fetal collecting duct cells [65]. The authors of the study suggested that during renal development the PC-1 multi-protein complex interacts with focal adhesions and/or adherens junction proteins to coordinate the assembly and disassembly of focal adhesions for proper ureteric bud migration and normal tubule epithelial differentiation [101]. Castelli et al. showed that cells that overexpressed PC-1 had enhanced adhesion rates, while cells that lacked PC-1 expression had reduced capacity to adhere to substrates [102]. They also found that PC-1 regulation of the activation status of FAK modulates the stability of the microtubules, as well as the actin cytoskeleton, impacting the turnover rate of focal adhesions in migrating cells. Overexpression of a PC-1 C-terminal fragment in CD cells stimulated the phosphorylation of FAK and paxillin, increased the association between FAK and paxillin, and stimulated the formation of focal adhesion complexes [103]. It may be reasoned that the loss of PC-1 during renal development causes to abnormal turnover of focal adhesions, leading to aberrant tubule formation and incomplete epithelial differentiation, hallmarks of initial cyst formation in ADPKD kidneys. PC-2 may also function in focal adhesions. In a yeast two-hybrid screen, the carboxyl termini of polycystin-2 and CD2AP, an adapter protein that functions in the assembly of focal adhesion complexes, were shown to be strong interacting partners [104].
5.2. The role of polycystin-1 as a mechanosensor of the ECM.
PC-1 has been classified as a mechanosensor based on its ability to interact with the ECM and the cell cytoskeleton through intermediate filaments [105]. It is generally thought that PC-1 and PC-2 form a receptor-channel complex involved in mechanosensation, possibly through the bending of primary cilia on the luminal surface of renal epithelial cells [7, 106]. Recent evidence suggests that the ECM, in addition to having a structural role, serves as a biomechanical signal for PC-1 that is critical for normal cell function [107]. It has been hypothesized that PC-1 enables the cell to sense the rigidity of the ECM and that the loss of this signal in PKD cells contributes to cystogenesis [107–109]. Nigro et al. showed that PC-1 negatively regulates cell contraction and activation of the Hippo signaling effector YAP in response to extracellular stiffness [107]. Loss of PC-1 activates YAP and its transcriptional target c-Myc, which promotes PKD pathogenesis. Yap activation involves the RhoA-ROCK signaling module and RhoA inhibition suppressed in vitro cyst formation of Pkd1-mutant kidney cells grown in a collagen matrix [109]. Previous work had shown that YAP is abnormally activated in cystic cells of human ADPKD and Pkd1-deficient mouse kidneys [110]. PKD mice were also found to have increased actomyosin contraction and inhibition of ROCK-dependent actomyosin contraction reduced renal YAP activity and PKD progression [107], further supporting the role of RhoA-ROCK-YAP-c-Myc signaling axis in PKD pathogenesis. Additional studies will be needed to delineate the role of PC-1 as a mechanosensor for ECM stiffness.
5.3. Role of polycystin-1 in cell-matrix and cell-cell interactions
PC-1 has a large N-terminal extracellular domain that contains multiple motifs that appear to be involved in cell-matrix and cell-cell interactions, including a leucine rich repeat (LRR), a C-type lectin domain, and Ig-like PKD repeats [3, 111]. In vitro assays showed that a fusion protein containing the LRR domain of PC-1 interacted with type I collagen, fibronectin, and laminin. Treatment with an LRR fusion protein reduced the proliferation of cultured cells, demonstrating a possible functional role of the LRR domain on the ability of PC-1 to interact with the ECM and regulate cell proliferation [112]. PC-1 has a conserved C-type lectin motif, which is commonly involved in Ca2+-dependent binding to carbohydrates [113]. A fusion protein containing an isolated C-type lectin domain from PC-1 was able to bind type IV collagen and laminin, two major components of basement membranes, in a Ca2+-dependent manner. This domain also interacted with type I and II collagens, in vitro. These data support the hypothesis that PC-1 participates in interactions between the cell and its basement membrane.
The extracellular domain of PC-1 contains 16 copies of a novel module called the “PKD domain.” These domains have high similarity to immunoglobulin and are likely adhesion domains [114]. The PKD domains develop strong Ca2+-independent homophilic interactions [115], suggesting that PC-1 may facilitate cell-cell interactions. Treatment with peptides derived from the PKD domain of PC-1 inhibited ureteric bud branching and the number of nephrons in embryonic kidneys ex vivo [116], consistent with a disruption of the PC-1 interactions. Furthermore, antibodies to these PKD domains disrupted cell-cell connections in MDCK cell monolayers. Taken together, these data support the hypothesis that PC1 mediates intercellular adhesion as well as cell-matrix adhesion. How this translates to cyst formation in ADPKD has yet to be determined.
6. Summary
Abnormal expression of ECM components, including collagens, laminin, and proteoglycans is a well-established feature of early cyst formation and progressive fibrosis in PKD kidneys. During the past 20 years, evidence has shown that cystic epithelial cells overexpress several integrins and ECM soluble factors, including matricellular proteins periostin and osteopontin. Binding of ECM and matricellular ligands to specific integrins activates ILK and FAK and downstream signaling pathways involved in tissue repair, EMT, cell proliferation and survival, and migration (Figure 3). Several studies have demonstrated that PC-1 interacts with components of focal adhesions and that PC-1 facilitates cell-cell and cell-matrix interactions. Future studies will be needed to further delineate these receptor-ligand pathways to better understand the influence of the microenvironment and to develop novel therapeutic approaches to inhibit their effects on cyst growth and fibrosis in PKD.
Figure 3. Proposed model for changes in extracellular matrix (ECM), focal adhesions, and integrin signaling in ADPKD.
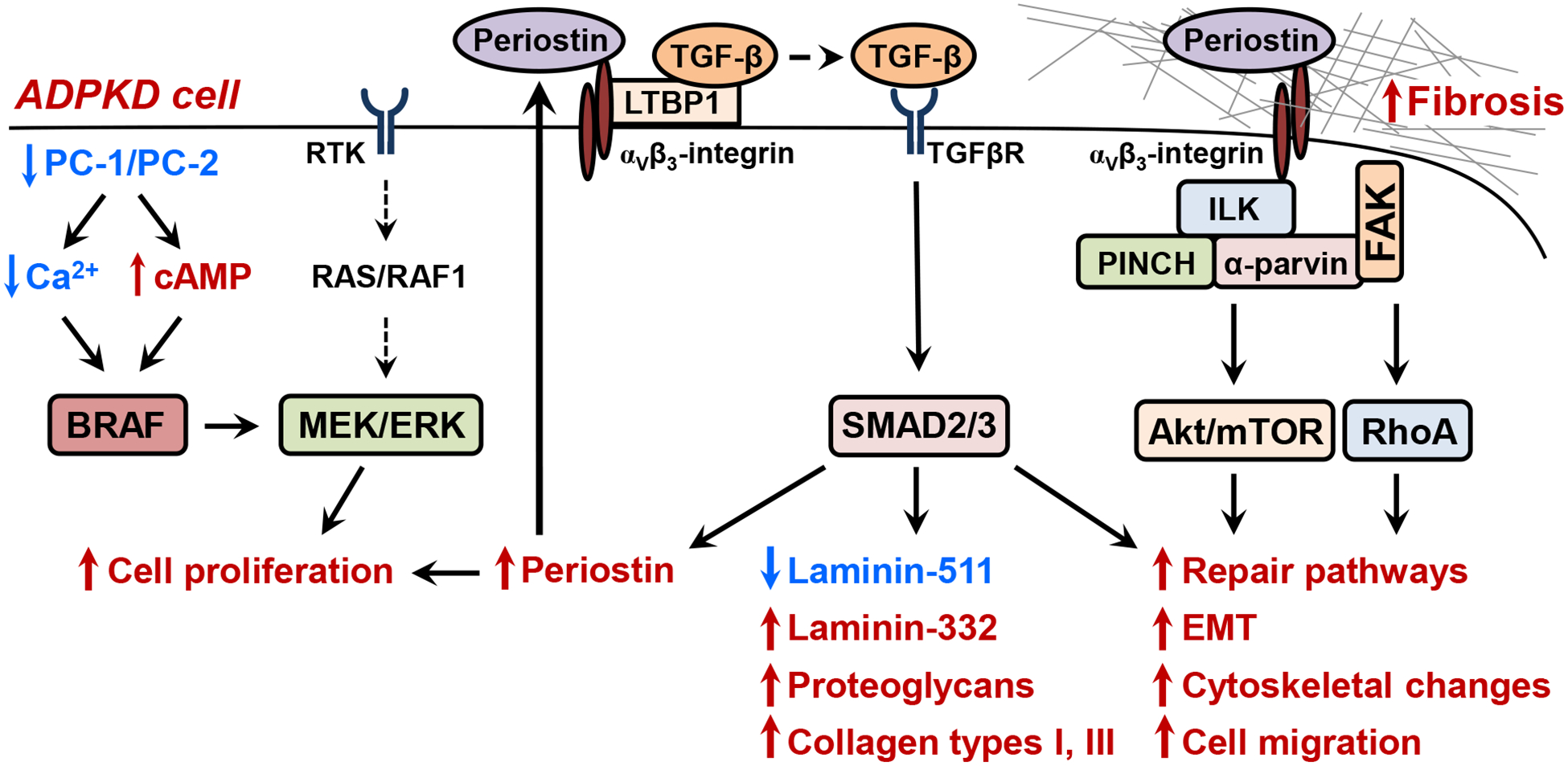
The functional loss of PC1-PC2 is thought to reduce intracellular Ca2+ homeostasis, lowering the activity of PDE1, a Ca2+ -dependent phosphodiesterase. Reduced PDE1 activity increases basal intracellular cAMP levels and causes a greater cAMP response to arginine vasopressin [15, 117]. Lower intracellular Ca2+ de-represses BRAF, a kinase upstream of the MEK/ERK pathway, leading to cAMP-dependent cell proliferation [8]. Cystic cells remain poorly differentiated and have abnormal expression of TGF-β and periostin, a matricellular protein involved in tissue repair [42]. TGF-β, the key cytokine for ECM production, causes overexpression of laminin −332 (with a concurrent reduction in laminin-511), proteoglycans, and collagen types I and II. Excessive ECM production contributes to interstitial fibrosis and cystogenesis [16, 20, 58, 118]. Periostin binding to αVβ3-integrins causes integrin bundling, formation of focal adhesions, and activation of focal adhesion kinase (FAK) [98]. Integrin activation stimulates integrin-linked kinase (ILK), a pseudokinase that is in a signaling complex with PINCH and α-Parvin. Periostin activation of the ILK-PINCH-Parvin complex and FAK stimulates pathways involved in tissue repair, including AKT/mTOR-dependent cell proliferation and survival, epithelial-mesenchymal transition (EMT), cytoskeletal rearrangement, and cell migration [88]. Periostin binding to αVβ3-integrin is also thought to release TGF-β from latent TGF-β binding protein 1 (LTBP1), and active TGF-β stimulates the SMAD pathway to increase ECM production and cause a further production of periostin, causing a feed-forward mechanism for renal fibrosis. Periostin has also been shown to activate lysyl oxidase, an enzyme that causes cross -linking of the collagen fibrils leading to fibrosis. Receptor tyrosine kinase (RTK); PINCH (particularly interesting new cysteine histidine-rich protein); Ras homology family member RhoA; Transforming growth factor-β (TGF-β) and its receptor (TGFβR).
Highlights.
Aberrant expression of extracellular matrix (ECM) and alterations in the basement membrane are early features of renal cyst development in polycystic kidney disease (PKD).
Abnormal expression of laminin-332 and its receptor α6β4-integrin promote cyst epithelial cell proliferation.
In PKD kidneys, cyst-lining cells overexpress periostin, a matricellular protein involved in tissue repair and fibrosis.
Periostin binding to αVβ3-integrin stimulates the integrin-linked kinase (ILK)-PINCH-Parvin signaling complex, leading to activation of tissue repair pathways, epithelial-mesenchymal transition (EMT) and proliferation of cyst epithelial cells.
7. Acknowledgements
The authors are grateful to Dr. James Calvet for helpful suggestions during the preparation of the manuscript. This work was supported by grants from the National Institutes of Health USA (R01 DK081579 and P30 DK106912).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
8. References
- [1].Grantham JJ, Clinical practice. Autosomal dominant polycystic kidney disease, N Engl J Med 359 (2008) 1477–85. [DOI] [PubMed] [Google Scholar]
- [2].Consortium EPKD, The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16, Cell 77 (1994) 881–894. [DOI] [PubMed] [Google Scholar]
- [3].Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, Gamble V, Harris PC, The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains, Nature genetics 10 (1995) 151–60. [DOI] [PubMed] [Google Scholar]
- [4].Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S, PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein, Science 272 (1996) 1339–42. [DOI] [PubMed] [Google Scholar]
- [5].Boletta A, Germino GG, Role of polycystins in renal tubulogenesis, Trends Cell Biol 13 (2003) 484–92. [DOI] [PubMed] [Google Scholar]
- [6].Kim S, Nie H, Nesin V, Tran U, Outeda P, Bai CX, Keeling J, Maskey D, Watnick T, Wessely O, Tsiokas L, The polycystin complex mediates Wnt/Ca(2+) signalling, Nature cell biology 18 (2016) 752–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J, Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells, Nature genetics 33 (2003) 129–37. [DOI] [PubMed] [Google Scholar]
- [8].Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP, Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype, J Biol Chem 279 (2004) 40419–30. [DOI] [PubMed] [Google Scholar]
- [9].Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, Torra R, Breuning M, Ravine D, Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group, Lancet 353 (1999) 103–7. [DOI] [PubMed] [Google Scholar]
- [10].Harris PC, Bae KT, Rossetti S, Torres VE, Grantham JJ, Chapman AB, Guay-Woodford LM, King BF, Wetzel LH, Baumgarten DA, Kenney PJ, Consugar M, Klahr S, Bennett WM, Meyers CM, Zhang QJ, Thompson PA, Zhu F, Miller JP, Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease, Journal of the American Society of Nephrology : JASN 17 (2006) 3013–9. [DOI] [PubMed] [Google Scholar]
- [11].Cornec-Le Gall E, Audrezet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, Charasse C, Whebe B, Renaudineau E, Jousset P, Guillodo MP, Grall-Jezequel A, Saliou P, Ferec C, Le Meur Y, Type of PKD1 mutation influences renal outcome in ADPKD, Journal of the American Society of Nephrology : JASN 24 (2013) 1006–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Grantham JJ, 1992 Homer Smith Award. Fluid secretion, cellular proliferation, and the pathogenesis of renal epithelial cysts, Journal of the American Society of Nephrology : JASN 3 (1993) 1841–57. [DOI] [PubMed] [Google Scholar]
- [13].Harris PC, Torres VE, Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease, J Clin Invest 124 (2014) 2315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Grantham JJ, Mangoo-Karim R, Uchic ME, Grant M, Shumate WA, Park CH, Calvet JP, Net fluid secretion by mammalian renal epithelial cells: stimulation by cAMP in polarized cultures derived from established renal cells and from normal and polycystic kidneys, Trans Assoc Am Physicians 102 (1989) 158–62. [PubMed] [Google Scholar]
- [15].Wallace DP, Cyclic AMP-mediated cyst expansion, Biochim Biophys Acta 1812 (2011) 1291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Joly D, Berissi S, Bertrand A, Strehl L, Patey N, Knebelmann B, Laminin 5 regulates polycystic kidney cell proliferation and cyst formation, J Biol Chem 281 (2006) 29181–9. [DOI] [PubMed] [Google Scholar]
- [17].Mangos S, Lam PY, Zhao A, Liu Y, Mudumana S, Vasilyev A, Liu A, Drummond IA, The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation, Disease models & mechanisms 3 (2010) 354–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu B, Li C, Liu Z, Dai Z, Tao Y, Increasing extracellular matrix collagen level and MMP activity induces cyst development in polycystic kidney disease, BMC Nephrol 13 (2012) 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Raman A, Parnell SC, Zhang Y, Reif GA, Dai Y, Khanna A, Daniel E, White C, Vivian JL, Wallace DP, Periostin overexpression in collecting ducts accelerates renal cyst growth and fibrosis in polycystic kidney disease, Am J Physiol Renal Physiol 315 (2018) F1695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wilson PD, Hreniuk D, Gabow PA, Abnormal extracellular matrix and excessive growth of human adult polycystic kidney disease epithelia, Journal of Cellular Physiology 150 (1992) 360–369. [DOI] [PubMed] [Google Scholar]
- [21].Song CJ, Zimmerman KA, Henke SJ, Yoder BK, Inflammation and Fibrosis in Polycystic Kidney Disease, Results Probl Cell Differ 60 (2017) 323–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Norman J, Fibrosis and progression of autosomal dominant polycystic kidney disease (ADPKD), Biochim Biophys Acta 1812 (2011) 1327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cuppage FE, Huseman RA, Chapman A, Grantham JJ, Ultrastructure and function of cysts from human adult polycystic kidneys, Kidney Int 17 (1980) 372–81. [DOI] [PubMed] [Google Scholar]
- [24].Frantz C, Stewart KM, Weaver VM, The extracellular matrix at a glance, J Cell Sci 123(Pt 24) (2010) 4195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kruegel J, Miosge N, Basement membrane components are key players in specialized extracellular matrices, Cell Mol Life Sci 67 (2010) 2879–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wilson PD, Schrier RW, Breckon RD, Gabow PA, A new method for studying human polycystic kidney disease epithelia in culture, Kidney Int 30 (1986) 371–8. [DOI] [PubMed] [Google Scholar]
- [27].Milutinovic J, Agodoa LY, Potential causes and pathogenesis in autosomal dominant polycystic kidney disease, Nephron 33 (1983) 139–44. [DOI] [PubMed] [Google Scholar]
- [28].Ebihara I, Killen PD, Laurie GW, Huang T, Yamada Y, Martin GR, Brown KS, Altered mRNA expression of basement membrane components in a murine model of polycystic kidney disease, Lab Invest 58 (1988) 262–9. [PubMed] [Google Scholar]
- [29].Jin H, Carone FA, Nakamura S, Liu ZZ, Kanwar YS, Altered synthesis and intracellular transport of proteoglycans by cyst-derived cells from human polycystic kidneys, Journal of the American Society of Nephrology : JASN 2 (1992) 1726–33. [DOI] [PubMed] [Google Scholar]
- [30].Calvet JP, Polycystic kidney disease: primary extracellular matrix abnormality or defective cellular differentiation?, Kidney Int 43 (1993) 101–8. [DOI] [PubMed] [Google Scholar]
- [31].Carone FA, Rowland RG, Perlman SG, Ganote CE, The pathogenesis of drug-induced renal cystic disease, Kidney Int 5 (1974) 411–21. [DOI] [PubMed] [Google Scholar]
- [32].Grantham JJ, Donoso VS, Evan AP, Carone FA, Gardner KD Jr., Viscoelastic properties of tubule basement membranes in experimental renal cystic disease, Kidney Int 32 (1987) 187–97. [DOI] [PubMed] [Google Scholar]
- [33].Vijayakumar S, Extracellular Matrix Abnormalities in Polycystic Kidney Disease, Novel Insights on Chronic Kidney Disease, Acute Kidney Injury and Polycystic Kidney Disease, Vijaykumar (Ed.) InTech, (2012). [Google Scholar]
- [34].Nagao S, Yamaguchi T, Kusaka M, Maser RL, Takahashi H, Cowley BD, Grantham JJ, Renal activation of extracellular signal-regulated kinase in rats with autosomal-dominant polycystic kidney disease, Kidney Int 63 (2003) 427–37. [DOI] [PubMed] [Google Scholar]
- [35].Yamaguchi T, Nagao S, Wallace DP, Belibi FA, Cowley BD Jr., Pelling JC, Grantham JJ, Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys, Kidney Int 63 (2003) 1983–1994. [DOI] [PubMed] [Google Scholar]
- [36].Candiano G, Gusmano R, Altieri P, Bertelli R, Ginevri F, Coviello DA, Sessa A, Caridi G, Ghiggeri GM, Extracellular matrix formation by epithelial cells from human polycystic kidney cysts in culture, Virchows Arch B Cell Pathol Incl Mol Pathol 63 (1992) 1–9. [DOI] [PubMed] [Google Scholar]
- [37].Schafer K, Bader M, Gretz N, Oberbaumer I, Bachmann S, Focal overexpression of collagen IV characterizes the initiation of epithelial changes in polycystic kidney disease, Exp Nephrol 2 (1994) 190–5. [PubMed] [Google Scholar]
- [38].Chea SW, Lee KB, TGF-beta mediated epithelial-mesenchymal transition in autosomal dominant polycystic kidney disease, Yonsei medical journal 50 (2009) 105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Togawa H, Nakanishi K, Mukaiyama H, Hama T, Shima Y, Sako M, Miyajima M, Nozu K, Nishii K, Nagao S, Takahashi H, Iijima K, Yoshikawa N, Epithelial-to-mesenchymal transition in cyst lining epithelial cells in an orthologous PCK rat model of autosomal-recessive polycystic kidney disease, Am J Physiol Renal Physiol 300 (2011) F511–20. [DOI] [PubMed] [Google Scholar]
- [40].Drummond IA, Polycystins, focal adhesions and extracellular matrix interactions, Biochim Biophys Acta 1812 (2011) 1322–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Calvet JP, Injury and development in polycystic kidney disease, Current opinion in nephrology and hypertension 3 (1994) 340–8. [DOI] [PubMed] [Google Scholar]
- [42].Wallace DP, Quante MT, Reif GA, Nivens E, Ahmed F, Hempson SJ, Blanco G, Yamaguchi T, Periostin induces proliferation of human autosomal dominant polycystic kidney cells through alphaV-integrin receptor, Am J Physiol Renal Physiol 295 (2008) F1463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Joly D, Morel V, Hummel A, Ruello A, Nusbaum P, Patey N, Noel LH, Rousselle P, Knebelmann B, Beta4 integrin and laminin 5 are aberrantly expressed in polycystic kidney disease: role in increased cell adhesion and migration, Am J Pathol 163 (2003) 1791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Song X, Di Giovanni V, He N, Wang K, Ingram A, Rosenblum ND, Pei Y, Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks, Hum Mol Genet 18 (2009) 2328–43. [DOI] [PubMed] [Google Scholar]
- [45].Schieren G, Rumberger B, Klein M, Kreutz C, Wilpert J, Geyer M, Faller D, Timmer J, Quack I, Rump LC, Walz G, Donauer J, Gene profiling of polycystic kidneys, Nephrol Dial Transplant 21 (2006) 1816–24. [DOI] [PubMed] [Google Scholar]
- [46].Frangogiannis NG, Matricellular proteins in cardiac adaptation and disease, Physiol Rev 92 (2012) 635–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kudo A, Periostin in fibrillogenesis for tissue regeneration: periostin actions inside and outside the cell, Cell Mol Life Sci 68 (2011) 3201–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Horiuchi K, Amizuka N, Takeshita S, Takamatsu H, Katsuura M, Ozawa H, Toyama Y, Bonewald LF, Kudo A, Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta, J Bone Miner Res 14 (1999) 1239–49. [DOI] [PubMed] [Google Scholar]
- [49].Takeshita S, Kikuno R, Tezuka K, Amann E, Osteoblast-specific factor 2: cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I, Biochem J 294 (Pt 1) (1993) 271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bornstein P, Sage EH, Matricellular proteins: extracellular modulators of cell function, Curr Opin Cell Biol 14 (2002) 608–16. [DOI] [PubMed] [Google Scholar]
- [51].Chiodoni C, Colombo MP, Sangaletti S, Matricellular proteins: from homeostasis to inflammation, cancer, and metastasis, Cancer metastasis reviews 29 (2010) 295–307. [DOI] [PubMed] [Google Scholar]
- [52].Kormann R, Kavvadas P, Placier S, Vandermeersch S, Dorison A, Dussaule JC, Chadjichristos CE, Prakoura N, Chatziantoniou C, Periostin Promotes Cell Proliferation and Macrophage Polarization to Drive Repair after AKI, Journal of the American Society of Nephrology : JASN 31 (2020) 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Okamoto H, Imanaka-Yoshida K, Matricellular proteins: new molecular targets to prevent heart failure, Cardiovasc Ther 30 (2012) e198–209. [DOI] [PubMed] [Google Scholar]
- [54].Walker JT, McLeod K, Kim S, Conway SJ, Hamilton DW, Periostin as a multifunctional modulator of the wound healing response, Cell Tissue Res 365 (2016) 453–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wallace DP, Periostin in the Kidney, Adv Exp Med Biol 1132 (2019) 99–112. [DOI] [PubMed] [Google Scholar]
- [56].Rousselle P, Scoazec JY, Laminin 332 in cancer: When the extracellular matrix turns signals from cell anchorage to cell movement, Semin Cancer Biol (2019). [DOI] [PubMed] [Google Scholar]
- [57].Shannon MB, Patton BL, Harvey SJ, Miner JH, A hypomorphic mutation in the mouse laminin alpha5 gene causes polycystic kidney disease, Journal of the American Society of Nephrology : JASN 17 (2006) 1913–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Vijayakumar S, Dang S, Marinkovich MP, Lazarova Z, Yoder B, Torres VE, Wallace DP, Aberrant expression of laminin-332 promotes cell proliferation and cyst growth in ARPKD, Am J Physiol Renal Physiol 306 (2014) F640–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Mathew S, Chen X, Pozzi A, Zent R, Integrins in renal development, Pediatr Nephrol 27 (2012) 891–900. [DOI] [PubMed] [Google Scholar]
- [60].Hu P, Luo BH, Integrin bi-directional signaling across the plasma membrane, J Cell Physiol 228 (2013) 306–12. [DOI] [PubMed] [Google Scholar]
- [61].Bissell MJ, Hall HG, Parry G, How does the extracellular matrix direct gene expression?, J Theor Biol 99 (1982) 31–68. [DOI] [PubMed] [Google Scholar]
- [62].Korhonen M, Ylanne J, Laitinen L, Virtanen I, The alpha 1-alpha 6 subunits of integrins are characteristically expressed in distinct segments of developing and adult human nephron, J Cell Biol 111 (1990) 1245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Rahilly MA, Fleming S, Differential expression of integrin alpha chains by renal epithelial cells, J Pathol 167 (1992) 327–34. [DOI] [PubMed] [Google Scholar]
- [64].Daikha-Dahmane F, Narcy F, Dommergues M, Lacoste M, Beziau A, Gubler MC, Distribution of alpha-integrin subunits in fetal polycystic kidney diseases, Pediatr Nephrol 11 (1997) 267–73. [DOI] [PubMed] [Google Scholar]
- [65].Wilson PD, Geng L, Li X, Burrow CR, The PKD1 gene product, “polycystin-1,” is a tyrosine-phosphorylated protein that colocalizes with alpha2beta1-integrin in focal clusters in adherent renal epithelia, Lab Invest 79 (1999) 1311–23. [PubMed] [Google Scholar]
- [66].Zeltner R, Hilgers KF, Schmieder RE, Porst M, Schulze BD, Hartner A, A promoter polymorphism of the alpha 8 integrin gene and the progression of autosomal-dominant polycystic kidney disease, Nephron Clin Pract 108 (2008) c169–75. [DOI] [PubMed] [Google Scholar]
- [67].Schnapp LM, Hatch N, Ramos DM, Klimanskaya IV, Sheppard D, Pytela R, The human integrin alpha 8 beta 1 functions as a receptor for tenascin, fibronectin, and vitronectin, J Biol Chem 270 (1995) 23196–202. [DOI] [PubMed] [Google Scholar]
- [68].Denda S, Reichardt LF, Muller U, Identification of osteopontin as a novel ligand for the integrin alpha8 beta1 and potential roles for this integrin-ligand interaction in kidney morphogenesis, Mol Biol Cell 9 (1998) 1425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Klingel R, Ramadori G, Schuppan D, Knittel T, Meyer zum Buschenfelde KH, Kohler H, Coexpression of extracellular matrix glycoproteins undulin and tenascin in human autosomal dominant polycystic kidney disease, Nephron 65 (1993) 111–8. [DOI] [PubMed] [Google Scholar]
- [70].Wilson PD, Burrow CR, Cystic diseases of the kidney: role of adhesion molecules in normal and abnormal tubulogenesis, Exp Nephrol 7 (1999) 114–24. [DOI] [PubMed] [Google Scholar]
- [71].Chen HC, Appeddu PA, Parsons JT, Hildebrand JD, Schaller MD, Guan JL, Interaction of focal adhesion kinase with cytoskeletal protein talin, J Biol Chem 270 (1995) 16995–9. [DOI] [PubMed] [Google Scholar]
- [72].Otey CA, Vasquez GB, Burridge K, Erickson BW, Mapping of the alpha-actinin binding site within the beta 1 integrin cytoplasmic domain, J Biol Chem 268 (1993) 21193–7. [PubMed] [Google Scholar]
- [73].Wu W, Kitamura S, Truong DM, Rieg T, Vallon V, Sakurai H, Bush KT, Vera DR, Ross RS, Nigam SK, Beta1-integrin is required for kidney collecting duct morphogenesis and maintenance of renal function, Am J Physiol Renal Physiol 297 (2009) F210–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lee K, Boctor S, Barisoni LM, Gusella GL, Inactivation of integrin-beta1 prevents the development of polycystic kidney disease after the loss of polycystin-1, Journal of the American Society of Nephrology : JASN 26 (2015) 888–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kreidberg JA, Donovan MJ, Goldstein SL, Rennke H, Shepherd K, Jones RC, Jaenisch R, Alpha 3 beta 1 integrin has a crucial role in kidney and lung organogenesis, Development 122 (1996) 3537–47. [DOI] [PubMed] [Google Scholar]
- [76].Qin S, Taglienti M, Nauli SM, Contrino L, Takakura A, Zhou J, Kreidberg JA, Failure to ubiquitinate c-Met leads to hyperactivation of mTOR signaling in a mouse model of autosomal dominant polycystic kidney disease, J Clin Invest 120 (2010) 3617–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Norris RA, Damon B, Mironov V, Kasyanov V, Ramamurthi A, Moreno-Rodriguez R, Trusk T, Potts JD, Goodwin RL, Davis J, Hoffman S, Wen X, Sugi Y, Kern CB, Mjaatvedt CH, Turner DK, Oka T, Conway SJ, Molkentin JD, Forgacs G, Markwald RR, Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues, J Cell Biochem 101 (2007) 695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Butcher JT, Norris RA, Hoffman S, Mjaatvedt CH, Markwald RR, Periostin promotes atrioventricular mesenchyme matrix invasion and remodeling mediated by integrin signaling through Rho/PI 3-kinase, Developmental biology 302 (2007) 256–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Cobo T, Viloria CG, Solares L, Fontanil T, Gonzalez-Chamorro E, De Carlos F, Cobo J, Cal S, Obaya AJ, Role of Periostin in Adhesion and Migration of Bone Remodeling Cells, PLoS One 11 (2016) e0147837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Conway SJ, Molkentin JD, Periostin as a heterofunctional regulator of cardiac development and disease, Curr Genomics 9 (2008) 548–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Jackson-Boeters L, Wen W, Hamilton DW, Periostin localizes to cells in normal skin, but is associated with the extracellular matrix during wound repair, Journal of cell communication and signaling 3 (2009) 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kanno A, Satoh K, Masamune A, Hirota M, Kimura K, Umino J, Hamada S, Satoh A, Egawa S, Motoi F, Unno M, Shimosegawa T, Periostin, secreted from stromal cells, has biphasic effect on cell migration and correlates with the epithelial to mesenchymal transition of human pancreatic cancer cells, International journal of cancer. Journal international du cancer 122 (2008) 2707–18. [DOI] [PubMed] [Google Scholar]
- [83].Norris RA, Moreno-Rodriguez R, Hoffman S, Markwald RR, The many facets of the matricelluar protein periostin during cardiac development, remodeling, and pathophysiology, J Cell Commun Signal 3 (2009) 275–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Watanabe T, Yasue A, Fujihara S, Tanaka E, PERIOSTIN regulates MMP-2 expression via the alphavbeta3 integrin/ERK pathway in human periodontal ligament cells, Archives of oral biology 57 (2012) 52–9. [DOI] [PubMed] [Google Scholar]
- [85].Chen G, Nakamura I, Dhanasekaran R, Iguchi E, Tolosa EJ, Romecin PA, Vera RE, Almada LL, Miamen AG, Chaiteerakij R, Zhou M, Asiedu MK, Moser CD, Han S, Hu C, Banini BA, Oseini AM, Chen Y, Fang Y, Yang D, Shaleh HM, Wang S, Wu D, Song T, Lee JS, Thorgeirsson SS, Chevet E, Shah VH, Fernandez-Zapico ME, Roberts LR, Transcriptional Induction of Periostin by a Sulfatase 2-TGFbeta1-SMAD Signaling Axis Mediates Tumor Angiogenesis in Hepatocellular Carcinoma, Cancer research 77 (2017) 632–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Li P, Oparil S, Novak L, Cao X, Shi W, Lucas J, Chen YF, ANP signaling inhibits TGF-beta-induced Smad2 and Smad3 nuclear translocation and extracellular matrix expression in rat pulmonary arterial smooth muscle cells, J Appl Physiol (1985) 102 (2007) 390–8. [DOI] [PubMed] [Google Scholar]
- [87].Ito T, Suzuki A, Imai E, Horimoto N, Ohnishi T, Daikuhara Y, Hori M, Tornado extraction: a method to enrich and purify RNA from the nephrogenic zone of the neonatal rat kidney, Kidney Int 62 (2002) 763–9. [DOI] [PubMed] [Google Scholar]
- [88].Wallace DP, White C, Savinkova L, Nivens E, Reif GA, Pinto CS, Raman A, Parnell SC, Conway SJ, Fields TA, Periostin promotes renal cyst growth and interstitial fibrosis in polycystic kidney disease, Kidney Int 85 (2014) 845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Elad N, Volberg T, Patla I, Hirschfeld-Warneken V, Grashoff C, Spatz JP, Fassler R, Geiger B, Medalia O, The role of integrin-linked kinase in the molecular architecture of focal adhesions, J Cell Sci 126(Pt 18) (2013) 4099–107. [DOI] [PubMed] [Google Scholar]
- [90].Li Y, Yang J, Dai C, Wu C, Liu Y, Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis, J Clin Invest 112 (2003) 503–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Zheng QM, Lu JJ, Zhao J, Wei X, Wang L, Liu PS, Periostin Facilitates the Epithelial-Mesenchymal Transition of Endometrial Epithelial Cells through ILK-Akt Signaling Pathway, Biomed Res Int 2016 (2016) 9842619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Cruet-Hennequart S, Maubant S, Luis J, Gauduchon P, Staedel C, Dedhar S, alpha(v) integrins regulate cell proliferation through integrin-linked kinase (ILK) in ovarian cancer cells, Oncogene 22 (2003) 1688–702. [DOI] [PubMed] [Google Scholar]
- [93].Hannigan GE, McDonald PC, Walsh MP, Dedhar S, Integrin-linked kinase: not so ‘pseudo’ after all, Oncogene 30 (2011) 4375–85. [DOI] [PubMed] [Google Scholar]
- [94].Wu C, Dedhar S, Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes, J Cell Biol 155 (2001) 505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Fukuda K, Knight JD, Piszczek G, Kothary R, Qin J, Biochemical, proteomic, structural, and thermodynamic characterizations of integrin-linked kinase (ILK): cross-validation of the pseudokinase, J Biol Chem 286 (2011) 21886–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Lange A, Wickstrom SA, Jakobson M, Zent R, Sainio K, Fassler R, Integrin-linked kinase is an adaptor with essential functions during mouse development, Nature 461 (2009) 1002–6. [DOI] [PubMed] [Google Scholar]
- [97].Wickstrom SA, Lange A, Montanez E, Fassler R, The ILK/PINCH/parvin complex: the kinase is dead, long live the pseudokinase!, EMBO J 29 (2010) 281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Raman A, Reif GA, Dai Y, Khanna A, Li X, Astleford L, Parnell SC, Calvet JP, Wallace DP, Integrin-Linked Kinase Signaling Promotes Cyst Growth and Fibrosis in Polycystic Kidney Disease, Journal of the American Society of Nephrology : JASN 28 (2017) 2708–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Cano-Penalver JL, Griera M, Serrano I, Rodriguez-Puyol D, Dedhar S, de Frutos S, Rodriguez-Puyol M, Integrin-linked kinase regulates tubular aquaporin-2 content and intracellular location: a link between the extracellular matrix and water reabsorption, FASEB journal : official publication of the Federation of American Societies for Experimental Biology (2014). [DOI] [PubMed] [Google Scholar]
- [100].Attwell S, Roskelley C, Dedhar S, The integrin-linked kinase (ILK) suppresses anoikis, Oncogene 19 (2000) 3811–5. [DOI] [PubMed] [Google Scholar]
- [101].Geng L, Burrow CR, Li HP, Wilson PD, Modification of the composition of polycystin-1 multiprotein complexes by calcium and tyrosine phosphorylation, Biochim Biophys Acta 1535 (2000) 21–35. [DOI] [PubMed] [Google Scholar]
- [102].Castelli M, De Pascalis C, Distefano G, Ducano N, Oldani A, Lanzetti L, Boletta A, Regulation of the microtubular cytoskeleton by Polycystin-1 favors focal adhesions turnover to modulate cell adhesion and migration, BMC Cell Biol 16 (2015) 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Joly D, Ishibe S, Nickel C, Yu Z, Somlo S, Cantley LG, The polycystin 1-C-terminal fragment stimulates ERK-dependent spreading of renal epithelial cells, J Biol Chem 281 (2006) 26329–39. [DOI] [PubMed] [Google Scholar]
- [104].Lehtonen S, Ora A, Olkkonen VM, Geng L, Zerial M, Somlo S, Lehtonen E, In vivo interaction of the adapter protein CD2-associated protein with the type 2 polycystic kidney disease protein, polycystin-2, J Biol Chem 275 (2000) 32888–93. [DOI] [PubMed] [Google Scholar]
- [105].Xu GM, Sikaneta T, Sullivan BM, Zhang Q, Andreucci M, Stehle T, Drummond I, Arnaout MA, Polycystin-1 interacts with intermediate filaments, J Biol Chem 276 (2001) 46544–52. [DOI] [PubMed] [Google Scholar]
- [106].Sharif-Naeini R, Folgering JH, Bichet D, Duprat F, Lauritzen I, Arhatte M, Jodar M, Dedman A, Chatelain FC, Schulte U, Retailleau K, Loufrani L, Patel A, Sachs F, Delmas P, Peters DJ, Honore E, Polycystin-1 and −2 dosage regulates pressure sensing, Cell 139 (2009) 587–96. [DOI] [PubMed] [Google Scholar]
- [107].Nigro EA, Distefano G, Chiaravalli M, Matafora V, Castelli M, Pesenti Gritti A, Bachi A, Boletta A, Polycystin-1 Regulates Actomyosin Contraction and the Cellular Response to Extracellular Stiffness, Sci Rep 9 (2019) 16640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Cruz NM, Song X, Czerniecki SM, Gulieva RE, Churchill AJ, Kim YK, Winston K, Tran LM, Diaz MA, Fu H, Finn LS, Pei Y, Himmelfarb J, Freedman BS, Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease, Nat Mater 16 (2017) 1112–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Cai J, Song X, Wang W, Watnick T, Pei Y, Qian F, Pan D, A RhoA-YAP-c-Myc signaling axis promotes the development of polycystic kidney disease, Genes Dev 32 (2018) 781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Happe H, van der Wal AM, Leonhard WN, Kunnen SJ, Breuning MH, de Heer E, Peters DJ, Altered Hippo signalling in polycystic kidney disease, J Pathol 224 (2011) 133–42. [DOI] [PubMed] [Google Scholar]
- [111].Weston BS, Malhas AN, Price RG, Structure-function relationships of the extracellular domain of the autosomal dominant polycystic kidney disease-associated protein, polycystin-1, FEBS Lett 538 (2003) 8–13. [DOI] [PubMed] [Google Scholar]
- [112].Malhas AN, Abuknesha RA, Price RG, Interaction of the leucine-rich repeats of polycystin-1 with extracellular matrix proteins: possible role in cell proliferation, Journal of the American Society of Nephrology : JASN 13 (2002) 19–26. [DOI] [PubMed] [Google Scholar]
- [113].Weston BS, Bagneris C, Price RG, Stirling JL, The polycystin-1 C-type lectin domain binds carbohydrate in a calcium-dependent manner, and interacts with extracellular matrix proteins in vitro, Biochim Biophys Acta 1536 (2001) 161–76. [DOI] [PubMed] [Google Scholar]
- [114].Bycroft M, Bateman A, Clarke J, Hamill SJ, Sandford R, Thomas RL, Chothia C, The structure of a PKD domain from polycystin-1: implications for polycystic kidney disease, EMBO J 18 (1999) 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Ibraghimov-Beskrovnaya O, Bukanov NO, Donohue LC, Dackowski WR, Klinger KW, Landes GM, Strong homophilic interactions of the Ig-like domains of polycystin-1, the protein product of an autosomal dominant polycystic kidney disease gene, PKD1, Hum Mol Genet 9 (2000) 1641–9. [DOI] [PubMed] [Google Scholar]
- [116].van Adelsberg J, Peptides from the PKD repeats of polycystin, the PKD1 gene product, modulate pattern formation in the developing kidney, Dev Genet 24 (1999) 299–308. [DOI] [PubMed] [Google Scholar]
- [117].Torres VE, Harris PC, Polycystic kidney disease: genes, proteins, animal models, disease mechanisms and therapeutic opportunities, Journal of internal medicine 261 (2007) 17–31. [DOI] [PubMed] [Google Scholar]
- [118].Taub M, Laurie GW, Martin GR, Kleinman HK, Altered basement membrane protein biosynthesis by primary cultures of cpk/cpk mouse kidney, Kidney Int 37 (1990) 1090–7. [DOI] [PubMed] [Google Scholar]