

Abstract
Cancer cells reprogram their metabolism to meet elevated energy demands and favor glycolysis for energy production. This boost in glycolytic flux supports proliferation, but also generates acid in the form of hydrogen ions that must be eliminated from the cytoplasm to maintain the alkaline intracellular pH (pHi) associated with transformation. To cope with acid production, tumor cells employ ion transport systems, including the family of sodium-hydrogen exchangers (NHEs). Here, we identify NHE7 as a novel regulator of pHi in pancreatic ductal adenocarcinoma (PDAC). We determine that NHE7 suppression causes alkalinization of the Golgi, leading to a build-up of cytosolic acid that diminishes tumor cell fitness mainly through the dysregulation of actin. Importantly, NHE7 knockdown in vivo leads to the abrogation of tumor growth. These results identify Golgi acidification as a mechanism to control pHi and point to the regulation of pHi as a possible therapeutic vulnerability in PDAC.
Keywords: NHE7, pH homeostasis, pancreatic cancer
Introduction
Cancer cells dramatically increase their glucose uptake in order to provide the necessary energy to fuel cell growth and survival (1). In untransformed cells, glucose is oxidized to yield ATP through aerobic metabolism, but tumor cells derive their ATP mainly through glycolysis, despite in many cases the availability of functional mitochondria. In this process, better known as the “Warburg effect”, cancer cells are able to produce the required ATP, but in turn also produce acid in the form of hydrogen ions (H+). Unlike normal tissues, tumors display a reversal of pH gradient where they maintain an alkaline intracellular pH (pHi) and a concomitant more acidic extracellular pH (2–4). Multiple aspects of tumor progression are thought to benefit from this pH gradient switch, including tumor growth, cell death evasion, and invasiveness. To maintain an alkaline pHi, tumor cells have evolved homeostatic mechanisms to clear protons from the cytoplasm. These mechanisms include ion transporters, such as sodium-hydrogen exchangers (NHEs), that act to regulate pHi through sodium-dependent proton secretion (5).
The NHE family consists of nine isoforms (NHE1–9) that export hydrogen in exchange for sodium (5). NHE isoforms are distributed across multiple cellular locales, including the plasma membrane, the Golgi apparatus, and endosomes, where they regulate cellular function through modulating organelle and cytoplasmic pH. We set out to characterize NHE expression in PDAC and found that NHE7 is upregulated in PDAC tumors and its expression levels correlate with patient survival and more aggressive disease subtypes. We find that NHE7 localizes to the trans-Golgi network (TGN) in PDAC cells, which is in accordance with previous studies (6–11), and that NHE7 knockdown compromises PDAC cell viability. We establish that NHE7 controls luminal acidification of the TGN and that glycosylation is generally unaffected in NHE7 knockdown cells, with the exception of a decline in sialylation, which is known to occur in the TGN. Despite these defects in sialylation, we find that the near-complete inhibition of sialyltransferases fails to impact PDAC cell viability. Interestingly, the failure to acidify the TGN caused by NHE7 suppression leads to a decrease in pHi, implicating the TGN as a repository for metabolic acid in cancer cells. This inability to maintain an alkaline pH elicits compromised proliferation and cell death that mainly occurs via the dysregulation of the actin cytoskeleton. Importantly, we determine that NHE7 plays an integral role in PDAC tumor maintenance. Our findings enhance our understanding of the mechanisms that regulate pH homeostasis in tumors and suggest that the targeting of organelle acidification might represent a novel therapeutic approach for cancer.
Results
PDAC displays increased NHE7 expression that correlates with poor prognosis.
To characterize NHEs in PDAC, we interrogated publicly available patient datasets. We found that of the nine known NHE isoforms, elevated NHE7 expression levels consistently correlated with poor prognosis (Fig. 1A and B; Supplementary Fig. S1A) and PDAC subtype analysis revealed that NHE7 expression is highest in the more aggressive tumor subtypes (Supplementary Fig. S1B; ref. (12–15)). Additionally, relative to normal pancreas or adjacent non-neoplastic tissue, NHE7 transcript levels were significantly higher in human PDAC tumors (Fig. 1C; Supplementary Fig. S1C). To investigate NHE7 expression at the protein level, we immunostained a tissue microarray containing PDAC specimens from 41 patients that underwent surgical resection. We found that NHE7 was expressed in all PDAC tumors and that 30/41 PDAC specimens displayed moderate or high levels of NHE7 protein (Fig. 1D; Supplementary Fig. S2). NHE7 protein expression was predominantly localized to the PDAC cells, with some detectable expression in the stroma.
Figure 1. NHE7 expression correlates with poor prognosis and is upregulated in PDAC.

A, Correlation between NHE isoform expression levels and prognosis in PDAC patients as assessed through the R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl).
B, Overall survival in PDAC patients with low and high NHE7 expression. P-values were calculated by Log-rank test. Hazard Ratio (HR) by the Mantel-Haenszel method.
C, NHE7 transcript levels in human PDAC tissues compared to normal pancreas or adjacent non-neoplastic tissue. Expression levels are graphed as box with whiskers from min to max. The P values were calculated by paired or unpaired, two-tailed t-test
D, Representative images of NHE7 immunohistochemical staining of a tissue microarray containing surgically resected human PDAC samples. Samples were stratified into groups based on staining intensity. Each group is presented with a group size out of a total of 41 human PDAC samples.
NHE7 is essential for PDAC cell viability.
To assess the role of NHE7 at a cellular level we examined the in vitro effects of NHE7 knockdown in a panel of PDAC cell lines. Via a crystal violet assay, we observed a substantial decrease in proliferation upon NHE7 knockdown in all the PDAC cell lines evaluated (Fig. 2A and B; Supplementary Fig. S3A). The compromised proliferative capacity of PDAC cells in the context of NHE7 suppression could be mediated by the induction of cell death; therefore, we assessed apoptosis and necrosis using Annexin V and propidium iodide (PI) staining. We found that NHE7 knockdown led to significant increases in the number of early apoptotic and late apoptotic cells, relative to the control (Fig. 2C). To better understand the dynamics of cell death upon NHE7 suppression, we performed a time course experiment and evaluated cell death utilizing Hoechst staining, which allows for the microscopic discrimination of dying cells by nuclear condensation (16). We determined that significant levels of increased cell death were first observable after four days of knockdown, with further enhancement by six days (Supplementary Fig. S3B). These cell death dynamics correlated with the reduction in cell numbers that were observed in the proliferation assays (Fig. 2A). To determine whether viability is controlled by NHE7 in untransformed cells, we knocked down NHE7 in normal immortalized human pancreatic nestin expressing (hTERT-HPNE) cells and normal pancreatic fibroblasts (NPF) (Fig. 2D and Supplementary Fig. S3C). Interestingly, proliferation in these untransformed cells was unaffected by NHE7 knockdown (Fig. 2E), suggesting that NHE7 might selectively regulate proliferation in cancer cells.
Figure 2. NHE7 suppression causes loss of viability in PDAC cells.

A, Proliferation for the indicated PDAC cell lines after transduction with the indicated short hairpins, as assessed by crystal violet staining.
B, NHE7 expression levels in MIA PaCa-2 and PANC-1 cells as assessed by western blot. αTubulin was used as loading control.
C, Cell death as assessed by Annexin V / PI staining in MIA PaCa-2 and PANC-1 cells after lentiviral transduction.
D, NHE7 expression levels in hTERT-HPNE and Normal Pancreatic Fibroblast cells as assessed by western blot. αTubulin was used as loading control.
E, Proliferation, as assessed by Syto-60 staining, for the indicated normal cell lines following transduction with the indicated short hairpins.
Data are presented as the mean ± s.e.m from at least three independent experiments. The P values were calculated by two-way ANOVA (A,E) or one-way ANOVA (C). NS = not significant, **P < 0.01, ***P < 0.001.
NHE7 regulates Golgi acidification.
Ectopic expression studies have revealed that NHE7 can localize to the trans-Golgi network (TGN), post-Golgi vesicles, the plasma membrane or endosomes, depending on the biological context (6–11). To examine the subcellular localization of endogenous NHE7, we performed immunofluorescence. We determined that in PDAC cells NHE7 predominantly localized to the TGN, as demonstrated by co-localization with Golgin-97 (Fig. 3A), an established TGN marker (17). While the majority of NHE7 protein localized to the TGN, there were some NHE7-positive puncta that were Golgin-97-negative, suggesting that NHE7 might also localize to other compartments, such as post-Golgi vesicles or endosomes. To assess whether NHE7 regulates the luminal pH of the TGN, we employed a ratiometric pH probe consisting of a TGN-targeting peptide derived from β1,4-galactosyltransferase (GalT) coupled to mCherry and EGFP tags, both of which face the lumen (18, 19). Co-localization assays with anti-Golgin-97 antibodies confirmed that the pH probe localizes to the TGN (Supplementary Fig. S4A). Moreover, co-localization between NHE7 and the pH probe further validated the TGN subcellular localization of NHE7 (Supplementary Fig. S4B). Due to the dependency of the EGFP fluorescent intensity on pH (pKa = 6.0; ref (20)) and the pH-insensitivity of mCherry (pKa < 4.5; ref (21)), we were able to verify the suitability of the probe to assess the luminal pH in live cells as shown by the linear correlation between pH and the EGFP/mCherry fluorescent intensity ratio (Supplementary Fig. S4C). Utilization of this pH probe revealed that upon the doxycycline-inducible knockdown of NHE7 (Supplementary Fig. S4D), the average luminal pH increased from pH 5.5 to pH 6, indicating impaired acidification of the TGN compartment relative to control (Fig. 3B–D; Supplementary Fig. S4E). Additionally, the TGN compartments in NHE7 knockdown cells appeared enlarged and there was a reduction in the overall number of discrete compartments. To further substantiate the role of NHE7 in acidifying the TGN, we used the drug 5-(N-Ethyl-N-isopropyl)-amiloride (EIPA), which targets multiple NHE family members, including NHE7 (10, 22). It is well-established that EIPA acidifies the cytoplasm, but when administered to MIA PaCa-2 cells we also observed an increase in TGN pH (Fig. 3E–G). These data corroborate the involvement of Na+/H+ exchange in TGN acidification.
Figure 3. NHE7 is localized to the trans-Golgi network and regulates luminal pH and glycan sialylation.
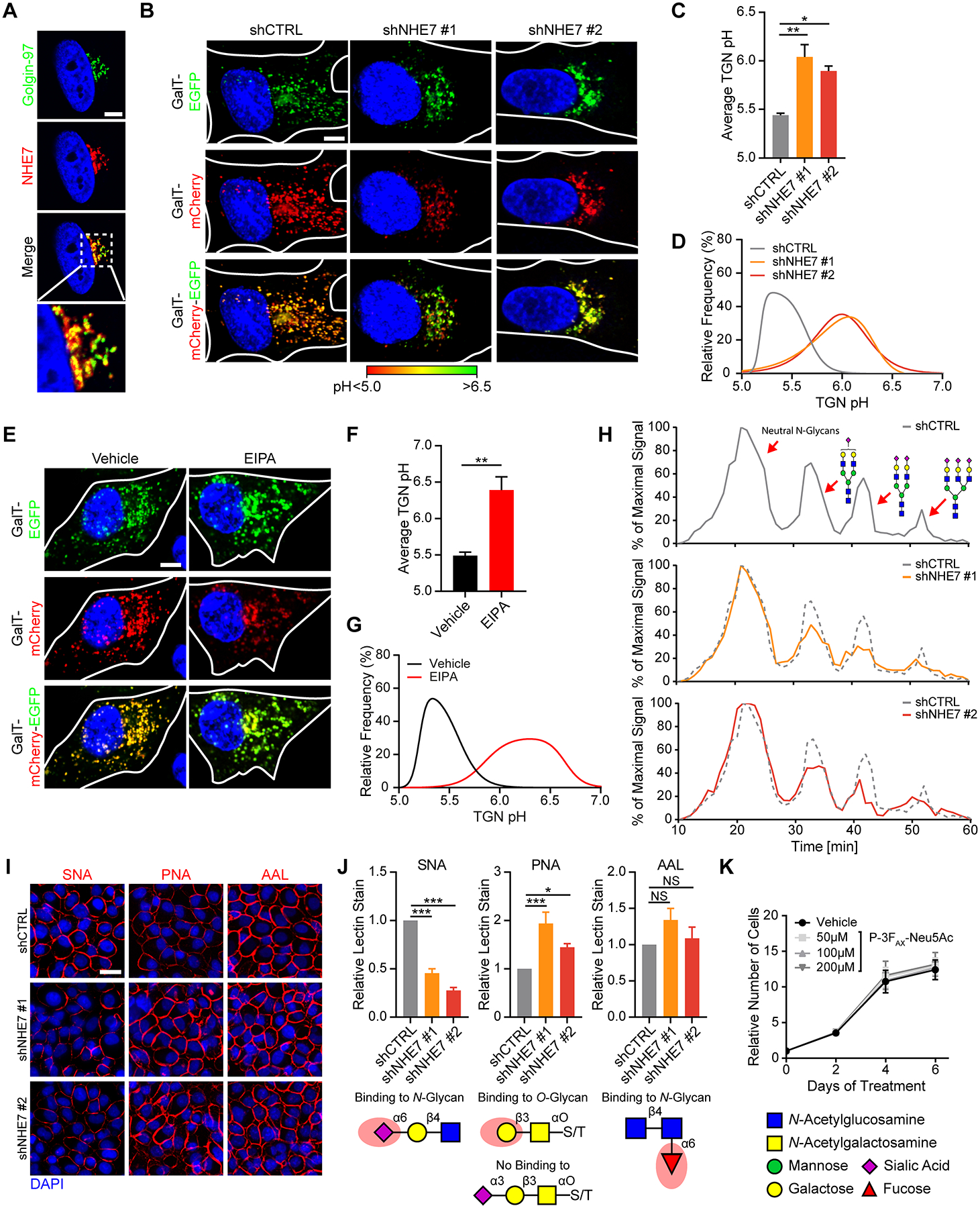
A, Representative images of NHE7 co-localization with the TGN marker Golgin-97 in MIA PaCa-2 cells, as assessed by immunofluorescence staining with anti-Golgin-97 (green) anti-NHE7 (red) antibodies. The nucleus is stained with DAPI (blue). Scalebar = 5 μm
B, Representative overlay images of EGFP (green) and mCherry (red) used to determine TGN luminal pH. The nucleus is stained with Hoechst 33342 (blue). Scalebar = 10 μm
C, Average TGN luminal pH as obtained through live-cell imaging of GalT-mCherry-EGFP-expressing MIA PaCa-2 cells four days after doxycycline-induced NHE7 knockdown.
D, Combined frequency distributions of TGN luminal pH from one of three independent experiments.
E, Representative overlay images of GalT-mCherry-EGFP-expressing MIA PaCa-2 cells after vehicle or EIPA treatment. Scalebar = 10 μm
F, Average TGN luminal pH after 30–60 min vehicle or EIPA treatment as determined by live-cell imaging of GalT-mCherry-EGFP-expressing MIA PaCa-2 cells.
G, Combined frequency distributions of TGN luminal pH after vehicle and EIPA administration in one of three independent experiments.
H, Representative HPLC analysis graphs obtained 3 days after transduction, with examples of possible mono-, di- and tri- sialylated N-glycan structures.
I, Representative images from cell surface lectin staining of MIA PaCa-2 cells, 4 days after transduction. Scalebar = 50 μm
J, Average cell surface lectin stain as obtained through quantification of signal intensities. The red shade on the glycan structures below indicates the lectin recognition site.
K, Growth curve of MIA PaCa-2 cells with the indicated treatments, as assessed by crystal violet staining.
Data are presented as the mean ± s.e.m from at least three independent experiments. The P values were calculated by one-way ANOVA (C, J) or unpaired, two-tailed t-test (F). NS = not significant, *P < 0.05, **P < 0.01, ***P < 0.001. For TGN pH measurements at least 10 ROIs per cell and a minimum of 20 cells were scored per experiment.
Sialylation is reduced by NHE7 suppression, but does not affect PDAC cell viability.
Since alkalinization of the TGN via NHE7 suppression might impair protein glycosylation (23), we initially examined the glycosylation status of the receptor tyrosine kinases (RTKs) EGFR and HER2, both well-characterized glycoproteins (24). We assessed glycosylation via western blot, and compared electrophoretic mobility of the RTKs in NHE7 knockdown cells relative to control cells. Inhibition of N-glycosylation by tunicamycin results in a mobility shift for both EGFR and HER2; however, no changes in electrophoretic mobility of these proteins were detected upon NHE7 knockdown (Supplementary Fig. S5A). Sialylation is a glycosylation event that specifically occurs in the trans cisternae of the Golgi and the TGN, resulting in the addition of negative charges to glycans (25). These negative charges can be exploited in high performance liquid chromatography (HPLC) to assess changes in glycan sialylation status. To analyze potential differences in N-glycan synthesis after NHE7 knockdown, cells were incubated with [2-3H] D-mannose to label newly synthesized N-glycans then subjected to separation via HPLC. We detected a reduction in charged sialylated glycans in NHE7 knockdown cells (Fig. 3H), as confirmed by neuraminidase digestion, which removes all sialic acids (Supplementary Fig. S5B). The reduction in glycan sialylation was further confirmed by cell surface staining with fluorescently-tagged Sambucus nigra lectin (SNA), which preferentially binds to sialic acids attached to terminal galactose on N-glycans (Fig. 3I and J). Concomitant with reduced sialylation, we also observed an increase in Peanut agglutinin (PNA) lectin staining, which recognizes Gal-β(1–3)-GalNAc but its binding is usually inhibited by sialic acid (Fig. 3I and J). Cell surface staining with the lectin AAL, which preferentially binds to α−1,6-linked fucose, a saccharide addition that occurs in the medial Golgi, was unaffected (Fig. 3I and J; ref. (26)). This lack of alteration in cell surface AAL staining suggests that protein trafficking from the Golgi to the plasma membrane is unaffected by NHE7 knockdown. Consistent with this notion, we did not detect changes in cell surface EGFR expression after NHE7 depletion (Supplementary Fig. S5C and S5D). To determine whether reduced sialylation affects PDAC cell viability and proliferation, we treated MIA PaCa-2 cells with the sialyltransferase-specific inhibitor P-3FAX-Neu5Ac (27). Similar to NHE7 knockdown, P-3FAX-Neu5Ac reduced SNA cell surface staining and increased staining of PNA (Supplementary Fig. S5E and S5F). As a control, we administered the N-glycosylation inhibitor tunicamycin, which reduces only N-glycan sialylation, while leaving O-glycan sialylation unaffected (Supplementary Fig. S5E and S5F). Despite these reductions in sialylation, we found that P-3FAX-Neu5Ac did not affect PDAC cell proliferation (Fig. 3K), suggesting that reduced sialylation is not responsible for the observed viability defects in NHE7 knockdown cells.
NHE7 depletion leads to deregulation of pHi homeostasis.
It is conceivable that an inability to transport H+ ions into an organelle like the Golgi could lead to an accompanying accumulation of cytosolic H+. Indeed, a disruption in the acidification of lysosomes in lung cancer cells has been shown to result in a concomitant pH reduction in the cytosol (28). To test whether suppression of TGN acidification by NHE7 knockdown can modulate cytosolic pH, we stained live PDAC cells with the pHrodo Red AM (PHRR) intracellular pH indicator. As pHi decreases, PHRR fluorescent intensity increases, and in conjunction with a standard curve, the extent of PHRR fluorescence can be utilized to quantitate pHi (Supplementary Fig. S6A–S6C). Using this approach, we determined that NHE7 knockdown in PDAC cells causes a significant decrease in pHi, as demonstrated by the increased fluorescent intensity of PHRR relative to control cells (Fig. 4A–C; Supplementary Fig. S6D). Interestingly, this effect might be specific to transformed cells since the pHi of HPNE cells was unaffected by NHE7 knockdown (Fig. 4D and E). If acidification of the TGN contributes to pH homeostasis in the cytosol, then loss of NHE7 expression might impair the ability of the PDAC cells to recover from acidification stress. To evaluate this possibility, we assessed pHi recovery upon intracellular acidification using an acute NH4+ acid-loading technique (29). Loading of the cells with NH4+ and the subsequent removal of all extracellular NH4+ causes the acidification of the cytosol through the intracellular sequestration of protons. In control cells, the addition of Na+ to the extracellular medium allows for NHE activity resulting in a gradual recovery of the pHi (Fig. 4F–H). In PDAC cells depleted of NHE7, this recovery stage was defective, confirming the role of NHE7 in cytosolic pH regulation upon acidification stress. To examine the link between the Golgi compartment and pHi homeostasis, we administered to the PDAC cells Brefeldin A (BFA), which blocks secretion and leads to the resorption of much of the Golgi apparatus into the endoplasmic reticulum (Supplementary Fig. S6E; ref (30)). Consistent with the acidification of the Golgi playing a pivotal role in pHi regulation, disruption of the Golgi via BFA resulted in a reduction in cytosolic pH (Fig. 4I and J). Altogether, these data demonstrate that the NHE7-mediated acidification of the Golgi contributes to the maintenance of an alkaline cytosol in PDAC cells.
Figure 4. NHE7 regulates cytosolic pH homeostasis.

A, Representative images of PHRR-loaded MIA PaCa-2 and PANC-1 cells four and six days, respectively, after lentiviral transduction. Scalebar = 50 μm
B, Average pHi as obtained through live-cell imaging of PHRR-loaded MIA PaCa-2 and PANC-1 cells.
C, Combined frequency distributions of pHi in PHRR-loaded MIA PaCa-2 and PANC-1 cells from one of three representative independent experiments.
D, Representative images of PHRR-loaded hTERT-HPNE cells six days after lentiviral transduction. Scalebar = 50 μm
E, Average pHi as assessed via quantification of live-cell imaged PHRR-loaded hTERT-HPNE cells.
F, Representative smoothed curve graph of pHi measurements during an acute NH4+ acid-load in BCECF-loaded MIA PaCa-2 cells.
G, Quantification of the pH recovery rate derived from the slope of the first four minutes of the recovery phase as indicated in e.
H, Representative images at different time points of an acute NH4+ acid-load in PHRR-loaded MIA PaCa-2 cells. Scalebar = 50 μm
I, Representative images of PHRR-loaded MIA PaCa-2 cells treated for six hours with brefeldin A or vehicle control. Scalebar = 50 μm
J, Quantification of the pHi in PHRR-loaded MIA PaCa-2 cells treated with brefeldin A or vehicle control.
Data are presented as the mean ± s.e.m from at least three independent experiments. The P values were calculated by one-way ANOVA (B,E,G) or unpaired, two-tailed t-test (J). NS = not significant, *P < 0.05, **P < 0.01, ***P < 0.001. For pHi measurements at least 75 cells per replicate were scored.
Loss of NHE7 expression reduces cell viability through the accumulation of actin stress fibers.
Cancer cells display a reversed pH gradient relative to normal cells, with pHi being constitutively more alkaline and the extracellular pH more acidic (2–4). One way that cytoplasmic alkalinity can drive transformative properties is through the regulation of actin cytoskeletal dynamics (2–4). Because the activities of actin polymerizing and disassembling proteins are highly dependent on cytosolic pH, we surmised that the regulation of pHi by NHE7 might be in turn affecting the actin cytoskeleton. To investigate this possibility, we stained NHE7 knockdown and control cells for filamentous actin (F-actin) using phalloidin. Interestingly, we observed a marked enhancement in F-actin staining upon NHE7 depletion, which coincided with Ser19-phosphorylated myosin light chain-2 (pMLC2) decoration (Fig. 5A and B), indicating the robust formation of actin stress fibers (31). In control cells, F-actin and pMLC2 staining were predominantly cortical. This accumulation of actin stress fibers in NHE7 knockdown cells suggested that actin depolymerizing proteins were inhibited. Indeed, in NHE7-depleted PDAC cells, we found increased levels of inhibitory Ser3-phosphorylated cofilin (Fig. 5C and D), a pH-sensitive actin depolymerizing factor whose inactivation is tightly linked to actin stress fiber maintenance (32–39). The mechanistic underpinnings of the pH-dependency of cofilin have been elegantly deciphered through structural biology (34–38). It has been shown that the actin severing and filament depolymerization activity of cofilin are more potent at alkaline pH. The basis for this more potent activity is due to a pH-dependent conformational change in the actin-cofilin interface. Thus, decreasing the pHi decreases the effective filament severing rate. The inability to disassemble actin stress fibers due to loss of cofilin activity is severely detrimental to cellular function and can cause nuclear abnormalities, such as morphological defects, multinucleation and a block in cytokinesis (39, 40). Relative to control cells, NHE7 knockdown cells displayed nuclear deformation, multinucleation, and increased DNA content (Supplementary Fig. S7A–S7D). LIM kinase 1 and 2 (LIMK) are known to negatively regulate cofilin activity through Ser3 phosphorylation (32, 33). We found that the LIMK inhibitor SR-7826 suppressed the cofilin phosphorylation and reduced the stress fiber formation associated with NHE7 depletion (Fig. 5E and F; Supplementary Fig. S7E). In addition, SR-7826 treatment partially rescued cell death in NHE7 knockdown cells, indicating that actin stress fiber maintenance through cofilin inactivation is partly responsible for the reduced viability observed upon NHE7 depletion (Fig. 5G).
Figure 5. NHE7 depletion-induced accumulation of actin stress fibers causes loss of viability.

A, F-actin (green) staining in MIA PaCa-2 and PANC1 cells four and six days, respectively, after doxycycline induction of the indicated short-hairpins. The nucleus is stained with DAPI (blue). Scalebar = 20 μm
B, F-actin (green) and Ser19-phosphorylated myosin light chain-2 (pMLC2, red) co-localization in MIA PaCa-2 cells. Scalebar = 20 μm
C, Ser3-phosphorylated cofilin (pCofilin) and total Cofilin levels in MIA PaCa-2 cells, as assessed by western blot. αTubulin was used as loading control.
D, Quantification of pCofilin levels relative to Cofilin from three independent experiments.
E, Ser3-phosphorylated cofilin (pCofilin) levels in MIA PaCa-2 cells transduced for 4 days and treated for 3 days with the LIMK inhibitor SR-7826.
F, F-actin (green) staining in MIA PaCa-2 cells after three days of vehicle or SR-7826 administration. Scalebar = 20 μm
G, Cell death as assessed by Hoechst staining after four days of vehicle or SR-7826 administration. Data normalized to shCTRL.
H, F-actin (green) staining in MIA PaCa-2 cells after three days of vehicle or (–)-blebbistatin treatment. Scalebar = 20 μm
I, Cell death assessed by Hoechst staining after four days of vehicle or (–)-blebbistatin treatment. Data normalized to shCTRL.
J, F-actin (green) staining in MIA PaCa-2 cells after three days of vehicle or monensin administration. Scalebar = 20 μm
K, Cell death as assessed by Annexin V / PI staining after four days of vehicle or monensin administration. Data normalized to shCTRL.
Data are presented as the mean ± s.e.m from at least three independent experiments. The P values were calculated by one-way ANOVA (D) or two-way ANOVA (G,I,K). NS = not significant, *P < 0.05, **P < 0.01, ***P < 0.001.
To further examine the involvement of actin stress fibers and aberrant actin-myosin contractility in the induction of cell death, we next performed rescue experiments with the non-muscle myosin II inhibitor (–)-blebbistatin. Similar to cofilin inhibition, (–)-blebbistatin administration rescued stress fiber formation and partially inhibited cell death in NHE7 knockdown cells (Fig. 5H and I). Ultimately, we investigated whether the stress fiber formation and loss of cellular viability upon NHE7 knockdown are specifically linked to the loss of Na+/H+ exchange activity. We performed rescue experiments using the Na+/H+ ionophore monensin. Monensin mediates Na+/H+ exchange independent of NHEs by allowing for the electroneutral exchange of Na+ and H+ ions across lipid membranes (41). Importantly, monensin administration rescued stress fiber formation and cell death caused by NHE7 knockdown (Fig. 5J and K). Altogether, these data show that NHE7 depletion can affect pH-dependent processes in the cytosol, such as actin polymerization, which then leads to the accumulation of contractile actin-myosin networks and the associated cellular abnormalities. In addition, the rescue experiments indicate that the accumulation of stress fibers and the reduction of cell viability upon NHE7 knockdown are linked to aberrant actin-myosin contractility; however, because the rescues by LIMK inhibition or (–)-blebbistatin were partial, it is possible that other cellular processes affected by NHE7 depletion might also be contributing to the observed cell death.
NHE7 depletion blocks tumor growth.
The above observations implicate NHE7 as an integral determinant of PDAC cell fitness; therefore, we sought to directly evaluate the role of NHE7 in tumor growth. To initially investigate the effects of NHE7 suppression in vivo, we injected MIA PaCa-2 cells subcutaneously into the flanks of immune-compromised mice shortly after NHE7 knockdown but prior to the observance of any in vitro cell death and assessed tumor formation and growth relative to control cells. In PDAC cells with depleted NHE7, we observed a complete loss of tumor formation (Supplementary Fig. S8A). To address whether NHE7 suppression could modulate tumor growth in established tumors, we employed an in vivo doxycycline-inducible knockdown system. NHE7 knockdown was initiated via doxycycline administration when tumors attained a volume of approximately 100 mm3. Relative to control tumors expressing a non-targeting control shRNA, NHE7 knockdown tumors halted their growth or regressed (Fig. 6A–C and Supplementary Fig. S8B). For tumors that did not completely regress, NHE7 knockdown was confirmed by qRT-PCR and immunohistochemistry (Fig. 6D and Supplementary Fig. S8C). Immunostaining of these same tumors revealed a reduction in proliferation and an increase in cell death, as assessed by Ki-67 or cleaved caspase-3 staining, respectively (Fig. 6D). Importantly, NHE7-depleted tumors showed increased F-actin staining, relative to the control tumors (Fig. 6E and F). These data suggest a model in which NHE7 depletion leads to an accumulation of actin filaments that compromises PDAC tumor growth (Fig. 6G).
Figure 6. NHE7 is required for tumor maintenance.
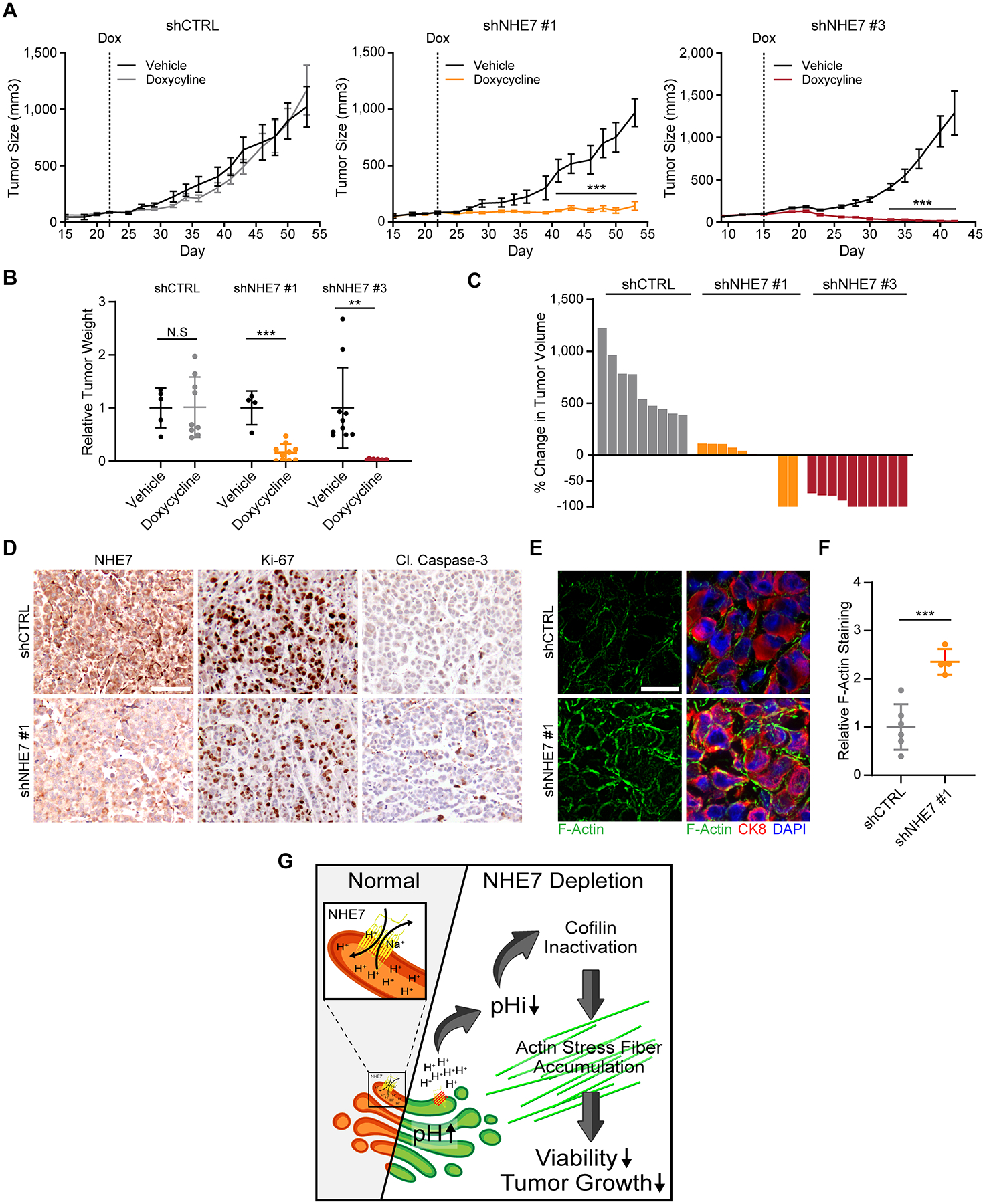
A, Heterotopic xenograft growth curves of Tet-pLKO-expressing MIA PaCa-2 cells in mice with or without a doxycycline-containing diet. The bar indicates size measurements where the Dox tumors were statistically different from the control tumors with P < 0.001.
B, Weight of extracted xenograft tumors at termination, normalized to their respective vehicle control.
C, Waterfall plot displaying the percent change in tumor volume after three weeks compared to seven days after the start of doxycycline administration.
D, Representative images of immunohistochemical staining of paraffin embedded tumors using the indicated antibodies. Scalebar = 100 μm
E, Representative images of phalloidin staining (green) and CK8 (red) immunofluorescence staining of frozen tumor sections. The nucleus is stained with DAPI (blue). Scalebar = 50 μm
F, Quantification of relative F-actin levels as assessed by phalloidin staining intensity within CK8-positive PDAC cells.
G, Graphical depiction of the proposed model for NHE7 depletion-induced loss of cell viability.
Data are presented as the mean ± s.e.m (A) or ± s.d (B, G). The P values were calculated by two-way ANOVA (A), one-way ANOVA (B) or unpaired, two-tailed t-test (B, D, G). NS = not significant, **P < 0.01, ***P < 0.001. For A, n = 5, 9, 4, 10, 10, and 10 tumors for the cohorts shCTRL – Veh, shCTRL – Dox, shNHE7 #1 – Veh, shNHE7 #1 – Dox, shNHE7 #3 – Veh, and shNHE7 #3 – Dox, respectively.
Discussion
Recent years have witnessed a renewed appreciation of the role that pH homeostasis plays in tumor progression (2–4). Our data provide evidence that NHE7 regulates Golgi luminal pH, and in so doing, is a critical regulator of pHi in PDAC. Our work supports the idea that intracellular organelles can act as repositories for metabolic acid in transformed cells. Indeed, STAT3 was recently identified to localize to lysosomes in A549 (lung carcinoma) and HeLa (cervical cancer) cells, where it associates with the vacuolar H+-ATPase to modulate lysosomal pH (28). Increased lysosome pH upon STAT3 depletion is accompanied by a decrease in pHi, suggesting that transfer of H+ from the cytosol into lysosomes might also play a role in pHi homeostasis. The Golgi accounts for approximately 3% of a HeLa cell by volume, while the cytosol accounts for about 55% (42, 43). Considering that we observe a differential of 0.5 pH units in either direction upon NHE7 depletion, and that the pH scale is logarithmic, this means that the acidification of the Golgi can more than account for the molar quantity of H+ ions that accumulate in the cytoplasm. Our data demonstrate that NHE7 is important in transporting H+ into the TGN, which is involved in assembling and secreting post-Golgi vesicles whose cargo is destined for the extracellular space. In this way, acidification of the Golgi may not only represent a repository for cytoplasmic acid, but also the means to expel H+ extracellularly. Whether NHE7, or more generally, TGN acidification, plays a role in lowering the pH of the tumor microenvironment is an interesting question that requires further scrutiny.
We establish that deregulation of pH homeostasis through the targeting of NHE7 is detrimental to cancer cell fitness and suppresses the growth of PDAC tumors. These findings open possibilities for the development of new therapeutic interventions. The targeting of NHE activity in cancer has been explored from the standpoint of more well-characterized NHE isoforms; however, current NHE inhibitors are pleiotropic and would block NHE activity irrespective of antiporter directionality (22). Our work on the particular depletion of NHE7 argues for the development of NHE7-specific inhibitors. Moreover, since NHE7 suppression in HPNE cells does not seem to affect viability or Golgi acidification, the role of NHE7 in pHi and the maintenance of the actin cytoskeleton might be a unique vulnerability in cancer cells. It is thought that the alkaline pHi in cancer cells is an important factor that drives dependency on glycolysis. For example, intracellular alkalinity in cancer cells stimulates the activity of phosphofructokinase-1 and other glycolytic enzymes, while inhibiting gluconeogenesis (44–46). In this way, the alkaline pHi associated with transformation helps to drive a metabolic shift and, as a consequence, PDAC tumor cells with increased glycolytic flux might require a ‘proton sink’ to maintain their cytosolic pH due to increased H+ production (47). Untransformed cells, such as HPNE cells, which have lower glycolytic capacity compared to KRAS-mutant cells, would not require such a mechanism for pH homeostasis (48). Hence, targeting NHE7 could be a specific approach to compromise the fitness of cancer cells and whether NHE7 is a bona fide target in PDAC certainly warrants further analysis.
Methods
Clinical data analysis
For screening of survival correlations, patient data was accessed through the R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl). Overall survival correlation with NHE7 expression levels was assessed using the following setting; cut_off modus = scan, minimal groupsize = 8. PDAC patient data for survival and expression analysis were collected from the following databases; GSE62452, GSE17891, GSE21501, GSE28735, GSE15471, GSE16515, GSE43795, GSE71729, E-MEXP-1121, E-MTAB-179, E-MTAB-6134, TCGA Pancreatic Cancer (PAAD) and Pancreatic Cancer (QCMG, AU). For survival analysis, samples were grouped in ‘high’ and ‘low’ based on maximum significance.
Cell culture
779E is a low passage patient-derived PDAC cell line established and kindly provided by Dr. A. M. Lowy (University of California, San Diego) and has been previously described (49). All other cell lines were obtained from ATCC. Cell lines were maintained under 5% CO2, 37°C in a humidified atmosphere in the presence of 100 units/ml penicillin/streptomycin. AsPC1 and BxPC-3 cells were cultured in RPMI, 10% fetal bovine serum (FBS), 20 mM HEPES, and 1 mM sodium pyruvate. All other cell lines were cultured in DMEM, 10% FBS, and 20 mM HEPES. For doxycycline-inducible expression cassettes, Tet System Approved FBS (#631101, Takara) was used. Media for Normal Pancreatic Fibroblasts was supplemented with 10 ng/ml hEGF (MilliporeSigma). Media for hTERT-HPNE cells was supplemented with 10 ng/ml hEGF and 750 ng/ml puromycin (AdipoGen). All cell lines in this study were maintained in culture no more than 6 weeks, have been authenticated via short tandem repeat fingerprinting no later than 2015 and were routinely tested for mycoplasma contamination using a PCR Mycoplasma Detection Kit (ABM). Cell culture media and supplements were purchased from Corning.
Growth curves
Cells were seeded on 48 well cell culture plates and transduced the next day. At the indicated time points, cells were fixed for 20 min with 4% paraformaldehyde. Subsequently, wells were washed by submersion in a water bath. For crystal violet staining, wells were aspirated and 0.5% crystal violet was added. After 20 min, crystal violet was aspirated and wells were thoroughly washed by submersion in a water bath. Plates were left to dry at 37°C and crystal violet was reconstituted in 10% acetic acid after which absorbance was measured at 590 nm using a VersaMax Microplate Reader (Molecular Devices). For SYTO 60 staining, wells were aspirated and incubated for 30 min at room temperature with 1 μM SYTO 60 (Thermo Fisher Scientific) in 0.2% Triton X-100 in PBS. Next, wells were washed with PBS and plates were imaged at 700 nm on an Odyssey CLx imager. Quantification of SYTO 60 staining was performed using the Image Studio Lite software.
Cell death assays
For Annexin V / PI staining, cells were seeded on 6 well cell culture plates. After 5 days of doxycycline treatment or stated otherwise, trypsinized cells and cells from cell culture media were collected, centrifuged for 3 min at 500 g, and washed in ice-cold PBS. Next, cells were resuspended in Annexin V Binding Buffer and used according to the FITC Annexin V Apoptosis Detection Kit I (BD Biosciences) instructions. Cells were analyzed using an LSRFortessa (BD Biosciences) at the Sanford Burnham Prebys Flow Cytometry Core. Cell death was analyzed using FCS Express 6 software. For Hoechst staining, cells were seeded on 48 well cell culture plates. After 5 days of doxycycline treatment or stated otherwise, cells were fixed with 4% paraformaldehyde and incubated for 30 min at room temperature with 0.05 μg/ml Hoechst 33342 (Cayman Chemical) and 0.1% Triton X-100.
Constructs, cloning, lentiviral production and transduction
The GalT-mCherry-EGFP biosensor was generated as followed; the fragment encoding for the 82aa TGN-targeting peptide from GalT was derived from mKeima-Red-Golgi-7 (Addgene plasmid #56014), by digestion with NheI and BamHI (Thermo Fisher Scientific). Subsequently, the insert was subcloned into mCherry-SEpHluorin (Addgene plasmid #32001) using NheI and BglII restriction enzymes. From the resulting construct, the SEpHluorin sequence was excised by BamHI and NotI and replaced for the EGFP sequence derived from pEGFP-N3 (Clontech) using the same restriction enzymes.
Tet-pLKO-puro (Addgene plasmid #21915) constructs for doxycycline-induced shRNA expression were generated according to supplier’s instructions. To obtain these constructs the following oligonucleotide sequences were used; shNHE7 #1 (TRCN0000043808), shNHE7 #2 (TRCN0000043811), and shNHE7 #3 (TRCN0000043810) from the TRC database, and shCTRL Forward 5’-CCGG CCTAAGGTTAAGTCGCCCTCG CTCGAG CGAGGGCGACTTAACCTTAGG TTTTTG-3’, shCTRL Reverse 5’-AATTCAAAAA CCTAAGGTTAAGTCGCCCTCG CTCGAG CGAGGGCGACTTAACCTTAGG-3’. pLKO.1-puro shCTRL was purchased from MilliporeSigma (SHC002). pLKO.1-puro shNHE7 #1, shNHE7 #2, and shNHE7 #2 were obtained from Dharmacon (TRCN0000043808, TRCN0000043811, TRCN0000043810, respectively) and share the same targeting sequence as their Tet-pLKO-puro counterparts. Recombinant lentiviral particles expressing the above shRNAs were produced from the Sanford Burnham Prebys Viral Vector Core.
For transduction, cells were incubated with serum-free media containing 8 μg/ml Polybrene (MilliporeSigma) and lentiviral particles (MOI = 8–32). After 8 hours, an equal volume of complete media was added and media was completely changed the day after. To obtain stably transduced cell lines, complete media containing 10 μg/ml puromycin was added after 48 hours transduction. Subsequently, cells were harvested and frozen or maintained in complete media supplemented with puromycin until further use.
Immunofluorescence
Cells seeded on acid-washed glass coverslips were washed quickly with PBS++ (10 mM phosphate buffer, 2.7 mM KCl, 137 mM NaCl, 1 mM CaCl2, 0.5 mM MgCl2) at 37°C and fixed in cytoskeleton fixation buffer (80 mM PIPES pH 6.8, 5 mM EGTA, 2 mM MgCl2, 4% PFA) for 10 min at 37°C. Glass slides containing frozen sections of xenograft tumor were processed at room temperature. After 10 min permeabilization at room temperature in 0.2% Triton X-100 in PBS, cells were blocked for 1 hour in blocking buffer (5% goat serum in PBS). Thereafter, cells were incubated for 4 hours at room temperature with primary antibodies or Acti-stain 488 phalloidin (#PHDG1-A, Cytoskeleton) diluted in blocking buffer. Next, cells were washed three times in PBS and incubated for 1 hour in blocking buffer containing secondary antibodies and 2 μg/ml DAPI (MilliporeSigma). Finally, cells were washed three times for 5 min in PBS and mounted on glass slides using fluorescence mounting medium (DAKO).
For morphology studies and xenograft samples, images were captured using an EVOS M5000 Cell Imaging System (Thermo Fisher Scientific) equipped with a Plan Fluorite 40x/0.75 NA air objective. Relative F-actin staining was assessed by determining the total F-actin area within CK8 positive cells using ImageJ software (National Institutes of Health). Co-localization studies were performed at the Sanford Burnham Prebys Cell Imaging Core, using a Zeiss LSM 710 laser scanning confocal microscope equipped with a Plan Apochromat 63X/1.4 NA oil immersion objective.
For immunofluorescence, the following primary antibodies were used; NHE7 (PA5–61843, Thermo Fisher Scientific), Golgin-97 (#13192 and #97537) and pMLC2 (#3671) from Cell Signaling Technologies, Giantin-Alexa Fluor 488 (#A488–114L, Biolegend), Lamin A (L1293) and αTubulin (T6074) from MilliporeSigma, and CK8 (Developmental Studies Hybridoma Bank, TROMA-I, TROMA-I was deposited to the DSHB by Brulet, P. / Kemler, R.). The secondary antibodies used were purchased from Thermo Fisher Scientific; Goat anti-Mouse IgG (H+L), Alexa Fluor Plus 488 (A32723) and Alexa Fluor 555 (A-21422), Goat anti-Rabbit IgG (H+L), Alexa Fluor 594 (A-11012) and Alexa Fluor Plus 647 (A32733), and Goat anti-Rat IgG (H+L), Alexa Fluor 594 (A-11007).
Reagents and small inhibitors
Doxycycline hyclate (2 μg/ml, Acros Organics). EIPA (25–75 μM, MilliporeSigma). Tunicamycin (10 μg/ml, MP Biomedicals). Brefeldin A (10 μg/ml, Cayman Chemical). Monensin (25 nM, Alfa Aesar). P-3FAX-Neu5Ac (Tocris) was used at 100 μM unless stated otherwise. SR-7826 (BioVision) and (–)-Blebbistatin (Biogems) were used at 3.33 μM, unless stated otherwise.
TGN luminal pH assessment
Stably Tet-pLKO-puro transduced MIA PaCa-2 cells were seeded on chambered #1.5 high performance cover glass (Cellvis) in the presence of doxycycline. After 48 hours, cells were transfected with GalT-mCherry-EGFP using Lipofectamine 2000 according to manufacturer’s instructions. On the day of the experiment, cells were washed and media was exchanged for EBSS without phenol red (MilliporeSigma). For pH controls, cells were incubated for 15 min with calibration buffers (130 mM KCl, 1 mM MgCl2, 10 μM Nigericin, 10 μM Valinomycin, 30 mM MES pH 5.5–6.5). Imaging was performed at 37°C and 5% CO2 using a Zeiss LSM 710 laser scanning confocal microscope equipped with an INC-2000 Incubator System (20/20 Technology Inc) and an EC Plan-Neofluar 40X/1.3 NA oil immersion objective at the Sanford Burnham Prebys Cell Imaging Core. EGFP and mCherry signals were collected simultaneously by 488 nm and 594 nm excitation and ratios were determined by ROI selection of fluorescent puncta using the ImageJ distribution Fiji.
HPLC analysis of N-glycosylation
On the day of the experiment, cells were grown for 8 hours with 25 μCi [2-3H]-D-mannose (20 Ci/mmol; American Radiolabeled Chemicals). Next, cells were washed twice and scraped in DPBS. Cell pellets were resuspended in 50 mM sodium phosphate buffer pH 7.5 containing EDTA-free protease inhibitor (Thermo Fisher Scientific), sonicated briefly and boiled for 10 min. After a second sonication and boil, DTT was added to the final concentration 40 mM. Deglycosylation was performed for 16 hours at 37°C in the presence of 100 U PNGase F. Released N-glycans were purified on TopTip 10–200 μl carbon columns (PolyLC Inc.,). All columns were prewashed using 1 × 200 μl 80 % acetonitrile solution with 0.1 % TFA and 2 × 200 μl 50 % acetonitrile solution with 0.05 % TFA, followed by equilibration using 2 × 200 μl water. Solvent was removed by 5 min centrifugation at 3500 rpm. Cell samples were centrifuged for 20 min at 14,000 rpm and resulting supernatant was loaded on the columns, which were subsequently washed four times with 200 μl of water. Glycans were eluted with 3 × 100 μl 50 % acetonitrile solution with 0.05 % TFA and dried by SpeedVac.
For desialylation, isolated N-glycans were treated with α2–3,6,8 neurminidase (New England Biolabs). The reaction was performed for 5 hours at 37°C and terminated by incubating samples for 15 min at 65°C. Desialylated N-glycans were then purified from the reaction mix using Carbon columns and dried.
For HPLC analysis, N-glycans were separated on amino column (Microsorb-MV 100 NH2 250 × 4.6 mm, 5 μm, Agilent Technologies), with 0.1 M ammonium formate pH 4.4 as solvent A and acetonitrile as solvent B. The separation method used a linear gradient of 65–53 % acetonitrile at flow rate of 0.6 ml/min for 20 min and a linear gradient of 53–0 % of acetonitrile at the same flow rate for 33 min. Separation temperature was 30°C. One-minute fractions were collected, and their radioactivity determined on a scintillation counter.
Cell surface staining
Four days after transduction, cells seeded on 24 or 48 well cell culture plates were washed with PBS and fixed for 10 min with 1% PFA in PBS. Next, cells were washed and blocked for 1 hour in 2% BSA/PBS after which cells were incubated for an additional hour with biotinylated SNA (2 μg/ml), PNA, or SNA (1 μg/ml) from Vector Laboratories. Cells were washed and incubated for 1 hour with Cy3-Streptavidin (Vector Laboratories) in 2% BSA/PBS containing DAPI. Ultimately, cells were washed and imaged on an EVOS M5000 Cell Imaging System equipped with a Plan Fluorite 20x/0.50 NA air objective. For EGFR staining, cells were blocked with 5% goat serum, incubated 4–16 hours with an extracellular EGFR targeting antibody (MA5–13269, Thermo Fisher Scientific), which was thereafter detected with Goat anti-Mouse IgG (H+L), Alexa Fluor 555.
Cytosolic pH assessment
MIA PaCa-2, PANC-1, and hTERT-HPNE cells were treated with the indicated inhibitor or transduced for 4, 6 and 6 days, respectively, after which cells were loaded with pHrodo Red AM (Thermo Fisher Scientific) according to manufacturer’s instructions. Next, cells were washed and incubated for 30 min at 37°C and 5% CO2 in EBSS without phenol red, to allow for the removal of nonhydrolyzed pHrodo Red AM. Thereafter, EBSS was refreshed and cells were imaged at 37°C and 5% CO2 on the EVOS FL Auto Imaging System (Thermo Fisher Scientific) equipped with the EVOS Onstage Incubator and a Plan Fluorite 20X/0.45 NA air objective. For pH controls, cells were incubated for 15 min with the Intracellular pH Calibration Buffer Kit (Thermo Fisher Scientific) or calibration buffers (130 mM KCl, 1 mM MgCl2, 10 μM Nigericin, 10 μM Valinomycin, 30 mM MOPS pH 6.5–8.0). Fluorescence intensities were determined using ROI selection of fluorescent cells using ImageJ software.
Acute acid load
MIA PaCa-2 cells were seeded on 24 or 48 well cell culture plates and transduced for 4 days. Next, cells on 24 and 48 well plates were loaded, respectively, with pHrodo Red AM and 1 μM BCECF AM (MilliporeSigma) in Live Cell Imaging Solution (Thermo Fisher Scientific). After 30 min at 37°C and 5% CO2, cells were washed and incubated with loading solution (50 mM NH4Cl, 70 mM choline chloride, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM glucose and 15 mM MOPS pH 7.4) for 30 min at 37°C in a CO2-free incubator. Before analysis, loading solution was refreshed. Next, media was replaced for acidification solution (120 mM choline chloride, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM glucose and 15 mM HEPES pH 7.0) and recovery solution (120 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM glucose and 15 mM HEPES pH 7.4). pHrodo Red AM-loaded cells were imaged using an EVOS M5000 Cell Imaging System equipped with a Plan Fluorite 20X/0.40 NA air objective. In between time-points, cells were returned to the CO2-free incubator. BCECF AM fluorescence was determined at 37°C using a Varioskan LUX multimode microplate reader (Thermo Fisher Scientific) with excitation at 440 nm and 510 nm while recording emission at 535 nm.
Xenograft studies
All animal experiments were performed under the Institutional Animal Care and Use Committee approved protocol. One million cells transduced for 2 days, or one and a half million stably transduced cells, were resuspended in 50% Matrigel (Corning) and injected subcutaneously into the flanks of female nude mice (Homozygous Fox1nu, Jackson Laboratory, 8 weeks old). Palpable tumors were used for electronic caliper measurement in two dimensions to estimate tumor volume according to the equation V = (L × W2)/2. For doxycycline administration, mice were enrolled when tumors reached approximately 100 mm3. Doxycycline was administered in the drinking water (2 mg/ml and 5% sucrose) and diet (Envigo, TD.01306). Vehicle control mice were given 5% sucrose drinking water and regular diet (Envigo, #2918). Tumor sizes were recorded by electronic caliper measurement and tumors were collected, weighted, and cut into pieces when reaching a volume of approximately 1000 mm3. For RNA extraction, samples were snap-frozen in liquid nitrogen. For IHC and IF, cross-section slices were rapidly frozen in optimal cutting temperature compound (Tissue-Tek).
Statistical analysis
Each experiment was repeated independently at least three times. Results are presented as the mean of independent experiments ± standard error of the mean (s.e.m), unless stated otherwise. Statistical analyses were performed using GraphPad Prism software (v8.2.0; GraphPad Software). Statistical significance between two groups was determined by unpaired or paired two-tailed Student’s t-test. For multiple-group and time-course comparison, one-way ANOVA and two-way ANOVA were used, respectively. A value of P < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Supplementary Material
Statement of significance.
NHE7 regulates cytosolic pH through Golgi acidification, which points to the Golgi as a ‘proton sink’ for metabolic acid. Disruption of cytosolic pH homeostasis via NHE7 suppression compromises PDAC cell viability and tumor growth.
Acknowledgements
We thank members of the Commisso laboratory for their helpful comments and discussions. We thank Dr. A. M. Lowy for providing the 779E cells. This work was primarily supported by a Department of Defense Career Developmental Award (W81XWH-17-10316), as well as a Pancreatic Cancer Action Network—AACR Career Development Award (15-20-25-COMM) and a NIH/NCI Research Project Grant (R01CA207189) to Cosimo Commisso. Financial support to Hudson H. Freeze was provided by The Rocket Fund and a National Institutes of Health grant (R01DK099551). Koen Galenkamp is the recipient of a TRDRP Postdoctoral Fellowship Award (T30FT0952). We thank the Sanford Burnham Prebys shared resources staff for assistance with histopathology sample preparation and lentivirus production. Sanford Burnham Prebys core services are supported by NCI Cancer Center Support grant P30 CA030199.
Financial support: This work was primarily supported by a Department of Defense Career Developmental Award (W81XWH-17-10316), as well as a Pancreatic Cancer Action Network—AACR Career Development Award, in memory of Skip Viragh (15-20-25-COMM) to Cosimo Commisso. Financial support to Hudson H. Freeze was provided by The Rocket Fund and a National Institutes of Health grant (R01DK099551). Koen Galenkamp is the recipient of a TRDRP Postdoctoral Fellowship Award (T30FT0952). Sanford Burnham Prebys core services are supported by NCI Cancer Center Support grant P30 CA030199.
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends in Biochemical Sciences. 2016;41(3):211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corbet C, Feron O. Tumour acidosis: from the passenger to the driver’s seat. Nature Reviews Cancer. 2017;17(10):577–93. [DOI] [PubMed] [Google Scholar]
- 3.Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nature Reviews Cancer. 2011;11(9):671–7. [DOI] [PubMed] [Google Scholar]
- 4.White KA, Grillo-Hill BK, Barber DL. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. Journal of Cell Science. 2017;130(4):663–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spugnini EP, Sonveaux P, Stock C, Perez-Sayans M, De Milito A, Avnet S, et al. Proton channels and exchangers in cancer. Biochimica Et Biophysica Acta-Biomembranes. 2015;1848(10):2715–26. [DOI] [PubMed] [Google Scholar]
- 6.Numata M, Orlowski J. Molecular cloning and characterization of a novel (Na+,K+)/H+ exchanger localized to the trans-Golgi network. Journal of Biological Chemistry. 2001;276(20):17387–94. [DOI] [PubMed] [Google Scholar]
- 7.Lin PJC, Williams WP, Luu Y, Molday RS, Orlowski J, Numata M. Secretory carrier membrane proteins interact and regulate trafficking of the organellar (Na+,K+)/H+ exchanger NHE7. Journal of Cell Science. 2005;118(9):1885–97. [DOI] [PubMed] [Google Scholar]
- 8.Lin PJC, Williams WP, Kobiljski J, Numata M. Caveolins bind to (Na+, K+)/H+ exchanger NHE7 by a novel binding module. Cellular Signalling. 2007;19(5):978–88. [DOI] [PubMed] [Google Scholar]
- 9.Fukura N, Ohgaki R, Matsushita M, Nakamura N, Mitsui K, Kanazawa H. A Membrane-Proximal Region in the C-Terminal Tail of NHE7 Is Required for Its Distribution in the Trans-Golgi Network, Distinct from NHE6 Localization at Endosomes. Journal of Membrane Biology. 2010;234(3):149–58. [DOI] [PubMed] [Google Scholar]
- 10.Milosavljevic N, Monet M, Lena I, Brau F, Lacas-Gervais S, Feliciangeli S, et al. The Intracellular Na+/H+ Exchanger NHE7 Effects a Na+-Coupled, but Not K+-Coupled Proton-Loading Mechanism in Endocytosis. Cell Reports. 2014;7(3):689–96. [DOI] [PubMed] [Google Scholar]
- 11.Khayat W, Hackett A, Shaw M, Ilie A, Dudding-Byth T, Kalscheuer VM, et al. A recurrent missense variant in SLC9A7 causes nonsyndromic X-linked intellectual disability with alteration of Golgi acidification and aberrant glycosylation. Human Molecular Genetics. 2019;28(4):598–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu SD, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nature Medicine. 2011;17(4):500–U140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nature Genetics. 2015;47(10):1168–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puleo F, Nicolle R, Blum Y, Cros J, Marisa L, Demetter P, et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology. 2018;155(6):1999–+. [DOI] [PubMed] [Google Scholar]
- 15.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–+. [DOI] [PubMed] [Google Scholar]
- 16.Crowley LC, Marfell BJ, Waterhouse NJ. Analyzing Cell Death by Nuclear Staining with Hoechst 33342. Cold Spring Harbor protocols. 2016;2016(9). [DOI] [PubMed] [Google Scholar]
- 17.Luke MR, Kjer-Nielsen L, Brown DL, Stow JL, Gleeson PA. GRIP domain-mediated targeting of two new coiled-coil proteins, GCC88 and GCC185, to subcompartments of the trans-Golgi network. Journal of Biological Chemistry. 2003;278(6):4216–26. [DOI] [PubMed] [Google Scholar]
- 18.Roth J, Berger EG. IMMUNO-CYTOCHEMICAL LOCALIZATION OF GALACTOSYLTRANSFERASE IN HELA-CELLS - CODISTRIBUTION WITH THIAMINE PYROPHOSPHATASE IN TRANS-GOLGI CISTERNAE. Journal of Cell Biology. 1982;93(1):223–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Llopis J, McCaffery JM, Miyawaki A, Farquhar MG, Tsien RY. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(12):6803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophysical Journal. 1997;73(5):2782–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp red fluorescent protein. Nature Biotechnology. 2004;22(12):1567–72. [DOI] [PubMed] [Google Scholar]
- 22.Masereel B, Pochet L, Laeckmann D. An overview of inhibitors of Na+/H+ exchanger. European Journal of Medicinal Chemistry. 2003;38(6):547–54. [DOI] [PubMed] [Google Scholar]
- 23.Kellokumpu S. Golgi pH, Ion and Redox Homeostasis: How Much Do They Really Matter? Frontiers in Cell and Developmental Biology. 2019;7:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Contessa JN, Bhojani MS, Freeze HH, Rehemtulla A, Lawrence TS. Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Research. 2008;68(10):3803–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varki A, Schnaar RL, Schauer R. Sialic Acids and Other Nonulosonic Acids In: rd, Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, et al. , editors. Essentials of Glycobiology. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; Copyright 2015–2017 by The Consortium of Glycobiology Editors, La Jolla, California. All rights reserved.; 2015. p. 179–95. [Google Scholar]
- 26.Stanley P, Taniguchi N, Aebi M. N-Glycans In: rd, Varki A, Cummings RD, Esko JD, Stanley P, Hart, et al. , editors. Essentials of Glycobiology. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; Copyright 2015–2017 by The Consortium of Glycobiology Editors, La Jolla, California. All rights reserved.; 2015. p. 99–111. [Google Scholar]
- 27.Rillahan CD, Antonopoulos A, Lefort CT, Sonon R, Azadi P, Ley K, et al. Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nature Chemical Biology. 2012;8(7):661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu B, Palmfeldt J, Lin L, Colaco A, Clemmensen KKB, Huang J, et al. STAT3 associates with vacuolar H+-ATPase and regulates cytosolic and lysosomal pH. Cell Research. 2018;28(10):996–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franchi A, Cragoe E Jr., Pouyssegur J. Isolation and properties of fibroblast mutants overexpressing an altered Na+/H+ antiporter. The Journal of biological chemistry. 1986;261(31):14614–20. [PubMed] [Google Scholar]
- 30.Klausner RD, Donaldson JG, Lippincottschwartz J. BREFELDIN-A - INSIGHTS INTO THE CONTROL OF MEMBRANE TRAFFIC AND ORGANELLE STRUCTURE. Journal of Cell Biology. 1992;116(5):1071–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pellegrin S, Mellor H. Actin stress fibres. Journal of Cell Science. 2007;120(20):3491–9. [DOI] [PubMed] [Google Scholar]
- 32.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, et al. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393(6687):805–9. [DOI] [PubMed] [Google Scholar]
- 33.Sumi T, Matsumoto K, Takai Y, Nakamura T. Cofilin phosphorylation and actin cytoskeletal dynamics regulated by Rho- and Cdc42-activated LIM-kinase 2. Journal of Cell Biology. 1999;147(7):1519–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yonezawa N, Nishida E, Sakai H. pH control of actin polymerization by cofilin. The Journal of biological chemistry. 1985;260(27):14410–2. [PubMed] [Google Scholar]
- 35.Yeoh S, Pope B, Mannherz HG, Weeds A. Determining the differences in actin binding by human ADF and cofilin. Journal of Molecular Biology. 2002;315(4):911–25. [DOI] [PubMed] [Google Scholar]
- 36.Pope BJ, Zierler-Gould KM, Kuhne R, Weeds AG, Ball LJ. Solution structure of human cofilin - Actin binding, pH sensitivity, and relationship to actin-depolymerizing factor. Journal of Biological Chemistry. 2004;279(6):4840–8. [DOI] [PubMed] [Google Scholar]
- 37.Wioland H, Jegou A, Romet-Lemonne G. Quantitative Variations with pH of Actin Depolymerizing Factor/Cofilin’s Multiple Actions on Actin Filaments. Biochemistry. 2019;58(1):40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blondin L, Sapountzi V, Maciver SK, Lagarrigue E, Benyamin Y, Roustan C. A structural basis for the pH-dependence of cofilin - F-actin interactions. European Journal of Biochemistry. 2002;269(17):4194–201. [DOI] [PubMed] [Google Scholar]
- 39.Kanellos G, Zhou J, Patel H, Ridgway RA, Huels D, Gurniak CB, et al. ADF and Cofilin1 Control Actin Stress Fibers, Nuclear Integrity, and Cell Survival. Cell Reports. 2015;13(9):1949–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiggan ON, Schroder B, Krapf D, Bamburg JR, DeLuca JG. Cofilin Regulates Nuclear Architecture through a Myosin-II Dependent Mechanotransduction Module. Scientific Reports. 2017;7:40953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huczynski A, Janczak J, Lowicki D, Brzezinski B. Monensin A acid complexes as a model of electrogenic transport of sodium cation. Biochimica Et Biophysica Acta-Biomembranes. 2012;1818(9):2108–19. [DOI] [PubMed] [Google Scholar]
- 42.Milo R, Phillips R. Cell biology by the numbers. New York, NY: Garland Science; 2016. xlii, 356 pages p. [Google Scholar]
- 43.Alberts B. Molecular biology of the cell. 4th ed New York: Garland Science; 2002. xxxiv, 1463, 86 p. p. [Google Scholar]
- 44.Cardone RA, Casavola V, Reshkin SJ. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nature Reviews Cancer. 2005;5(10):786–95. [DOI] [PubMed] [Google Scholar]
- 45.Alfarouk KO, Verduzco D, Rauch C, Muddathir AK, Adil HHB, Elhassan GO, et al. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience. 2014;1(12):777–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swietach P. What is pH regulation, and why do cancer cells need it? Cancer and Metastasis Reviews. 2019;38(1–2):5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hardie RA, van Dam E, Cowley M, Han TL, Balaban S, Pajic M, et al. Mitochondrial mutations and metabolic adaptation in pancreatic cancer. Cancer & Metabolism. 2017;5:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ju HQ, Ying HQ, Tian T, Ling JH, Fu J, Lu Y, et al. Mutant Kras- and p16-regulated NOX4 activation overcomes metabolic checkpoints in development of pancreatic ductal adenocarcinoma. Nature Communications. 2017;8:14437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Villarino N, Signaevskaia L, van Niekerk J, Medal R, Kim H, Lahmy R, et al. A screen for inducers of bHLH activity identifies pitavastatin as a regulator of p21, Rb phosphorylation and E2F target gene expression in pancreatic cancer. Oncotarget. 2017;8(32):53154–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.