

SUMMARY:
Poly-ADP-ribose-polymerase inhibitors (PARPi) are promising in BRCA2-altered prostate cancer. Data were presented on PARPi efficacy in prostate cancers with alterations in other DNA damage repair genes which suggest low response rates in ATM-, CHEK2-, CDK12-altered tumors and promising results in PALB2-, RAD51B-, FANCA-, and BRIP1-altered tumors.
In this issue of Clinical Cancer Research, Abida and colleagues report response to the PARPi rucaparib in patients with metastatic castration resistant prostate cancer (mCRPC) harboring deleterious alterations in non-BRCA DNA Damage Repair (DDR) genes.1 The data provides early evidence that prostate tumors with ATM, CDK12 and CHEK2 alterations may have limited response to rucaparib, while tumors with alterations in PALB2, FANCA, BRIP1 and RAD51B may benefit from PARP inhibition.1
Several studies suggest that PARPi are effective in prostate cancer tumors with BRCA2 alterations.2 The mechanism of action of PARPi in tumors is typically attributed to impaired double strand DNA damage repair - homologous recombination (HR) repair deficient tumors, but likely involves additional mechanisms such as 1) trapping of the PARP1 – enzyme, which regulates several DNA repair processes; 2) inhibition of base excision, critical to single-strand DNA repair; 3) activation of error prone non-homologous end joining repair, important in double strand DNA repair; and 4) inhibition of DNA repair protein recruitment (e.g. BARD1-BRCA1 complex).3 Multiple non-BRCA genes mediate HR repair (e.g. ATM, CHEK2 and others), so many have been included as candidate biomarkers for PARPi sensitivity. However, data to date has been limited by small numbers and technical differences.2
To begin to address this, Abida, et al. evaluated response to rucaparib in patients with mCRPC and non-BRCA DDR gene alterations in the phase 2 TRITON2 study. Eligibility included patients with disease progression after second-generation androgen receptor-targeted therapy and taxane-based chemotherapy AND who were identified to have germline and/or somatic mutations in selected DDR genes (BRCA1, BRCA2, ATM, BARD1, BRIP1, CDK12, CHEK2, FANCA, NBN, PALB2, RAD51, RAD51B, RAD51C, RAD51D, or RAD54L). Whole blood was used for germline testing and somatic sequencing was performed on circulating tumor DNA (ctDNA) or on tumor tissue by central or local laboratory. This ad-hoc analysis included 78 patients whose tumors were found to have alterations in non-BRCA DDR genes. Response was defined as partial or complete radiographic response per RECIST criteria or PSA50 response (decline of serum Prostate Specific Antigen by 50% from baseline). Overall rates of response among patients with ATM, CHEK2 and CDK12 alterations were low: 2/49 (ATM), 2/12 (CHEK2), 1/15 (CDK12). Interestingly, the proportion of patients without evidence of radiographic progression (labeled clinical benefit) at 6 and 12 months suggests that rucaparib may stabilize disease in these patients. Responses were observed in 2/2 patients with PALB2, 1/4 with FANCA, 1/2 with BRIP1, 1/1 with RAD51B mutations, small, but intriguing numbers.
These findings are consistent with previously reported data: 1/5 patients with FANCA-, 4/7 with PALB2-, 2/19 with ATM-, and 0/20 with CDK12 -alterations had RECIST or PSA50 response to PARPi in TOPARP-B trial.2 In the PROfound study, patients with RAD51B alterations treated with PARPi had a 6-month median radiographic progression free survival (rPFS) benefit compared to physician choice therapy arm, and similar rPFS was observed between two arms in patients with ATM alterations.2 Similarly 0/6 patients with ATM alterations had PSA50 response in a retrospective study.2 As a field, we must be careful to draw conclusions from small numbers in each analysis, however, in aggregate, some convincing trends are emerging.
The authors of this manuscript acknowledge several limitations.1 First, there was no requirement for central laboratory confirmation of reported alterations. Some, but not all, sequencing results were confirmed by central laboratory, increasing the chance of false positive results for eligibility. Second, clonal hematopoietic (CH) variants may introduce interference in ctDNA studies and mistakenly attributed to tumor-specific alterations.4 Among genes included for eligibility in this study, alterations in ATM are more common in CH and thus may contribute to variant origin misclassification.4 Third, loss of heterozygosity (LOH) and tumor sequencing data were not available for several patients, including the patients with ATM alterations who had response to rucaparib. It is possible that non-ATM tumor alterations contributed to PARPi responses in these patients even though their eligibility was due to an ATM alteration.
This study addresses important knowledge gaps by characterizing response to PARPi in tumors with relatively rare alterations in DDR genes. It is especially timely as we anticipate Food and Drug Administration approval of PARPi in prostate cancer based on the positive phase 3 PROfound study and other eagerly anticipated trials such as TRITON3, GALAHAD and TALAPRO-1.2 Understanding the efficacy of PARPi in prostate tumors with non-BRCA DDR gene alterations is essential before widespread clinical use makes the task more challenging. The data presented by Abida, et al may help generate hypotheses about mechanisms of response and resistance, and refine predictive biomarkers for PARPi use for patients with prostate cancer, including in subsequent preclinical and clinical trials.
A better understanding of which patients benefit the most from PARPi and which DDR gene mutations could be the best predictors of response to PARPi will help refine precision oncology in prostate cancer therapy. A notable example is that not all BRCA2-mutated tumors respond to PARPi1,2 and conversely, in the TOPARP-A study, two patients without evidence of DDR gene mutations achieved response to PARPi, suggesting additional mechanisms of PARPi sensitivity beyond those specifically tested.2 Sensitivity to PARPi is hypothesized to be determined by functional HR deficiency (HRD), such that relying solely on gene mutations to predict PARPi sensitivity may overlook other mechanisms resulting in loss of HR function, e.g. DNA hypermethylation. Loss of HR repair function, regardless of the cause, is thought to lead to characteristic genomic changes such as HRD mutational signature and genomic instability. HRD mutational signature is characterized by a more-or-less equal representation of all possible single base substitutions and all 96 mutant nucleotide contexts.5 With the data and specimens from Abida, et al. and other PARPi studies, examining the downstream consequences of HR loss and the associated genomic footprints with clinical response may facilitate more accurate prediction of response to PARPi in the future (Figure 1).
Figure 1.
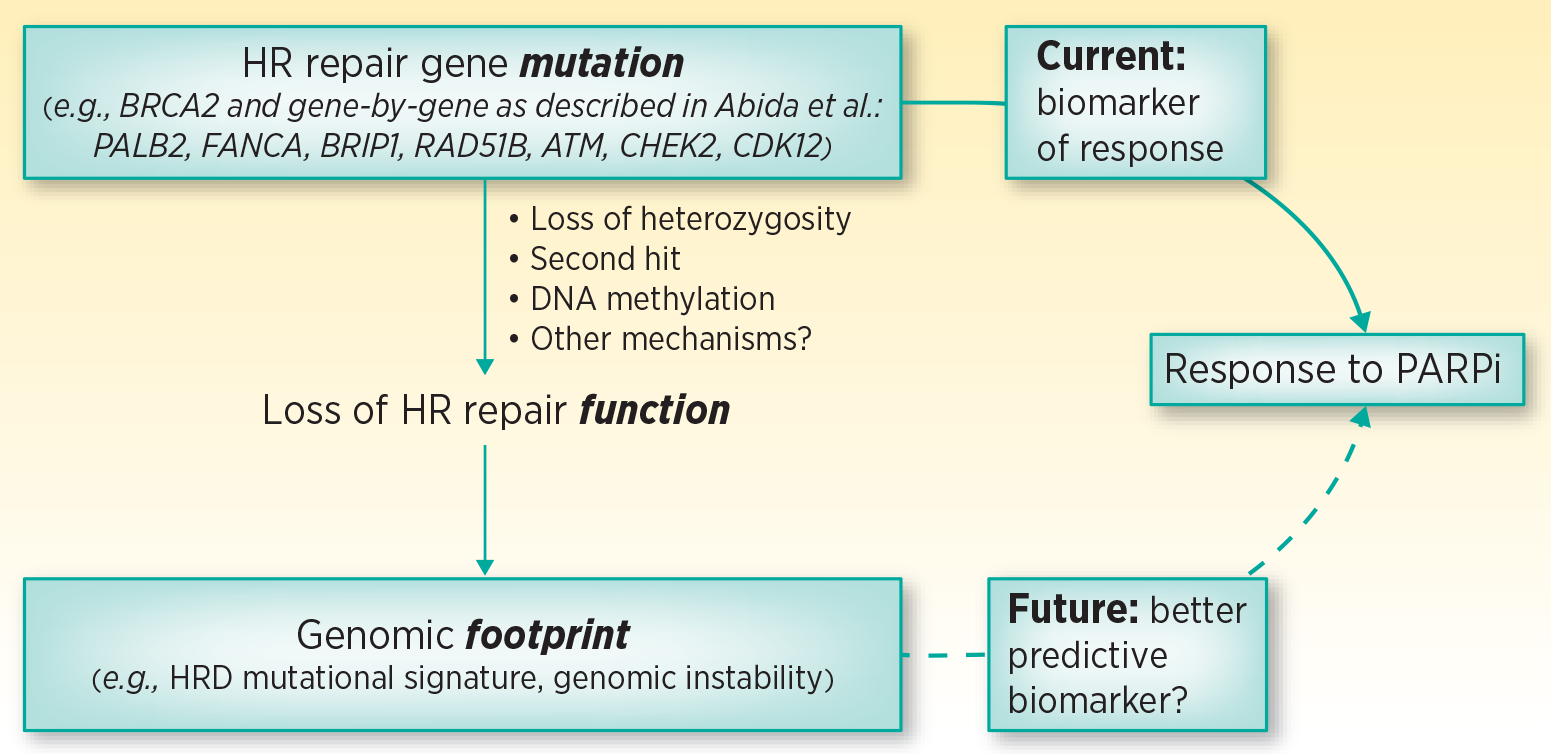
Predictive biomarkers for PARPi efficacy. Currently, alterations in HR repair genes are used to predict loss of HR repair function and to select prostate cancer patients most likely to respond to PARPi, potentially overlooking other causes of HR function loss. HR repair function loss consequences (i.e. genomic footprint) may be a better predictive biomarker for response to PARPi.
HR- homologous recombination; HRD – homologous recombination deficiency; PARPi - Poly-ADP-ribose-polymerase inhibitors
We applaud the authors in reporting their findings of response to PARPi in tumors with non-BRCA DDR gene alterations. To further strengthen the available data for patients whose tumors carry these and other rare alterations and the response to PARPi and other treatments, a concerted team approach is needed. Assembling greater numbers of patients with rare mutations will allow more robust investigation of clinical response, mutational signatures of specific gene alterations, mechanisms of resistance such as reversion mutations, and opportunities for model systems to dissect mechanisms. This will further refine predictive biomarkers, stratify appropriate patients to PARPi therapy and improve the precision of our targeted therapies.
Acknowledgments
We gratefully acknowledge funding support from the Prostate Cancer Foundation (to H. H. Cheng), National Cancer Institute (T32CA009515 to A. O. Sokolova, P50 CA097186–16A1 [PNW Prostate SPORE] to H. H. Cheng and E. Y. Yu, and P30 CA015704 to H. H. Cheng and E. Y. Yu), and Brotman Baty Institute (to H. H. Cheng).
Abbreviations list:
- CH
Clonal Hematopoietic
- ctDNA
circulating tumor DNA
- DDR
DNA Damage Repair
- HR
Homologous Recombination
- HRD
Homologous Recombination Deficiency
- LOH
loss of heterozygosity
- mCRPC
metastatic Castration Resistant Prostate Cancer
- PARPi
Poly-ADP-Ribose-Polymerase inhibitors
- PSA
Prostate Specific Antigen
- rPFS
radiographic Progression Free Survival
Footnotes
Conflict of interest disclosure statement: Dr. Evan Yu reports following potential conflicts of interest: Abbvie – Personal fees; Advanced Accelerator Applications – Personal fees; Amgen- Personal fees; Astrazeneca- Personal fees; Bayer- Grant & personal fees; Clovis – Personal fees; Dendreon- Grant & personal fees; Janssen- Personal fees; Merck- Grant, personal fees; Pharmacyclics- Personal fees; Sanofi-Genzyme – Personal fees; Seattle Genetics- Grant & personal fees; Pharmacycles – Personal fees and research; QED – Personal fees; Taiho – Research; Daiichi Sankyo – Research; EMD Serono – Personal fees; Tolmar – Personal Fees; Incyte – Personal fees. Dr. Heather Cheng reports the following potential conflicts of interest: Clovis: research grant to institution, Color Genomics: research grant to institution, Janssen: research grant to institution, Medivation: research grant to institution, Sanofi: research grant to institution. Dr. Sokolova declares no potential conflicts of interest.
References:
- 1.Abida W Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sokolova AO & Cheng HH Genetic Testing in Prostate Cancer. Curr. Oncol. Rep 22, 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott CL, Swisher EM & Kaufmann SH Poly (ADP-ribose) polymerase inhibitors: recent advances and future development. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 33, 1397–1406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ptashkin RN et al. Prevalence of Clonal Hematopoiesis Mutations in Tumor-Only Clinical Genomic Profiling of Solid Tumors. JAMA Oncol. 4, 1589–1593 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexandrov LB et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]