

Abstract
Chimeric antigen receptor (CAR) T cell therapy has been acclaimed as a revolution in cancer treatment following the impressive results in hematological malignancies. Unfortunately, in patients with solid tumors, objectives responses to CAR T cells are still anecdotal, and important issues are driven by on-target but off-tumor activity of CAR T cells and by the extremely complex biology of solid tumors. Here, we will review the recent attempts to challenge the therapeutic impediments to CAR T cell therapy in solid tumors. We will focus on the most promising strategies of antigen targeting to improve tumor specificity and address the tumor heterogeneity, efforts to circumvent the physical barriers of the tumor architecture such as subverted tumor vasculature, impediments of CAR T cell trafficking and immune suppressive microenvironment.
Keywords: solid tumors, adoptive cell therapy, CAR T cells
INTRODUCTION
Chimeric antigen receptors (CARs) are antigen receptors resulting from the fusion of a single-chain variable fragment (scFv) of a monoclonal antibody (mAb) with the ζ-chain of the T cell receptor complex and one or two costimulatory moieties(1–3). Upon expression in T cells, CARs allow Major Histocompatibility Complex (MHC)-unrestricted recognition of surface antigens expressed by tumor cells, T cell activation, proliferation and tumor cytolysis. MHC-unrestricted antigen recognition of CAR T cells overcomes the downregulation of Human Leucocyte Antigen molecules frequently orchestrated by cancer cells to escape immune recognition, while the scFv-mediated antigen recognition enables T cells to target also non-protein epitopes, widening the repertoire of actionable targets in cancer immunotherapy, and expanding the potentials of adoptive cell therapy in solid tumors pioneered by the use of tumor infiltrating lymphocytes (TILs) in melanoma(4). As briefly summarized in Table 1, CAR T cells may have several strengths as compared to TILs for the treatment of solid tumors. Although CAR T cells have been a clinical breakthrough in some hematologic malignancies(5–7), the development of such strategy for solid tumors is still in its infancy. As illustrated in Table 2, objective responses in patients with solid tumors treated with CAR T cells are scant, whereas on-target but off-tumor toxicities remain a great concern(8–32). The modest activity of CAR T cells in solid tumors as compared to B cell malignancies can be largely attributed to the intrinsic biologic differences between the two cancerous entities. Extreme heterogeneity of antigen expression and antigen-sharing with vital organs, presence of physical barriers for immune cell trafficking and penetration, together with the development of an immune suppressive microenvironment are general features of most of the non-hematological malignancies. In the present article, we provide a concise overview of the approaches that have been implemented to adapt CAR T cells to the complex pathophysiology of solid tumors taking particular attention to the strategies that are reaching the clinical investigation.
Table 1. Key features of TILs versus CAR T cells for adoptive cell therapy in solid tumors.
Green background indicates the strengths whereas red background indicates the weaknesses of the two immunotherapy approaches.
| TILs | CAR T cells |
|---|---|
| Difficult manufacturing (insufficient availability of tumor tissue or modest T cell tumor infiltration) | Easy manufacturing regardless of tumor localization and tumor burden |
| Invasive surgical procedures to obtain tumor tissue for T cell isolation | Non-invasive procedures (leukapheresis) for T cell isolation |
| Long selection and expansion process that can lead to T cell exhaustion | Relatively short manufacturing process |
| Lack of T cell costimulation | T cell costimulation is engineered within the CAR in second and third generation molecules |
| Autologous use only | Possible engineering strategies to produce “off-the-shelf” T cell products |
| HLA-restricted recognition of the tumor cells that can lead to tumor escape (e.g. alteration in antigen processing and presenting mechanisms in tumor cells, HLA downregulation) | Recognize antigens in HLA-unrestricted manner |
| Can target both surface and intracellular antigens | Can only target surface antigens, even if scFvs targeting peptide presented in the MHC have been developed |
| Can provide simultaneously multiple target specificity | Up-to-now it is not feasible to target more than two antigens simultaneously |
| No on-target toxicity | Possible on-target but off-tumor toxicity |
Table 2.
Summary of the safety and activity data from published studies on CAR T cell therapy in solid tumors.
| Author (year) | Type of study | Patients treated | Target | Tumor type(s) | Costimulation | Route | Lymphodepletion | On-target off-tumor toxicity | Objective responses | ORR n (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Morgan RA et al. (2010)(10) | Case report | 1 | HER2 | Colorectal cancer | CD28+4–1BB | Yes | ARDS (fatal) | Not applicable | Not applicable | |
| Ahmed N et al. (2015)(14) | Phase I/II | 19 | HER2 | Sarcoma | CD28 | i.v. | No | No | Not observed | 0/19 (0) |
| Ahmed N et al. (2017)(25) | Phase I | 17 | HER2 | Glioblastoma | Virus-specific T cells+CD28 | i.v. | No | No | 1 PR | 1/17 (6) |
| Feng K et al. (2018)(27) | Phase I | 11 | HER2 | Biliary tract cancer Pancreatic cancer | 4–1BB | i.v. | Yes | Liver enzymes increase | 1 PR | 1/11 (9) |
| Katz SC et al (2015)(15) | Phase I | 6 | CEA | Liver metastases | CD28 | hepatic artery infusion | No | No | Not observed | 0/6 (0) |
| Zhang C et al. (2017)(24) | Phase I | 10 | CEA | Colorectal cancer | CD28 | i.v. | Yes | No | 2 PR | 2/10 (20) |
| Thistlethwaite FC et al. (2017)(26) | Phase I | 14 | CEA | Colorectal cancer Gastro-esophageal cancer Pseudomyxoma peritonei Pancreatic cancer | - | i.v. | Yes | Transient acute respiratory toxicity | Not observed | 0/14 (0) |
| Katz SC et al. (2019)(32) | Phase I | 6 | CEA | Liver metastases | CD28+use of selective internal radiation therapy (SIRT) | hepatic artery infusion | No | No | 1 metabolic CR | 1/6 (17) |
| Louis CU et al. (2011)(11) | Phase I | 19 | GD2 | Neuroblastoma | Virus-specific T cells | i.v. | No | No | 3 CR | 3/19 (16) |
| Gargett T et al. (2016)(17) | Case report | 4 | GD2 | Melanoma | CD28+OX40 | i.v. | No/Yes | No | Not observed | Not applicable |
| Beatty GL et al. (2014)(13) | Case report | 2 | Mesothelin | Pleural mesothelioma Pancratic cancer | 4–1BB | i.v./intratumoral | No | No | 1 PR | Not applicable |
| Beatty GL et al. (2018)(30) | Phase I | 6 | Mesothelin | Pancreatic cancer | 4–1BB | i.v. | No | No | Not observed | 0/6 (0) |
| Brown CE et al. (2015)(16) | Pilot study | 3 | IL13Ra2 | Glioblastoma | - | Intracranial | No | Neurologic event | Not observed | 0/3 (0) |
| Brown CE et al. (2016)(19) | Case report | 1 | IL13Rα2 | Glioblastoma | 4–1BB | Intracranial | No | No | 1 CR | Not applicable |
| Feng K et al (2016)(21) | Phase I | 11 | EGFR | Non-small cell lung cancer | 4–1BB | i.v. | No/Yes | Serum lipase increase | 2 PR | 2/11 (18) |
| Feng KC et al. (2017)(22) | Case report | 1 | EGFR/CD133 | Cholangiocarcinoma | 4–1BB | i.v. | No | Liver enzymes increase (aEGFR), rash and mucositis (aCD133) | 1 PR | Not applicable |
| O’Rourke DM et al. (2017)(28) | Case report | 10 | EGFRvIII | Glioblastoma | 4–1BB | i.v. | No | No | Not observed | Not applicable |
| Goff SL et al. (2019)(31) | Phase I | 18 | EGFRvIII | Glioblastoma | CD28+4–1BB | i.v. | Yes | ARDS (one treatment-related death) | Not observed | 0/18 (0) |
| Kershaw MH et al. (2006)(8) | Phase I | 14 | α-folate receptor | Ovarian cancer | - | i.v. | No | No | Not observed | 0/14 (0) |
| Park JR et al. (2007)(9) | Phase I | 6 | CD171 | Neuroblastoma | - | i.v. | No | Lymphopenia, neutropenia, anemia, pneumonitis | 1 PR | 1/6 (17) |
| Lamers CH et al. (2013)(12) | Phase I | 12 | CAIX | Renal cell carcinoma | - | i.v. | No | Liver enzymes increase | Not observed | 0/12 (0) |
| Junghans RP et al. (2016)(18) | Phase I | 5 | PSMA | Prostate cancer | - | Yes | No | 2 PR | 2/5 (40) | |
| You F et al. (2016)(20) | Case report | 1 | MUC1 | Seminal vesicle cancer | CD28+4–1BB | Intratumoral | No | No | Not observed | Not applicable |
| Hege KM et al. (2017)(23) | Phase I | 16 | TAG-72 | Colorectal cancer | - | i.v./hepatic artery infusion | No | Retinal artery occlusion | Not observed | 0/16 (0) |
| Tchou J et al. (2017)(29) | Phase 0 | 6 | c-MET | Breast cancer | 4–1BB | Intratumoral | No | No | Not observed | 0/6 (0) |
IDENTIFICATION OF TARGETABLE ANTIGENS IN SOLID TUMORS
On-target but off-tumor activity
The success of CAR T cells targeting the pan-B cell marker CD19 in B-cell malignancies was predicated on the tolerability of the protracted B-cell aplasia caused by the sustained persistence of CAR T cells(7). In sharp contrast, the development of CAR T cells targeting solid tumors has been hampered by the anticipated and intolerable toxicity that can be caused by CAR T cells targeting antigens expressed by tumor cells but shared with normal tissues. Severe toxicities caused by on-target but off-tumor antigen recognition by CAR T cells have been reported in clinical studies. In a phase I study of anti-CAIX CAR T cells in metastatic renal cell carcinoma, the expression of CAIX on the bile duct epithelial cells caused dose-limiting liver toxicity(12). In a second study, the infusion of CAR T cells targeting HER2 caused fatal acute respiratory distress syndrome due to recognition of lung epithelia cells expressing low levels of HER2(10). It remains an open question if antigens overexpressed on the cell surface of tumor cells in solid tumors and at low levels in some normal tissues can be safely targeted by CAR T cells. Some clinical data and preclinical models suggest that a therapeutic window may be achievable taking in consideration which antigen epitopes are targeted by CAR T cells, the density of antigen expression and the affinity of the scFvs used to generate CARs. For example, HER2-specific CAR T cells targeting different epitopes can significantly reduce the activity of CAR T cells in normal tissues without compromising tumor recognition likely due to different accessibility to the epitope by the scFv in normal tissue as compared to cancerous cells(14). Preclinical observations suggest that higher antigen density expression in the tumor as compared to normal tissues can be exploited to effectively eliminate tumor cells, while sparing normal cells(33,34). Similarly, tuning the affinity of scFvs may also preferentially trigger CARs upon binding to tumor cells expressing high levels of the antigen, sparing normal cells expressing physiologic levels of the antigen, as preclinically showed for both HER2 and EGFR(35,36). Solid tumors can express aberrant splicing isoforms and molecules characterized by subverted protein glycosylation, thus targeting tumor-specific splicing variants or tumor-specific glycosylation sites represents another strategy to overcome on-target but off-tumor activity in solid tumors(28,37,38). In order to increase the tumor specificity, CAR T cells can also be engineered to express combinatorial antigen-sensing systems that trigger T cell activation only when two tumor-specific antigens are simultaneously present on the cell surface, as in the case of the integration of combinatorial targeting and splitting signal [38] or ON-switch strategies (39,40).
Heterogeneity of antigen expression in solid tumors
The second critical aspect of the identification of targetable antigens in solid tumors concerns the high heterogeneity of antigen expression at both intra and intertumoral level leading to tumor escape due to both antigen loss and clonal evolution. Targeting more than one antigen already represents the next generation CAR T cells in B-cell malignancies upon the evidence that both leukemia and lymphoma cells can lose the expression of the CD19 targeted epitope after treatment with CD19-specific CAR T cells(41). Consistently, CAR T cells engineered to recognize multiple antigens in solid tumors such as EGFR, HER2 and IL13Rα2 have been tested in pre-clinical models(42). Alternatively, CAR T cells can be combined with epigenetic drugs that can upregulate the expression of target antigens, as reported for GD2 upregulation in Ewing Sarcoma upon pharmacological inhibition of EZH2(43).
OVERCOME PHYSICAL BARRIERS IN SOLID TUMORS
Immune cell trafficking and infiltration of peripheral tissues is regulated by a complex signaling network and physical processes, which are significantly subverted in tumors. Tumor blood vessels are characterized by abnormalities in endothelial junctions and expression of adhesion and extravasation molecules that limit an efficient T cell recruitment(44). Moreover, the chemokine network orchestrating immune cell migration and the stroma including fibroblasts and myeloid cells are highly dysregulated in solid tumors further impairing the access of T cells within the tumor bed(45).
Local delivery of CAR T cells
Local drug delivery is one of the most exploited strategy to overcome biological barriers in solid tumors especially in malignancies characterized by locoregional aggressiveness (Figure 1, panel A). Local CAR T cell delivery in solid tumors in addition to maximizing the accumulation of CAR T cells at the tumor site, may also improve the safety profile, limiting their systemic biodistribution and, consequently, the access to vital organs sharing the expression of targeted antigens. Brain tumors are a typical example where local CAR T cell delivery has high clinical potential. Complete tumor regression was reported in a patient with highly aggressive recurrent glioblastoma with multifocal leptomeningeal disease upon infusion of anti-IL13Rα2 CAR T cells into the resected tumor cavity and into the ventricular system(19). Intracranial delivery of CAR T cells was effective also in a mouse model of HER2+ breast cancer with brain metastasis(46).
Figure 1. Strategies to overcome physical barriers in solid tumors.
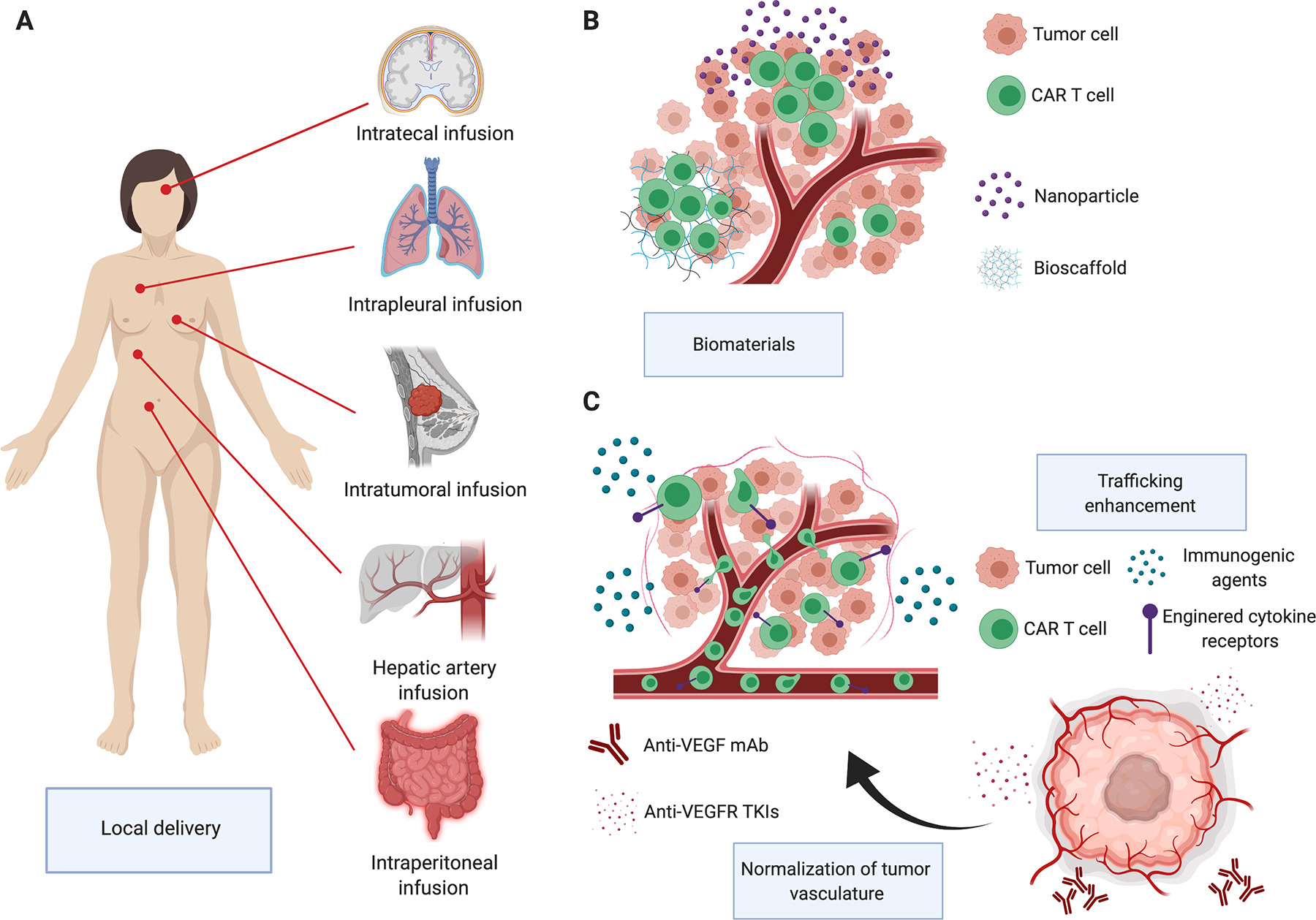
Local T cell delivery can maximize the accumulation of CAR T cells at the tumor site and may also improve the safety profile (panel A). Biomaterials can be used to enhance the persistence and functionality of locally delivered CAR T cells (panel B). CAR T cells can be combined with antiangiogenic drugs in the effort of normalizing the intratumoral blood flow or with immunomodulating agents or engineered with cytokine receptors to enhance the trafficking (panel C).
Malignancies with a pleural or peritoneal spreading are also particularly attractive for the local delivery of CAR T cells. As a proof of concept, intravenous injection of anti-MUC1 CAR T cells caused their redistribution mainly to the liver and spleen, with a trafficking circuit comprising axillary, retroperitoneal and popliteal lymph nodes, but with modest localization in the peritoneal cavity(47). In contrast, CAR T cells infused intraperitoneally localized and persisted throughout the peritoneal cavity with limited extraperitoneal distribution causing improved antitumor activity in mouse models of colorectal cancer peritoneal carcinomatosis and ovarian cancer as compared to systemic administration(33,48). Liver-limited metastatic disease is another interesting setting for local delivery of CAR T cells. In a phase I clinical trial, anti-CEA CAR T cells delivered through percutaneous hepatic artery infusion in 5 patients with CEA-positive colorectal cancer liver metastases were safe and promoted sustained stabilization of disease in one patient(15). Other proof-of-concept clinical studies with promising data of antitumor activity were conducted with locally delivered anti-cMET CAR T cells in patients with metastatic breast cancer and with anti-mesothelin CAR T cells administered intrapleurally in patients with primary malignant pleural mesothelioma or secondary metastatic disease(29,49). In line with the effort to develop local delivery of CAR T cells is the parallel development of implantable biomaterials to manipulate cell and tissue properties and to enhance persistence of locally delivered cells (Figure 1, panel B). Encapsulating or embedding CAR T cells in implantable biopolymer scaffolds resulted in a better persistence and antitumor effects in mouse models of pancreatic cancer and melanoma(50). Furthermore, biopolymer scaffolds can not only accommodate cells but also be loaded with costimulatory molecules, cytokines and small molecules that can further enhance the antitumor activity of CAR T cells or target the TME(50).
Overcome the subverted tumor vasculature
The first obstacle for CAR T cells in reaching the tumor bed upon intravenous infusion is represented by the subverted tumor blood and lymphatic vasculature. To overcome the aberrant tumor vasculature, CAR T cells can be combined with antiangiogenic drugs in the effort of normalizing the intratumoral blood flow (Figure 1, panel C). Administration of the anti-VEGF agent bevacizumab boosted the tumor infiltration and anti-tumor activity of CAR T cells targeting the GD2 antigen in an orthotopic xenograft model of neuroblastoma(51). Similarly, sub pharmacologic concentrations of the tyrosine-kinase inhibitor with anti-VEGFR activity sorafenib enhanced the infiltration and antitumor activity of CAR T cells targeting GPC3 in a mouse model of hepatocellular carcinoma(52). In contrast, pharmacologic concentrations of sorafenib showed inhibitory effects on CAR T cell proliferation and cytotoxic activity suggesting that combinatorial strategies involving CAR T cells and kinase inhibitors with broad targeted specificity should be carefully investigated preclinically to avoid any deleterious effects on CAR T cells(53). T cells can also be directly engineered to target the tumor vasculature via targeting of the VEGF receptors(54). A phase I/II clinical study ( NCT01218867) targeting VEGFR2 via CAR T cells in patients with metastatic solid tumors has been conducted at National Cancer Institute. Treatment was well tolerated and even if no significant antitumor responses were observed, the study paves the possibility to develop a dual targeting strategy attacking both tumor vasculature and tumor cells.
Enhance trafficking of CAR T cells at the tumor site
CAR T cells show homing to lymphoid tissues upon intravenous infusion in patients with B cell malignancies, while trafficking outside the lymphatic system and into solid tumors remains suboptimal. Several strategies are currently under investigation in the effort to induce an effective trafficking of CAR T cells at the tumor site (Figure 1, panel C). Classical cytotoxic drugs can have different immunomodulatory effects, and the choice of the right combination of cytotoxic drugs to be used within the lymphodepletion regimen prior to CAR T cell administration may promote CAR T cell trafficking(55). Doxorubicin and IL12 showed synergistic effects in promoting tumor homing of TILs in a xenograft mouse model of melanoma(56). Combination of doxorubicin and IL12 induces the release of the chemokines CXCL9 and CXCL10 binding to the receptor CXCR3, which is highly expressed in T cells, promote their migration to the tumor site. As alternative to chemotherapy, other biological agents can be used to alter the TME to support a more favorable milieu to T cell trafficking and persistence.
Oncolytic viruses selectively infect, lyse and replicate in malignant cells, whilst leaving non-malignant cells unaffected(57). These viruses thus represent an appealing platform to promote CAR T cell migration and survival within the TME(58). Similarly, physical modifications of the tumor architecture that can be caused by local mild hyperthermia can promote the recruitment and effector function of CAR T cells(59). It is also possible to engineer CAR T cells to express the receptors of chemokines that are highly expressed by tumor cells. For example, some tumor cells produce the chemokine CCL2, but its receptor CCR2 is expressed at low levels on ex vivo expanded T cells. Promoting the expression of CCR2b in CAR T cells have been demonstrated to increase the trafficking of CAR T cells to the tumor(60). Finally, after ex vivo manipulation, CAR T cells also show reduced ability of degrading the extracellular matrix due to the lack of expression of heparanase, an enzyme that degrades the heparan sulfate proteoglycans. CAR T cells engineered to re-express heparanase effectively degrade the extracellular matrix, infiltrate the tumor and exert more profound antitumor activity(61).
The field of CAR engineering has been so far dominated by the expression of CARs in activated T lymphocytes, but other cell subsets can be redirected against tumor cells via CAR gene transfer. In particular for the clinical setting of solid tumors, natural killer (NK) cells and classical natural killer T (NKT) cells, also known as invariant NKTs, possess unique properties such as such as enhanced trafficking at the tumor site and CD1d restricted cytotoxic activity for NKT cells and innate cytotoxicity activity against tumor cells for NK cells. Both NK cells and NKTs can be genetically manipulated to express CARs and acquire antigen specificity, while maintaining their innate properties(62,63). Phase I clinical studies with CAR-engineered NKTs or NK cells are currently ongoing ( NCT03294954, NCT03579927, NCT03056339).
COPING WITH THE TUMOR MICROENVIRONMENT
The TME is a complex network that comprehends the extracellular matrix and several non-malignant cells such as fibroblasts, macrophages and myeloid-derived suppressor cells (MDSCs) that contribute to tumor progression and immune evasion (Figure 2). The expression of inhibitory molecules, also known as inhibitory immune checkpoints, by tumor and stromal cells is one of the most important mechanisms impairing T cell effector function. The neutralization of inhibitory immune checkpoints can be carried out by either combining CAR T cells with the systemic administration of immune checkpoint inhibitors or knocking down inhibitor receptors such as PD1 in CAR T cells(64,65). Both strategies are currently under clinical investigation, and at the moment it is difficult to anticipate if selective blockade of inhibitory immune checkpoints in CAR T cells achieved by knocking down of PD1 is advantageous as compared to a more generalized blockade effect achieved with the systemic administration of immune checkpoint inhibitors. An alternative strategy to counteract the PD1/PD-L1 axis is to revert the inhibitory signal of PD1 in T cells coupling PD1 extracellular domain with the signaling domain of costimulatory molecules such as CD28(66). Phase I clinical trials with CRISPR-Cas9 mediated PD1 gene-knockdown in anti-mesothelin CAR T cells or with a combination of anti-mesothelin CAR T cells and pembrolizumab are actually ongoing ( NCT03545815, NCT02414269), as well as phase I studies with CAR T cells administered in combination with nivolumab alone or with nivolumab and ipilimumab in patients with glioblastoma ( NCT04003649, NCT03726515).
Figure 2. Strategies to counteract the immune suppressive tumor microenvironment.
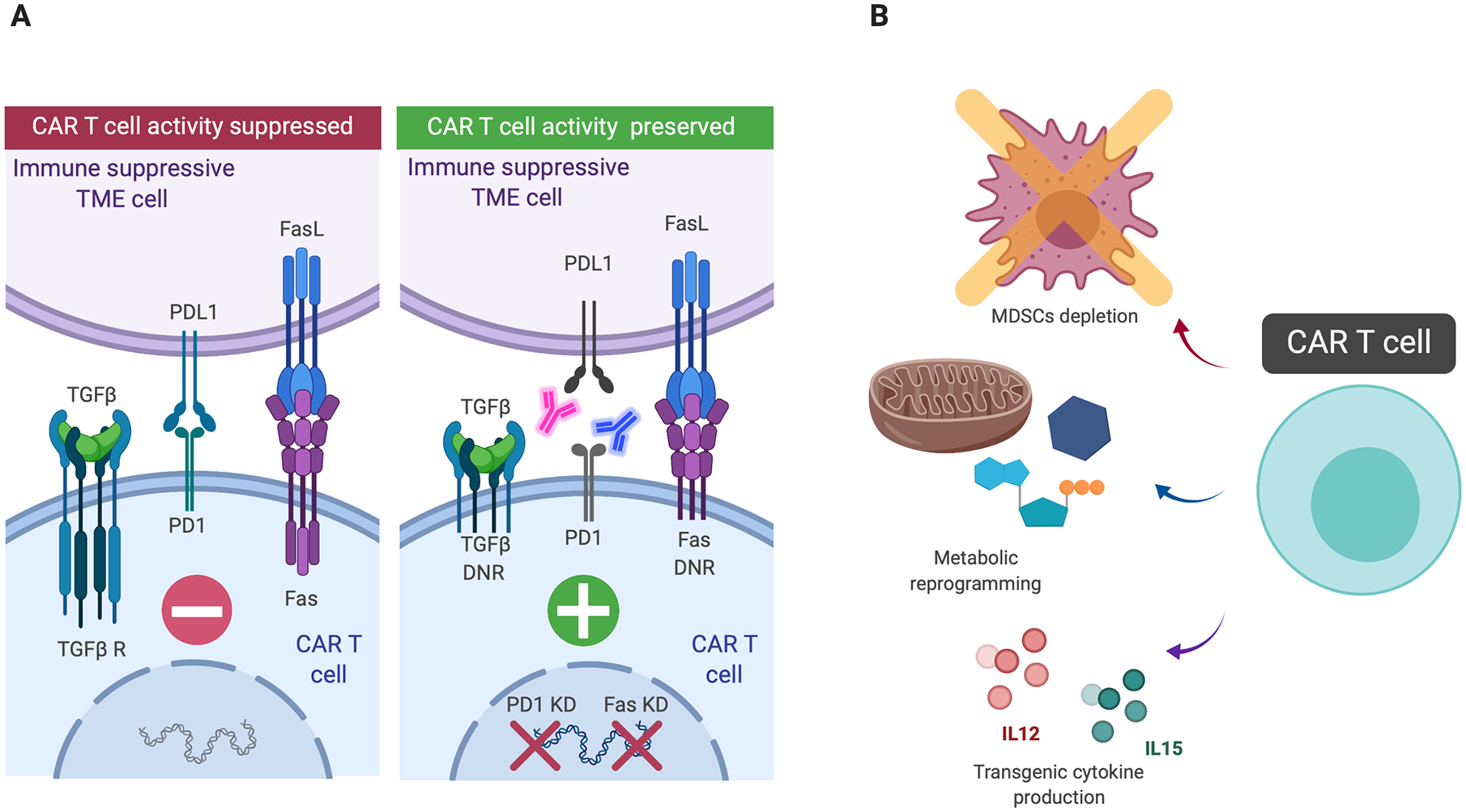
Neutralization of inhibitory mechanisms can be carried out by either knocking down specific receptors in CAR T cells such as PD1 or Fas, by engineering CAR T cells to express dominant negative receptors (DNRs) and by combining CAR T cells with the systemic administration or transgenic production of immune checkpoint inhibitors (panel A). Other strategy to overcome the immune suppressive tumor microenvironment consist in myeloid-derived suppressor cell (MDSCs) depletion, metabolic reprogramming of CAR T cells and transgenic cytokine production (panel B).
Physiologically, the interaction between the receptor Fas (CD95) on T cells and its ligand FasL (CD95L) represents an immune homeostasis mechanism rapidly leading to T cell apoptotic death. FasL can be overexpressed in the TME as an immune-escape mechanism and the disruption of the Fas/FasL pathway by engineering CAR T cells to express a dominant negative receptor, as well as by Fas knockdown, showed promising results in pre-clinical models(67,68).
One of the main soluble inhibitory factors within the TME is the transforming growth factor-β (TGF-β), produced by cancer cells as well as by regulatory T cells (Tregs), fibroblasts, macrophages and platelets that inhibits T cell proliferation and effector function. TGF-β signaling in T cells can be blocked by engineering T cells to express a dominant-negative TGF-β receptor and this strategy has been already safely tested in a clinical study using virus specific T cells(69). A similar approach can be applied to CAR T cells and is actually under clinical investigations in patients with metastatic prostate cancer using anti-PSMA CAR T cells ( NCT03089203).
The TME is also characterized by nutrient deprivation and the presence of catabolites that impair persistence and effector function of T cells. Metabolic reprogramming is emerging as a strategy to enhance the functionality of CAR T cells within the metabolically disrupted TME of solid tumors, and genetic manipulations of glucose or acetate metabolisms in adoptively transferred T cells showed promising results in preclinical models(70,71).
Among the immunosuppressive catabolites that are enriched in the TME, adenosine has been well characterized. Adenosine inhibits effector functions by engaging the cell-surface receptor A2AR in T cells. A2AR blockade showed the potential to profoundly increase CAR T cell efficacy in mouse models of HER2+ solid tumors, particularly when combined with PD-1 blockade(72).
Cellular components of the TME such as MDSCs are considered major players in hampering immune responses. Depleting MDSCs with a nanoformulation of auroyl-modified gemcitabine improved CAR T cell efficacy in pre-clinical models of solid tumors(73). MDSCs can also be effectively targeted by blocking GM-CSF, and the combination of anti-GM-CSF agents with anti-CEA CAR T cells was effective in a mouse model of colorectal cancer with liver metastases(74). Engineering T cells to release cytokines that reshape the TME, such as IL12 may reduce the inhibitory effects of M2-type tumor-associated macrophages and Tregs. Unfortunately, clinical experience with TILs engineered to express IL12 in response to NFAT in melanoma patients did not prevent the occurrence of the toxic effects caused by IL12 in human subjects(75). However, a clinical trial with anti-MUC16 CAR T cells engineered to secrete IL-12 is currently ongoing in patients with MUC16-positive solid tumors ( NCT02498912).
Cytokine such as IL15 that do not have direct effects on the TME may however play a critical role in sustaining expansion, survival and function of CAR T cells and partially compensate the inhibitory effects of the TME(76). IL-15 expressing CAR T cells are currently under investigation in patients with liver cancer targeting glypican-3 ( NCT04093648) and in patients with neuroblastoma targeting GD2 ( NCT03721068).
CONCLUSION
Targeting solid tumors with CAR T cells in the clinical setting remains challenging. Since its first conception in late eighties, the strategy of using chimeric molecules as functional receptors with antibody-type specificity to arm T cells against solid tumors has clearly evolved. Nowadays CAR T cell-based therapy encompasses sophisticated genetic engineering techniques and combinatorial approaches to counteract biological barriers, tumor heterogeneity and immunosuppressive properties of the TME. Nevertheless, further hypothesis-driven research is needed to speed up the preclinical development and fill the gap in the clinic between CAR T cell-based treatment in hematological malignancies and solid tumors. Moreover, new strategies are rapidly emerging for the redirection of T cells against solid tumors with the premise of a better safety profile, as in the case of CD3-bispecific engagers (BiTEs) that allow cytotoxic killing of tumor cells by patients T cells without ex-vivo manipulation(77). Optimal strategies to pursue among TILs, CAR T cells and BiTEs are likely to be tailored to different clinical settings and tumor types.
As outlined in this review article, only translational, cooperative and interdisciplinary efforts including clinical oncologists, immunologists and biomedical engineers would succeed in decoding and effectively implement strategies to address the complex pathophysiology of solid tumors and immune surveillance.
Financial support:
This work was supported in part by Alex’s Lemonade Stand Foundation (G.D.), Department of Defense W81XWH-161-0500 (G.D.), National Institute of Heath R01-CA193140-03, R21-CA229938-01A1 (G.D.) and U01CA239258 (B.S.).
Footnotes
Conflict of interest disclosure statement:
Drs Dotti and Savoldo hold patents in the field of T cell engineering and have sponsor research agreements with Bluebird Bio, Cell Medica and Bellicum Pharmaceutical. Dr. Dotti serves in the scientific advisory board of MolMed S.p.A and Bellicum Pharmaceutical. All the other authors declare no conflict of interest.
REFERENCES
- 1.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004;18(4):676–84. [DOI] [PubMed] [Google Scholar]
- 2.Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol 1998;161(6):2791–7. [PubMed] [Google Scholar]
- 3.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A 1993;90(2):720–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol 2008;26(32):5233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J Clin Oncol 2015;33(6):540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 2013;5(177):177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371(16):1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 2006;12(20 Pt 1):6106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther 2007;15(4):825–33. [DOI] [PubMed] [Google Scholar]
- 10.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010;18(4):843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011;118(23):6050–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol 2006;24(13):e20–e2. [DOI] [PubMed] [Google Scholar]
- 13.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2014;2(2):112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol 2015;33(15):1688–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, et al. Phase I Hepatic Immunotherapy for Metastases Study of Intra-Arterial Chimeric Antigen Receptor-Modified T-cell Therapy for CEA+ Liver Metastases. Clin Cancer Res 2015;21(14):3149–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin Cancer Res 2015;21(18):4062–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, et al. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol Ther 2016;24(6):1135–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Junghans RP, Ma Q, Rathore R, Gomes EM, Bais AJ, Lo AS, et al. Phase I Trial of Anti-PSMA Designer CAR-T Cells in Prostate Cancer: Possible Role for Interacting Interleukin 2-T Cell Pharmacodynamics as a Determinant of Clinical Response. Prostate 2016;76(14):1257–70. [DOI] [PubMed] [Google Scholar]
- 19.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med 2016;375(26):2561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.You F, Jiang L, Zhang B, Lu Q, Zhou Q, Liao X, et al. Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated by modified Anti-MUC1 chimeric antigen receptor transduced T cells. Sci China Life Sci 2016;59(4):386–97. [DOI] [PubMed] [Google Scholar]
- 21.Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci 2016;59(5):468–79. [DOI] [PubMed] [Google Scholar]
- 22.Feng KC, Guo YL, Liu Y, Dai HR, Wang Y, Lv HY, et al. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J Hematol Oncol 2017;10(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer 2017;5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA(+) Metastatic Colorectal Cancers. Mol Ther 2017;25(5):1248–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol 2017;3(8):1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother 2017;66(11):1425–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng K, Liu Y, Guo Y, Qiu J, Wu Z, Dai H, et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell 2018;9(10):838–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017;9(399): eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol Res 2017;5(12):1152–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beatty GL, O’Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of Mesothelin-specific Chimeric Antigen Receptor T cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018. doi S0016-5085(18)30323-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J Immunother 2019;42(4):126–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katz SC, Hardaway J, Prince E, Guha P, Cunetta M, Moody A, et al. HITM-SIR: phase Ib trial of intraarterial chimeric antigen receptor T-cell therapy and selective internal radiation therapy for CEA(+) liver metastases. Cancer Gene Ther 2019. doi 10.1038/s41417-019-0104-z. [DOI] [PubMed] [Google Scholar]
- 33.Du H, Hirabayashi K, Ahn S, Kren NP, Montgomery SA, Wang X, et al. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell 2019;35(2):221–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin Cancer Res 2019;25(8):2560–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res 2015;75(17):3505–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res 2015;75(17):3596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 2015;15(9):540–55. [DOI] [PubMed] [Google Scholar]
- 38.Di C, Syafrizayanti, Zhang Q, Chen Y, Wang Y, Zhang X, et al. Function, clinical application, and strategies of Pre-mRNA splicing in cancer. Cell Death Differ 2019;26(7):1181–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015;350(6258):aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol 2013;31(1):71–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov 2015;5(12):1282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol Ther Nucleic Acids 2013;2:e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kailayangiri S, Altvater B, Lesch S, Balbach S, Gottlich C, Kuhnemundt J, et al. EZH2 Inhibition in Ewing Sarcoma Upregulates GD2 Expression for Targeting with Gene-Modified T Cells. Mol Ther 2019;27(5):933–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res 2010;70(15):6171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013;19(11):1423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2(+) Breast Cancer Metastasis to the Brain. Clin Cancer Res 2018;24(1):95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parente-Pereira AC, Burnet J, Ellison D, Foster J, Davies DM, van der Stegen S, et al. Trafficking of CAR-engineered human T cells following regional or systemic adoptive transfer in SCID beige mice. J Clin Immunol 2011;31(4):710–8. [DOI] [PubMed] [Google Scholar]
- 48.Katz SC, Point GR, Cunetta M, Thorn M, Guha P, Espat NJ, et al. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther 2016;23(5):142–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adusumilli PS. A phase I clinical trial of malignant pleural disease treated with regionally delivered autologous mesothelin-targeted CAR T cells: Safety and efficacy. 2019 AACR Annual Meeting; 2019. [Google Scholar]
- 50.Smith TT, Moffett HF, Stephan SB, Opel CF, Dumigan AG, Jiang X, et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J Clin Invest 2017;127(6):2176–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bocca P, Di CE, Caruana I, Emionite L, Cilli M, De AB, et al. Bevacizumab-mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2-CAR T cells in a human neuroblastoma preclinical model. Oncoimmunology 2017;7(1):e1378843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu X, Luo H, Shi B, Di S, Sun R, Su J, et al. Combined Antitumor Effects of Sorafenib and GPC3-CAR T Cells in Mouse Models of Hepatocellular Carcinoma. Mol Ther 2019;27(8):1483–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gargett T, Fraser CK, Dotti G, Yvon ES, Brown MP. BRAF and MEK inhibition variably affect GD2-specific chimeric antigen receptor (CAR) T-cell function in vitro. J Immunother 2015;38(1):12–23. [DOI] [PubMed] [Google Scholar]
- 54.Chinnasamy D, Yu Z, Theoret MR, Zhao Y, Shrimali RK, Morgan RA, et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest 2010;120(11):3953–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015;28(6):690–714. [DOI] [PubMed] [Google Scholar]
- 56.Hu J, Sun C, Bernatchez C, Xia X, Hwu P, Dotti G, et al. T-cell Homing Therapy for Reducing Regulatory T Cells and Preserving Effector T-cell Function in Large Solid Tumors. Clin Cancer Res 2018;24(12):2920–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parato KA, Senger D, Forsyth PA, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer 2005;5(12):965–76. [DOI] [PubMed] [Google Scholar]
- 58.Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res 2014;74(18):5195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen Q, Hu Q, Dukhovlinova E, Chen G, Ahn S, Wang C, et al. Photothermal Therapy Promotes Tumor Infiltration and Antitumor Activity of CAR T Cells. Adv Mater 2019;31(23):e1900192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother 2010;33(8):780–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med 2015;21(5):524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014;124(18):2824–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Altvater B, Landmeier S, Pscherer S, Temme J, Juergens H, Pule M, et al. 2B4 (CD244) signaling via chimeric receptors costimulates tumor-antigen specific proliferation and in vitro expansion of human T cells. Cancer Immunol Immunother 2009;58(12):1991–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res 2017;23(9):2255–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol Ther 2017;25(9):2214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res 2016;76(6):1578–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dotti G, Savoldo B, Pule M, Straathof KC, Biagi E, Yvon E, et al. Human cytotoxic T lymphocytes with reduced sensitivity to Fas-induced apoptosis. Blood 2005;105(12):4677–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yamamoto TN, Lee PH, Vodnala SK, Gurusamy D, Kishton RJ, Yu Z, et al. T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J Clin Invest 2019;129(4):1551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bollard CM, Tripic T, Cruz CR, Dotti G, Gottschalk S, Torrano V, et al. Tumor-Specific T-Cells Engineered to Overcome Tumor Immune Evasion Induce Clinical Responses in Patients With Relapsed Hodgkin Lymphoma. J Clin Oncol 2018;36(11):1128–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015;162(6):1217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015;162(6):1229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest 2017;127(3):929–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sasso MS, Lollo G, Pitorre M, Solito S, Pinton L, Valpione S, et al. Low dose gemcitabine-loaded lipid nanocapsules target monocytic myeloid-derived suppressor cells and potentiate cancer immunotherapy. Biomaterials 2016;96:47–62. [DOI] [PubMed] [Google Scholar]
- 74.Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA, et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother 2015;64(7):817–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res 2015;21(10):2278–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, Savoldo B. Eradication of Neuroblastoma by T Cells Redirected with an Optimized GD2-Specific Chimeric Antigen Receptor and Interleukin-15. Clin Cancer Res 2019. doi 1078-0432.CCR-18-1811. [DOI] [PubMed] [Google Scholar]
- 77.Slaney CY, Wang P, Darcy PK, Kershaw MH. CARs versus BiTEs: A Comparison between T Cell-Redirection Strategies for Cancer Treatment. Cancer Discov 2018;8(8):924–34. [DOI] [PubMed] [Google Scholar]