

Abstract
Studies comparing the prognostic role of RUNX1 mutations (RUNX1mut) in acute myeloid leukemia (AML) and acute myeloid leukemia-with myelodysplasia-related changes (AML-MRC) are limited. Our study examines the genetic profile of 118 RUNX1mut AML patients including 57 AML with RUNX1mut and 61 AML-MRC with RUNX1mut and 100 AML, NOS patients with wild type RUNX1 (RUNX1wt). Results revealed that AML-MRC patients with RUNX1mut had shorter median overall survival (OS) (11 ± 3.3 months) when compared to AML with RUNX1mut (19 ± 7.1 months) and AML, NOS with RUNX1wt (not reached) (p = .001). The most common concurrent mutations observed in AML-MRC with RUNX1mut patients were DNMT3A, SRSF2, ASXL1, and IDH2 while in AML with RUNX1mut patients were ASXL1, SRSF2, TET2, IDH2, and DNMT3A. ASXL1 and TET2 mutations appeared to adversely affect OS in AML-MRC, but not in AML with RUNX1mut. Concurrent RUNX1/DNMT3A mutations, in contrast had negative impact on OS in AML with RUNX1mut, but not in AML-MRC with RUNX1mut.
Keywords: RUNX1, AML, AML-MRC, NGS
Introduction
Acute myeloid leukemia (AML) is a clonal proliferation of immature myeloid precursors with a variety of clinical and immunophenotypic presentations depending on the inherited and acquired genetic aberrations [1,2]. AML is an aggressive disease with a 5-year overall survival (OS) rate of 27.4% according to the National Cancer Institute, Surveillance, Epidemiology, and End Results Program (SEER) [3]. Emerging evidence shows that the clinical prognosis of AML largely depends on several key factors including the patient’s age, coexisting comorbidities, and the precise disease sub-classification based on morphology, cytogenetics, and molecular signatures as identified by the World Health Organization (WHO) [1,4,5]. In addition to recurrent chromosomal aberrations, somatic mutations in RUNX1, NPM1, FLT3, TP53, KMT2A/MLL, and CEBPA genes are critical in risk stratification of AML patients [2,6].
As one of the key transcriptional factors, RUNX1 plays an important role in the regulation of normal hematopoietic stem cells differentiation [7]. Therefore, translocation or intragenic mutation of RUNX1 leads to impaired hematopoiesis and ultimately leukemia [8–11]. Two large cohort studies have shown that somatic RUNX1 mutations (RUNX1mut) are frequently identified in myelodysplastic syndromes (MDS) and therapy-related AML (t-AML) [12,13]. Additionally, approximately 10% of patients with de novo AML, not otherwise specified (AML-NOS), were shown to have RUNX1mut, particularly patients with intermediate-risk and/or non-complex cytogenetics [11,14,15]. Furthermore, AML patients with RUNX1mut are shown to be more resistant to induction chemotherapies and associated with inferior survival outcomes when compared to AML, NOS with RUNX1wt [1,11,14].
AML with RUNX1mut is a new provisional entity in the 2017 WHO classification of Tumors of Hematopoietic and Lymphoid Tissue [5,16], distinguishing it from AML with recurrent genetic abnormalities, AML with myelodysplasia-related changes (AML-MRC) and t-AML [4,16]. However, studies comparing the clinical impact of RUNX1mut and its associated genetic landscape in AML-MRC and AML with RUNX1mut are very limited. This is clinically important for accurate risk stratification and personalized treatment approach for AML patients. Therefore, our study aimed to investigate the clinical outcomes in AML with RUNX1mut and AML-MRC with RUNX1mut and identify potential cooperative mutations and cytogenetic abnormalities that may have prognostic significance in these specific subgroups of AML.
Methods and material
Patient population
This study was approved by the Institutional Review Board (IRB) and Scientific Review Committee of H. Lee Moffitt Cancer Center and Research Institute (Moffitt). Between 2012 and 2017, a total of 118 AML patients harboring RUNX1mut were detected by next-generation sequencing (NGS) using the Illumina Trusight myeloid sequencing panel (54 genes). The patients with RUNX1mut were further categorized as AML with RUNX1mut (n = 57) or AML-MRC with RUNX1mut (n = 61) (Table 1). A total of 100 AML, NOS patients with RUNX1wt were included as a control group after matching for age, sex, bone marrow (BM) biopsy, and NGS test period. Clinical parameters including diagnosis, complete blood count (CBC), molecular data, and treatment regimen were collected. t-AML and AML with recurrent genetic aberrations beyond RUNX1 (PML-RARA, RUNX1/RUNX1T1, CBFB-MYH11, BCR-ABL1 etc.) were excluded. Patients with known congenital hematological malignancies were also excluded. Germline studies for RUNX1 mutation were not performed in this study.
Table 1.
Patient demographics and frequency of genetic aberrations in patients with AML with RUNX1mut, AML-MRC with RUNX1mut, and AML, NOS with RUNX1wt.
| Characteristics | AML with RUNX1mut (n = 57) | AML-MRC with RUNX1mut (n = 61) | AML, NOS with RUNX1wt (n = 100) | p-Value |
|---|---|---|---|---|
| Median age (range, years) | 67 (21–86) | 72 (31–92) | 60.9 (19–85) | >.05 |
| Gender (Male: Female) | 1.85 | 2.21 | 1.16 | >.05 |
| CBC data (median) | ||||
| WBC, × 109/L (range) | 4.5 (0.6–103.2) | 3.8 (0.7–134.1) | 7.1 (0.9–248) | N/A |
| ANC, × 109/L (range) | 0.9 (0–20.7) | 2.1 (0–23.8) | 1.0 (0–24.9) | |
| Hemoglobin, g/dL (range) | 8.5 (6.6–12.2) | 8.7 (7.4–12.1) | 9.1 (4.5–13.2) | |
| Platelets, × 109/L (range) | 41 (0.5–136) | 43 (2–597) | 56 (8–351) | |
| Prior MDS, MDS/MPN, MPN | 20.8 (18–95.5) | 22 (18–59) | 50.5 (13.2–94) | |
| Prior t-MDS | 0 (0%) | 42 (68.8%) | 0 (0%) | |
| Cytogenetics | (n = 51) | (n = 60) | (n = 98) | |
| Normal karyotype | 28 (54.9%) | 25 (41.7%) | 60 (61.2%) | .05639 |
| <3 abnormalities | 15 (29.4%) | 27 (45%) | 25(25.5%) | .03496* |
| ≥3 abnormalities | NA | 11(18.3%) | NA | NA |
| −5/del(5q) | 0 (0%) | 4 (6.7%) | 0 (0%) | N/A |
| −7/del(7q) | 0 (0%) | 8 (13.3%) | 5 (5.1%) | .0072* |
| +8 | 6 (11.8%) | 7 (11.7%) | 3 (3.1%) | .063618 |
| +13 | 3 (5.9%) | 2 (3.3%) | 1 (1.0%) | .676687 |
| −17/del(17p) | 0 (0%) | 1 (1.7%) | 1 (1.0%) | N/A |
| −20/del(20q) | 0 (0%) | 5 (8.3%) | 4 (4.1%) | N/A |
| Somatic mutations | (n = 57) | (n = 61) | (n = 100) | |
| ASXL1 | 18 (31.6%) | 23 (37.7%) | 18 (18%) | .016166* |
| DNMT3A | 19 (33.3%) | 11 (18.0%) | 29 (29%) | .146216 |
| IDH2 | 10 (17.5%) | 11 (18.0%) | 12 (12%) | .49133 |
| SRSF2 | 16 (28.1%) | 18 (29.5%) | 4 (4%) | <.00001* |
| TET2 | 5 (8.8%) | 16 (26.2%) | 20 (20%) | .048484* |
| BCOR | 5 (8.8%) | 2 (3.3%) | 5 (5%) | .406604 |
| BCORL | 4 (7.0%) | 3 (4.9%) | 3 (3%) | .506597 |
| CBL | 0 (0%) | 4 (6.6%) | 2 (2%) | N/A |
| IDH1 | 7 (12.3%) | 6 (9.8%) | 9 (9%) | .803816 |
| ETV6 | 6 (10.5%) | 4 (6.6%) | 0 (0%) | N/A |
| EZH2 | 3 (5.3%) | 3 (4.9%) | 0 (0%) | N/A |
| FLT3 | 7 (12.3%) | 0 (%) | 1 (1%) | N/A |
| GATA2 | 3 (5.3%) | 1 (1.6%) | 1 (1%) | .21159 |
| KMT2A | 2 (3.5%) | 1 (1.6%) | 2 (2%) | .692851 |
| NRAS | 7 (12.3%) | 4 (6.6%) | 8 (8%) | .555879 |
| PHF6 | 3 (5.3%) | 4 (6.6%) | 3 (3%) | .555413 |
| SF3B1 | 0 (0%) | 4 (6.6%) | 0 (%) | N/A |
| SETBP | 2 (3.5%) | 0 (0%) | 2 (2%) | N/A |
| STAG2 | 2 (3.5%) | 2 (2.3%) | 0 (0%) | N/A |
| TP53 | 3 (5.35) | 4 (6.6%) | 1 (1%) | .144823 |
| U2AF1 | 6 (10.5%) | 6 (9.83%) | 9 (9%) | .950672 |
| WT1 | 3 (5.3%) | 0 (0%) | 2 (2%) | N/A |
| ZRSR2 | 1 (1.76%) | 3 (4.9%) | 1 (1%) | .474143 |
| Othersa | 1 (1.8%) | 1 (1.6%) or less | 1 (1%) or less | .90689 |
| Number of mutations | ||||
| <3 mutations | 17 (29.8%) | 12 (19.6%) | 50 (59.5%) | .000267* |
| ≥3 mutations | 40 (70.2%) | 49 (80.3%) | 34 (40.5%) | <.00001 * |
| No mutations | – | – | 16 | N/A |
| Treatment | ||||
| Induction chemotherapy | 25 (43.9%) | 10 (16.3%) | 44 (44%) | .000733* |
| Hypomethylation agentsb | 7 (12.2%) | 23 (37.7%) | 11 (11%) | .000049* |
| Allo-SCT | 15 (26.3%) | 13 (21.3%) | 29 (29%) | .559621 |
| Other | 10 (17.5%) | 15 (24.5%) | 16 (16%) | .384509 |
Allo-SCT: allogeneic hematopoietic stem cell transplant; ANC: absolute neutrophil count; CBC: complete blood count; MDS: myelodysplastic syndromes; MPN: myeloproliferative neoplasm; t-MDS: therapy-related MDS; WBC: white blood cells. AML: acute myeloid leukemia; AML-MRC: acute myeloid leukemia with myelodysplasia-related changes; NOS: not otherwise specified.
Other somatic mutations include CSF3R, CEBPA, JAK2, KIT, ZRSR2, KDM6A, PDGFR, NOTCH1, ABL1, TRX, FBXW7, and RAD21.
Hypomethylation agents include azacitidine and decitabine.
Indicates statistical significance.
Conventional karyotyping/fluorescence in situ hybridization (FISH)
Routine cytogenetic analysis using standard trypsin-Giemsa banding technique by Laboratory Corporation of America (Burlington, NC, USA) was performed on all patients with sufficient BM aspirate specimen and reported in accordance with an International System for Human Cytogenetic Nomenclature, 2016 (ISCH 2016, Karger). FISH probe sets designed for MDS [del(5q)/−5, del(7q)/−7, 8, del(17p)/−17, and del(20q)/−20] and AML [t(8;21)/þRUNX1-RUNX1T1, t(15;17)/PML-RARA, and inv(16)/CBFB-MYH11] were conducted based on the manufacturer’s instructions (Vysis, Downers Grove, IL, USA) at Moffitt Cancer Center.
NGS and mutation analysis
NGS was performed as previously described [17]. The panel used in our study encompasses a total of 54 genes including ABL1, ASXL1, ATRX, BCOR, BCORL1, BRAF, CALR, CBL, CBLB, CBLC, CDKN2A, CEBPA, CSF3R, CUX1, DNMT3A, ETV6/TEL, EZH2, FBXW7, FLT3, GATA1, GATA2, GNAS, HRAS, IDH1, IDH2, IKZF1, JAK2, JAK3, KDM6A, KIT, KRAS, MLL, MPL, MYD88, NOTCH1, NPM1, NRAS, PDGRA, PHF6, PTEN, PTPN11, RAD21, RUNX1, SETBP1, SF3B1, SMC1A, SMC3, SRSF2, STAG2, TET2, TP53, U2AF1, WT1, and ZRSR2. The filter setting for variant detection was set to identify any somatic, non-synonymous DNA mutations of single nucleotide variants (SNVs), insertions, and deletions. Fifty nanograms of DNA were extracted using the Qiagen AllPrep DNA extraction kit (Qiagen, Germantown, MD, USA) and quantitated by NanoDrop spectrophotometry (Agilent, Santa Clara, CA, USA). The samples were sequenced using an Illumina NextSeq 500 sequencer (Illumina, San Diego, CA, USA), with variant detection by the PierianDx bioinformatics pipeline. The variant filters were set at 5% variant allele frequency (VAF) for SNV and at 10% for insertions/deletions (indels). Any variants that met the threshold of the filters were automatically placed into a high confidence group. A second low-confidence group was established for specimens that did not meet the desired filter thresholds. In these cases, individual variants were manually interrogated by an integrated genomic viewer (IGV). If a mutation was clinically significant, it was then manually added to the final molecular pathology report. All RUNX1 mutations described were categorized as levels 1–4 variants excluding known germline polymorphisms or level 5 variants. Given that all the patients developed adult-onset AML, no additional tissue was harvested to test for RUNX1 germline mutation.
Statistical analysis
The Chi-square or Fisher exact test was used for continuous variables and Chi-squared test for categorical variables. Both tests encompassed baseline demographic data, laboratory, and BM biopsy findings and disease features. Kaplan–Meier method was used for estimated overall survival (OS) analysis and log-rank test was used for comparison of median OS. The OS was calculated from the time of AML diagnosis.
Results
Clinical characteristics of AML with RUNX1mut and AML-MRC with RUNX1mut
Of the 118 patients harboring RUNX1mut, 57 patients (48.3%) were classified as AML with RUNX1mut and the remaining 61 patients (51.7%) were categorized as AML-MRC with RUNX1mut according to 2017 WHO classification. Patients with AML with RUNX1mut had a median age of 67 years (range, 21–86) and were predominantly male (M:F ratio 1.85) while patients with AML-MRC with RUNX1mut had a median age of 72 years (range, 31–92) also showing male predominance (M:F ratio 2.21). Patients with AML, NOS with RUNX1wt had a median age of 60.9 years (range, 19–85) with a M:F ratio of 1.16. Table 1 summarizes the clinicopathologic features of these three cohorts. Among 57 patients with AML with RUNX1mut, missense mutations constituted 38.6% RUNX1mut followed by frameshift (36.8%) and nonsense (24.6%) mutations. Among 61 AML-MRC patients with RUNX1mut, 45.9% had missense mutations followed by frameshift (41.0%) and nonsense (13.1%) mutations. These numbers were not statistically different between these two groups (p>.05). In this study, patients with AML with RUNX1mut or AML, NOS with RUNX1wt were more likely to receive induction chemotherapy than their AML-MRC with RUNX1mut counterparts. Approximately 20–30% of AML patients from each cohort subsequently received allogeneic stem cell transplantation (Allo-SCT) (Table 1).
Concurrent mutations in AML with RUNX1mut and AML-MRC with RUNX1mut
The most frequent mutations in AML with RUNX1mut were DNMT3A (33.3%), ASXL1 (31.6%), SRSF2 (28.1%), and IDH2 (17.5%); while in AML-MRC with RUNX1mut the most common mutations were ASXL1 (37.7%), SRSF2 (29.5%), TET2 (26.2%), IDH2 (18%), and DNMT3A (18%). Other mutations were also identified in these cohorts with VAF ranging from less than 1 to 10% (Table 1). Of note, FLT3-ITD were more commonly seen in the patients with AML with RUNX1mut as compared to AML, NOS with RUNX1wt (7/62 versus 1/100, p = .003).
Clinical outcomes in AML with RUNX1mut and AML-MRC with RUNX1mut
AML-MRC patients with RUNX1mut demonstrated shorter median OS (18.9 ± 1.9 months) when compared to patients with AML with RUNX1mut (25.1 ± 2.8 months) or RUNX1wt AML (39.5 ± 4.0 months) (p = .001) (Figure 1). When RUNX1mut patients were stratified by concurrent gene mutations, TET2 appeared to adversely affect OS in AML-MRC with RUNX1mut (RUNX1, 21.9 ± 3.0 months versus RUNX1/TET2, 9.7 ± 2.2 months, p = .009) (Figure 2(A)), but not in AML with RUNX1 mut (RUNX1, 26.3 ± 3.1 months versus RUNX1/TET2, 12.4 ± 3.2 months, p = .123) (Figure 2(B)). Concurrent DNMT3A mutation also had a negative impact on OS in AML with RUNX1mut (RUNX1, 29.8 ± 3.5 months versus RUNX1/DNMT3A, 14.7 ± 3.5 months, p = .002), but not in AML-MRC with RUNX1mut (RUNX1, 20.7 ± 2.9 months versus RUNX1/DNMT3A, 12.3 ± 2.1 months, p = .099) (Figure 3(A,B)). Concurrent IDH1 mutation was associated with worse clinical outcome in AML-MRC with RUNX1mut patients (RUNX1, 19.5 ± 2.5 months versus RUNX1/IDH1, 2.6 ± 1.4 months, p = .008), but it did not affect OS in patients with AML with RUNX1mut (RUNX1, 26.4 ± 3.1 months versus RUNX1/IDH1, 15.6 ± 7.8 months, p = .168); however, there were only seven patients with IDH1 in AML-MRC and seven patients in AML with RUNX1mut cohorts. The combination of RUNX1/ASXL1 mutations did show a slightly worse statistically significant clinical outcome in AML patients with RUNX1mut (RUNX1, 27.8 ± 3.6 months versus RUNX1/ASXL1, 19.4 ± 4.2 months, p = .203) (Figure 4(A)). There was a difference in clinical outcome in AML-MRC patients with RUNX1mut (RUNX1, 20.3 ± 3.3 months versus RUNX1/ASXL1, 16.8 ± 3.5 months, p = .51) (Figure 4(B)), but this did not reach statistical significance. Other concurrent gene mutations (ETV6, EZH2, FLT3, IDH2, NRAS, SRSF2, TP53, and U2AF1, etc.) showed no effect on OS in AML with RUNX1mut or AML-MRC with RUNX1mut cohorts. Additionally, the presence of three or more gene mutations (including RUNX1) significantly decreased OS in patients with AML with RUNX1mut (19.5 ± 2.9 months, versus 36.3 ± 4.2 months p = .004) (Figure 5(A)); while having no significant impact on OS in patients with AML-MRC with RUNX1mut (17.8 ± 2.4 months versus 24.9 ± 7.5 months, p = .508) (Figure 5(B)).
Figure 1.
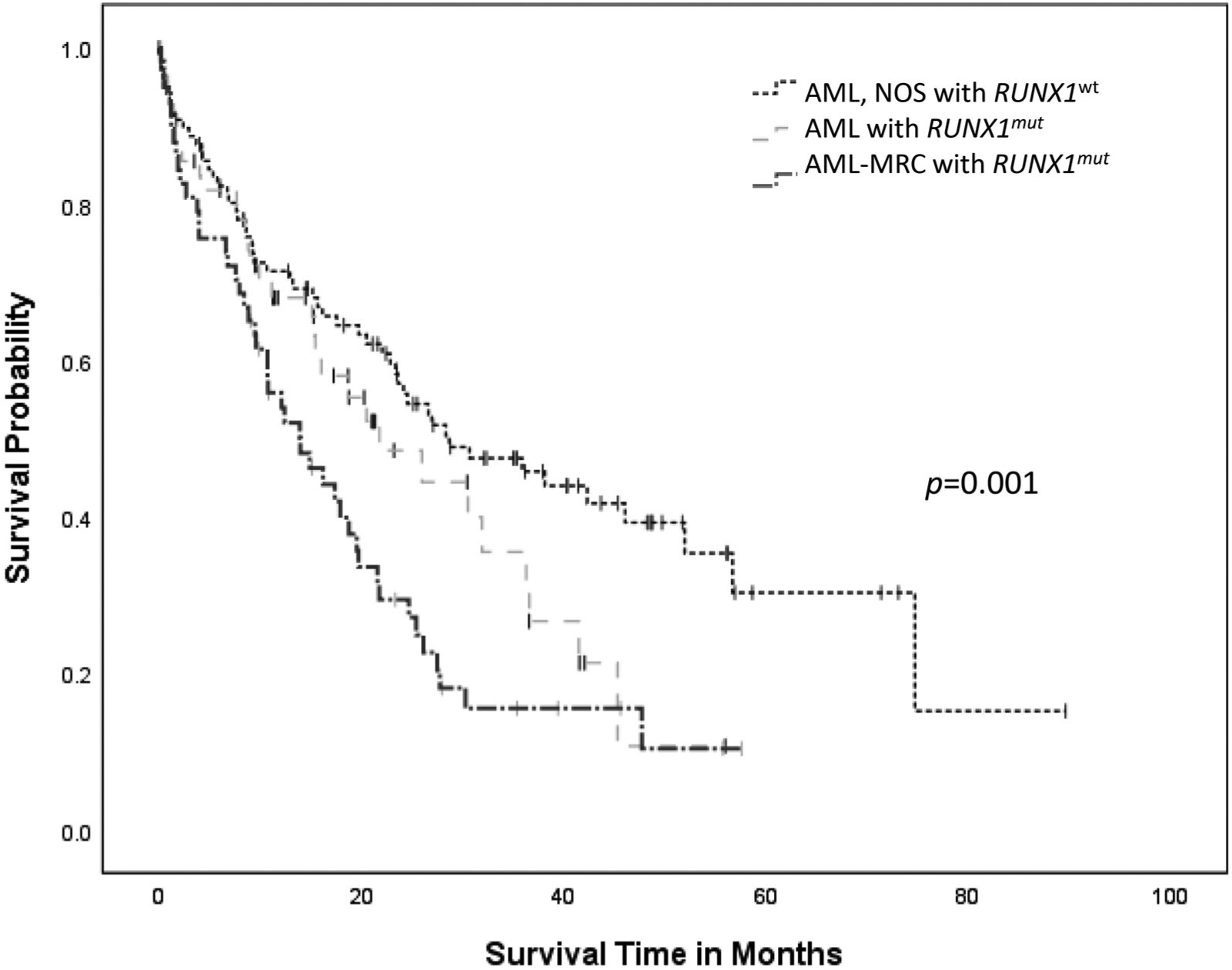
Overall survival curves for cases of AML with RUNX1mut, AML-MRC with RUNX1mut and AML, NOS with RUN1Xwt.
Figure 2.
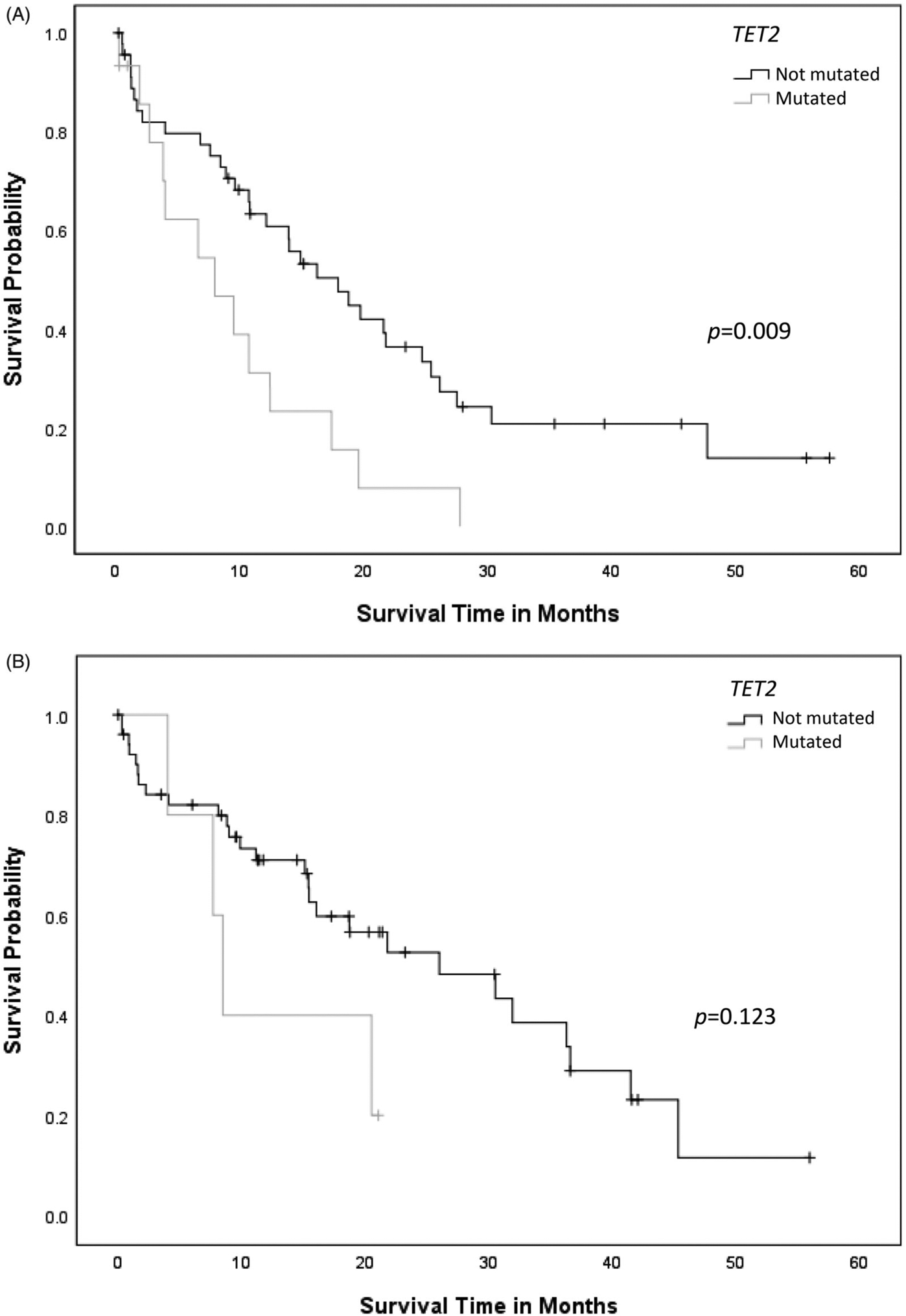
(A) Overall survival curves for cases of AML-MRC with concurrent RUNX1/TET2 mutation compared to AML-MRC cases with RUNX1 mutation alone. (B) Overall survival curves for cases of AML with RUNX1mut with or without concurrent TET2 mutation.
Figure 3.
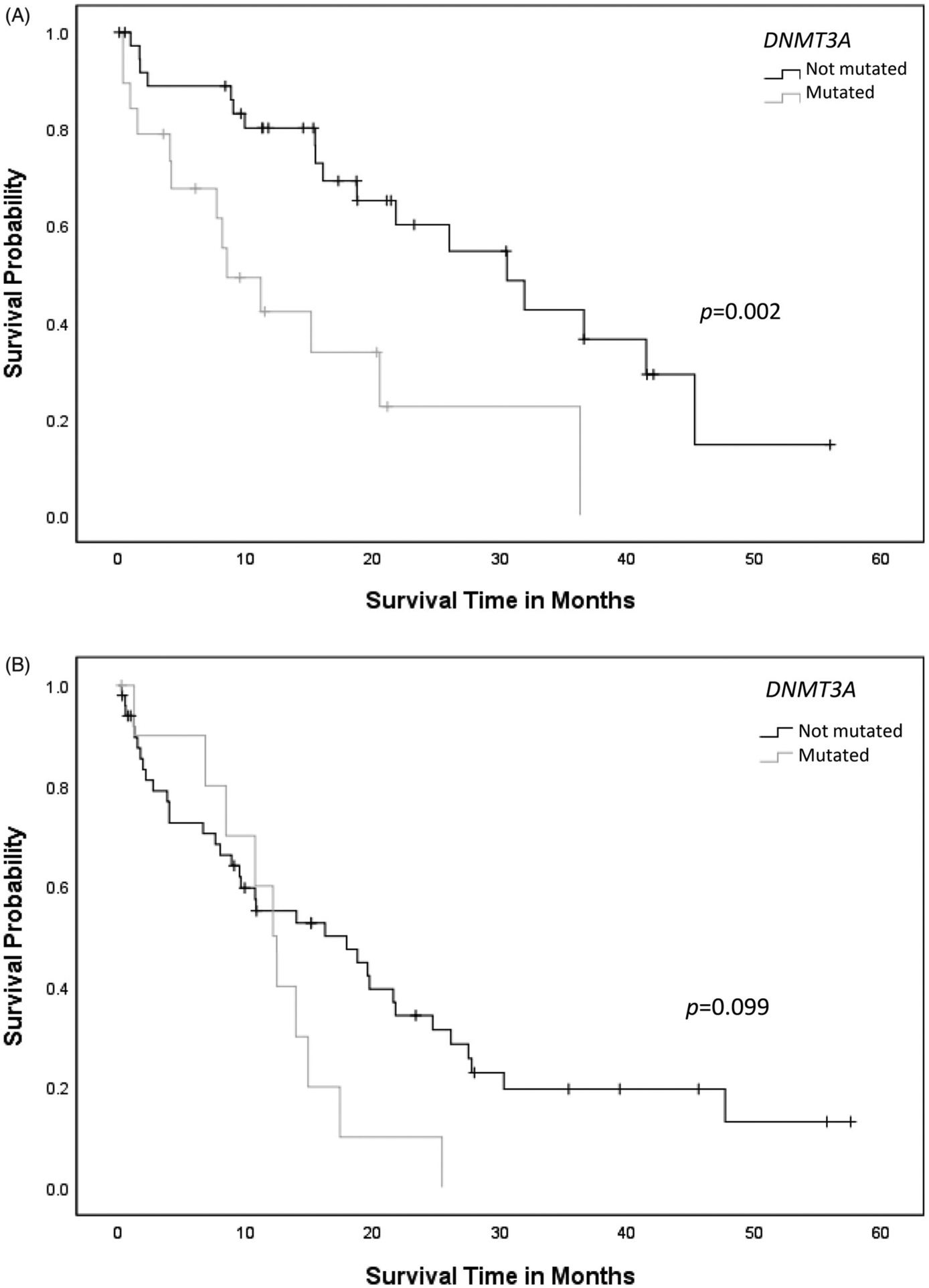
(A) Overall survival curves for cases of AML with RUNX1mut with or without concurrent DNMT3A mutation. (B) Overall survival curves for cases of AML-MRC with concurrent RUNX1/DNMT3A mutation compared to cases with RUNX1 mutation alone.
Figure 4.
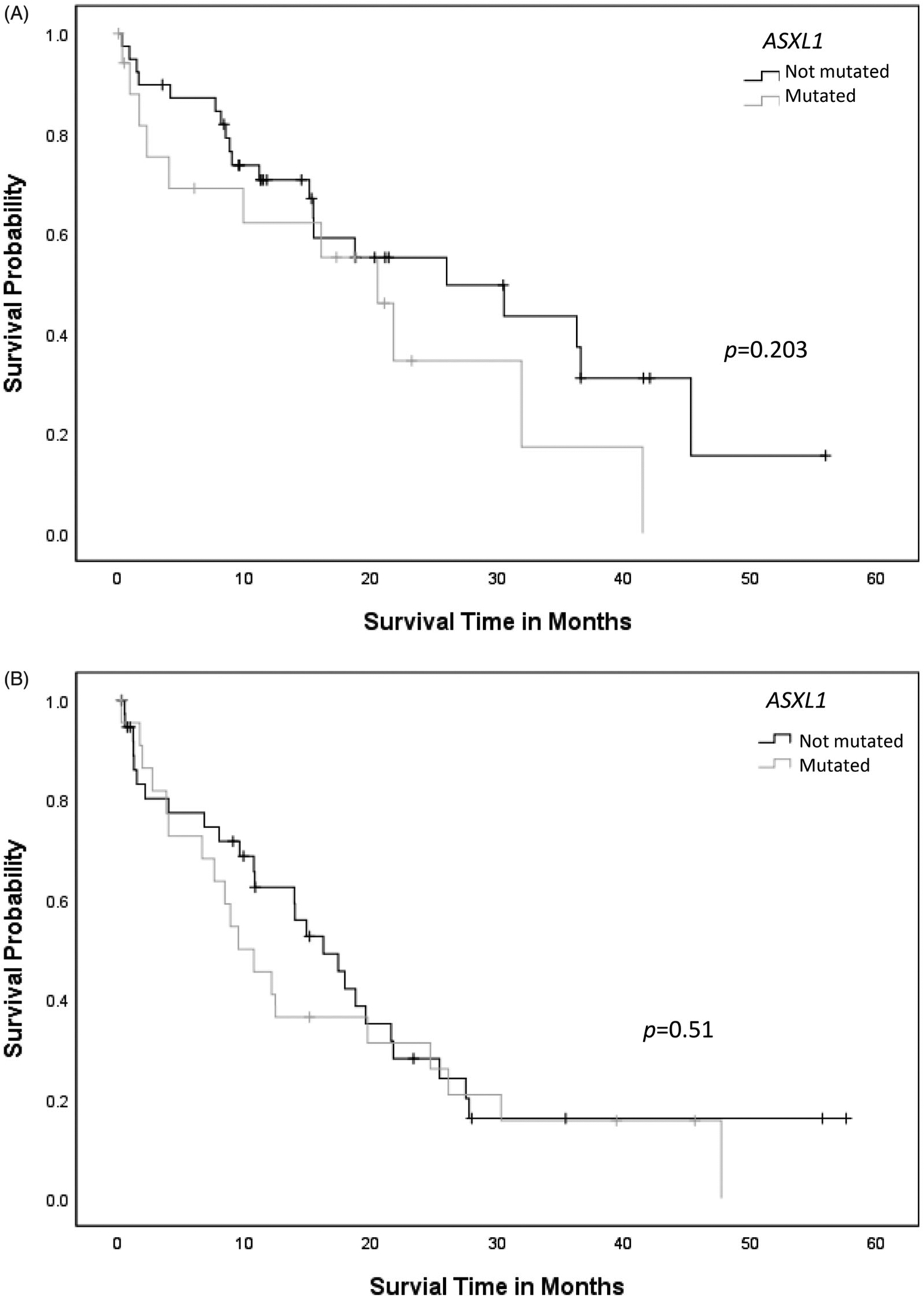
(A) Overall survival curves for cases of AML with RUNX1mut with or without concurrent ASXL1 mutation. (B) Overall survival curves for cases of AML-MRC with concurrent RUNX1/ASXL1 mutation compared to cases with RUNX1 mutation alone.
Figure 5.
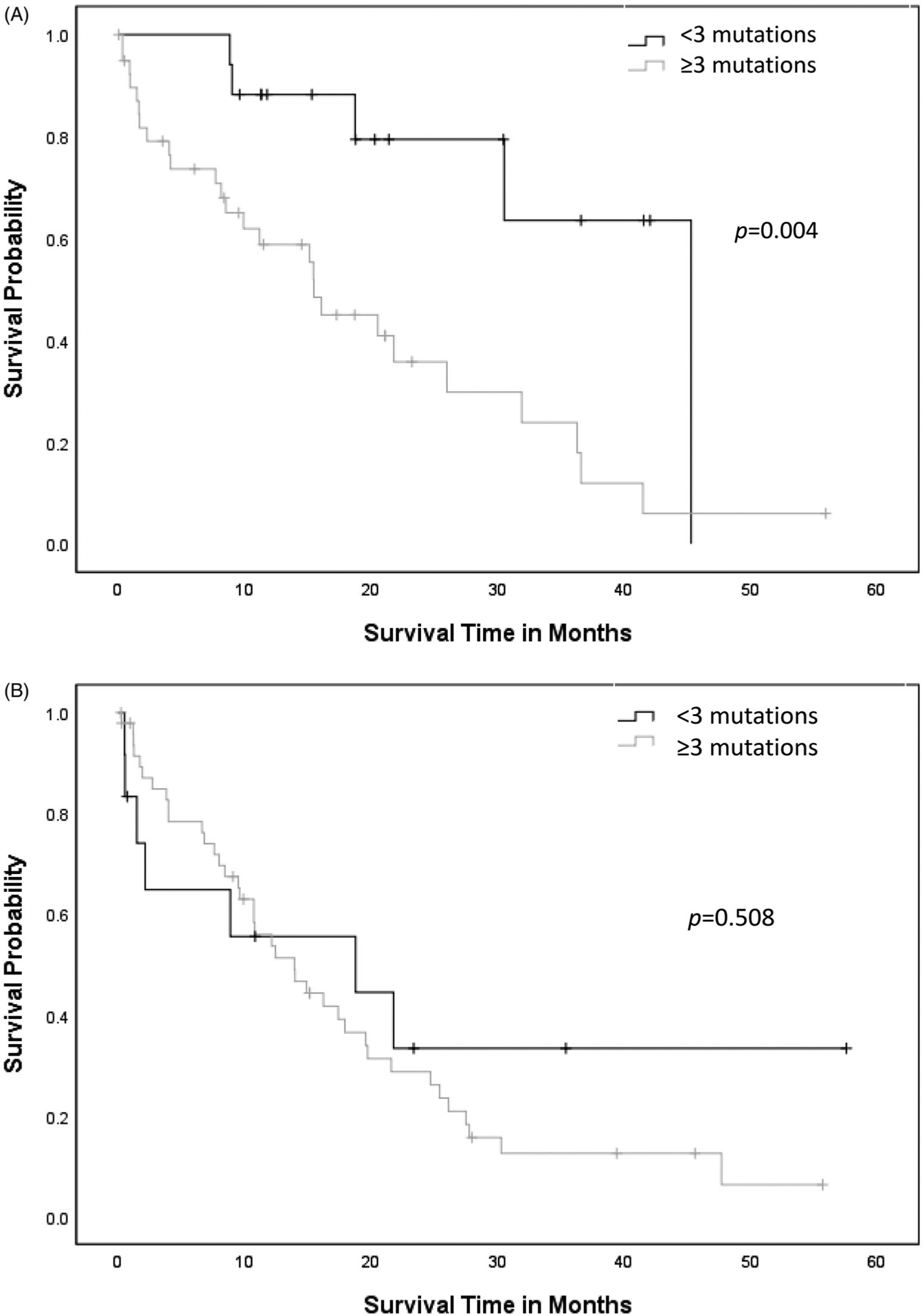
(A) Overall survival curves for cases of AML with RUNX1mut with three or more mutations versus cases with fewer than three mutations. (B) Overall survival curves for cases of AML-MRC with three or more mutations versus fewer than three mutations (including RUNX1).
Clinical impact of cytogenetic alterations in AML with RUNX1mut and AML-MRC with RUNX1mut
Conventional karyotyping data was retrieved in 51 of 57 patients with AML with RUNX1mut, 60 of 61 patients with AML-MRC with RUNX1mut, and 98 of 100 AML, NOS with RUNX1wt patients. Patients with normal karyotype were more commonly observed in the AML regardless of RUNX1 mutation status (AML with RUNX1mut and AML, NOS with RUNX1wt) when compared to AML-MRC with RUNX1mut (p < .01) (Table 1). Monosomy 7 or del(7q) was more commonly identified in AML-MRC with RUNX1mut (13%) compared to the other two groups (0.0% in AML with RUNX1mut and 5.1% in AML, NOS with RUNX1wt, p = .0072). The prevalence of trisomy 8 was similar in AML with RUNX1mut (11.8%) and AML-MRC with RUNX1mut (11.7%), while trisomy 13 appeared in approximately 6% of patients with AML with RUNX1mut and in 3.3% of AML-MRC with RUNX1mut patients.
Discussion
Studies have shown that RUNX1mut are associated with poor prognosis in AML [5,18,19]. However, whether clinical outcome of AML-MRC with RUNX1mut is worse than AML with RUNX1mut is yet unknown and there has been debate regarding prognostic impact of multilineage dysplasia in AML patients with RUNX1mut. Similar to a previous study reported by Haferlach’s group [20], mild morphologic dysplasia in hematopoietic precursor did not adversely affect the clinical outcome in our patient with AML with RUNX1mut or AML-MRC with RUNX1mut (data not shown). However, we observed that AML-MRC with RUNX1mut is associated with inferior OS compared to AML with RUNX1mut. This observation is clinically important since poor treatment response and inferior survival outcome associated with RUNX1mut could be further compromised in AML-MRC patients [21,22]. Although CPX-351 was recently approved as a front-line therapy in AML-MRC patients and hypomethylating agents are commonly used in AML patients who are not eligible for the intensive chemotherapy [23–27], there is still lack of data regarding the optimal treatment in this unique group of AML-MRC with RUNX1mut patients. Future studies are warranted to further improve survival outcomes in this poor prognostic group.
Additionally, recent studies have shown that ASXL1, RAS, FLT3-ITD, and IDH1/2 mutations frequently concur in patients with AML with RUNX1mut [21]. Also, the incidence of ASXL1 mutation was higher in RUNX1mut versus AML, NOS with RUNX1wt patients with normal karyotype (36% versus 8%) [22] and Gaidzik et al. reported that in addition to aforementioned genes, EZH2, SRSF2, SF3B1, STAG2, PHF6, and BCOR mutations frequently concur in AML with RUNX1mut patients [14]. Similar to these studies, we observed a relatively high incidence of mutations in SRSF2, TET2, DNMT3A, ASXL1, IDH1/2, EZH2, BCOR, PHF6, STAG, and FLT3-ITD genes in RUNX1 mutant patients, either AML with RUNX1mut or AML-MRC with RUNX1mut group. In our study, some of these co-mutations appear prognostically important; RUNX1/TET2 co-mutations were associated with worse OS compared to RUNX1mut alone in AML-MRC while concurrent RUNX1/DNMT3A mutations were associated with worse OS in patients with AML with RUNX1mut than AML-MRC with RUNX1mut. Concurrent RUNX1/ASXL1 mutations also showed negative clinical impact on OS in patients with AML with RUNX1mut and RUNX1/IDH2 co-mutations were associated with worse clinical outcome than RUNX1mut alone in AML-MRC patients, but not in AML with RUNX1mut. However, these observations require cautious interpretation because of the limited number of patients included from a single institution, requiring further validation with independent patient cohorts.
Recent studies have demonstrated that AML with RUNX1mut has a unique cytogenetic signature [20,28]. Trisomy 13 is reportedly strongly associated with RUNX1 mutation in AML (9%, 13/152) [20,29]. However, the presence of trisomy 13 was extremely low in our study appearing only in 5.9% and 3.3% of patients with AML with RUNX1mut and AML-MRC with RUNX1mut, respectively. Following normal karyotype, trisomy 8 was the most common aberration making up 11.8% of AML with RUNX1mut and 11.7% of AML-MRC with RUNX1mut.
Although RUNX1 mutations have been identified in 10–33% of MDS and AML patients [18,19,30,31], the proportion of germ-line versus somatic mutations is largely unknown. Patients with germ-line RUNX1 mutations develop early onset MDS/AML with a median age of 33 years at diagnosis whereas sporadic MDS/AML patients typically present later time in life with median age of 65–68 years [32,33]. A recent study showed that the carriers of RUNX1 germ-line mutations are typically asymptomatic, but have a high cumulative risk of developing a clonal myeloid neoplasm by age 50, suggesting that secondary mutations are necessary to trigger MDS/AML [34,35]. The median age of patients in the groups in our study was 60–72 years old and only six patients were diagnosed before 35 years. This suggests that the majority of patients in our cohort likely have RUNX1 somatic mutations, but this is inconclusive given the lack of germ-line mutation analysis.
Despite the need for studies to further elucidate the association between AML-MRC and RUNX1mut to predict poor clinical outcome, our study provides some insight to potential points of clinical interest. We have demonstrated that patients with AML-MRC with RUXN1mut are a unique poor prognostic group and certain co-mutations and/or cytogenetics could further affect their clinical outcomes. This warrants routine investigation by a comprehensive myeloid mutation panel to have better risk stratification and identify those that may benefit from future targeted therapies.
Funding
This work was supported in part by a research grant from the Graduate Medical Education (GME) at the University of South Florida (S.Y. and L.Z.), a Research Training Award for Fellow (RTAF) from the American Society of Hematology (S.Y.), and by NIH [grant K08 CA237627] (S.Y.).
Footnotes
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373:1136–1152. [DOI] [PubMed] [Google Scholar]
- [2].Suguna E, Farhana R, Kanimozhi E, et al. Acute myeloid leukemia: diagnosis and management based on current molecular genetics approach. Chddt. 2018; 18(3):199–207. [DOI] [PubMed] [Google Scholar]
- [3].Scaffidi C, Schmitz I, Krammer PH, et al. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem. 1999;274(3):1541–1548. [DOI] [PubMed] [Google Scholar]
- [4].Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127(20):2391–2405. [DOI] [PubMed] [Google Scholar]
- [5].Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumors of haematopoietic and lymphoid tissues. Geneva, Switzerland: WHO Press; 2017. [Google Scholar]
- [6].Dohner H, Eslev E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Swiers G, de Bruijn M, Speck NA. Hematopoietic stem cell emergence in the concepts and the role of Runx1. Int J Dev Biol. 2010;54(6–7):1151–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sakurai M, Kunimoto H, Watanabe N, et al. Impaired hematopoietic differentiation of RUNX1-mutated induced pluripotent stem cells derived from FPD/AML patients. Leukemia. 2014;28(12):2344–2354. [DOI] [PubMed] [Google Scholar]
- [9].Mangan JK, Speck NA. RUNX1 mutations in clonal myeloid disorders: from conventional cytogenetics to next generation sequencing, a story 40 years in the making. Crit Rev Oncog. 2011; 16(12):77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Imai Y, Kurokawa M, Izutsu K, et al. Mutations of the AML1 gene in myelodysplastic syndrome and their functional implications in leukemogenesis. Blood. 2000;96(9):3154–3160. [PubMed] [Google Scholar]
- [11].Gaidzik VI, Bullinger L, Schlenk RF, et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. JCO. 2011;29(10):1364–1372. [DOI] [PubMed] [Google Scholar]
- [12].Harada H, Harada Y, Niimi H, et al. High incidence of somatic mutations in the AML1/RUNX1 gene in myelodysplastic syndrome and low blast percentage myeloid leukemia with myelodysplasia. Blood. 2004; 103(6):2316–2324. [DOI] [PubMed] [Google Scholar]
- [13].Osato M Point mutations in the RUNX1/AML1 gene: another actor in RUNX leukemia. Oncogene. 2004; 23(24):4284–4296. [DOI] [PubMed] [Google Scholar]
- [14].Gaidzik VI, Teleanu V, Papaemmanuil E , for the German-Austrian Acute Myeloid Leukemia Study Group (AMLSG), et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia. 2016; 30(11):2282–2282. [DOI] [PubMed] [Google Scholar]
- [15].Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Arber DA, et al. WHO classification of tumors of haematopoietic and lymphoid tissues. Geneva, Switzerland: WHO Press; 2017. [Google Scholar]
- [17].Yun S, Sharma R, Chan O, et al. Prognostic significance of MYC oncoprotein expression on survival outcome in patients with acute myeloid leukemia with myelodysplasia related changes (AML-MRC). Leuk Res. 2019;84:106194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schnittger S, Dicker F, Kern W, et al. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood. 2011;117(8):2348–2357. [DOI] [PubMed] [Google Scholar]
- [19].Tang J-L, Hou H-A, Chen C-Y, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009; 114(26):5352–5361. [DOI] [PubMed] [Google Scholar]
- [20].Haferlach T, Stengel A, Eckstein S, et al. The new provisional WHO entity ‘RUNX1 mutated AML’ shows specific genetics but no prognostic influence of dysplasia. Leukemia. 2016;30(10):2109–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Khan M, et al. Clinical outcomes and co-occurring mutations in patients with RUNX1-mutated acute myeloid leukemia. Int J Mol Sci. 2017;18:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mendler JH, Maharry K, Radmacher MD, et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. JCO. 2012; 30(25):3109–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dombret H, Gardin C. An update of current treatments for adult acute myeloid leukemia. Blood. 2016; 127(1):53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Quintas-Cardama A, Ravandi F, Liu-Dumlao T, et al. Epigenetic therapy is associated with similar survival compared with intensive chemotherapy in older patients with newly diagnosed acute myeloid leukemia. Blood. 2012;120:4840–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gupta N, Miller A, Gandhi S, et al. Comparison of epigenetic versus standard induction chemotherapy for newly diagnosed acute myeloid leukemia patients 60 years old. Am J Hematol. 2015;90(7): 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lancet JE, et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J Clin Oncol. 2018;36:2684–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yun S, Vincelette ND, Abraham I, et al. Targeting epigenetic pathways in acute myeloid leukemia and myelodysplastic syndrome: a systematic review of hypomethylating agents trials. Clin Epigenet. 2016; 8(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Greif PA, Konstandin NP, Metzeler KH, et al. RUNX1 mutations in cytogenetically normal acute myeloid leukemia are associated with a poor prognosis and up-regulation of lymphoid genes. Haematologica. 2012;97(12):1909–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Silva FP, Lind A, Brouwer-Mandema G, et al. Trisomy 13 correlates with RUNX1 mutation and increased FLT3 expression in AML-M0 patients. Haematologica. 2007;92(8):1123–1126. [DOI] [PubMed] [Google Scholar]
- [30].Chen C-Y, Lin L-I, Tang J-L, et al. RUNX1 gene mutation in primary myelodysplastic syndrome–the mutation can be detected early at diagnosis or acquired during disease progression and is associated with poor outcome. Br J Haematol. 2007; 139(3):405–414. [DOI] [PubMed] [Google Scholar]
- [31].Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].West AH, Godley LA, Churpek JE. Familial myelodysplastic syndrome/acute leukemia syndromes: a review and utility for translational investigations. Ann NY Acad Sci. 2014;1310(1):111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Forman D, Stockton D, Møller H, et al. Cancer prevalence in the UK: results from the EUROPREVAL study. Ann Oncol. 2003;14(4):648–654. [DOI] [PubMed] [Google Scholar]
- [34].Churpek JE, Pyrtel K, Kanchi K-L, et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood. 2015; 126(22):2484–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Owen C, Barnett M, Fitzgibbon J. Familial myelodysplasia and acute myeloid leukaemia–a review. Br J Haematol. 2007;140(2):123–132. [DOI] [PubMed] [Google Scholar]