

Abstract
DNA forms conformational states beyond the right-handed double-helix; however, the functional relevance of these non-canonical structures in the brain remains unknown. We show that, in the prefrontal cortex of mice, the formation of one such structure, Z-DNA, is involved in the regulation of extinction memory. Z-DNA is formed during fear learning, and reduced during extinction learning, which is mediated, in part, by a direct interaction between Z-DNA and the RNA editing enzyme Adar1. Adar1 binds to Z-DNA during fear extinction learning which leads to a reduction in Z-DNA at sites where Adar1 is recruited. Knockdown of Adar1 leads to an inability to modify a previously acquired fear memory and blocks activity-dependent changes in DNA structure and RNA state; effects that are fully rescued by the introduction of full-length Adar1. These findings suggest a novel mechanism of learning-induced gene regulation dependent on both proteins which recognize DNA structure, and the state.
Introduction
It is often assumed that DNA only encodes information in its nucleotide sequence. This is partly because when Watson and Crick proposed their double-helix model of DNA, they described a right-handed form of DNA, now called B-DNA, and implied a single static conformation. Thus, other models, including Linus Pauling’s “incorrect” structural model of DNA that is now termed P-DNA, were relegated to the theoretical dustbin. Thus, there are in fact a wide repertoire of DNA structures, which may possess unique functions. Thought of in this way, DNA, much like its RNA and protein products, may also encode information through dynamic changes in its secondary structure.
To explore this possibility we chose to focus on one specific structural configuration, known as Z-DNA1,2, as well as Adar1, because its p150 isoform possesses a Zα domain that enables binding to Z-DNA. In general, Z-DNA structure deviates from classical models of DNA, termed B-DNA3,4, by forming a left-hand turn of the double helix and expanding the distance and electrostatic charge between the nucleotide bases5. In addition, it has been suggested that the genome, through the formation of Z-DNA, maintains some rudimentary chemical memory of its previous structure occurrence6. This change in DNA structure, coupled to alterations in chromatin state, could then impact the threshold for future gene activation7. In fact, the formation and stability of this particular DNA structure depends on changes in the concentration of salt8 and calcium9, as well as DNA modification10, all of which are known to occur under physiological conditions, and are required for memory formation11.
These observations led us to question whether Z-DNA states influence the threshold for experience-dependent transcriptional activity, which itself is critical for at least the first stage of memory consolidation12. Given that Adar1 recognizes Z-DNA in vitro13, and potentially alters the formation of Z-DNA14,15, we hypothesized that Adar1-mediated changes in DNA structure states are critical for learning-induced gene expression in the adult brain and that this impacts the formation of fear extinction memory, an important form of memory that is dependent on rapid changes in gene expression16 and on behavioral flexibility following extinction learning17. In confirmation of this hypothesis we observed that Z-DNA is dynamic in response to fear extinction learning, with manipulation of Z-DNA by doxorubicin impairing extinction when infused into the infralimbic PFC. Further, recognition of Z-DNA by Adar1 also occurred during extinction and is critical for extinction learning as knockdown of Adar1 both modifies Z-DNA state and extinction learning.
Results
Adar1 is induced in activated neurons during fear extinction learning.
To test the hypothesis that the Z-DNA reader Adar1 is involved in memory formation, we first examined Adar1 mRNA expression in response to fear extinction training. We found that fear extinction led to a rapid and transient increase in Adar1 mRNA expression in the adult prefrontal cortex (PFC) (Figure 1a). To better understand the functional role of Adar1 in the formation of fear extinction memory, we examined Adar1 mRNA and protein levels in neurons selectively activated by fear extinction learning. In this experiment, we used fluorescence-activated cell sorting (FACS) on neurons labeled with the neuronal nuclei marker (NeuN) and activity-regulated cytoskeleton-associated protein (Arc; see Figure 1b). The NeuN marker allows for differentiation of non-neurons from neurons, and the Arc marker differentiates between recently activated neurons and quiescent neurons18. We found that fear extinction-trained mice exhibited significantly higher levels of Adar1 mRNA expression in activated neurons (high Arc) than in quiescent neurons (low in Arc) from the same animal (Figure 1c). This effect was selective for Adar1 in the PFC, as the expression of ZBP1, which contains a Z-DNA binding domain, and Braca1, which contains a Z-DNA forming region, were not affected (Supplementary Figure 1a–c). Adar1 p150 and p110 isoforms are known to differentially localize in the cell, with the former translocating between the cytoplasm and the nucleus, and the latter being expressed primarily in the cytoplasmic compartment19. We therefore assessed the activation-induced dynamics of these proteins by comparing the nucleus/cytoplasm ratio, which revealed a significant increase in the nuclear expression of the Adar1 p150 isoform following fear extinction learning (Figure 1e,f).
Figure 1. Adar1 binds to DNA and targets DNA repetitive elements in response to behavioral training.

a, Naïve home-cage control mice compared to those fear trained (FC), and exposed to a novel context following fear conditioning (retention control: RC) or fear conditioned followed by 10CS (EXT 10), 30 CS (EXT 30) or 60CS extinction (EXT 60) led to a significant increase in Adar1 mRNA (n = 6–10 animals per group, ANOVA, F5,39 = 3.97, p = 0.005, Dunnett post-hoc test relative to naïve, 10CS *p=0.012). b, Representative fluorescence-activated cell sorting plot showing the separation between neurons and non-neurons based on NeuN, and the gating of Arc levels into high and low c, RNA transcript levels for EXT 10 animals which are either high in the neuronal marker NeuN, and activity-regulated cytoskeleton-associated expression (EXT ARC +ve) or high in the neuronal marker NeuN and low in Arc (EXT ARC -ve) are significantly different, n = 4 per group, t = 2.706, d.f. = 6, *p<0.05. d,There was also a significant elevation in the nuclear:cytoplasmic ratio of the Adar1 p150 isoform (n = 3–6 protein samples per group, ANOVA, F5,15 = 4.06, p = 0.0157, Dunnett’s post-hoc test relative to naïve, 10CS was significantly increased *p=0.026), e, but no significant change in the nuclear:cytoplasmic ratio for the p110 isoform (n = 3–6 protein samples per group, ANOVA, F5,15 = 1.24, p = 0.3375). Experience-dependent distribution of Adar1 binding within PFC neurons of mice that were either subjected to fear conditioning training followed by exposure to a novel context (retention control: RC) or fear conditioning followed by 10 presentations of the conditioned stimulus (EXT) f, Mapping of Adar1 peaks from the ChIP-seq data revealed that the peaks were distributed equally across the genome g, USC genome browser annotated DNA repetitive elements including long-terminal repeats (LTR), SINEs, LINEs and other elements 1500 base pairs +/− to the identified Adar1 peaks.
Fear extinction learning leads to increased Adar1 occupancy at DNA repetitive elements.
In order to determine where in the genome Adar1 occurs, we next performed ADAR1 ChIP-seq was then performed on activated (high Arc) and quiescent (low Arc) neurons derived from the PFC following fear extinction learning. After high stringency filtering of reads and statistical corrections for multiple comparisons, a significant increase in Adar1 binding was observed at 2150 sites (Supplementary Table 1), with Adar1 being selectively enriched at 122 genomic loci following fear extinction training. There appeared to be no clear bias in the genomic distribution of Adar1 binding (Figure 1g). However, our analysis revealed significant enrichment at DNA repetitive elements, including SINEs and LINEs proximal to peaks of Adar1 occupancy in all groups (Figure 1h). Furthermore, CTCF prediction software (http://insulatordb.uthsc.edu/) identified several putative sites (Supplementary Table 2) and structural prediction software (https://nonb-abcc.ncifcrf.gov/apps/site/default) indicated that 70% of sites exhibiting differential Adar1 enrichment had a high probability of forming alternative DNA structures. Together, these findings suggest a potential link between Adar1 and the recognition of dynamic DNA structure states.
In an independent cohort of animals, we infused either a scrambled control or Adar1 shRNA into the infralimbic PFC followed by an assessment of Adar1 binding to DNA in regions proximal to the promoter of 9 candidate genes: neurexin 3 (Nrxn3), carbonic anhydrase 5a, mitochondrial (Car5a), integrator complex subunit 2 (Ints2), synaptosomal-associated protein 47 (Snap47), calcium voltage-gated channel auxiliary subunit alpha 2 delta 2 (Cacna2d2), eukaryotic translation initiation factor 4 gamma 3 (Eif4g3), regulator of chromosome condensation (Rcc1), sidekick cell adhesion molecule 1 (Sdk1) and glutamate ionotropic receptor kainate type subunit 2 (Grik2). All of the selected candidates are abundantly expressed in the brain, with Nrxn3 having been shown to be critical for fear-related learning20. In control animals, there was a general increase in Adar1 binding following fear extinction training with 10 and 30 conditioned stimulus (CS) exposures (Supplementary Figure 2), an effect that was not observed with Adar1 knockdown (Supplementary Figure 3).
Adar1 knockdown impairs memory flexibility
To assess the functional consequence of Adar1 binding to DNA, we infused an Adar1 shRNA into the infralimbic PFC (Supplementary Figure 4a) and validated its effect on both Adar1 mRNA and protein levels (Supplementary Figure 4b–c). In animals trained in the presence of Adar1 shRNA (Figure 2a) there was no effect on within-trial fear extinction learning (Supplementary Figure 4d). Retention control animals that were infused with a scrambled control virus and exposed to context B, exhibited significantly higher freezing scores at tests 1 and 2 compared to fear extinction trained animals treated with scrambled control. In contrast, there was no difference between these retention control animals and fear extinction-trained animals treated with Adar1 shRNA (Figure 2b–d). Together, these data suggest three possibilities: 1) Adar1 knockdown enhances the consolidation of the original fear, 2) Adar1 knockdown blocks the formation of fear extinction memory or 3) Adar1 knockdown impairs stimulus-independent forgetting.
Figure 2. Adar1 knockdown impairs memory updating following extinction training.
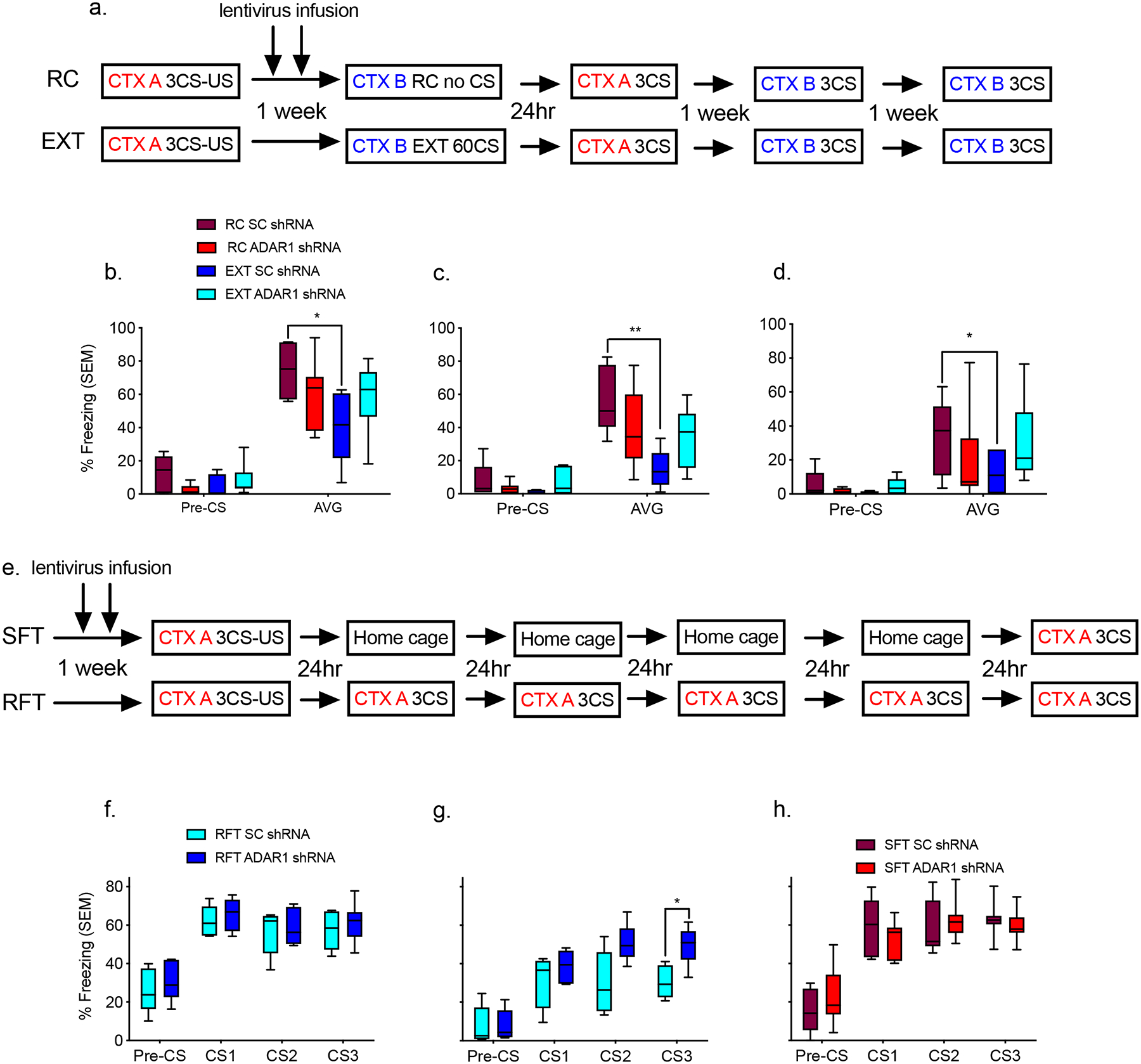
a, Schematic of the behavioral protocol used to test the effect of lentiviral-mediated knockdown of Adar1 in the PFC on fear extinction memory. CTX, context; CS, conditioned stimulus; 3CS, a test with three conditioned stimuli; CS1, the first conditioned stimulus; CS2 the second conditioned stimulus; CS3 the third conditioned stimulus; US, unconditioned stimulus; EXT, extinction session by repeated presentations of CS; RC retention control session in which no CS was presented b, Animals treated with scrambled control virus, fear conditioned and exposed to a novel context (RC SC shRNA) had significantly higher freezing scores than scrambled control animals that were fear conditioned followed by extinction (EXT SC shRNA), but this was blocked in animals that were treated with Adar1 shRNA and extinction trained (EXT Adar1 shRNA; n=5–8 animals per group from two combined independent cohorts, repeated two-way ANOVA, F1,21 = 67.17, ***p<.0001; Dunnett’s post-hoc tests: RC SC vs. EXT SC, * p = 0.0462, RC SC vs EXT Adar1 p = 0.3369, RC SC vs RC Adar1 p = 0.4407. c, This effect held at the second (repeated two-way ANOVA, F1,21 = 85.21, *p<.0001; Dunnett’s post-hoc tests: RC SC vs. EXT SC, ** p = 0.0065. RC SC vs EXT Adar1 p = 0.1568. RC SC vs RC Adar1 p = 0.2560), d, and third test (two-way ANOVA, F1,21 =27.56, *p<.0001; Dunnett’s post-hoc tests: RC SC vs. EXT SC, * p = 0.0404, RC SC vs EXT Adar1 p = 0.9994, RC SC vs RC Adar1 p = 0.6229). e, Schematic of the behavioral protocol used to test the effect of lentiviral-mediated knockdown of Adar1 on memory stability. f, There was no significant difference between SC and Adar1 shRNA animals subjected to repeated fear tests (RFT) on the first day of testing. g, However, at day 5 there was a significant difference between the SC and Adar1 shRNA (n = 5–7 animals per group from two combined independent cohorts, ANOVA, F1,9 = 5.571 *p<0.05; Bonferonni post-hoc test: SC vs Adar1 CS3 *p = 0.0326). There was also no significant difference between h, SC and Adar1 shRNA animals subjected to a single fear test (SFT) on day 5.
To clarify which putative mechanism accounted for the effect of Adar1 knockdown on fear extinction memory, a new cohort of animals was infused with either scrambled control or Adar1 shRNA. The animals were then subjected to fear conditioning consisting of pairing three CS to three unconditioned stimuli (US) in the form of shocks, followed by either five days of repeated three CS tests in context A, or a single fear test in context A on the fifth day (Figure 2e). Control and Adar1-treated animals did not differ during fear acquisition (Supplementary Figure 4f–h). There was also no significant difference in terms of fear expression in context A at 24 hour post-test, suggesting that Adar1 knockdown did not alter either the acquisition or consolidation of the original fear memory (Figure 2f). However, following five fear tests scrambled control treated animals froze significantly less than Adar1 shRNA-treated animals (Figure 2g). This group difference was not observed when scramble control and Adar1-treated animals were only tested once after five days, indicating that repeated exposure to the stimuli, not the mere passage of time, was required for these differences to emerge (Figure 2h). These data therefore demonstrate that Adar1 knockdown did not alter passive forgetting mechanisms, or fear consolidation, but instead disrupted the formation of fear extinction memory.
Adar1 is critical for updating DNA structure states
To further examine the activity-dependent relationship between Adar1 and Z-DNA we enhanced the formation of Z-DNA using spermidine21, or blocked it with doxorubicin, a drug which reduces the conversion of B to Z-DNA22, and then assessed Adar1 DNA binding in primary cortical neurons under conditions of KCl-induced depolarization. Blocking Z-DNA formation with doxorubicin led to a reduction in Adar1 binding (Supplementary Figure 5d,e). Subsequently, a global inverse relationship between Adar1 and Z-DNA during fear extinction learning was observed, such that when Adar1 transcript and protein levels were high (Figure 1 a–f), Z-DNA was decreased (Data not shown). This relationship was also observed at candidate gene loci selected from the Adar1 ChIP-seq dataset. Specifically, there were comparable levels of Z-DNA at retention control and 10 conditioned stimuli extinction, which then decreased following 30 or 60 conditioned stimuli training during which time Adar1 occupancy increased at all sites (Figure 3f–i). Z-DNA levels were also increased immediately following fear conditioning (1h), but not at later time points (2–5h) (Figure 3c–f), during which time there was very little Adar1 binding and no significant group differences (Supplementary Figure 2h,i). Together, the findings suggest that the induction of Z-DNA is associated with fear learning, which is Adar1-independent. This formation of Z-DNA then appears to serve as a signal to promote Adar1 binding. The experience-dependent recruitment of Adar1 may then subsequently lead to the assembly of chromatin remodelling/transcriptional complexes that mediate activity-induced gene expression and subsequent editing of those transcripts (Figure 3e).”
Figure 3. Z-DNA is a critical modulator of activity-dependent transcription.
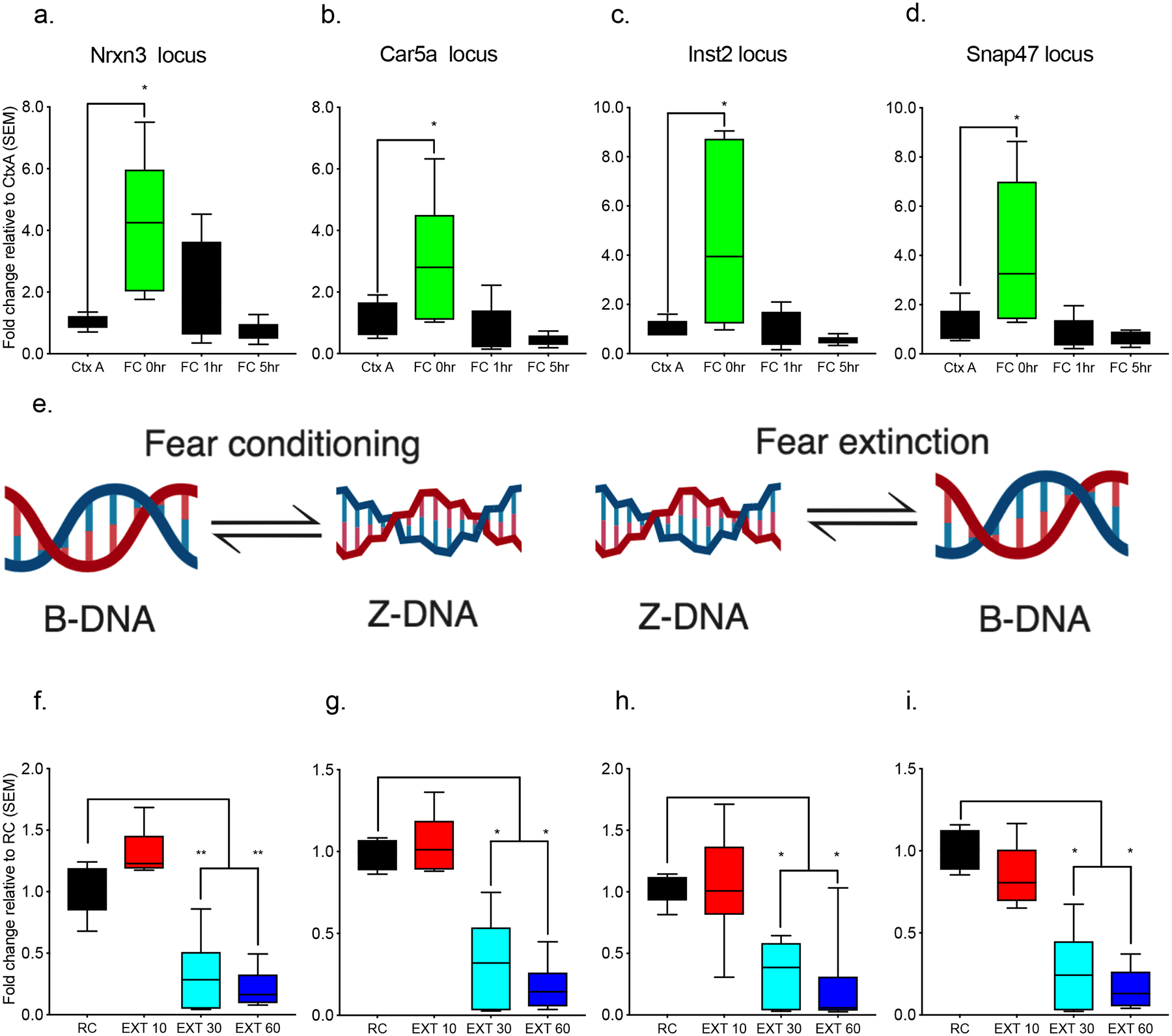
Animals that were exposed to the fear conditioning context but not conditioned (Ctx A) compared to those that were fear conditioned and euthanized immediately after training (FC O h), 1 h after (FC 1h), or 5 h after (FC 5h) exhibited significantly increased Z-DNA at the a, Nrxn3 locus (n = 5–6 animals per group from a single cohort, ANOVA, F3,18 = 5.983, *p<0.01 Dunnett’s posthoc tests: Ctx A vs FC 0h p = 0.0083, Ctx A vs FC 1h p = 0.4994, Ctx A vs FC 5h 0.9694), b, Car5a locus (ANOVA, F3,18 = 5.419, *p<0.01 Dunnett’s post-hoc tests: Ctx A vs FC 0h p = 0.0344, Ctx A vs FC 1h p = 0.9087, Ctx A vs FC 5h p = 0.6681) c, Inst2 (ANOVA, F3,18 = 5.459, *p<0.01; Dunnett’s post-hoc tests: Ctx A vs FC 0h p = 0.0142, Ctx A vs FC 1h p = 0.999, Ctx A vs FC 5h p = 0.9496), and f, Snap47 locus (ANOVA, F3,18 = 5.214, *p<0.01, Dunnett’s post-hoc tests: Ctx A vs FC 0h p = 0.0209, Ctx A vs FC 1h p = 0.9807, Ctx A vs FC 5h p = 0.9316). d, Model depicting the transition from B-DNA to Z-DNA during fear conditioning and from Z-DNA to B-DNA during fear extinction. There was also a significant alteration in Z-DNA when comparing fear-trained animals exposed to a novel context following fear conditioning (retention control: RC) to fear-conditioning followed by 10CS (EXT 10), 30CS (EXT 30), or 60CS extinction (EXT 60). Specifically, Z-DNA significantly decreased at the e, Nrxn3 locus (n = 6–8 animals per group from a single cohort, ANOVA F3,23 = 39.48 ****p<0.0001, Dunnett’s post-hoc tests: RC vs EXT 10 p = 0.0879 RC vs EXT 30 ****p <0.0001, RC vs EXT 60 <0.0001), i, Car5a locus (ANOVA F(3,22) = 32.94 ****p<0.0001, Dunnett’s post-hoc tests: RC vs EXT 10 p = 0.9031 RC vs EXT 30 ****p <0.0001, RC vs EXT 60 ****p<0.0001), f, Inst2 locus (ANOVA, F3,25 = 13.88, ****p<0.0001, Dunnett’s post-hoc tests: RC vs EXT 10 p = 0.9920, RC vs EXT 30 p = 0.0009, RC vs EXT 60 p = 0.0002), and g, Snap47 locus (ANOVA, F3,23 = 34.88, ****p<0.0001, Dunnett’s post-hoc tests: RC vs EXT 10 p = 0.3363, RC vs EXT 30 ****p < 0.0001, RC vs EXT 60 ****p<0.0001).
DNA structure states are critical for memory flexibility
To determine whether Z-DNA formation prior to Adar1 binding is required for fear memory, we next infused doxorubicin into either the prelimbic or infralimbic PFC prior to behavioral training. A wide range of doses (0.2–20μg) infused into the infralimbic PFC prior to fear learning had no significant effect on fear acquisition (Supplementary Figure 6a). Similarly, there was no effect of doxorubicin on memory when tested in context A 24h later, suggesting no influence on the consolidation of fear memory (Supplementary Figure 6b,c). Following extinction training, all groups exhibited significantly less freezing relative to controls in context B (Supplementary Figure 6d). However, although the saline- and high dose (20 μg) doxorubicin-treated groups which underwent extinction exhibited reduced freezing compared to retention controls in context A, the low (0.2 μg) and medium (2 μg) dose doxorubicin-treated mice did not, suggesting that the modification of the original trace was impaired (Supplementary Figure 6d).
Similarly, when saline or the medium dose of doxorubicin was infused into the infralimbic PFC prior to extinction, there was no effect on the acquisition or consolidation of extinction memory as both groups exhibited significantly lower freezing compared to retention controls in context B (Supplementary Figure 6e–g). Interestingly, the saline-treated extinction group showed less freezing in context A compared to retention controls, an effect not observed in the medium dose group, which further suggests an inability to modify the original trace (Supplementary Figure 6g). When the same dose of doxorubicin was infused into the prelimbic PFC during fear learning, fear acquisition and fear consolidation were equally unaffected (Supplementary Figure 6h–i). Both extinction-trained saline- and doxorubicin-treated animals had significantly lower freezing scores than retention controls during post-extinction testing in context A or context B, suggesting that the formation of Z-DNA is not critical for the acquisition or consolidation of fear learning, but instead plays a role in memory flexibility.
Adar1 disinhibits Z-DNA, thereby promoting flexible transcription
We next explored how Adar1 binding and Z-DNA modulation are functionally linked through their relationship with gene expression. We used a Z-DNA specific antibody in a ChIP assay to identify accessible DNA sequences capable of forming Z-DNA from targets identified by Adar1 binding. The expression of mRNA derived from DNA repetitive elements mirrored the profile of Z-DNA. For both fear conditioning and extinction, sequences capable of forming Z-DNA were accessible in regions proximal to protein coding gene promoters, as expected for a transcriptionally driven process (see Table 1). In addition, there was an increase in RNA polymerase II (RNA polII) occupancy at all candidate loci during extinction, as well as a significant reduction during fear training (Supplementary Figure 7a–h), which mirrored the observation of a significant reduction in Z-DNA during extinction and an increase in Z-DNA during fear learning (Figure 3b–k). The significant decrease in accessible Z-DNA-forming sequences could reflect the inability to detect Z-DNA due to the presence of Adar protein (see Supplementary Figure 8d), a change in chromatin structure making the DNA inaccessible, or the loss of negative supercoiling necessary to maintain Z-DNA formation associated with the assembly of active transcriptional complexes.
Table 1.
Relationship between Z-DNA and transcription
Correlations are between the amount of Z-DNA enrichment at Adar1 binding sites and transcript levels of exon expression from targets proximal to these sites.
| Group | ||||
|---|---|---|---|---|
| Fear conditioned | Extinction trained | |||
| Gene target | Correlation | P-value | Correlation | P-value |
| Car5a | 0.5671 | 0.0176 | −0.3522 | 0.1391 |
| Snap47 | 0.2885 | 0.2614 | −0.4733 | 0.0261 |
| Inst2 | 0.2368 | 0.0559 | 0.4632 | 0.0820 |
| Nrxn3 | −0.1343 | 0.6073 | 0.3582 | 0.1444 |
Control experiments in which Z-DNA assays were performed in the presence or absence of proteinase K indicated that while Adar1 protein may partially obscure the ability to detect Z-DNA, a reduction was still observed (see Supplementary Figure 8e). In addition, an exploration of the variance within the retention control group revealed a significant correlation between the freezing of these retention control animals at the third CS of fear training and Z-DNA levels following test the next day (Pearsons R = 0.71 p<0.05). Together this further supports the hypothesis that Z-DNA flexibility is related to behavioral flexibility.
With respect to the Nrxn3 locus, Z-DNA initially increased, followed by a significant increase in Adar1 binding and a reduction in Z-DNA, and increased expression of the upstream transcript (Figure 4a–d). Importantly, Adar1 knockdown led to a significant increase in Z-DNA, which correlated with a reduction in Adar1 binding and a further reduction in Nrxn3 RNA expression (Figure 4e–g). We found that full-length Adar1 (p110 and p150 isoforms) was able to rescue the observed reduction in Z-DNA during extinction, whereas Zα mutants (N175A/Y179A) which block its ability to bind DNA, or Adar1 with a mutated RNA editing domain (E861A), could not (Supplementary Figure 8e). Z-DNA may therefore serve as a negative regulator of transcription, which is normally disinhibited by Adar1, implicating genomic updating by dynamic changes in Z-DNA though Adar1 binding as a potential mechanism underlying the gene expression associated with memory flexibility.
Figure 4. Adar1-Z-DNA interaction at the Nrxn3 locus modulates the expression of transcripts derived from a non-coding region enriched with SINEs.
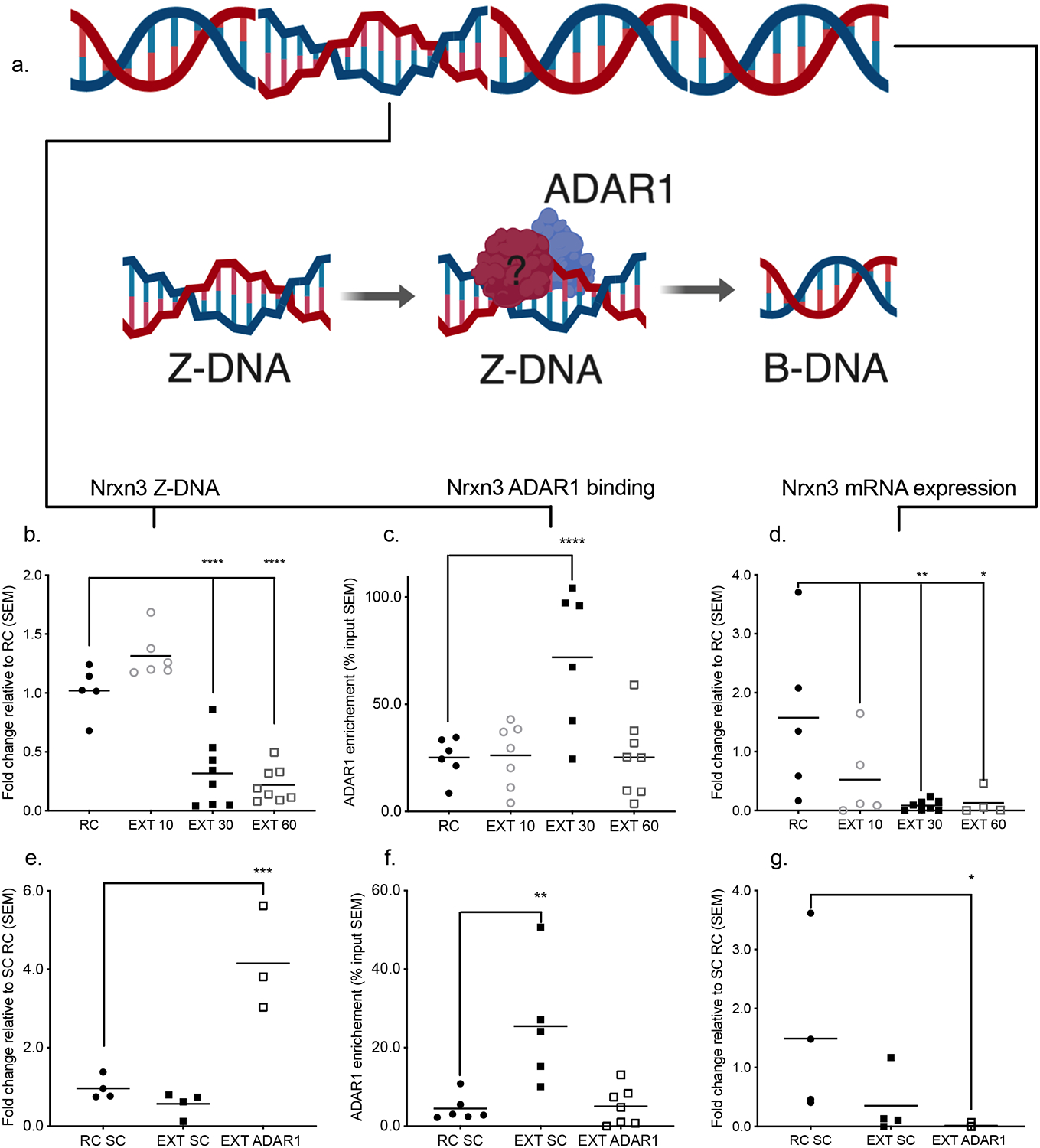
a, Model depicting the recognition and resolution of Z-DNA by Adar1 binding b, There was a significant reduction in Z-DNA when comparing fear-trained animals exposed to a novel context following fear conditioning (retention control: RC) to those fear-conditioned followed by 10CS (EXT 10), 30CS (EXT 30), or 60CS extinction (EXT 60) for the Nrxn3 locus (n = 5–8 animals per group from a single cohort, ANOVA F3,23 = 39.48 ****p<0.0001, Dunnett’s post-hoc tests: RC vs EXT 10 p = 0.0879 RC vs EXT 30 ****p <0.0001, RC vs EXT 60 ****p<0.0001). Further there was a significant increase in c, Adar1 at the 30 CS EXT time point (n = 5–8 animals per group from a single cohort, ANOVA F3,23 = 8.130 ***p<0.001, Dunnett’s post-hoc tests: RC vs EXT 10 p = 0.9993 RC vs EXT 30 ****p <0.0001, RC vs EXT 60 p > 0.9999). d, In response to extinction training, the Nrxn3 transcript was significantly decreased (n = 5–8 animals per group from a single cohort, ANOVA, F3,18 = 4.636 *p<0.01, Dunnett’s post-hoc tests: RC vs EXT 10 p = 0.0938 RC vs EXT 30 **p = 0.0066, RC vs EXT 60 *p = 0.0251), e, When these tests were conducted with Adar1 shRNA-treated animals, Z-DNA was significantly increased (n = 4 animals per group from a single cohort ANOVA, F2,8 = 24.96 ***p<0.001, Dunnett’s post-hoc tests: RC SC vs EXT SC 10 p = 0.6690 RC SC vs EXT Adar1 ***p = 0.0007), f, Adar1 shRNA also blocked the significant increase in Adar1 binding (ANOVA, F2,15 = 9.798 **p<0.01, Dunnett’s post-hoc tests: RC SC vs EXT SC **p = 0.0027 RC SC vs EXT Adar1 p = 0.9897), g, Adar1 shRNA-treated animals had a significantly lower level of Nrxn3 transcript, (ANOVA, F2,10 = 3.345 p = 0.0772, Fisher’s LSD post-hoc tests: RC SC vs EXT SC p = 0.0956 RC SC vs EXT Adar1 *p = 0.0307).
RNA editing at SINEs and LINEs require Adar1 binding to DNA
Because Adar1 is critically involved in RNA editing we next investigated whether ADAR1 binding at DNA repetitive elements also impacts RNA editing of transcripts derived from those loci. We examined all editing sites in RNA expressed in response to fear or fear extinction learning using our previously published FACS-sorted RNA sequencing dataset18 and examined whether these sites of RNA editing align with annotated SINE/LINEs and Adar1 DNA binding. We discovered a total of 496 genes in which RNA editing had occurred in at least 75% of the high arc samples (Figure 5a). A GO-term analysis revealed enrichment for genes related to the post-synaptic membrane, synapse, post-synaptic density and dendritic spines (Figure 5b). In addition, the selective increase in RNA editing implied behaviorally specific control over RNA editing, as 49% of edited genes were unique to the extinction trained animals, 42% unique to retention controls, and 9% of sites overlapped between the two groups. Finally, our RNA editing data showed 80% (97/122) overlap with Adar1 DNA binding, as well as an overrepresentation of SINE/LINE elements at these sites (Figure 5c,d). In fact, the distance between Adar1 DNA binding peak and DNA repetitive elements in Nrxn3, Car5a, Inst2 and Snap47 was tight, ranging from 0 to 278bp (Supplementary Table 3).
Figure 5. RNA editing enriched at SINEs and LINEs in activated neurons.
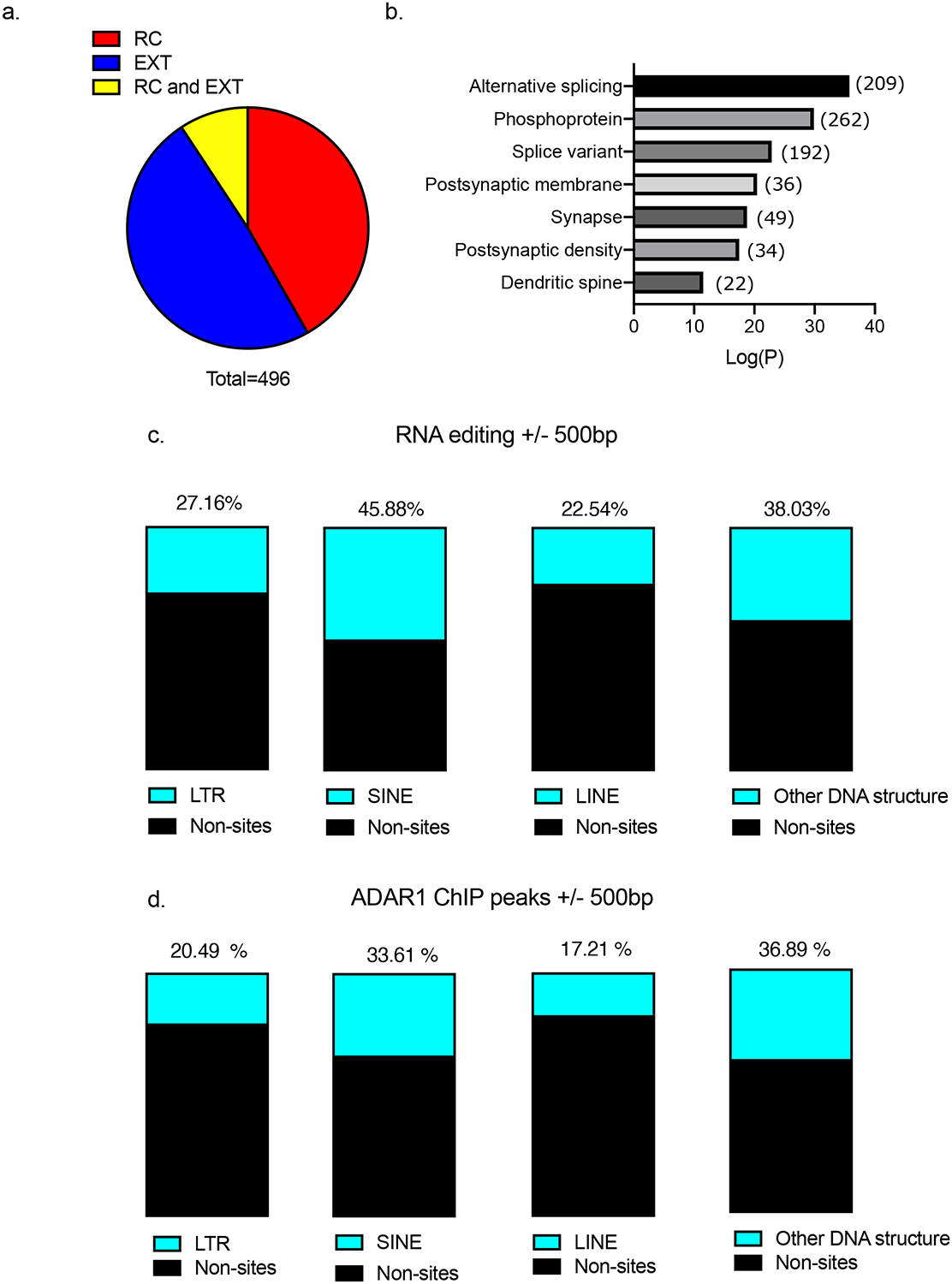
a, Distribution of RNA editing for the 496 significantly enriched targets; 49% of edited genes were specific to EXT, 42% were specific to RC, and 9% of sites occurred in the same genes for RC and EXT b, GO-term analysis of identified RNA edited genes found categories associated with both splicing and neuronal regulation c, Percent of structural elements within 500bp of RNA editing site d, Percent of structural elements within 500bp of the Adar1 DNA binding sites.
Gel electrophoresis followed by sequencing confirmed the expression of transcripts derived DNA repetitive elements that overlapped with Adar1 binding sites. Using DNA repetitive element prediction software (http://sines.eimb.ru/), several SINE/LINEs were detected within regions occupied by Adar1. Furthermore, the expression of transcripts from these regions was altered in response to fear and fear extinction learning (Figure 6a–h). Given that, polymerase III is known to drive the expression of non-long terminal repeat retrotransposons such as SINEs23, we next performed polymerase III (PolIII) ChIP at the same sites and found, for example, that a significant increase in PolIII occupancy at the Nrxn3 locus occurred in response to extinction learning. Although Adar1 knockdown disrupted this effect, the reintroduction of full-length Adar1 led to a complete rescue of PolIII recruitment following fear extinction learning (Supplementary Figure 6i–l).
Figure 6. Editing of RNA derived from SINEs and LINEs requires Adar1 binding to DNA.
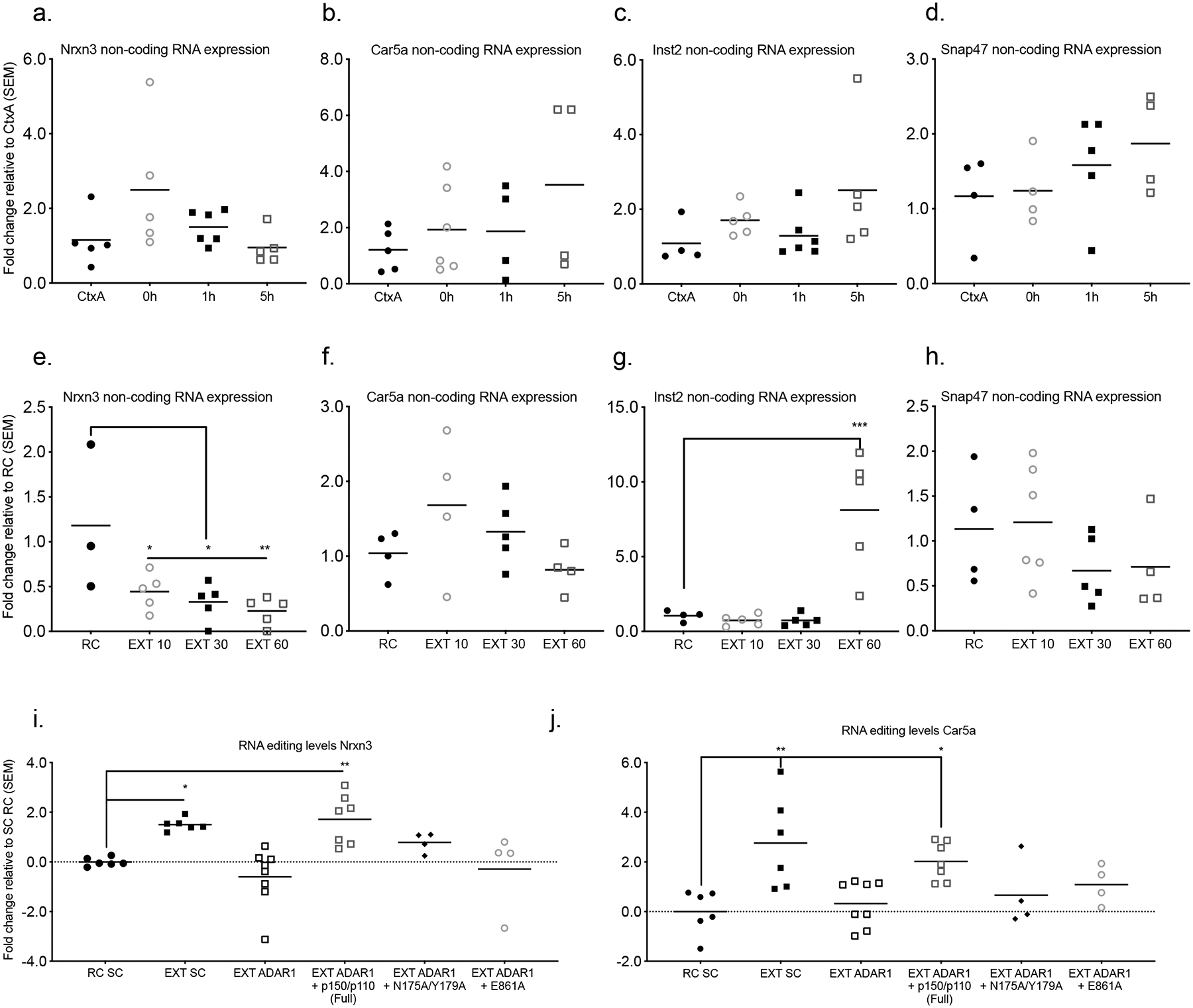
RNA expression at the sites of Adar1 binding outside the exons did not reach statistical significance for animals that were exposed to the fear conditioning context but not conditioned (Ctx A) compared to those that were fear conditioned and euthanized immediately after training (FC O h), 1 h after (FC 1h), or 5 h after (FC 5h) fear learning in the a, Nrxn locus (n = 4–6 animals per group from a single cohort, ANOVA, F3,17 = 0.0946), b, Car5a locus (ANOVA, F3,14 = 0.3320), c, Inst2 locus (ANOVA, F3,16 = 0.1590), or d, Snap47 locus (ANOVA, F3,13 = 0.3756). Fear-trained animals exposed to a novel context following fear conditioning (retention control: RC) compared to fear conditioning followed by 10CS (EXT 10), 30CS (EXT 30), or 60 CS extinction training (EXT 60) instead showed a significant reduction of expression at the e, Nrxn3 locus (ANOVA, F3,14 = 5.016, *p < 0.05. Dunnett’s post-hoc tests: RC vs EXT 10 *p = 0.0342, RC vs EXT 30 p = 0.0145, RC vs EXT 60 p = 0.0069), as well as a significant increase in RNA expression at the g, Inst2 locus (ANOVA, F3,15 = 15.15, ****p < 0.0001. Dunnett’s post-hoc tests: RC vs EXT 10 p = 0.9925, RC vs EXT 30 p = 0.9924, RC vs EXT 60 ***p = 0.0004). However, the f, Car5a (n = 3–5 animals per group from a single cohort, ANOVA, F3,13 = 1.812, p = 0.1946) and h, Snap47 locus (ANOVA, F(3,15) = 1.233, p = 0.3326) did not reach statistical significance. Extinction-trained animals injected with scrambled control shRNA (EXT SC), Adar1 shRNA (EXT Adar1), Adar1 shRNA and full-length Adar1 (EXT Adar1 + FL), Adar1 shRNA and Zα mutant (EXT Adar1 + N175A/Y179A) or Adar1 shRNA and RNA editing mutant (EXT Adar1 + E861A) were compared to animals subjected to fear conditioning and injected with scrambled control shRNA (RC SC) and assessed for RNA editing with Endo V treatment. Significant increases in RNA editing levels were observed at the Nrxn3 locus i, (ANOVA, F5,29 = 7.27, ***p < 0.001. Dunnett’s post-hoc tests RC SC vs EXT SC *p = 0.0333, RC SC vs EXT Adar1, p = 0.6325, RC SC vs EXT Adar1 + FL, **p = 0.0092, RC SC vs EXT Adar1 + N175A/Y17 p = 0.5532, RC SC vs EXT Adar1 + E861A p = 0.9848), as well as the j, Car5a locus (ANOVA, F5,29 = 5.337, **p<0.01 Dunnett’s post-hoc tests: RC SC vs EXT SC **p = 0.0012, RC SC vs EXT Adar1, p = 0.9797, RC SC vs EXT Adar1 + FL, *p = 0.0159, RC SC vs EXT Adar1 + N175A/Y17 p = 0.8424, RC SC vs EXT Adar1 + E861A p = 0.4666).
In order to validate the relationship between Adar1 binding to DNA and RNA editing, we took advantage of Endo V, an enzyme that recognizes and cuts inosine in RNA, followed by qPCR. We observed a significant increase in the editing of transcripts derived from sites of increased Adar1 DNA occupancy in response to fear extinction learning, an effect that was blocked by Adar1 knockdown. Importantly, simultaneous introduction of full-length Adar1 rescued the observed effect of Adar1 knockdown on both Z-DNA and RNA editing, whereas Zα (N175A/Y179A) and RNA editing (E861A) mutants did not (Figure 6i,j). Together, these findings strongly suggest a dual function of Adar1, which supports the hypothesis that an interaction between Z-DNA and Adar1 may be required for co-transcriptional RNA editing.
Memory flexibility requires both Adar1 Z-DNA binding and RNA editing domains
To determine whether the observed impairment in fear extinction is dependent on both Adar1 DNA binding and RNA editing, three variants of Adar1 were introduced into the infralimbic PFC following fear training. Full-length Adar1 (p150 and p110 isoforms), Adar1 with a mutated Zα domain (N175A/Y179A) which blocks its ability to bind DNA, or Adar1 with a mutated RNA editing domain (E861A) that blocks its ability to RNA edit was introduced against an Adar1 knockdown background and compared to scrambled control animals subjected to either retention control or extinction conditions (Figure 7a). At the first test, all groups that had been exposed to extinction training showed lower freezing scores than control animals, except those treated with Adar1 shRNA (Figure 7b–d). On the second and third tests, only the scrambled control extinction and Adar1 + Full length extinction group had significantly lower freezing scores. Our findings therefore indicate that both the Zα DNA binding domain and the RNA editing domain are required for memory flexibility. However, we have not differentiated between RNA editing performed primarily in the nucleus by the p150 isoform or that by the p110 isoform in the cytoplasm.
Figure 7. Fear extinction requires both Adar1 Z-DNA binding and RNA editing domains.

a, Schematic of the behavioral protocol used to test the effect of lentiviral-mediated knockdown of Adar1 and its restoration using either Adar1 shRNA and full-length Adar1 + ADAR1 shRNA + N175A/Y17, or Adar1 shRNA + E861A, on extinction learning. CTX, context; CS, conditioned stimulus; US, unconditioned stimulus. b, Animals treated with scrambled control virus, then fear conditioned and exposed to a novel context (RC SC) had significantly higher freezing scores than animals that were fear conditioned followed by extinction training and scrambled control virus treatment (EXT SC), Adar1 shRNA + full-length p150/p110 Adar1 (EXT Adar1 + FL), Adar1 shRNA + N175A/Y17 (EXT Adar1 + N175A/Y17), or Adar1 shRNA + E861A (EXT Adar1 + E861A), but this was blocked in animals that were treated with Adar1 shRNA (EXT Adar1; n= 6–8 animals per group from two independent cohorts, repeated two-way ANOVA, F5,60 = 5.353, ***p<0.001; Dunnet’s post-hoc tests; RC SC vs. EXT SC, **** p < 0.0001, RC SC vs EXT Adar1 p = 0.0697, RC SC vs EXT Adar1 + FL **p = 0.0022, RC SC vs EXT Adar1 + N175A/Y17 **p = 0.0044, RC SC vs EXT Adar1 + E861A *p = 0.0334). c, During the second test, only the EXT SC and EXT Adar1 + FL groups were significantly different from the RC SC groups (n=11 biologically independent animals per group, repeated two-way ANOVA, F5,60 = 3.861 **p<0.01; Dunnet’s post-hoc tests; RC SC vs. EXT SC, * p = 0.0192, RC SC vs Adar1 EXT p = 0.3487, RC SC vs Adar1 + FL EXT ***p = 0.0009, RC SC vs EXT Adar1 + N175A/Y17 p = 0.4766, RC SC vs EXT Adar1 + E861A p = 0.2542). d, Similarly, during the third test only the EXT SC and EXT Adar1 + FL groups were significantly different from the RC SC group, (n = 9–12 animals per group from two independent cohorts, repeated two-way ANOVA, F5,60 = 2.736, *p<0.05; Dunnet’s post-hoc tests; RC SC vs EXT SC, *p = 0.0130, RC SC vs EXT Adar1 p = 0.4897, RC SC vs EXT Adar1 + FL **p = 0.0019, RC SC vs EXT Adar1 + N175A/Y17 p = 0.3247, RC SC vs EXT Adar1 + E861A p = 0.3153). Animals treated with scrambled control virus, then fear conditioned and exposed to a novel context (SC RC) had significantly higher freezing scores than animals that were fear conditioned followed by extinction and treatment with scrambled control virus (SC EXT), Adar1 shRNA and p150 (EXT Adar1 + p150), or Adar1 p110 (EXT p110) on the e, first (two-way ANOVA, F6,41 = 5.527, ***p<0.001; Dunnet’s post-hoc tests; RC SC vs. EXT SC, *** p = 0.0001, RC SC vs EXT ADAR1 p = 0.0976, RC SC vs EXT Adar1 + p150 **p = 0.0021, RC SC vs EXT Adar1 + p110 p = 0.5993, RC SC vs EXT p150 p = 0.874, RC SC vs EXT p110 *p = 0.0249) and f, second test (two-way ANOVA, F6,41 = 6.731, ****p<0.0001; Dunnet post-hoc; RC SC vs. EXT SC, **** p < 0.0001, RC SC vs EXT Adar1 p = 0.0625, RC SC vs EXT Adar1 + p150 **p = 0.0122, RC SC vs Adar1 + p110 p = 0.2284, RC SC vs EXT p150 p = 0.9997, RC SC vs EXT p110 **p = 0.0020). g, However only the EXT SC group had significantly lower freezing than RC SC group on the third test (two-way ANOVA, F6,41 = 1.966, p = 0.0929; Sidak post-hoc test; RC SC vs. EXT SC, * p = 0.046).
To address which isoform contributes to the observed behavioral variance a second rescue experiment was then performed in which either the p150 isoform (EXT Adar1 + p150) or the p110 (EXT Adar1 + p110) was restored following Adar1 knockdown in extinction trained animals, and compared with control groups that underwent extinction training and overexpressed either the p150 isoform (EXT p150) or the p110 isoform (EXT p110). At the first and second tests, freezing scores for the scrambled control extinction, extinction Adar1 + p150, and extinction p110 were significantly lower than control mice (Figure 7e,f). On the third test, only the scrambled control extinction had significantly lower freezing score than control mice (Figure 7g). Thus, the p150 isoform was sufficient to restore extinction in the presence of Adar1 knockdown. Further, the p110 isoform had a minimal effect on extinction behavior by itself while p150 overexpression impaired fear extinction, indicating there may be a critical window of optimal expression for the p150 isoform in regulating the formation of fear extinction memory.
Discussion
We have discovered that Z-DNA forms in the PFC of C57 mice. Subsequently, this mark maintains an inverse relationship with gene expression, and coordinates the editing of transcripts derived from putative SINE/LINE elements in response to fear extinction learning. In addition, this structural mark and related downstream processes appear to be critical for the flexibility of fear memory following extinction learning. We have also found that Adar1 interacts with over 100 genomic regions in activated neurons in a learning-induced manner, and that this triggers a rapid reduction in Z-DNA, which is followed by increased RNA editing. Importantly, these effects are abolished following Adar1 knockdown. Finally, the ability of Adar1 to modulate the flexibility of fear memory is dependent on both the DNA binding and RNA editing domains within the Adar1 p150 isoform.
Over the years, a very strong case has been made for the existence of Z-DNA and its role in many biological processes, particularly transcriptional regulation, where Z-DNA has been linked to both transcriptional activation24 and inhibition25. Mechanistically, this change in DNA structure is thought to be coupled to gene expression by either the recruitment of transcription factors, such as PolII for enhancement, or the direct blockade of the transcriptional factors and machinery leading to transcriptional inhibition26–30. Similarly, we found that Z-DNA was both positively and negatively correlated to gene expression in response to learning, with positive correlations tending to occur during fear conditioning when Z-DNA was formed, and negative correlations being observed in the same locus following fear extinction training when Z-DNA was reduced. When the formation of Z-DNA was blocked by doxorubicin, the modification of the original trace following extinction was impaired, with no direct effect on the consolidation of fear or fear extinction memory. These findings suggest that Z-DNA formation may be a reversible mechanism which dictates memory flexibility by marking the genome for subsequent recruitment of recognition factors, such as Adar117.
The idea that RNA editing enzymes and other factors interact with DNA structure dates back to original observations made by Rich and colleagues that the Zα domain of Adar1, co-crystalized with an alternative left-handed DNA structure1,2, and stabilizes this Z-DNA state31,32. However, Adar1 binding to DNA was minimal during the formation of Z-DNA, suggesting that under these conditions Adar1 does not coordinate the formation of Z-DNA33. Instead, DNA methylation20–24, local salt changes18, or endogenous synthesis and release of polyamines34 may contribute to the formation and stabilization of Z-DNA at these sites. Overall, it may only be through the ability of Z-DNA to modulate downstream events such as gene expression or RNA editing that its functional importance for cellular memory can be observed. Changes in DNA structure may therefore lower the threshold for future transcriptional activation or local RNA editing by concentrating modulatory effectors to particular genomic loci.
Although Adar1 is not directly related to enzymatically regulated Z-DNA formation, full-length Adar1 is required to reduce Z-DNA during fear extinction. To do this, Adar1 may recruit a helicase or some other DNA repair enzyme to cut and release the torsion created by Z-DNA35,36, as well as the release of poised PolII, as has been observed with topoisomerase II beta (Top2ß) during fear learning37,38. This process could explain how damage in regions of DNA, assuming a Z-conformation39, and the role of Adar1 as a transcriptional activator are functionally linked. When Z-DNA is present it may block transcriptional elongation40, but resolution of Z-DNA or DNA breaks at Z-DNA sites could release PolII and allow transcription to proceed. Thus, genomic loci with a high probability to form Z-DNA, which have recently been described as “flipons”7, and proposed to represent a class of dynamic DNA structures that convert their thermodynamic entropy into the information entropy of RNA41, may therefore act as a digital transcriptional control mechanism across the genome.
A pathway involving LINEs and SINEs in the regulation of higher-order chromatin structure is also plausible based on our data, and the work of Lu and colleagues42, indicating the relationship between these two molecular features. Together, our findings more generally suggest that Adar1, and other proteins containing putative Z-DNA binding domains, may modulate transcription by targeting the reversibility of Z-DNA-forming genomic regions associated with critically important neuronal genes, such as c-fos, corticotropin-releasing hormone, and neurofibromin 1, during particular behavioral epochs24,43–45.
Beyond transcription, the downstream consequences of Z-DNA formation appear to be as important as its formation and resolution, as we have found that a reduction in Adar1-mediated editing of previously uncharacterized RNAs derived from SINE/LINE elements impairs fear extinction. Furthermore, both the editing and Zα domains are required for RNA editing and behavioral flexibility, as restoring the Zα domain without the editing domain is not as effective as the restoration of both. Furthermore, the p150 isoform, which contains the Zα subunit, is sufficient to rescue extinction memory, and this effect was not observed with p110. Although p150 appears to be critical for fear extinction, this does not rule out the possibility that p110 may be involved in establishing fear memory or other aspects of the memory, such as its maintenance or retrieval in other contexts. Taken together, these findings suggest that Adar1 DNA binding and RNA editing are interdependent and temporally linked, such that DNA structure changes occur first, followed by binding and RNA editing by Adar1.
The model of co-transcriptional RNA editing mediated by Adar1 to regulate behavioral flexibility is well supported by the data; however, there are several limitations. For example, not all Adar1 sites identified by sequencing appear to be related to Z-DNA. There may be additional DNA structures or motifs that also recruit Adar1, such as the G-quadruplex structure, which has been shown to be related to Adar1 in HeLa cells46. Furthermore, not all genomic loci bound by Adar1 align with annotated genes, which are subject to RNA editing, suggesting an unknown functional influence of Adar1 in these regions. In addition, our experiments were performed with lentivirus knockdowns rather than transgenic animals, which may have led to baseline activation of the immune system. Finally, although we focused on mechanisms surrounding transcription and RNA editing, Adar1 has been shown to influence active translation and can do so without its RNA editing domain47; it can also induce the interferon response48. Hence, these processes may also contribute, in part, to the observed changes in behavioral performance
In summary, we have demonstrated that Adar1 shows a strong bias toward structurally rich genomic regions in activated neurons and binds to DNA in an experience-dependent manner. The recognition and modification of Z-DNA by Adar1 is required for memory flexibility such that the removal of this domain impairs the ability of fear extinction to overcome previously established fear memories. Our data support the emerging idea that RNA modification is required for learning, and include dynamic changes in DNA structure as an important upstream factor in the regulation of transcriptional activity underlying flexible memory states.
Methods
Materials and Methods
Animals
9–11 week-old C57BL/6 male mice were housed four per cage, maintained on a 12h light/dark schedule, and allowed free access to food and water. To allow for identification of behavioral outliers, animals were transferred to pair-housed conditions and split with a plexiglass divider at least 24h prior to training. All testing was conducted during the light phase in red-light-illuminated testing rooms. All animal use and training, including the use of embryos, followed protocols approved by the Animal Ethics Committee of the University of Queensland and in accordance with the Australian Code for the Care and Use of Animals for Scientific Purposes (8th edition, revised 2013).
Primary cortical neurons
Cortical tissue was isolated from embryonic day 16 embryos. Primary cortical neurons were isolated by removing the skull and meninges with fine-tipped tweezers. Cells were then mixed into a medium solution comprising Neurobasal medium (GIBCO 21103) containing 5% serum, B27 supplement (GIBCO 17504–044) and 0.5–1% Penicillin-Streptomycin (GIBCO 15140), made homogenous with gentle pipetting. The cells were then passed through a 40 micrometer cell strainer (BD Falcon 352340) and plated onto 6-well cell culture dishes coated with poly-L- ornithine (Sigma P2533) at a density of 1 million cells per well.
RNA and DNA extraction
Both cultured cells and tissue from mice were extracted and then placed in PBS. Gentle pipetting was used for in vitro preparations to generate a homogenous solution. Tissue was prepared by dounce homogenization in 500 μl of PBS and RNA/DNA was extracted using Trizol. For RNA, the Trizol reagent was used according to the manufacturer’s instructions (Invitrogen). DNA extraction was carried out using the DNeasy Blood & Tissue Kit (Qiagen) with RNAse A (5 prime), RNAse H and RNAse T1 treatment (Invitrogen). Both extraction protocols were conducted according to the manufacturer’s instructions. The concentration of DNA or RNA was measured by Qubit assay (Invitrogen).
qRT-PCR
1μg of RNA was used for cDNA synthesis prepared from the Quantitect Reverse Transcription Kit according to the manufacture’s protocol (Qiagen). Quantitative PCR was then performed on a RotorGeneQ (Qiagen) real-time PCR cycler with SYBRGreen Master mix (Qiagen), using primers for target genes and beta actin or phosphoglycerate kinase as an internal control. The threshold cycle for each sample was chosen from the linear range and converted to a starting quantity by interpolation from a standard curve run on the same plate for each set of primers. All mRNA levels were normalized for each well relative to the internal control using the ΔΔCT method, and each PCR reaction was run in duplicate for each sample and repeated at least twice.
Western blot
Individual wells from cultured cells and tissue from mice were collected as described above. The homogenate was then fractionated with a NXTRACT CelLytic™ NuCLEAR™ Extraction Kit (Sigma) according to the manufacturer’s instructions. Following this, samples were prepared on ice (to a final volume of 40 microliters) in a mixture of Laemmli sample buffer and 2-mercaptoethanol (BioRad) and then vortexed and denatured for 10 min at 70°C. Gels were run and proteins transferred onto nitrocellulose membrane (Hybond-ECL, Amersham). The membrane was blocked with Odyssey Blocking Buffer (LiCore P/N 927–40100) for 1 h at room temperature, washed with Trizma-buffered saline with 1% Triton-X-100 (TBST) for 5 min (3x) and incubated with 5ml of mouse monoclonal antibody (Adar1 1:1000; Santa Cruz sc-73408 B) in blocking buffer for 24 hours at 4°C. The membrane was washed with TBST (3x), incubated for 1 h with IRDye 680 rabbit anti-mouse secondary antibody (1:10000; LI-COR) in TBST, and washed in TBST for 10 min (3x). Optical density readings of the blots were made using the LI-COR analysis system.
Lentiviral delivery and behavioral analysis
Double cannulae (PlasticsOne) were implanted in the anterior posterior plane, along the midline, into the infralimbic prefrontal cortex (ILPFC). The injection locations were centred at +1.8 mm in the anterior-posterior plane (AP), and −2.8 mm in the dorsal-ventral plane (DV). Animals were then separated into single housing and given at least one week to recover before being behaviorally trained. Following the first day of training a total volume of two microliters of lentivirus per hemisphere was infused via two one microliter injections over a 48h period. Mice were first fear conditioned, followed by 2 lentiviral infusions 24 hours post-fear conditioning, and, after a one-week of incubation, the mice were either extinction trained or just exposed to context B for an equivalent period of time (see Figure 2a). In brief, this training consisted of two contexts (A and B). Both conditioning chambers (Coulbourn Instruments) had two transparent walls and two stainless steel walls with a steel grid floors (3.2 mm in diameter, 8 mm centers); however, the grid floors in context B were covered by a flat white plastic transparent surface. Context A was also sprayed with a dilute lemon smell, and context B was sprayed with a dilute vinegar smell to minimize context generalization. Cameras within the boxes captured movement and were processed automatically with a freezing measurement program (FreezeFrame). The training protocol consisted of 120 s pre-fear-conditioning incubation, followed by three pairings of a 120 s, 80dB, 16,000 Hz tone (CS) co-terminating with a 1 sec (2 min intervals), 0.7 mA foot shock, or unconditioned stimulus (US). Mice were matched into equivalent treatment groups based on freezing during the third training CS. For extinction, mice were exposed in context B in which they habituated to the chamber for 2 min, after which the extinction training (EXT) comprised 10, 30 or 60 non-reinforced 120 s CS presentations (5-s intervals). For the retention control (RC) animals, context exposure was performed following fear conditioning, but without presentation of the tones. For the retention tests, all mice were returned to either context A or B and following a 2 min acclimation (used to minimize context generalization), freezing was assessed during three 120s CS presentations (120 s intertrial interval). Memory was inferred by the percentage of time spent freezing during the tests.
Adar1 and Adar1 scrambled control (SC) knockdown lentiviral constructs
Lentiviral plasmids were generated by inserting either Adar1 or Adar1 SC shRNA using the following sequences or Adar1: GATCCCCGCCGAGTCAGTGTTTATGATTTTCAAGA GAAATCATAA ACACTGACTCGGCTTTTTTC Adar1 SC: GATCCCCAGTTCATTAG GCTAACGTATTTCAAG AGAATACGTTAGCCTAATGAACTTTTTTTC immediately downstream of the human H1 promoter in a modified FG12 vector (FG12H1, derived from the FG12 vector originally provided by David Baltimore, CalTech). Lentivirus was prepared and maintained according to previously published protocols49.
Adar1, Adar Za mutant and catalytically inactive Adar1;
Murine Adar1, Adar1 Za-mutant (N175A/Y179A) and editing dead mutant (E861A) were generated by gene synthesis and mutants introduced by gBlocks (IDTDna). All plasmids were sequence verified. The cDNA was cloned into the pLVX vector (Clontech).
Immunohistochemistry
Following the end of testing mice were euthanized with 100mg/kg ketamine, after which 20ml of saline was pumped through the circulatory system followed by 4% paraformaldehyde to fix the tissue. The brains were then placed in 30% sucrose for a minimum 24h prior to cryostat slicing. Sectioning at 40μm was performed using a cryostat and sections were mounted on Menzel-Glaser Superfrost Plus microscope slides. Briefly, the sections were incubated for 1–2h in blocking buffer, after which primary antibodies (Adar1 and GFP ab6556) were added and the slides incubated at 4°C overnight. The slides were then washed 3 times with PBS containing 0.02% Tween 20 (PBS-T), after which secondary antibodies were added. The slides were then incubated at room temperature for 2h, washed 3 times with PBS-T, and cover-slipped with ProLong Gold Antifade Mountant with DAPI (P36931). The sections were imaged on a slide scanning confocal microscope.
FACS.
The procedure for sorting activated neurons for cDNA preparation and ChIP-seq was modified from published protocols18. Briefly, following sample preparation for FACS the identified population of neurons, as indicated by their high intensity in the 488nm channel, was further split into two populations which had intensity in the 647nm Arc channel above the upper part of the non-NeuN population (high Arc) or below it (low Arc; see Figure 1b). 250,000 cells from each sample was then taken and four biological replicates were pooled to make one for further processing to reach at least the 1 million cells required for reliable chromatin immunoprecipitation.
Chromatin and carrier ChIP immunoprecipitation (ChIP)
Standard ChIP was performed following modification of the Invitrogen ChIP kit protocol. Lysate from cells or tissue was fixed in 1% formaldehyde and cross-linked cell lysates were sheared by Covaris in 1% SDS lysis buffer to generate chromatin fragments with an average length of 300bp. For samples not being subjected to sequencing, 1 million yeast (kindly provided by Micheal Kobor) per sample were spiked in prior to fixation to enhance antibody-target interactions when the cell count was low50. Following shearing, the chromatin was then immunoprecipitated using the Adar1 antibody preconjugated to biotin (sc-73408 B), anti-Z DNA antibody (ab2079), anti-RNA polymerase II CTD repeat YSPTSPS (phospho S5) antibody (ab5131), anti-POLR3A antibody (ab96328), or normal rabbit IgG (Santa Cruz), overnight at 4°C. Protein-DNA-antibody complexes were then precipitated with Dynabeads Protein G (Thermofisher 10003D) or Dynabeads MyOne Streptavidin C1 (Thermofisher 65001) for 1h at 4°C, followed by three washes in low salt buffer, and three washes in high salt buffer for protein G beads, and 3 washes in biotin wash buffer, followed by 2 washes in PBS. The precipitated protein-DNA complexes were eluted from the antibody with 1% SDS and 0.1M NaHCO3 and incubated for 4h at 65C in proteinase K. Following proteinase K digestion, phenol-chloroform extraction, and ethanol precipitation, the samples were subjected to qPCR using primers specific for 200bp segments corresponding to the target regions. For Z-DNA control experiments the lysate was split into two equal volumes and half was treated with the standard protocol as described above. The other half followed the same protocol but during the Proteinase K step no enzyme was added.
ChIP-Seq data analysis
Bioinformatics analysis was performed as previously reported18. After removing the duplicate reads, low mapping quality reads and paired-end reads that were not properly aligned, MACS2 (v.2.1.1.20160309) were used to call peaks for each sample using the parameter setting ‘callpeak -t SAMPLE -c INPUT -f BAMPE–keep-dup = all -g mm -p 0.05 -B’. Peak summits identified by MACS2 from all samples were collected to generate a list of potential binding sites. Custom PERL script was then applied to parse the number of fragments (hereafter referred to as counts) that covered the peak summit in each sample. Each pair of properly paired-end aligned reads covering the peak summit represented one count. The total counts in each sample were normalized to 20 million before comparison among samples. The potential binding sites were kept if they met all of the following conditions: (1) the sites were not located in the Mus musculus (house mouse) genome assembly GRCm38 (mm10) empirical blacklists and (2) the normalized counts in all three biological replicates in one group were larger than in its input sample, and the normalized counts in at least two replicates were more than twofold larger than in the normalized input count.
RNA editing analysis
STAR (version 2.7.1a) was used to perform the two-pass alignment against the mouse reference genome (mm10). Picard (version 2.9.0) was then applied to add read groups and mark duplicates. RNA-Seq variant calling was performed using the GATK (version 3.8) pipeline, including modules ‘SplitNCigarReads’ with the setting of ‘-rf ReassignOneMappingQuality -RMQF 255 -RMQT 60 -U ALLOW_N_CIGAR_READS’, ‘HaplotypeCaller’ with the setting of ‘-stand_call_conf 20.0’, and ‘VariantFiltration’ with the setting of ‘-window 35 -cluster 3 -filterName FS -filter “FS > 30.0” -filterName QD -filter “QD < 2.0”‘. To remove false positives originated from random PCR/sequencing errors, only loci with A->G conversions existing in at least two samples were kept for further analyses.
RNA editing assay
Following extraction of RNA, 500ng per sample was either combined with 10 units of Endo V (NEB M0305S) in 1X NEBbuffer 4 (NEB #B7004), or an equivalent volume of ultra-pure water and 1X NEBbuffer 4. According to the manufacturer’s protocol, both reactions were incubated at 37°C for 1h, followed by 65°C for 5 min to inactivate. RNA was further purified by Zymo RNA clean concentrator kit and then amplified with the RT enzyme, buffer, and random primer mix from Qiagen (Cat No. 205313) at 42°C for 30 min, and 95°C for 3 min. Following this, cDNA samples were subjected to standard qPCR comparing Endo V-treated to -untreated for each target.
Statistical Analyses
No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications18,49. In all cases where an unpaired t-test was employed, we opted for a two-tailed test. For behavioral analysis, freezing (the absence of all non-respiratory movement) was rated during all phases by automated digital analysis system, using a 5-sec instantaneous time sampling technique. The percentage of observations with freezing was calculated for each mouse, and data represent mean ± SEM freezing percentages for groups of mice during specified time bins. Total session means were analyzed with one-way ANOVA for the behavioral data in Suppl. Fig 4. In experiments using doxorubicin and viral manipulation, all data analysis was performed by two-way ANOVA followed by Dunnett’s posthoc tests where appropriate. For all qPCR and western blots one-way ANOVAs were performed followed by Dunnett’s posthoc tests, except for Figure 4 panel g where Fisher’s LSD was used. For behavioral studies, no blinding to group allocation since the groups were counterbalanced based on fear conditioning. During behavioral tests, the data were captured and analysed using a fully automatic analysis program. For IHC, FACS and Sequencing, the investigators were blind to group allocation during data collection and analysis.
Data availability
All data and code used to generate the data is available by reasonable request to the authors.
Supplementary Material
Acknowledgements:
The authors gratefully acknowledge grant support from the NIH (R01MH105398-TWB) and the NHMRC (GNT1145172 and GNT1160823-TWB), the ARC (GNT190101078-XL), the Westpac Future Scholars program (ELZ, LJL and SUM) and postgraduate scholarships from NSERC (PRM) and the University of Queensland (PRM, ELZ, LJL, DB, JY, and SUM). We would also like to thank Ms. Rowan Tweedale for helpful editing of the manuscript.
Footnotes
Accession codes: Data from paper can be accessed at, PRJNA545193.
Competing interests
The authors declare no competing interests.
References
- 1.Schwartz T, Rould MA, Lowenhaupt K, Herbert A & Rich A Crystal structure of the Zα domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science (80-.). 284, 1841–1845 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Herbert A et al. The Zα domain from human ADAR1 binds to the Z-DNA conformer of many different sequences. Nucleic Acids Res. 26, 3486–3493 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crick FHC & Watson JD The Complementary Structure of Deoxyribonucleic Acid. Proc. R. Soc. A Math. Phys. Eng. Sci 223, 80–96 (1954). [Google Scholar]
- 4.Franklin RE & Gosling RG The structure of sodium thymonucleate fibres. I. The influence of water content. Acta Crystallogr. 6, 673–677 (1953). [Google Scholar]
- 5.Wang AHJ et al. Molecular structure of a left-Handed double helical DNA fragment at atomic resolution. Nature 282, 680–686 (1979). [DOI] [PubMed] [Google Scholar]
- 6.Pohl FM Hysteretic behaviour of a Z-DNA-antibody complex. Biophys. Chem 26, 385–390 (1987). [DOI] [PubMed] [Google Scholar]
- 7.Herbert A A Genetic Instruction Code Based on DNA Conformation. Trends in Genetics (2019). doi: 10.1016/j.tig.2019.09.007 [DOI] [PubMed] [Google Scholar]
- 8.Haniford DB & Pulleyblank DE Facile transition of poly[d(TG)·d(CA)] into a left-handed helix in physiological conditions. Nature 302, 632–634 (1983). [DOI] [PubMed] [Google Scholar]
- 9.Dobi A & Agoston DV Submillimolar levels of calcium regulates DNA structure at the dinucleotide repeat () n. Chem. Biochem 95, 5981–5986 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Behe M & Felsenfeld G Effects of methylation on a synthetic polynucleotide: the B--Z transition in poly(dG-m5dC).poly(dG-m5dC). Proc. Natl. Acad. Sci. U. S. A 78, 1619–1623 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marshall P & Bredy TW Cognitive neuroepigenetics: the next evolution in our understanding of the molecular mechanisms underlying learning and memory? npj Sci. Learn 1, 16014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGaugh JL Memory - A century of consolidation. Science 287, 248–251 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Schade M et al. The solution structure of the Zalpha domain of the human RNA editing enzyme ADAR1 reveals a prepositioned binding surface for Z-DNA. Proc. Natl. Acad. Sci. U. S. A 96, 12465–12470 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee YM et al. NMR investigation on the DNA binding and B-Z transition pathway of the Zα domain of human ADAR1. Biophys. Chem 172, 18–25 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Lee A-R et al. NMR Dynamics Study Reveals the Zα Domain of Human ADAR1 Associates with and Dissociates from Z-RNA More Slowly than Z-DNA. ACS Chem. Biol 428, acschembio.8b00914 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Rudenko A et al. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron (2013). doi: 10.1016/j.neuron.2013.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marshall PR & Bredy TW Neuroepigenetic mechanisms underlying fear extinction: emerging concepts. Psychopharmacology (Berl). (2018). doi: 10.1007/s00213-018-5084-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X et al. The DNA modification N6-methyl-2’-deoxyadenosine (m6dA) drives activity-induced gene expression and is required for fear extinction. Nat. Neurosci (2019). doi: 10.1038/s41593-019-0339-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nie Y, Zhao Q, Su Y & Yang JH Subcellular Distribution of ADAR1 Isoforms Is Synergistically Determined by Three Nuclear Discrimination Signals and a Regulatory Motif. J. Biol. Chem (2004). doi: 10.1074/jbc.M312753200 [DOI] [PubMed] [Google Scholar]
- 20.Keum S et al. A Missense Variant at the Nrxn3 Locus Enhances Empathy Fear in the Mouse. Neuron (2018). doi: 10.1016/j.neuron.2018.03.041 [DOI] [PubMed] [Google Scholar]
- 21.Thomas TJ, Gunnia UB & Thomas T Polyamine-induced B-DNA to Z-DNA conformational transition of a plasmid DNA with (dG-dC)(n) insert. J. Biol. Chem 266, 6137–6141 (1991). [PubMed] [Google Scholar]
- 22.Van Helden PD The effect of adriamycin on Z-DNA formation and DNA synthesis. Nucleic Acids Res. 11, 8415–8420 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dieci G, Fiorino G, Castelnuovo M, Teichmann M & Pagano A The expanding RNA polymerase III transcriptome. Trends in Genetics (2007). doi: 10.1016/j.tig.2007.09.001 [DOI] [PubMed] [Google Scholar]
- 24.Champ PC, Maurice S, Vargason JM, Camp T & Ho PS Distributions of Z-DNA and nuclear factor I in human chromosome 22: A model for coupled transcriptional regulation. Nucleic Acids Res. 32, 6501–6510 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naylor LH & Clark EM D(TG)n·d(CA)n sequences upstream of the rat prolactin gene form z-DNA and inhibit gene transcription. Nucleic Acids Res. 18, 1595–1601 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rich A & Zhang S Z-DNA: The long road to biological function. Nature Reviews Genetics 4, 566–572 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Shin SI et al. Z-DNA-forming sites identified by ChIP-Seq are associated with actively transcribed regions in the human genome. DNA Res. 23, 477–486 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang G & Vasquez KM Non-B DNA structure-induced genetic instability. Mutat. Res. - Fundam. Mol. Mech. Mutagen 598, 103–119 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Harteis S & Schneider S Making the bend: DNA tertiary structure and protein-DNA interactions. International Journal of Molecular Sciences 15, 12335–12363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rohs R et al. The role of DNA shape in protein-DNA recognition. Nature 461, 1248–1253 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berber I et al. Spectroscopic characterization of a DNA-binding domain, Zα, from the editing enzyme, dsRNA adenosine deaminase: Evidence for left-handed Z-DNA in the Zα-DNA complex. Biochemistry 37, 13313–13321 (1998). [DOI] [PubMed] [Google Scholar]
- 32.Bae S, Kim D, Kim KK, Kim YG & Hohng S Intrinsic Z-DNA is stabilized by the conformational selection mechanism of Z-DNA-binding proteins. J. Am. Chem. Soc (2011). doi: 10.1021/ja107498y [DOI] [PubMed] [Google Scholar]
- 33.Bae S et al. Energetics of Z-DNA binding protein-mediated helicity reversals in DNA, RNA, and DNA-RNA duplexes. J. Phys. Chem. B (2013). doi: 10.1021/jp409862j [DOI] [PubMed] [Google Scholar]
- 34.Antrup H & Seiler N On the turnover of polyamines spermidine and spermine in mouse brain and other organs. Neurochem. Res 5, 123–143 (1980). [DOI] [PubMed] [Google Scholar]
- 35.Paleček E Local supercoil-stabilized DNA structure. Crit. Rev. Biochem. Mol. Biol 26, 151–226 (1991). [DOI] [PubMed] [Google Scholar]
- 36.Rahmouni AR & Wells RD Stabilization of Z DNA in vivo by localized supercoiling. Science (80-.). 246, 358–363 (1989). [DOI] [PubMed] [Google Scholar]
- 37.Madabhushi R et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 161, 1592–1605 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X et al. The DNA repair associated protein Gadd45 regulates the temporal coding of immediate early gene expression within the prelimbic prefrontal cortex and is required for the consolidation of associative fear memory. J. Neurosci 2024–18 (2018). doi: 10.1523/JNEUROSCI.2024-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang G, Christensen LA & Vasquez KM Z-DNA-forming sequences generate large-scale deletions in mammalian cells. PNAS 103, 2677–82 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ditlevson JV et al. Inhibitory effect of a short Z-DNA forming sequence on transcription elongation by T7 RNA polymerase. Nucleic Acids Res. (2008). doi: 10.1093/nar/gkn136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prigogine I Time, structure, and fluctuations. Science (80-.). (1978). doi: 10.1126/science.201.4358.777 [DOI] [PubMed] [Google Scholar]
- 42.Lu JY et al. L1 and B1 repeats blueprint the spatial organization of chromatin. biorxiv 1–42 (2019). doi: 10.1101/802173 [DOI] [Google Scholar]
- 43.Oh D-B, Kim Y-G & Rich A Z-DNA-binding proteins can act as potent effectors of gene expression in vivo. Proc. Natl. Acad. Sci 99, 16666–16671 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rothenburg S et al. A PKR-like eukaryotic initiation factor 2 kinase from zebrafish contains Z-DNA binding domains instead of dsRNA binding domains. Proc. Natl. Acad. Sci 102, 1602–1607 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wölfl S, Martinez C, Rich a & Majzoub J. a. Transcription of the human corticotropin-releasing hormone gene in NPLC cells is correlated with Z-DNA formation. Proc. Natl. Acad. Sci. U. S. A 93, 3664–3668 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang HJ et al. Novel interaction of the Z-DNA binding domain of human ADAR1 with the oncogenic c-myc promoter G-quadruplex. J. Mol. Biol 426, 2594–2604 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Feng S et al. Alternate rRNA secondary structures as regulators of translation. Nat. Struct. Mol. Biol (2011). doi: 10.1038/nsmb.1962 [DOI] [PubMed] [Google Scholar]
- 48.Liddicoat BJ et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science (80-.). 349, 1115–1120 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin Q et al. The brain-specific microRNA miR-128b regulates the formation of fear-extinction memory. Nat. Neurosci 14, 1115–1117 (2011). [DOI] [PubMed] [Google Scholar]
- 50.O’Neill LP, VerMilyea MD & Turner BM Epigenetic characterization of the early embryo with a chromatin immunoprecipitation protocol applicable to small cell populations. Nat. Genet 38, 835–841 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and code used to generate the data is available by reasonable request to the authors.