

Abstract
Clinical response rates after adoptive cell therapy (ACT) are highly correlated with in vivo persistence of the infused T cells. However, antigen-specific T cells found in tumor sites are often well-differentiated effector cells with limited persistence. Central memory CD8+ T cells, capable of self-renewal, represent desirable ACT products. We report here that exposure to a histone deacetylase inhibitor (HDACi) and IL21 could reprogram differentiated human CD8+ T cells into central memory-like T cells. De-differentiation of CD8+ T cells was initiated by increased H3 acetylation and chromatin accessibility at the CD28 promoter region. This led to IL21-mediated pSTAT3 binding to the CD28 region, and subsequent upregulation of surface CD28 and CD62L (markers of central memory T cells). The reprogrammed cells exhibited enhanced proliferation in response to both IL2 and IL15, and a stable memory-associated transcriptional signature (increased Lef1 and Tcf7). Our findings support the application of IL21 and HDACi for the in vitro generation of highly persistent T cell populations that can augment the efficacy of adoptively transferred T cells.
Keywords: Adoptive cell therapy, reprogramming, memory CD8+ T cells, IL21, HDACi
Introduction
Immune-based strategies are effective in mediating durable clinical responses in cancer patients. Adoptive cell therapy (ACT), a form of immunotherapy that involves the administration of ex vivo activated and expanded autologous natural or gene-engineered tumor-specific T lymphocytes, can induce remission in patients with B cell malignancies and metastatic melanoma (1,2). Clinical response rates are highly correlated with persistence of the infused T cells in vivo (3,4), with the proliferative potential being the key determinant in persistence (5,6). However, antigen-specific T cells found in peripheral blood and tumor sites are often well-differentiated effector, effector memory, and sometimes terminal effector cells with limited proliferative ability (7). Several studies suggest that inhibiting terminal differentiation of tumor-reactive CD8+ T cells during activation and expansion ex vivo will lead to an increase in their persistence and anti-tumor effect in vivo (8,9). Central memory CD8+ T cells, which express high amounts of CD28 and CD62L, represent desirable products for ACT due to their capacity for self-renewal and persistence in vivo (10). Therefore, a means to de-differentiate tumor-specific effector T cells to a more highly replicative phenotype of central memory cells would improve the clinical effectiveness of adoptive cellular therapy.
CD8+ T cell effector/memory differentiation is governed by transcriptional and epigenetic regulation (11,12). Genome-wide studies indicate dynamic gene expression changes and different epigenetic landscapes, including DNA methylation and histone modifications, among T cell subsets (naïve, memory, or effector CD8+ T cells) (11,12). Acetylation of the N-terminal tails of histone H3 and H4 associates with “open” or permissive chromatin states for gene transcription, thus facilitating expression of genes in memory CD8+ T cells (13). Inhibition of histone deacetylase (HDAC)-mediated removal of acetylation from histones increases histone acetylation , thus histone deacetylase inhibitors (HDACi) allow the persistence of open chromatin states and alter chromatin accessibility, resulting in commensurate changes in gene expression (14). Reduced histone acetylation in CD8+ T-cells correlates with functional exhaustion, which can be reversed by in vitro treatment with the HDACi, valproic acid (15). Therefore, in vitro manipulation of the epigenetic program has the potential to alter CD8+ T-cell phenotype and function.
CD8+ T cell differentiation can also be influenced by γC cytokines (IL2, IL7, IL15, and IL21), which play unique and overlapping roles in CD8+ T cell development and differentiation (16). Our previous studies comparing the effect of these γC cytokines on the generation of tumor-antigen specific CTLs in vitro shows that IL21 has the unique ability to enrich for CD28hi CTLs that exhibit enhanced persistence and upregulated expression of central-memory surface markers such as CD28, CD127 and CCR7, following adoptive transfer (17,18). IL21 functions through phosphorylation of signal transducer and activator of transcription (STAT)-3 and, to a lesser extent, STAT1 and STAT5 (16). Here, we showed that IL21 regulated CD28 expression at the transcriptional level specifically through pSTAT3 binding to the CD28 promoter region in primary human CD8+ T-cells. We found that IL21 exerted these effects primarily on naïve rather than memory or effector CD8+ T cells. As an explanation for this observation, we postulated that naïve CD8+ T cells possess higher histone acetylation at the CD28 locus compared to antigen-specific effector CD8+ T cells or CD45RA+ effector memory (TEMRA) cells, which are known to have low CD28 on the cell surface (19), suggesting epigenetic regulation of CD28 and possibly other central memory markers. We postulated that the reduced histone acetylation at the CD28 locus in effector cells limited the accessibility of the upstream binding sites to IL21, which induced pSTAT3 to bind and drive CD28 expression. Therefore, treatment of effector/effector memory cells with HDACi may recapitulate the relaxed chromatin state of naïve T cells and allow IL21 mediated modulation of differentiation. In this study, we investigated the molecular mechanism of CD28 enhancement by IL21 in primary human naïve CD8+ T cells and also evaluated the effect of HDACi and/or IL21 on human effector/effector memory CD8+ T cells ex vivo.
Materials and Methods
Human peripheral blood mononuclear cells (PBMCs) and patient tumor sample acquisition
PBMCs from fifteen healthy donors 18 years of age or older (Exclusion criteria: less than 120 pounds/54.43 kg, nursing or pregnant females, autoimmune disease diagnosis, history of cancer) were collected by leukapheresis. PBMCs from three STAT3 genotyped Job’s syndrome patients were isolated from whole blood using Ficoll density gradient centrifugation. Briefly, whole blood samples mixed with phosphate buffer saline (PBS) were loaded on top of Ficoll-Paque and centrifuged at 400g for 20 minutes without braking. PBMCs at the interface of Ficoll-Paque and plasma were retrieved and washed three times with PBS. PBMCs were stored in liquid nitrogen until use. Tumor samples were acquired from patients with stage IIIc and stage IV melanoma undergoing surgery at UT MD Anderson Cancer Center, immediately followed by tumor infiltrating lymphocytes (TIL) generation. All human sample collection was performed with informed written consent and approved by the institutional review board of UT MD Anderson Cancer Center.
Cell culture and rapid expansion protocol (REP)
The medium for cytotoxic T lymphocyte (CTL) lines was RPMI1640, 10% FBS, 4 mM Glutamine, and 2-Mercaptoethanol. TILs were cultured in 50% AIM-V, 50% TIL complete medium which contained RPMI1640, 10% human AB serum, 10mM HEPES, and 2-Mercaptoethanol. Lymphoblastoid cell lines (LCLs) (obtained from the Beckman Research Institute of the City of Hope in 2002) were cultured in RPMI1640 containing 10% FBS and 1% penicillin/streptomycin. Mel 526 melanoma tumor cell line (gift from M. Lotze, University of Pittsburgh, Pittsburgh, PA in 2002) was cultured in RPMI1640, 10%FBS, 4 mM Glutamine, 1xNon-essential Amino Acids, 1mM Sodium Pyruvate, and 1% penicillin/streptomycin. No authentication was performed in the past year. The cell lines were tested for mycoplasma using a PCR-based test and used within one month after thawing cryopreserved stock vials. For REP, CTL lines or TILs were expanded using 30ng/mL anti-CD3 (OKT3, Miltenyi Biotec, 170-076-116) and 200x irradiated allogeneic PBMCs and LCLs as feeder cells. The cultures were fed with IL2 at 50 U/ml every 3 days. IL21 (30ng/ml, PeproTech, 200–21) or HDACi SAHA (1–5μM, Sigma Aldrich, SML0061) or Panobinostat (1–3nM, Selleckchem, S1030) was added on day 0, 4 and 7 if included in the expansion. After 14 days, expanded cells were subjected to further analyses.
Generation of human tumor-antigen-specific CTL lines
Tumor antigen-specific (MART1, NY-ESO-1) CTL lines were generated as described before (20). Briefly, adherent PBMCs were cultured in AIM-V media (Life Technologies) containing IL4 (500 U/ml) and GM-CSF (800 U/ml) for 6 days followed by maturation using IL1β (2 ng/ml), IL6 (1000 U/ml), TNFα (10 ng/ml) and prostaglandin E2 (PGE2; 1 μg/ml) for at least 24 hours. Mature DCs were harvested and pulsed with 40μg/ml synthetic tumor-antigen-specific peptide (PolyPeptide Group) in the presence of 3 μg/ml β2-microglobulin (Scripps Laboratories) in PBS containing 1% human serum albumin (Life Technology) for 4 hours at room temperature. Peptide-pulsed, irradiated mature DCs were mixed with PBMCs (PBMC:DC=40:1). 30 ng/ml IL21 was added immediately following co-culture initiation for IL21-priming group. The second round of stimulation, following the same protocol except for 10μg/ml synthetic peptide, occurred on day 7. IL2 (10 U/ml; R & D Systems) and IL7 (5 ng/ml; R & D Systems) were added on day 8 to support further expansion of antigen-specific CD8+ T cells. On day 14, following two rounds of stimulation, the cells were harvested for subsequent analysis. Tumor-antigen-specific CD8+Tetramer+ cells were sorted using Aria II (as described below) and expanded using REP with IL2 at 50U/mL every three days.
Generation of tumor infiltrating lymphocytes (TILs)
TILs were generated as described previously (21). Melanoma tumor samples were cut into 1–2 mm3 pieces. Each fragment was maintained in one well of 24-well culture plates in RPMI1640 with 10% human AB serum, 6000U/mL IL2 (Proleukin, Novartis), 10mM HEPES, 10mM Glutamine, and 10mM penicillin/streptomycin for 2–5 weeks.
Activation of CTL lines
Tumor-antigen-specific CTL lines were activated with 1μg/mL cognate-peptide pulsed mature irradiated DCs at DC:CTL=1:10 or together with IL21 (30ng/mL) or SAHA (1–5μM) for 1–4 days before analysis.
Polyclonal stimulation of CD8+ T cells
Naïve CD8+ T cells (CD8+CD45RA+CCR7+) were flow cytometry-sorted using Aria II (as described below) or were isolated using the EasySep™ Human Naïve CD8+ T Cell Enrichment Kit (StemCell, 19158) according to the manufacturer’s instructions. In some experiments, total CD8+ T cells were negatively selected using EasySep™ Human CD8+ T Cell Enrichment Kit (StemCell, 19053) according to the manufacturer’s instructions. The purity of the naïve or total CD8+ T cells was greater than 95% as determined by flow cytometry. CD8+ T cells were activated using Dynabeads® Human T-Activator CD3/CD28 for T-Cell Expansion and Activation (Life Technologies, 11132D) at a bead:cell ratio of 1:1 or together with 30ng/mL human IL21 (Peprotech, 200–21). At the indicated time points, T cells were harvested and beads were removed using a magnet before downstream analysis according to the manufacturer’s instructions.
Flow Cytometry
All flow antibodies were obtained from Biolegend. PE-conjugated custom tetramers were generated by the Immune Monitoring Lab at the Fred Hutchinson Cancer Research Center (Seattle, WA). Cells were stained with antibodies against CD8 (344714), CD28 (302906), CD62L (304826), CCR7 (353220), CD27 (356404), CD127 (351326), CD45RO (304210) or CD132 (338606), PD-1 (367404), or Tim-3 (345027). For surface staining, the cells were stained with antibodies and/or Tetramer-PE in FACS staining buffer (PBS/1%FBS) for 15–30 minutes at 4 degrees. After washing, cells were analyzed or sorted by flow-cytometry. For cytokine and cytotoxic granule staining, M27+ CTL lines were reactivated with M526 tumor cells at the effector: tumor ratio of 5:1 for 16 hours in the presence of Brefeldin A (Life Technology). Intracellular cytokine staining was performed using Intracellular Fixation and Permeabilization Buffer Set (ThermoFisher, 88-8824-00). Briefly, cells were stained with Live/dead fixable Aqua (ThermoFisher, L34957) as described above, followed by fixation for 15 minutes at 4 degrees and washing, then the cells were stained using antibodies against IFNγ (502512), TNFα (502926), IL2 (500326), perforin (353304) and granzyme B (515408) in permeabilization buffer for 30 minutes at 4 degrees. All FACS data were acquired via an LSR II or Novocyte flow cytometer and analyzed via FlowJo software (Tree Star, Inc.).
Chromium release assay (CRA)
Tumor target cells were labeled with 100μCi Cr51 for 2 hours. After washing, labeled tumor target cells were plated at 2,000 cells/well of 96-well V-bottom plates (4–6 repeats) and incubated with 20,000 antigen-specific CTL lines at the effector: tumor ratio of 10:1 for 4 hours. The negative control was labeled tumor target cells without effector T cells, and the positive control was tumor target cells incubated with Trypan lysis buffer (0.4% Trypan blue, 10% Nonidet P40). Then 30μl supernatants from each well were collected and the Cr51 amount in the supernatants was measured with MicroBeta2 Microplate counter (PerkinElmer) and the killing efficiency was calculated as % killing =100% × (sample average – average of negative control) / (average of positive control – average of negative control).
Quantitative RT-PCR
CD28+CD62L+ and CD28−CD62L− T cells were sort-purified using Aria II. Total RNA was purified using the Qiagen RNeasy kit (Qiagen, 74106) according to the manufacturer’s instructions and RNA concentration was measured using NanoDrop spectrophotometer (Thermo Fisher, ND-2000). Equal amount of RNA was used to synthesize first strand cDNA using M-MLV Reverse Transcriptase (Thermo Fisher, 28025013) according to the manufacturer’s instructions. Real-time PCR was performed with cDNA equal to 4ng RNA per 20μl reaction in duplicates using CFX96 Touch system (Bio-rad Laboratories, 1855195) and iQ SYBR green real-time PCR kit (Bio-rad Laboratories, 1708882) according to the manufacturer’s instructions. The housekeeping gene RPL13A was used for normalization and the 2–ΔΔCT method was used to determine the relative mRNA gene expression fold change. The following primers were used: CD28 forward: GTTCCCCTCACACTTCGGGT, reverse: ATGGGCGACTGCTTCACCAAA; Sell forward: ATGGAACGATGACGCCTGCC, reverse: GGCCTCCAAAGGCTCACACT; Ccr7 forward: CAAGCTGTCCTGTGTGGGCA, reverse: CGCTCAAAGTTGCGTGCCTG; Il7r forward: GCACGATGTAGCTTACCGCCA, reverse: GGATCCATCTCCCCTGAGCTA; Lef1 forward: CACACCCGTCACACATCCCA, reverse: TGGGAAAACCAGCCAAGAGGTG; Tcf7 forward: TGCAGCTATACCCAGGCTGG, reverse: CCTCGACCGCCTCTTCTTC; Tbx21 forward: CAACACAGGAGCGCACTGGA, reverse: GTGTTGGAAGCGTTGCAGGC; Eomes forward: GCCCACGTCTACCTGTGCAA, reverse: GGGCAGTGGGATTGAGTCCG; Prdm1 forward: AGGGCACACGTTTTGGACCC, reverse: GACACGCAGCCAGGTTTTGC; Id2 forward: CCTGTCCTTGCAGGCTTCTGA, reverse: AGGTCCATTCAACTTGTCCTCCT; and RPL13A forward: CCTCAAGGTCGTGCGTCTGA, reverse: TCCACGTTCTTCTCGGCCTG.
Metabolism assays
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using Seahorse XF Cell Mito Stress Test Kit (Agilent Technologies, 103015–100) according to the manufacturer’s instruction. Specifically, the assay was performed in XF RPMI media (nonbuffered RPMI 1640 containing 10 mM glucose, 2 mM L-glutamine, and 1 mM sodium pyruvate) under basal conditions and in response to 2 μM oligomycin, 1 μM carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), and 0.5 μM rotenone/antimycin A with Seahorse XFe96 Analyzer (Agilent Technologies). Mitochondrial mass was measured by staining the cells in 100nM MitoTracker Green FM (Cell Signaling Technology, 9074S) for 30 minutes, followed by flow cytometry analysis.
Human shRNA Knockdown
GIPZ shRNA scrambled negative control, STAT1 shRNA (V2LHS_86643, V3LHS_352188), STAT3 shRNA (V3LHS_376018, V3LHS_641819) were obtained from Dharmacon through Functional Genomics Core of UT MD Anderson Cancer Center. Total CD8+ T cells were isolated and transfected with 5μg scrambled negative control, STAT1 shRNA, or STAT3 shRNA using Amaxa human T cell Nucleofector Kit (Lonza, VPA-1002) according to the manufacturer’s instructions. Transfected cells were rested for 1–2 days and live GFP+ cells were sort-purified using the BD FACS Aria II for immunoblot analysis or were stimulated for 7 days as previously described before further analysis.
Western Blot analysis
Equal numbers of cells were lysed in 2x SDS loading buffer and loaded for immunoblot analysis with different antibodies (Cell Signaling, pSTAT3 (Y705): 9145, STAT3: 12640, pSTAT1 (Y701): 7649, STAT1: 14994, pSTAT5 (Y694): 4322, STAT5: 25656, H3: 4499, anti-rabbit-IgG-HRP: 7074S; Millipore, AcH3: 06–599). Anti–β-actin-HRP was from Santa Cruz Biotech (sc-47778 HRP). Proteins were resolved using SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked with 5% BSA/PBS for one hour at room temperature and then incubated with primary antibodies in 5% BSA/PBS at 4 degrees overnight. After washing with 0.1% Tween-20/PBS for three times, the membrane was probed with HRP-conjugated secondary antibodies for one hour at room temperature, followed by washing three times. Then the membrane was incubated with enhanced chemiluminescence reagent and exposed to X-ray films, which were developed using an automatic film processor. β-actin was used as the loading control for all immunoblot experiments. The results were scanned and quantified using ImageJ and normalized to the density of actin in the corresponding samples.
Chromatin immunoprecipitation (ChIP)
ChIP was performed using a ChIP Assay Kit (Millipore, 17–295) according to the manufacturer’s instructions. Quantitative PCR was performed with primers: CD28 promoter proximal STAT sites: forward TCTGCTGGATTTCAAGCACCC, reverse GACTGCAGCATTTCACACAGG; distal STAT sites: forward TGCTTGCACGTAGAATGGGT, reverse GGATGGGGACAGGTTGTGTC; transcription start site: forward AACCCTAGCCCATCGTCAG, reverse AACACATTGCCCTATTACAGC. Rabbit IgG was used as a negative control.
Statistical Analysis
Graphical presentation and statistical analysis of the data were performed using GraphPad Prism (Version 7 and 8, GraphPad software, San Diego, CA). Data are displayed as mean ± SEM. Results between experimental groups were compared using ANOVA followed by multiple comparisons test or Student’s t test as described in the figure legends. p<0.05 was considered statistically significant.
Results
IL21 upregulated CD28 expression on activated human naïve CD8+ T cells
CD28 is a pivotal costimulatory molecule for naïve T cell activation and memory T cell function (22). Our previous studies comparing the effects of various γC cytokines on the generation of tumor-antigen specific CTLs in vitro find that IL21 has the unique ability to enrich for CD28hi CTLs that exhibit enhanced persistence and improve patient clinical responses after adoptive transfer (17,23). To investigate the molecular mechanisms of IL21-induced CD28 expression, melanoma antigen recognized by T cells (MART1 or M27)-specific CTLs were generated in the absence or presence of IL21 as previously described (17,20). Consistent with our previous findings (17), M27-specific CTLs (Supplemental Fig. 1A) generated with IL21 displayed significantly higher CD28 expression than cells generated in the absence of IL21 (Fig. 1, A and B). In addition, a greater proportion of these cells exhibited the central memory phenotype CD45RO+CCR7+ (Supplemental Fig. S1). IL21 priming also enhanced the expression of CD27, CD127, and CD62L (Supplemental Fig. S1), which are highly expressed on human naïve and stem cell-like/central memory CD8+ T cells (19). Our previous studies found that IL21 exerted its effects primarily on human naïve CD8+ T cells (5,17), thus to corroborate the above findings, sort-purified human naïve CD8+ T cells (CD45RA+CCR7+) from healthy donors were activated with anti-CD3/CD28 beads in the absence or presence of IL21. Consistent with antigen-specific CTLs (Fig. 1, A and B) (17), surface expression of CD28 (Fig. 1, C and D) as well as CD127, CD62L and CCR7 (Supplemental Fig. S1) was significantly increased in the IL21-treated cells. In line with enhanced CD28 protein expression, increased CD28 mRNA expression was consistently detected in M27-specific CTLs generated in the presence of IL21 (Fig. 1E) and in IL21-treated anti-CD3/CD28 activated human naïve CD8+ T cells (Fig. 1F). Together these results indicated that IL21 upregulated CD28 mRNA expression to increase CD28 surface expression.
Fig. 1. IL21 upregulated CD28 expression in activated human naïve CD8+ T cells.
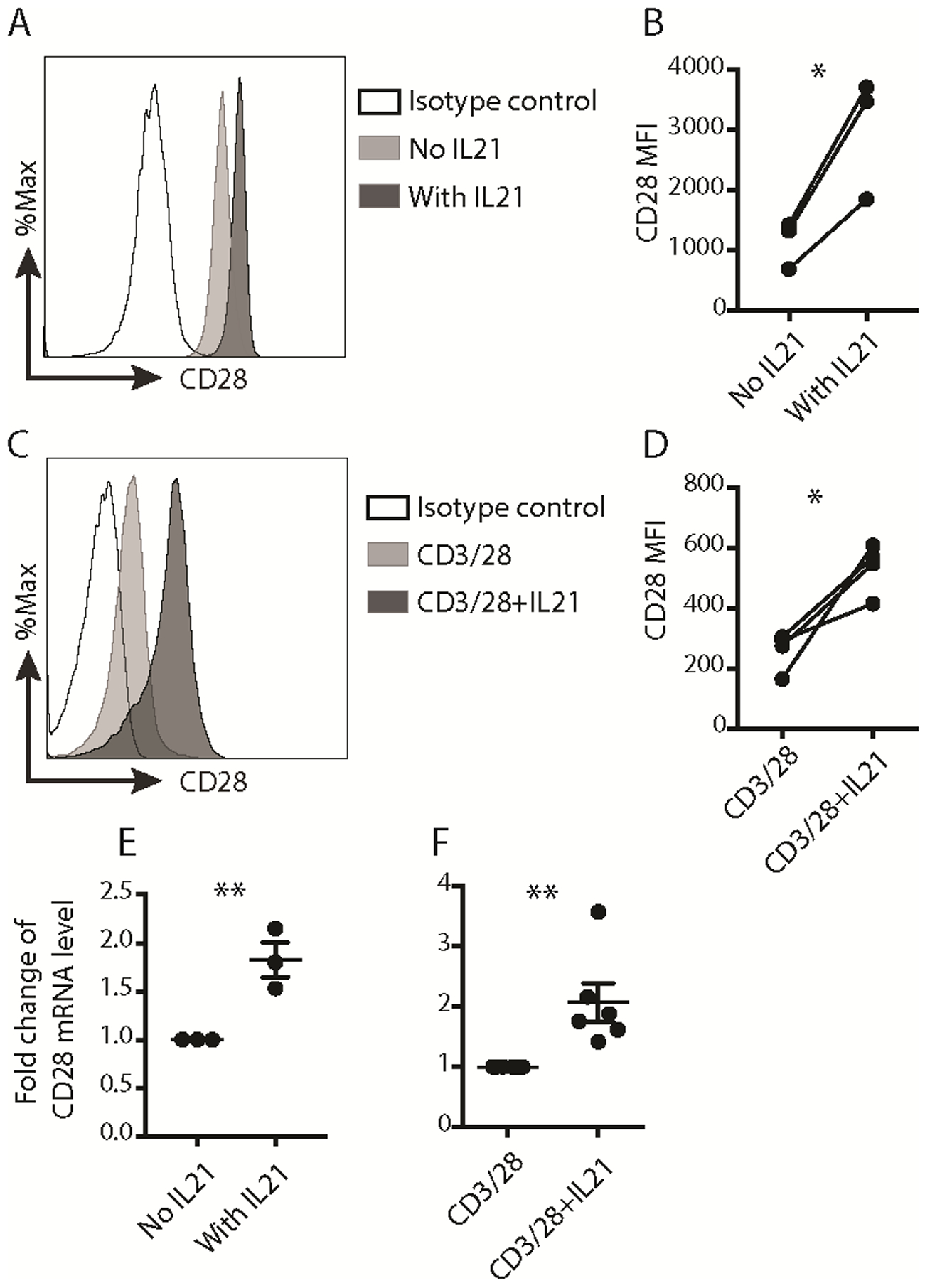
(A) Representative histogram of CD28 surface level on M27-specific CD8+ T cells and the isotype control antibody was used as a negative staining control. (B) MFI of CD28 protein expression on the surface of M27-specific CD8+ T cells. (n=3, * p < 0.05, paired t test). MFI: mean fluorescence intensity. (C) Representative histogram of CD28 surface expression on human naïve CD8+ T cells stained on day 7 after activation. The isotype control antibody was used as a negative staining control. (D) MFI of CD28 protein expression on the surface of CD8+ T cells activated with the indicated conditions for 7 days. (n=4, * p < 0.05, paired t test). (E) The quantitative RT-PCR results of CD28 mRNA in sort-purified M27-specific CD8+ T cells generated with or without IL21. The expression in cells expanded without IL21 was set as 1. (n=3, mean ± SEM, ** p < 0.01, unpaired t test). (F) The quantitative RT-PCR results of CD28 mRNA in human CD8+ T cells activated with the indicated conditions for 7 days. The expression in cells activated with anti-CD3/CD28 beads for 7 days was set as 1. (n=6, mean ± SEM, ** p < 0.01, unpaired t test). Results of quantitative RT-PCR for CD28 gene were normalized to RPL13A. The results in A and C were representative out of 3 (A) or 4 (C) independent experiments using cells from different healthy donors. The results in B, D, E and F were pooled from 3 (B, E), 4 (D) or 6 (F) independent experiments using cells from different healthy donors.
STAT3 activation was required for IL21-mediated enhancement of CD28 expression
IL21 functions through activation of Janus-activated kinase 1 (JAK1) and JAK3 and subsequent phosphorylation of signal transducer and activator of transcription (STAT)-3 and, to a lesser extent, STAT1 and STAT5 (24). Thus, we examined the phosphorylation of STAT1, STAT3 and STAT5 in naïve CD8+ T cells under our culture conditions. Human naïve CD8+ T cells from healthy donors were activated with anti-CD3/CD28 beads in the absence or presence of IL21. IL21 stimulation induced STAT1 and STAT3 phosphorylation but weak STAT5 phosphorylation 30 minutes after activation (Supplemental Fig. S2A). Since IL21 mainly induced STAT1 and STAT3 activation, we aimed to elucidate whether STAT1 and/or STAT3 activation was essential for CD28 upregulation by IL21. To examine the role of STAT3, we made use of peripheral blood mononuclear cells (PBMCs) from Job’s syndrome patients. Job’s syndrome (also known as hyper-IgE syndrome, characterized by abnormally high amounts of immunoglobulin E (IgE) in the blood) can be caused by diminished STAT3 functions due to dominant negative mutations in the STAT3 gene (25,26). Total CD8+ T cells were isolated from PBMCs of healthy donors or Job’s syndrome patients and activated as described above. IL21 increased the expression of CD28 at both the protein and mRNA levels in CD8+ T cells from healthy donors. However, IL21-mediated enhancement of CD28 expression was completely abrogated in cells from Job’s syndrome patients (Fig. 2, A–C). These results indicated that STAT3 activity was essential for the upregulation of CD28 expression by IL21 in activated human CD8+ T cells.
Fig. 2. STAT3 activation was essential for IL21-induced CD28 upregulation.
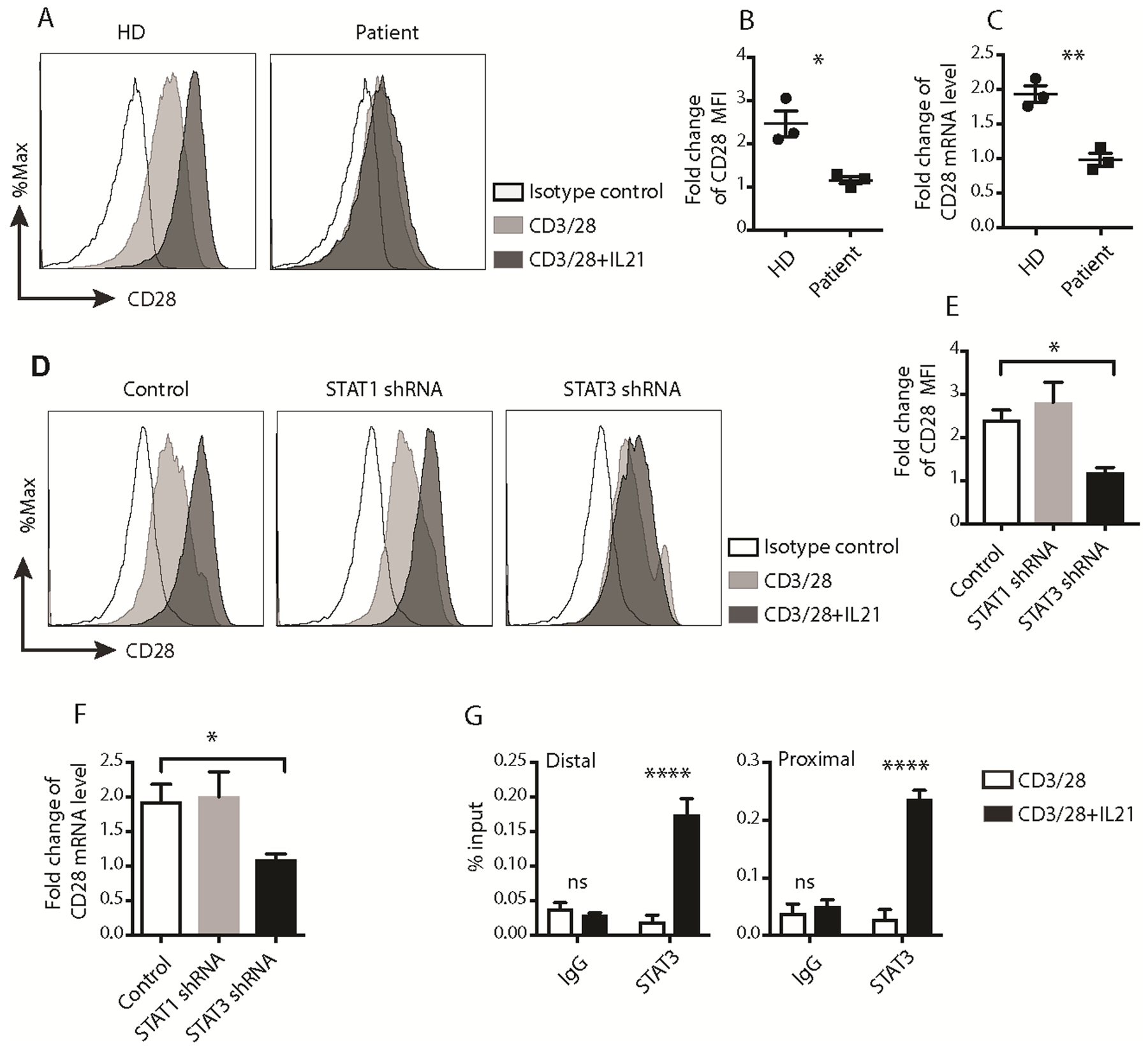
(A) Representative histograms of CD28 surface expression on activated human CD8+ T cells from healthy donors or Job’s syndrome patients. HD: healthy donor. (B) Fold change of CD28 MFI, which is presented as fold of MFI of cells activated with anti-CD3/CD28 and IL21 over MFI of cells activated with only anti-CD3/CD28. (n=3, mean ± SEM, * p < 0.05, unpaired t test). (C) Fold change of quantitative RT-PCR results of CD28 mRNA in human CD8+ T cells from healthy donors or Job’s syndrome patients activated with anti-CD3/CD28 or together with IL21 for 7 days. The expression in cells from healthy donor or Job’s syndrome patient activated with anti-CD3/28 beads alone for 7 days was set as 1. (n=3, mean ± SEM, ** p < 0.01, unpaired t test). (D) Representative histograms of CD28 surface expression on human CD8+ T cells transfected with control, STAT1 or STAT3 shRNAs and activated with the indicated conditions for 7 days. (E) Fold change of CD28 MFI on the surface of negative control (Control) or STAT-knockdown CD8+ T cells activated with the indicated conditions for 7 days. Data is presented as fold of MFI of cells activated with anti-CD3/CD28 and IL21 over MFI of cells activated with only anti-CD3/CD28. (n=6, mean ± SEM, * p < 0.05, one-way ANOVA). (F) Fold change of quantitative PCR results of CD28 mRNA in human CD8+ T cells transfected with control, STAT1 or STAT3 shRNAs and activated with anti-CD3/CD28 or together with IL21 for 7 days. The expression in cells activated with CD3/28 beads alone for 7 days was set as 1. (n=4, mean ± SEM, * p < 0.05, one-way ANOVA). (G) Representative ChIP results of STAT3 binding to the proximal and distal STAT sites on the human CD28 promoter. The results were normalized to the percentage of the input amount. (n=3, mean ± SEM, **** p < 0.0001, two-way ANOVA). The results were representative (A, D, G) or pooled from 3 (B, C), 4 (F), or 6 (E) independent experiments using cells from different donors.
To affirm our findings and also assess the role of STAT1 in IL21-induced CD28 upregulation, different shRNA constructs targeting various regions of the human STAT1 or STAT3 genes were used to knockdown STAT1 or STAT3 expression in untreated human CD8+ T cells. Total STAT1 and STAT3 expression showed that STAT1 or STAT3 shRNA specifically and efficiently decreased expression of their respective proteins (Supplemental Fig. S2B). As shown in Fig. 2 D–F, compared to control cells, IL21-induced CD28 protein and mRNA upregulation was diminished in STAT3 shRNA-transfected and activated CD8+ T cells, but not in STAT1 shRNA-transfected and activated cells. These results supported the critical role of STAT3, but not STAT1, in IL21-induced upregulation of CD28 expression in activated human CD8+ T cells. Consistent with our findings, a previous study of STAT3 mutant, STAT1 mutant, and IL21R mutant patient cells indicates that IL21/STAT3, but not STAT1, is required for differentiation of CD8+ central memory (CD45RA−CCR7+) and effector memory (CD45RA−CCR7−) cells in vivo (27).
To delineate the molecular mechanism by which STAT3 mediated IL21-induced upregulation of CD28 expression in activated human CD8+ T cells, we analyzed the human CD28 promoter and identified several consensus STAT sites clustered in the proximal and distal part of the CD28 promoter. ChIP assays showed significantly increased enrichment of STAT3 at both proximal and distal CD28 promoter regions in cells activated with anti-CD3/CD28 and IL21, relative to anti-CD3/CD28 treatment alone (Fig. 2G). Collectively, these results suggested that IL21-activated STAT3 binds to the human CD28 promoter to promote CD28 transcription.
IL21-induced CD28 correlated with histone H3 acetylation of CD8+ T cells
Our previous and current studies demonstrated that IL21 uniquely enhanced CD28 expression on naïve CD8+ T cells following activation (Fig. 1) (17). However, we did not observe the induction of CD28 by IL21 on MART1 (M27)-specific effector CD8+ T cells, generated via the Endogenous T Cell (ETC) approach and expanded in vitro by Rapid Expansion Protocol (REP) (1), activated with their cognate peptide-pulsed mature dendritic cells (Fig. 3, A and B). Since IL21 functions mainly through the phosphorylation of STAT3 (Fig. 2) (14), we compared IL21-induced STAT3 phosphorylation in naïve and effector CD8+ T-cells and found them to be comparable (Fig. 3C), suggesting the inability of IL21 to increase CD28 expression on effector CD8+ T cells was not due to absence of IL21 signaling, but to lack of access of pSTAT3 to its binding sites.
Fig. 3. IL21-induced CD28 correlated with histone H3 acetylation of CD8+ T cells.
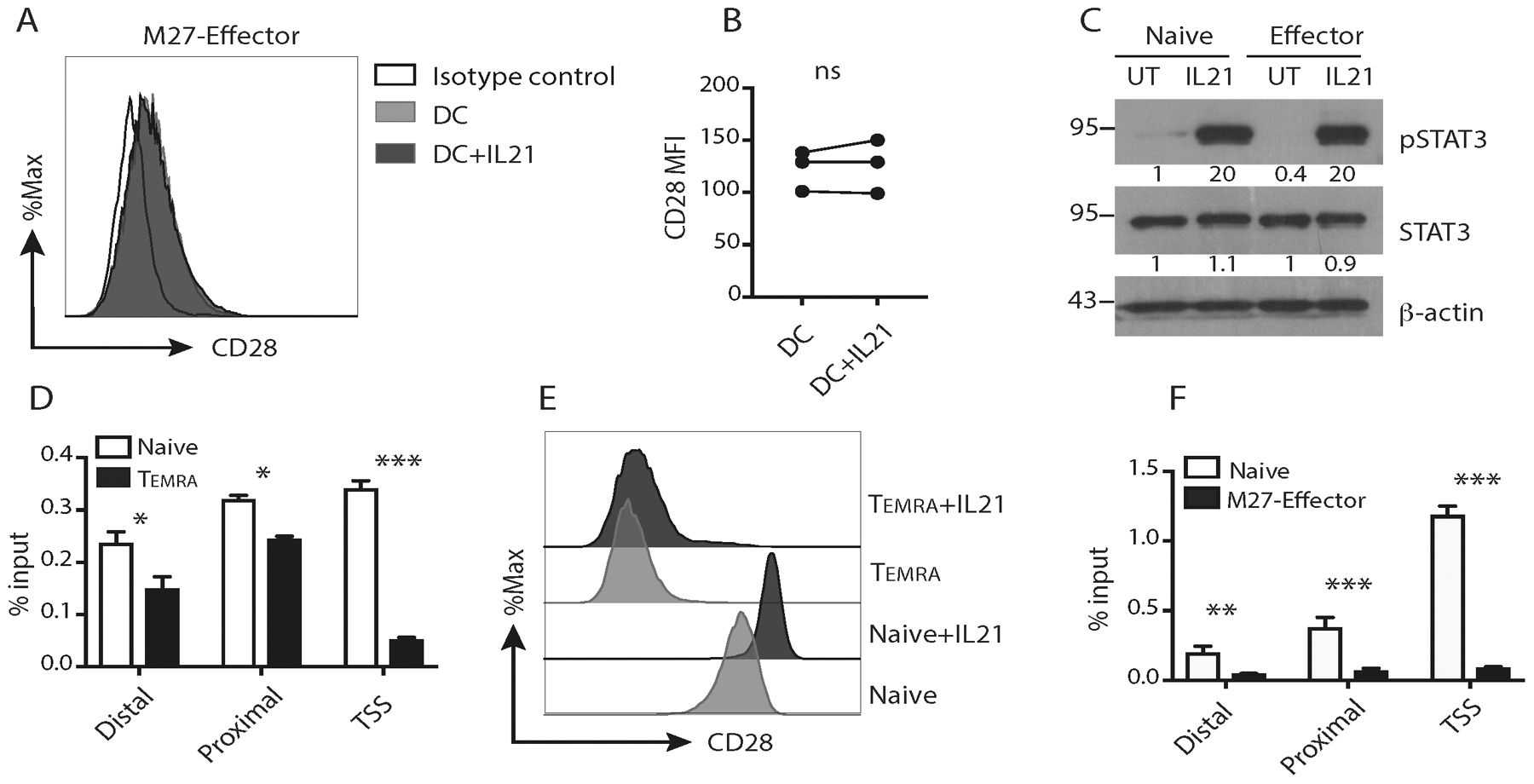
(A-B) Representative histogram and summary of CD28 expression on M27-specific effector CD8+ T cells activated with M27-pulsed mature dendritic cells in the presence or absence of IL21 for 4 days. Isotype antibody was used as a negative staining control. [n=3; ns: not significant; paired t test]. (C) Representative western blot results of IL21-induced pSTAT3 in naïve and M27-specific effector CTLs. STAT3 and β-actin were used as loading controls. The bands were quantified using ImageJ and normalized to the density of actin in the corresponding samples. Molecular weight is indicated in kilodaltons. UT: untreated. (D) Representative ChIP results of H3 acetylation on the CD28 promoter comparing Naïve to CD45RA+EM (TEMRA) CD8+ T cells. The results were normalized to the percentage of the input amount. TSS: transcription start site. [n=3; mean ± SEM; * p<0.05, *** p<0.001; two-way ANOVA]. (E) Representative histogram of CD28 expression on naïve and TEMRA CD8+ T cells activated with anti-CD3/CD28 or together with IL21 for 4 days. (F) Representative ChIP results of H3 acetylation on the CD28 promoter comparing Naïve to M27-specific effector CD8+ T cells. The results were normalized to the percentage of the input amount. [n=3; mean ± SEM; ** p<0.01, *** p<0.001; two-way ANOVA]. The results were representative out of two (C, E, F) or three (A, D), or pooled from 3 (B) independent experiments using cells from different donors.
Chromatin accessibility and gene expression can be regulated by histone acetylation. To determine whether histone acetylation correlated with CD28 expression, chromatin immunoprecipitation (ChIP) was performed on naïve (CD45RA+CCR7+) and TEMRA effector memory (CD45RA+CCR7−) CD8+ T cells, which have high and low CD28 expression, respectively (19). In line with their high CD28 expression, naïve CD8+ T cells showed increased acetylated histone H3 (AcH3) around the distal and proximal STAT3 binding sites on the promoter and around the transcription start site (TSS) of the CD28 gene, compared to TEMRA CD8+ T cells (Fig. 3D). In line with our previous results, IL21 treatment minimally upregulated CD28 expression in activated TEMRA CD8+ T cells (Fig. 3E). Similarly, MART1 (M27)-specific CD28neg effector CD8+ T cells displayed significantly decreased AcH3 on the CD28 locus, compared to naïve CD8+ T cells (Fig. 3F). These results indicated that CD28 transcription was regulated by histone acetylation, which correlated with the differential induction of CD28 by IL21 in naïve and effector CD8+ T cells. Therefore, modulation of AcH3 could allow IL21-mediated CD28 upregulation in effector CD8+ T cells.
SAHA allowed IL21 to upregulate CD28 expression in effector CD8+ T cells
Our above findings indicated that CD28 transcription was regulated by histone acetylation, which suggested that the reduction of histone acetylation may have led to CD8+ T cell differentiation and loss of naïve/central memory marker expression. We hypothesized that increasing histone acetylation through the use of an HDACi would reverse CD8+ T cell differentiation. Since IL21 significantly enhances CD28 expression on naïve CD8+ T cells (Fig. 1) (17), which have higher histone acetylation (Fig. 3D), we reasoned that the combination of HDACi and IL21 would have a synergistic effect on CD28 expression. To test our hypothesis, we first chose to assess the effect of a clinically available compound, Suberoylanilide Hydroxamic Acid (SAHA, Vorinostat), a broad histone deacetylase inhibitor (HDACi), that is approved to treat cutaneous T-cell lymphoma and has been used in clinical trials to treat other diseases (28). Titration studies to determine the effective dose showed that SAHA at concentrations of 1μM or greater could augment AcH3 levels in effector CD8+ T cells (Supplemental Fig. S2C).
To assess the effect of SAHA/IL21 on CD28 expression in the context of a more physiologic, antigen-specific (in contrast to non-specific polyclonal) stimulation, we evaluated this effect on MART1 (M27)-specific effector/effector memory cells generated using peptide-pulsed autologous dendritic cells (1). MART1 (M27)-specific effector CD8+ T cells (CD45RO+, CD28-neg, CD62L-neg) were generated following iterative cycles of in vitro stimulation, tetramer-guided sorting of M27-specific CTL and expansion to uniformity (> 95% MART-1-speicifc effector CTL). First, we evaluated the effect of SAHA on pSTAT3 binding to the CD28 promoter region. As anticipated, SAHA treatment significantly increased AcH3 expression on the promoter and TSS region of CD28 gene (Fig. 4A). In correlation with increased AcH3 expression, SAHA treatment increased IL21-induced pSTAT3 binding to the CD28 promoter (Fig. 4B). Next, M27-specific effector CD8+ T cells were left untreated or pretreated with SAHA for 24 hours, followed by activation with M27-pulsed mature dendritic cells in the presence or absence of SAHA/IL21 for 4 days. SAHA and IL21 together significantly enhanced CD28 expression (Fig. 4, C and D), demonstrating the cooperative effect of SAHA and IL21 on CD28 expression. These results demonstrated that SAHA treatment increased AcH3 expression and chromatin accessibility at the CD28 region in M27-specific effector CD8+ T cells, thus allowing IL21-activated STAT3 to bind to its promoter sites and induce CD28 expression.
Fig. 4. SAHA allowed IL21 to upregulate CD28 expression in effector CD8+ T cells.
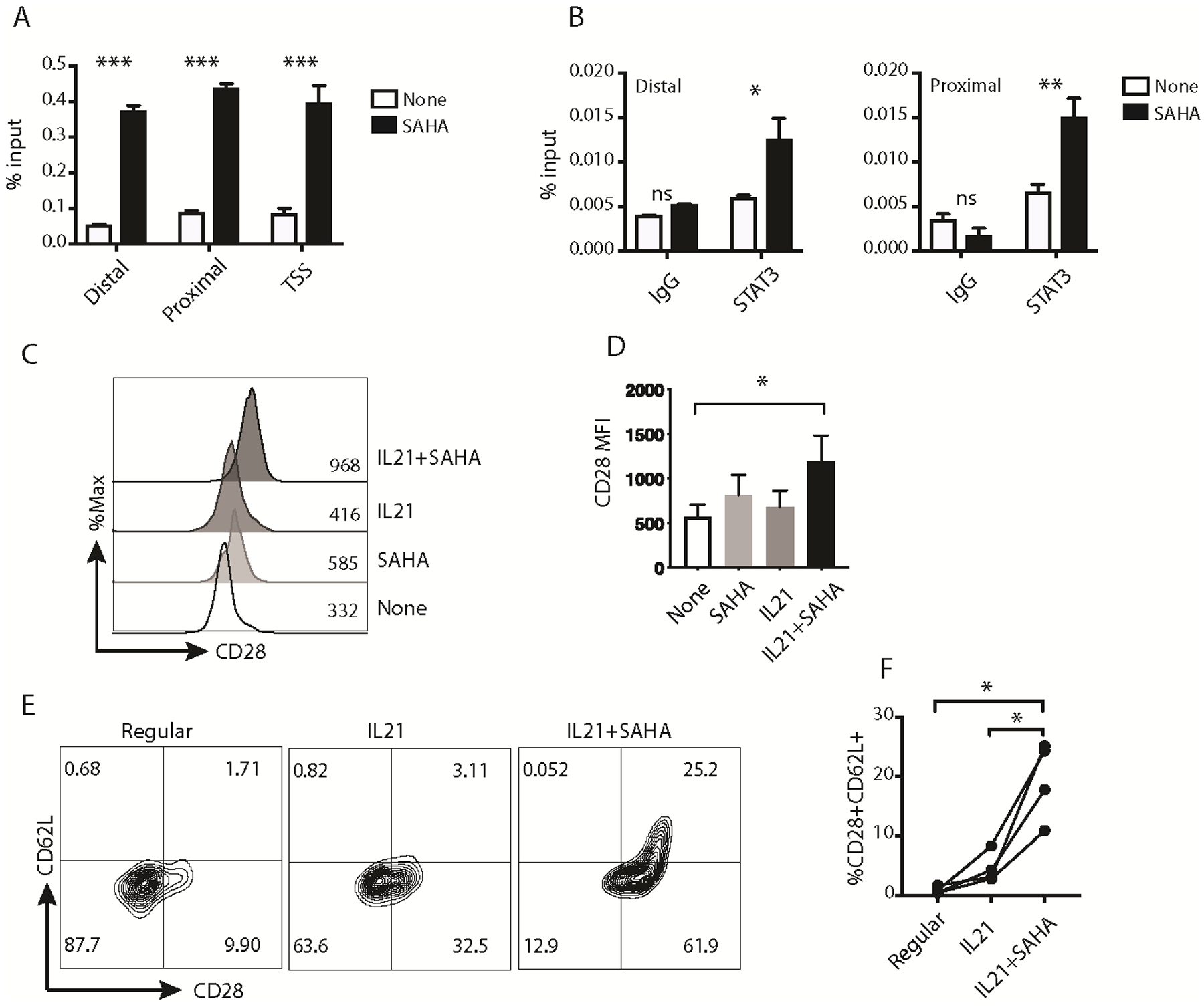
(A) Representative ChIP results of H3 acetylation on the CD28 promoter for M27-specific effector CD8+ T cells left untreated (None) or treated with SAHA for 24 hours. [n=3; mean ± SEM; *** p<0.001; two-way ANOVA]. (B) Representative ChIP results of STAT3 binding to the CD28 promoter for M27-specific effector CD8+ T cells left untreated or treated with SAHA for 24 hours, followed by IL21 stimulation for 30 minutes. [n=3; mean ± SEM; ns: not significant, * p<0.05, ** p<0.01; two-way ANOVA]. (C) Representative histogram of CD28 expression on activated CTLs treated with the indicated conditions for 4 days. The numbers inside the histogram graph show the representative CD28 MFI for each condition. (D) MFI of CD28 on CTLs from independent experiments (n=6; mean ± SEM; * p<0.05; one-way ANOVA, comparing IL21+SAHA to the other conditions). (E) Representative plots of CD28 and CD62L expression on TILs expanded with the indicated conditions for 2 weeks. The numbers within the plots annotate the percentage of cells in each quadrant. (F) Percentage of CD28+CD62L+ cells in TILs expanded with the indicated conditions from independent experiments (n=4; * p<0.05; one-way ANOVA). The representative results out of two (B), three (A), four (E), or six (C) independent experiments are shown.
IL21 and SAHA synergized to upregulate CD28 and CD62L expression
To assess the effect of SAHA/IL21 in the translational setting, we evaluated this program on tumor-infiltrating lymphocytes (TIL). Adoptive transfer of TIL cells for the treatment of patients with metastatic melanoma, and other TIL+ solid tumors, involves extraction of infiltrating lymphocytes from tumor biopsies, in vitro treatment with high dose IL2, in vitro expansion with a Rapid Expansion Protocol (REP) and then infusion of ex vivo expanded TILs following high dose lymphodepletion conditioning (2). Although TIL therapy has shown some success in the treatment of metastatic melanoma patients, many patients do not respond to TIL therapy, partly due to limited persistence of the infused cells (2). CD8+ T cells in TIL products are usually well-differentiated effector, effector memory, and terminal effector cells with reduced proliferative ability (7). To examine the possible de-differentiating effect of an HDAC inhibitor/IL21 combination, TILs were untreated or pretreated with SAHA for 24 hours, then subjected to regular REP (irradiated PBMC and LCL cells, anti-CD3 and IL2), or REP with IL21/SAHA. Compared to regular REP or REP with IL21 alone, SAHA and IL21 given in combination during REP increased CD28 and CD62L expression (Fig. 4, E and F), two markers highly expressed on naïve and central memory T cells (19). At the end of REP, CCR7 and CD127 surface expression was not detected on the TILs and the expression of exhaustion markers (PD-1 and Tim3) was comparable between regular REP and REP with IL21/SAHA (Supplemental Fig. S3). These results suggested that, similar to the results in M27-effector CD8+ T cells, SAHA treatment increased AcH3 expression and chromatin accessibility at the CD28 region in TILs, thus allowing IL21-activated STAT3 to bind to its promoter sites and induce CD28 expression and phenotypic evidence of de-differentiation of effector CD8+ T cells.
IL21 and Panobinostat (Pano) cooperated to induce central-memory-like T cells
Since the cytotoxicity of SAHA limited its application in ACT, we screened other pharmacologically available HDACi (Supplemental Fig. S4A) and found that Panobinostat (LBH589, Pano) had an effect similar to that of SAHA but with minimal cytotoxicity. Panobinostat increased AcH3 expression at 0.5 nM or higher doses (Supplemental Fig. S4B). We initially investigated the effect of Panobinostat on TILs in REP at a small scale and compared pretreatment (pretreating cells with Panobinostat for 24 hours followed by rapid expansion with Pano and IL21) with co-treatment (adding Panobinostat and IL21 when starting cell expansion). Since these two strategies had comparable effects in inducing a CD28+CD62L+ cell population for TILs (Supplemental Fig. S5A and S5B), the co-treatment scheme was followed in subsequent studies for simplicity.
To examine the clinical applicability of Panobinostat (Pano) in the setting of antigen-specific ACT we used MART1 (M27)-specific effector/effector memory cells generated as previously described (1). Cells were expanded using four different protocols (regular, adding IL21 alone, adding Pano alone, or IL21 + Pano). Although adding Panobinostat alone to REP slightly reduced the overall yield, fold-expansion was similar for the other three conditions (Supplemental Fig. S5C). IL21/Pano treatment resulted in increased expression of CCR7, CD27, CD127, CD28 and CD62L at day 7 of REP (Supplemental Fig. S6). Although the cells downregulated the expression of CCR7 and CD127 at the end of REP (Supplemental Fig. S6), addition of Panobinostat consistently induced a CD28+CD62L+ cell population that was further enhanced when combined with the addition of IL21 (Fig. 5, A and B). We posit that Panobinostat, like SAHA, enabled STAT3 and other transcription factors/cofactors to access binding sites and induce CD28 and CD62L expression.
Fig. 5. IL21 and Panobinostat (Pano) cooperated to induce CD28+CD62L+ cells.
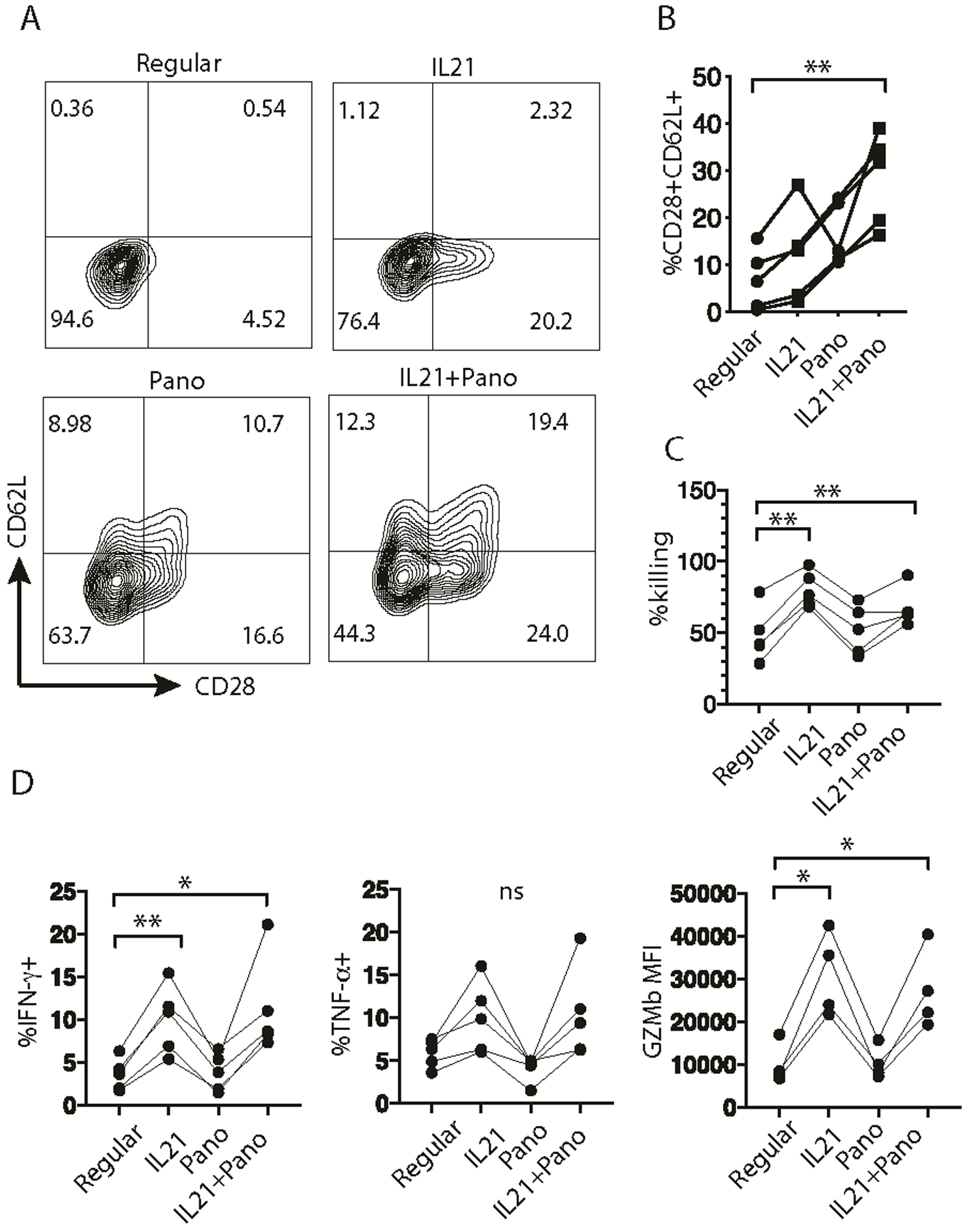
(A) Representative plots of CD28 and CD62L expression on CTLs expanded with the indicated conditions for 2 weeks. The numbers within the plots annotate the percentage of cells in each quadrant. (B) Percentage of CD28+CD62L+ cells in CTLs expanded with the indicated conditions from independent experiments (n=5; ** p<0.01; one-way ANOVA, as compared to CTLs expanded with the regular protocol). (C) Percentage of target tumor cell killing by CTLs expanded with the indicated condition from independent experiments (n=5; ** p<0.01; one-way ANOVA). (D) Percentage of IFN-γ+ or TNF-α+ cells and GZMb MFI of CTLs expanded with the indicated condition from independent experiments GZMb: granzyme B. (n=5, * p<0.05, ** p<0.01; ns: not significant, one-way ANOVA). The representative results out of five (A) independent experiments are shown.
To assess the clinical applicability of this strategy, we tested tumor killing ability of the expanded cells ex vivo. The cells expanded with IL21 in the presence or absence of Panobinostat displayed dramatically increased tumor killing capability (Fig. 5C), which correlated with significantly enhanced IFNγ and Granzyme B production (Fig. 5D). The production of TNFα, IL2, and perforin was not significantly changed (Fig. 5D and Supplemental Fig. S7).
T cell metabolic states can influence their effector functions (29,30). While memory CD8+ T cells rely on fatty acid oxidation for energy, naïve CD8+ T cells mainly utilize glucose oxidative phosphorylation (OXPHOS) and dramatically increase glucose uptake and glycolysis shortly after activation, which is essential for the effector function of CD8+ T cells (29,31). Inhibition of AKT promotes generation of T cells with memory phenotype, which exhibit enhanced fatty-acid oxidation (32) or reduced glycolysis (33). Thus, to test whether IL21/Pano scheme affects the metabolism of the expanded CTLs, the metabolic states of the cells expanded with regular protocol or together with IL21/Pano were evaluated by using an extracellular flux analyzer. The cells expanded with these two schemes showed similar basal and maximal OCR (oxygen consumption rate to measure mitochondrial oxidation rate) and ECAR (extracellular acidification rate to measure glycolysis rate) (Supplemental Fig. S8A). In addition, mitochondrial mass was comparable between these two groups (Supplemental Fig. S8B), suggesting that IL21/Pano treatment in REP did not change the glycolysis and mitochondrial oxidation of the expanded cells.
Central memory function associated with the IL21/Pano-induced CD28+CD62L+ population was evaluated by the ability of these central memory-like T cells to undergo homeostatic proliferation in response to IL7 and IL15 (14). ETC cells expanded with four different protocols (regular, adding IL21 alone, adding Pano alone, or IL21 + Pano) were labeled with CFSE and cultured with IL2, IL7 or IL15 for 5 days. IL7 did not induce cell division, likely due to low expression of CD127 expression (Supplemental Fig. 6). Cells expanded in the presence of IL21 exhibited enhanced IL2- and IL15-induced proliferation (Fig. 6A). Adding Panobinostat alone to REP increased cell proliferation in response to IL15 but not to IL2. The cells expanded with the combination of IL21 and Panobinostat exhibited greater proliferative responses to IL2 and IL15 than any other cohort (Fig. 6A). Since IL2 and IL15 share CD132 (γC) and CD122 receptor subunits (14), we assessed CD132 and CD122 expression on the surface of these cells. Treatment with IL21 +/− Panobinostat led to significantly increased surface CD132 expression (Fig. 6, B and C), which could have contributed to their increased self-renewal to IL2 and IL15.
Fig. 6. IL21/Panobinostat-expanded CTLs display central memory-like characteristics in vitro.
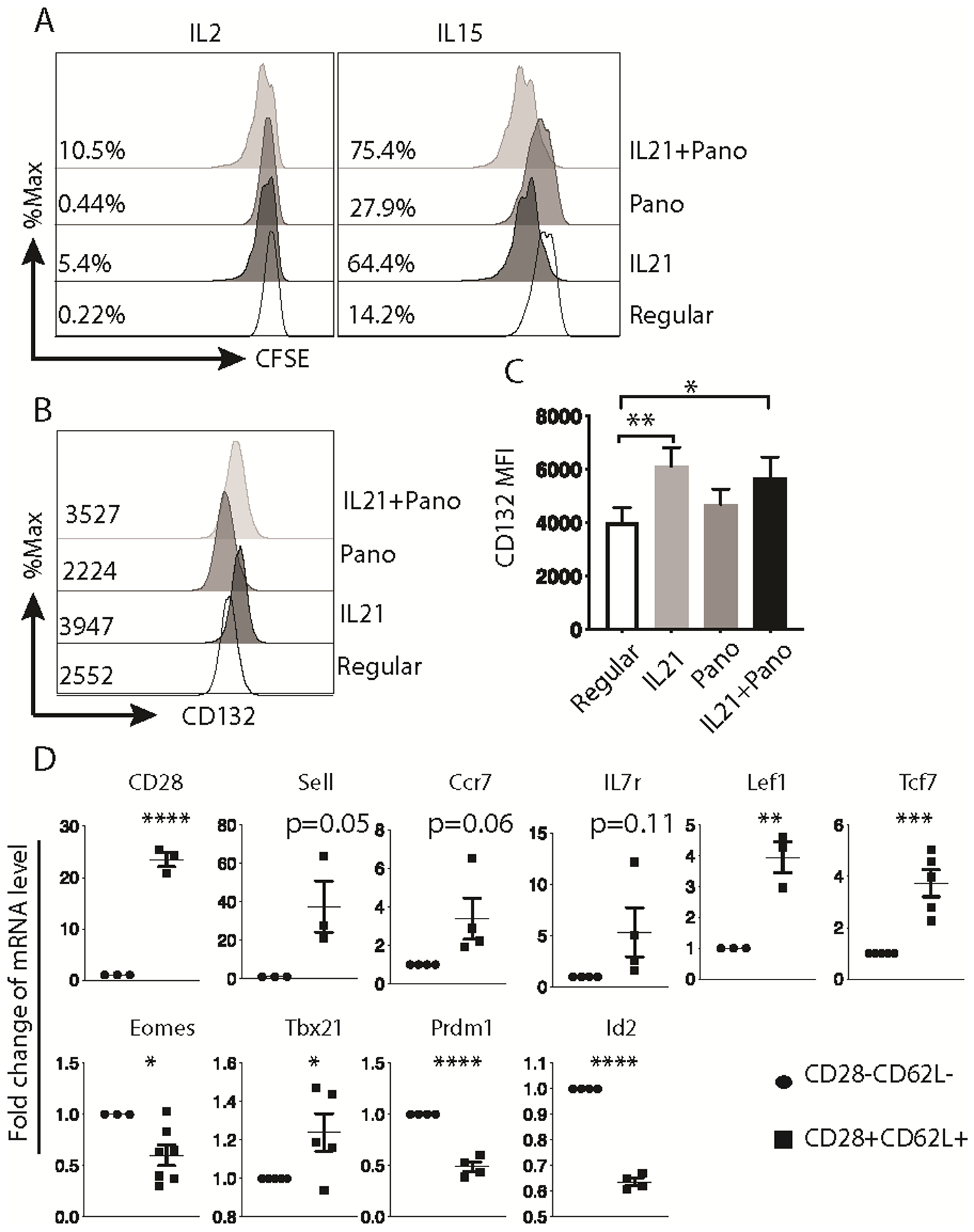
(A) Proliferation of expanded CTLs when treated with either IL2 or IL15 indicated by CFSE dilution. The numbers indicate the percentage of cells divided 2 times or more in 5 days. (B) Representative histogram of CD132 (γC) expression on CTLs expanded with the indicated conditions. The numbers show the representative MFI of CD132 in each condition. (C) Summary of CD132 MFI on CTLs expanded with the indicated conditions from independent experiments (n=6; mean ± SEM; * p<0.05, ** p<0.01; one-way ANOVA, as compared to CTLs expanded with the regular protocol). (D) The results of mRNA gene expression in CD28−CD62L− and CD28+CD62L+ cells sorted from CTLs expanded with IL21 and Panobinostat. Gene expression was normalized to housekeeping gene RPL13A expression. The expression in CD28−CD62L− cells was set as 1. (n=3–7; mean ± SEM; * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001, two-tailed t test). The representative results out of two (A) or six (B) independent experiments are shown. The results in C and D were pooled from more than 3 independent experiments.
To further confirm the central-memory-like properties of HDACi/IL21 treated CTL, expression of relevant differentiation genes was assessed. The central memory-associated transcriptional signature (Lef1hi, Tcf7hi), known to play a key role in central memory/stem cell memory CD8+ T cell differentiation (34–36), was found to be highly expressed among CD28+CD62L+ cells generated by the combination of Panobinostat and IL21 treatment (Fig. 6D). In addition, naïve/memory associated genes Ccr7 and Il7r showed the trend of upregulation in CD28+CD62L+ cells. The transcription factors T-bet, eomesodermin (Eomes), B lymphocyte-induced maturation protein-1 (Blimp1), and inhibitor of DNA binding 2 (ID2) have essential roles in effector and memory T cell formation, and their expression is increased in differentiated CD8+ T cells (37,38). Tbx21 expression was increased and Eomes, Prdm1, Id2 expression was decreased in CD28+CD62L+ cells (Fig. 6D). Altogether, these gene signatures supported the central-memory-like features of IL21/Pano-reprogrammed CD28+CD62L+ cells.
Discussion
In this study, we demonstrated that the combination of IL21 and HDACi may be used to re-program effector cells to become less differentiated, central memory-like T cells with high replicative capacity. T cell differentiation is regulated by epigenetic mechanisms (12), thus in vitro manipulation of epigenetic modifications has the potential to alter CD8+ T-cell phenotype and function. Our previous and current studies demonstrated that CD28 was upregulated by IL21 activated STAT3 in naïve CD8+ T cells but not in effector/effector memory cells (17). We attribute this partly to decreased acetylated histone H3 (AcH3) on the promoter and around the transcription start site of CD28 gene in effector CD8+ T cells, which reduced chromatin accessibility and obviated IL21 induced pSTAT3 to bind to its sites and induce CD28 expression. We demonstrated in human T cells that by increasing histone acetylation, the HDACi SAHA treatment allows IL21-activated STAT3 to drive CD28 expression in effector CD8+ T cells and leads to epigenetic reprogramming of effector T cells into memory T cells. STAT3 is critical in the development of human central memory T cells (39) and the IL21/IL-10/STAT3 signaling cascade promotes expression of transcription factors critical during the effector to memory transition of activated murine CD8+ T cells (40).
Genome-wide studies reveal unique gene expression profiles among different CD8+ T cell subsets (11,37). Specifically, Lef1 and Tcf7, essential in central memory/stem cell memory CD8+ T cell differentiation, are highly expressed in naïve and stem/central memory CD8+ T cells but their expression decreases in differentiated effector/effector memory cells (34–37). CD28+CD62L+ T cells from IL21+Pano-expanded ETCs expressed dramatically higher Lef1 and Tcf7 mRNA than CD28−CD62L− cells, which suggested that CD28+CD62L+ T cells were central-memory-like cells and that IL21+Pano treatment de-differentiated effector CD8+ T cells through upregulation of T cell factor family (Tcf1 and Lef1). We do not exclude other target gene expression changes, and whole transcriptomic analysis is warranted to further elucidate the molecular mechanism of IL21+Pano-mediated de-differentiation of effector CD8+ T cells.
Central memory T cells exhibit superior in vivo self-renewal that occurs through homeostatic proliferation (16). IL2 is routinely administered to patients treated with ACT to encourage in vivo proliferation of the infused cells, and IL15 promotes in vivo homeostatic proliferation of memory T cells. IL21+Pano-expanded CD8+ T cells had enhanced proliferation in response to IL2 and IL15 in vitro. Addition of Panobinostat did not significantly affect tumor-killing ability of ETCs expanded with IL21 (Fig. 5C). These results suggested that IL21+Panobinostat-expanded CD8+ T cells would proliferate, persist and exert their tumor-killing function well in vivo after transfusion to patients, leading to enhanced patient response. Indeed, in a mouse model of chronic viral infection (LCMV) CD8+ T-cells treated with the HDACi valproic acid in vitro survive and maintain increased effector function in vivo after adoptive transfer, resulting in better protection against challenge with L. monocytogenes expressing the viral epitope GP33–41 (15).
Overall, we have demonstrated the potential application of IL21+Pano approach to two clinically-relevant ACT modalities: ETC and TIL (1). The evidence for persistence of these de-differentiated cells in vivo would need ACT clinical trials to track the infused cells. Nevertheless, our study has demonstrated a translatable approach to generate less-differentiated ACT product that would lead to improved clinical outcome.
Supplementary Material
Acknowledgements
We acknowledge the help from South Campus Flow Cytometry & Cell Sorting Core of UT MD Anderson Cancer Center, which is supported by NCI P30CA016672.
Financial support: This work was supported by the Parker Institute for Cancer Immunotherapy (C.Y.) and by a Stand Up To Cancer-Cancer Research Institute Cancer Immunology Dream Team Translational Cancer Research grant SU2C-AACR-DT1012 (C.Y.). Stand Up To Cancer is a division of the Entertainment Industry Foundation. Grants are administered by the American Association for Cancer Research. H.S.L. and S.S.W. were supported by NIH NIAID RO1AI109294 (S.S.W.).
Footnotes
Conflict of interest: C.Y. is a consultant/advisory board member for Adaptive Biotechnologies and Immatics. A patent application based on this manuscript has been filed (J.W. and C.Y.). No potential conflicts of interest were disclosed by the other authors.
References
- 1.Yee C The use of endogenous T cells for adoptive transfer. Immunological reviews 2014;257(1):250–63 doi 10.1111/imr.12134. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nature reviews Cancer 2008;8(4):299–308 doi 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. Journal of immunology 2004;173(12):7125–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou J, Shen X, Huang J, Hodes RJ, Rosenberg SA, Robbins PF. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. Journal of immunology 2005;175(10):7046–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chapuis AG, Desmarais C, Emerson R, Schmitt TM, Shibuya K, Lai I, et al. Tracking the Fate and Origin of Clinically Relevant Adoptively Transferred CD8(+) T Cells In Vivo. Science immunology 2017;2(8) doi 10.1126/sciimmunol.aal2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2008;26(32):5233–9 doi 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu R, Forget MA, Chacon J, Bernatchez C, Haymaker C, Chen JQ, et al. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: current status and future outlook. Cancer J 2012;18(2):160–75 doi 10.1097/PPO.0b013e31824d4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang S, Ji Y, Gattinoni L, Zhang L, Yu Z, Restifo NP, et al. Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer immunology, immunotherapy : CII 2013;62(4):727–36 doi 10.1007/s00262-012-1378-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Waart AB, van de Weem NM, Maas F, Kramer CS, Kester MG, Falkenburg JH, et al. Inhibition of Akt signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood 2014;124(23):3490–500 doi 10.1182/blood-2014-05-578583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proceedings of the National Academy of Sciences of the United States of America 2005;102(27):9571–6 doi 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moskowitz DM, Zhang DW, Hu B, Le Saux S, Yanes RE, Ye Z, et al. Epigenomics of human CD8 T cell differentiation and aging. Science immunology 2017;2(8) doi 10.1126/sciimmunol.aag0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez RM, Suarez-Alvarez B, Lavin JL, Mosen-Ansorena D, Baragano Raneros A, Marquez-Kisinousky L, et al. Epigenetic Networks Regulate the Transcriptional Program in Memory and Terminally Differentiated CD8+ T Cells. Journal of immunology 2017;198(2):937–49 doi 10.4049/jimmunol.1601102. [DOI] [PubMed] [Google Scholar]
- 13.Chandele A, Joshi NS, Zhu J, Paul WE, Leonard WJ, Kaech SM. Formation of IL-7Ralphahigh and IL-7Ralphalow CD8 T cells during infection is regulated by the opposing functions of GABPalpha and Gfi-1. Journal of immunology 2008;180(8):5309–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qu K, Zaba LC, Satpathy AT, Giresi PG, Li R, Jin Y, et al. Chromatin Accessibility Landscape of Cutaneous T Cell Lymphoma and Dynamic Response to HDAC Inhibitors. Cancer cell 2017;32(1):27–41 e4 doi 10.1016/j.ccell.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang F, Zhou X, DiSpirito JR, Wang C, Wang Y, Shen H. Epigenetic manipulation restores functions of defective CD8(+) T cells from chronic viral infection. Molecular therapy : the journal of the American Society of Gene Therapy 2014;22(9):1698–706 doi 10.1038/mt.2014.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nature reviews Immunology 2009;9(7):480–90 doi 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Bleakley M, Yee C. IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. Journal of immunology 2005;175(4):2261–9. [DOI] [PubMed] [Google Scholar]
- 18.Chapuis AG, Thompson JA, Margolin KA, Rodmyre R, Lai IP, Dowdy K, et al. Transferred melanoma-specific CD8+ T cells persist, mediate tumor regression, and acquire central memory phenotype. Proceedings of the National Academy of Sciences of the United States of America 2012;109(12):4592–7 doi 10.1073/pnas.1113748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who’s who of T-cell differentiation: human memory T-cell subsets. European journal of immunology 2013;43(11):2797–809 doi 10.1002/eji.201343751. [DOI] [PubMed] [Google Scholar]
- 20.Park J, Talukder AH, Lim SA, Kim K, Pan K, Melendez B, et al. SLC45A2: A Melanoma Antigen with High Tumor Selectivity and Reduced Potential for Autoimmune Toxicity. Cancer immunology research 2017;5(8):618–29 doi 10.1158/2326-6066.CIR-17-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ritthipichai K, Haymaker CL, Martinez M, Aschenbrenner A, Yi X, Zhang M, et al. Multifaceted Role of BTLA in the Control of CD8(+) T-cell Fate after Antigen Encounter. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23(20):6151–64 doi 10.1158/1078-0432.CCR-16-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nature immunology 2005;6(4):331–7 doi 10.1038/ni1179. [DOI] [PubMed] [Google Scholar]
- 23.Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Science translational medicine 2013;5(174):174ra27 doi 10.1126/scitranslmed.3004916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spolski R, Leonard WJ. Interleukin-21: a double-edged sword with therapeutic potential. Nature reviews Drug discovery 2014;13(5):379–95 doi 10.1038/nrd4296. [DOI] [PubMed] [Google Scholar]
- 25.Renner ED, Torgerson TR, Rylaarsdam S, Anover-Sombke S, Golob K, LaFlam T, et al. STAT3 mutation in the original patient with Job’s syndrome. The New England journal of medicine 2007;357(16):1667–8 doi 10.1056/NEJMc076367. [DOI] [PubMed] [Google Scholar]
- 26.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 2007;448(7157):1058–62 doi 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 27.Ives ML, Ma CS, Palendira U, Chan A, Bustamante J, Boisson-Dupuis S, et al. Signal transducer and activator of transcription 3 (STAT3) mutations underlying autosomal dominant hyper-IgE syndrome impair human CD8(+) T-cell memory formation and function. J Allergy Clin Immunol 2013;132(2):400–11 e9 doi 10.1016/j.jaci.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. American journal of translational research 2011;3(2):166–79. [PMC free article] [PubMed] [Google Scholar]
- 29.Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. The Journal of experimental medicine 2015;212(9):1345–60 doi 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ganeshan K, Chawla A. Metabolic regulation of immune responses. Annual review of immunology 2014;32:609–34 doi 10.1146/annurev-immunol-032713-120236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013;153(6):1239–51 doi 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer research 2015;75(2):296–305 doi 10.1158/0008-5472.CAN-14-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klebanoff CA, Crompton JG, Leonardi AJ, Yamamoto TN, Chandran SS, Eil RL, et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight 2017;2(23) doi 10.1172/jci.insight.95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeannet G, Boudousquie C, Gardiol N, Kang J, Huelsken J, Held W. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proceedings of the National Academy of Sciences of the United States of America 2010;107(21):9777–82 doi 10.1073/pnas.0914127107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nature medicine 2009;15(7):808–13 doi 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willinger T, Freeman T, Herbert M, Hasegawa H, McMichael AJ, Callan MF. Human naive CD8 T cells down-regulate expression of the WNT pathway transcription factors lymphoid enhancer binding factor 1 and transcription factor 7 (T cell factor-1) following antigen encounter in vitro and in vivo. Journal of immunology 2006;176(3):1439–46. [DOI] [PubMed] [Google Scholar]
- 37.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nature medicine 2011;17(10):1290–7 doi 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monaco G, Lee B, Xu W, Mustafah S, Hwang YY, Carre C, et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep 2019;26(6):1627–40 e7 doi 10.1016/j.celrep.2019.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siegel AM, Heimall J, Freeman AF, Hsu AP, Brittain E, Brenchley JM, et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity 2011;35(5):806–18 doi 10.1016/j.immuni.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity 2011;35(5):792–805 doi 10.1016/j.immuni.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.