

Abstract
The utilization of chiral transient directing groups (TDGs) has recently emerged as a promising approach for developing Pd(II)-catalyzed enantioselective C(sp3)−H activation reactions. However, this strategy is particularly challenging because the stereogenic center present on the TDG is often far from the C–H bond. Additionally, the TDG covalently attached to the substrate and the free TDG are both capable of coordinating to Pd(II) centers, which can result in a mixture of reactive complexes that may lead to opposite asymmetric induction. To date, the single example of TDG-enabled enantioselective C(sp3)−H activation is limited to the functionalization of benzylic C–H bonds. Herein we report the first example of a Pd(II)-catalyzed enantioselective β-C(sp3)−H arylation reaction of aliphatic ketones using a chiral transient directing group. A chiral trisubstituted cyclobutane is efficiently synthesized from a mono-substituted cyclobutane via sequential C–H arylation reactions, demonstrating the ability of this method to access structurally complex products from simple starting materials. The use of an electron-deficient pyridone ligand is also crucial for the observed high enantioselectivity. Interestingly, employing different silver salts can reverse the enantioselectivity in the C(sp3)−H arylation reaction. These key mechanistic findings will provide insight for future development of Pd(II)-catalyzed enantioselective C(sp3)−H activation reactions with chiral TDGs.
Keywords: pyridone ligand, arylation, C–H activation, transient directing group, palladium
Graphical Abstract
A Pd(II)-catalyzed enantioselective C(sp3)−H arylation of ketones using an α-amino acid as a chiral transient directing group is reported. The 3-nitro-5-trifluoromethyl-2-pyridone was identified as an effective ligand, serving as an acetate surrogate to accelerate C–H bond cleavage in the transient directing group strategy. The combination of an electron-deficient 2-pyridone ligand with different silver salts offers an effective method for controlling the rate-limiting steps which enables the high enantioselectivity and yield of this reaction.
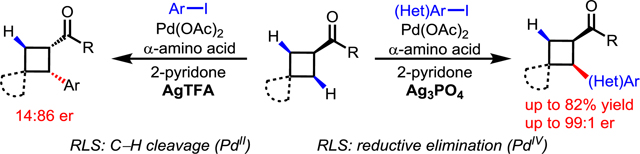
Introduction
Transition metal-catalyzed enantioselective activation of prochiral C(sp3)–H bonds has emerged as a valuable avenue for asymmetric catalysis. Notably, a wide range of Pd(II)-catalyzed enantioselective C–H activation/C–C bond and C–heteroatom bond forming reactions have been realized in the past decade.[1] Two fundamental strategies have been established for achieving intermolecular catalytic asymmetric C–H activation reactions. The first involves the design and development of various chiral bidentate ligands,[2–7] including mono-N-protected amino acids (MPAA),[2] quinolines,[3] oxazolines,[4] and amines[5] (Scheme 1a). The second strategy employs chiral transient directing groups attached to substrates via a reversible imine linkage (Scheme 1b).[8,9] The transient directing groups have a dual role: (1) to direct C–H activation through a reversible linkage with aldehydes, ketones or amines, and (2) to induce chirality by generating chiral Pd(II) intermediates.
Scheme 1.

Two strategies for Pd(II)-catalyzed directed asymmetric C(sp3)–H activation
The employment of chiral transient directing groups allows for enantioselective C−H activation without the need to install, and later remove, a directing group. Nonetheless, this strategy still has intrinsic limitations. In C–H activation reactions utilizing well-established chiral ligands bearing privileged acetyl-protected amino groups (NHAc), the ligand plays a crucial role in the C−H cleavage transition state.[2–5] However, in reactions instead employing chiral TDGs, another external base will be needed to perform this function (Scheme 1). Secondly, the stereogenic center in the transient directing groups is distal from the site of C−H bond cleavage, rendering the asymmetric induction more difficult. Finally, both the free TDG and the TDG-substrate adduct could coordinate with Pd(II) center and lead to a mixture of reactive and unreactive complexes. Despite extensive efforts on developing non-enantioselective C–H activation reactions using this TDG strategy,[8] asymmetric variants are limited to benzylic C(sp3)−H (Scheme 2a)[9] and C(sp2)–H bonds.[10].
Scheme 2.

Enantioselective C(sp3)‒H arylation using a transient directing group strategy
Cyclobutanes are prevalent motifs in naturally occurring alkaloids and important synthetic intermediates for the synthesis of biologically active molecules.[11,12] Although progress has been made on enantioselective functionalization of cyclobutyl carboxylic acids and amides using chiral NHAc-containing ligands,[2c,e,4c] the C–H activation of cyclobutyl ketones has not yet been realized. We envision that the chiral TDG strategy via a reversible imine linkage could fill this significant gap in enantioselective C–H activation. Herein, we report the first example of an enantioselective C–H arylation of aliphatic ketones using an α-amino acid as a chiral transient directing group responsible for chiral induction. Both the electron-deficient 2-pyridone ligand and Ag3PO4 additive are crucial in order to attain high enantioselectivity (Scheme 2b). Mechanistic experiments using deuterium incorporation indicate that the use of different Ag salts might switch the rate-limiting step by impacting the reductive elimination, hence reversing the enantioselectivity.
Results and Discussion
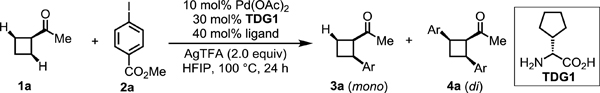
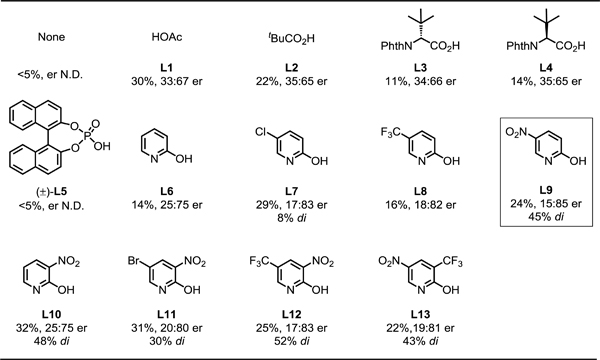
On the basis of our laboratory’s previous β-C−H arylation of ketones with aryl iodides,[8e,9a] we began our investigation by conducting the enantioselective β-C−H arylation of cyclobutyl ketone 1a with methyl 4-iodobenzoate 2a in the presence of Pd(OAc)2, D-cyclopentyl glycine TDG1 and AgTFA (Table 1). Under our previous reaction conditions which utilize OAc as an X-type ligand that can accelerate the C−H activation step, only 30% yield of the desired product with 33:67 er was observed. We envisioned that using a bulkier X-type ligands to accelerate the C(sp3)−H bond cleavage would enchance the enantioselectivity. First, we tried PivOH L2, N-protected amino acids L3 and L4, and phosphoric acid L5, as these ligands have been previously utilized to promote enantioselective C–H activation.[1] However, none of these ligands increased the enantioselectivity, presumably because they failed to replace OAc/OTFA and coordinate to the palladium catalyst. Therefore, we moved on to stronger binding X-type ligands. 2-Pyridones were recently identified as exceptionally efficient ligands that can accelerate C−H activation.[13,14] We evaluated various ligands of this type and were encouraged to find that electron-deficient pyridone ligands (L7–L13) not only promote the C–H activation to afford a higher yield of diarylation product but also increase the enantioselectivity up to 15:85 er, with 5-nitropyridone L9 generating the highest enantioselectivity. The enantioselectivity of this reaction could be further optimized to 14:86 er by adding 1.5 equiv of TFA (see the Supporting Information for optimization of reaction conditions).
Table 1.
 |
|---|
 |
Conditions: 1a (0.2 mmol, 2.0 equiv), methyl 4-iodobenzoate (1.0 equiv), Pd(OAc)2 (10 mol %), TDG1 (30 mol %), ligand (40 mol %), AgTFA (2.0 equiv), HFIP (0.6 mL), 100 °C, under air, 24 h.
Yield determined by 1H NMR analysis of the crude product using CH2Br2 as internal standard.
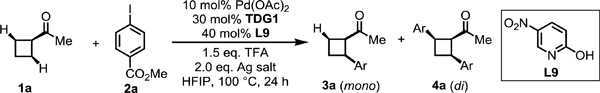
Although we found pyridone ligands are capable of promoting the C–H activation reaction, the yield and enantioselectivity of the desired product 3a required further improvement. With the best pyridone ligand in the presence of AgTFA identified, we next evaluated other Ag salts with the ligand L9 (Table 2). A control experiment without a silver salt additive afforded no desired product, indicating that silver salts are crucial to this reaction. Notably, most of the silver salts evaluated, such as AgOAc, Ag2CO3, and Ag2O, afforded moderate to good enantioselectivities (15:85–10:90 er), with the exception of AgNO3 or AgF. Surprisingly, a reversal of enantioselectivity (96:4 er) of the desired product was observed when Ag3PO4 was employed. Moreover, the undesired di-arylation product was also reduced. A moderate and reversed enantioselectivity (75:25 er) of the desired product was observed when K3PO4 and AgTFA was employed. Lastly, control experiment show both Ag3PO4 and the pyridone ligand are essential for the observed reversal of the enantioselectivity (Table 2).
Table 2.
 |
|---|
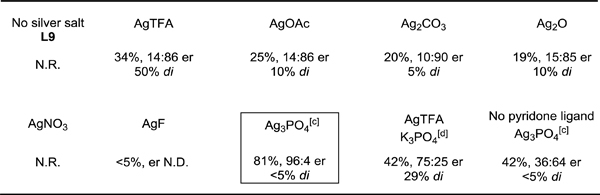 |
Conditions: 1a (0.2 mmol, 2.0 equiv), methyl 4-iodobenzoate (1.0 equiv), Pd(OAc)2 (10 mol %), TDG1 (30 mol %), L9 (40 mol %), TFA (1.5 equiv), silver salt (2.0 equiv), HFIP (0.6 mL), 100 °C, under air, 24 h.
Yield determined by 1H NMR analysis of the crude product using CH2Br2 as internal standard. [c] Ag3PO4 (1.0 equiv). [d] K3PO4 (1.0 equiv).
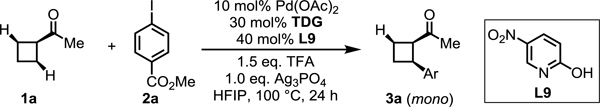
With the best silver salt in the presence of L9 established, we next systematically evaluated a series of chiral transient directing groups (Table 3). We found that substituents at the α-position of free amino acids has a significant effect on the enantioselectivity. Bulkier substituents at the α-position of amino acids afforded higher enantioseletivities (TDG3 vs TDG8, TDG3 vs TDG6). Although TDG6 generates the highest enantioselectivity, a lower yield was delivered presumably because of sluggish imine formation. Therefore, we selected TDG3 as the optimal transient directing group, which affords the best yield and a high enantioselectivity of 82% and 97:3 er, respectively. The control experiment with TDG4 (ent-TDG3) was also conducted to verify the role of an amino acid as a chiral transient directing group to predominantly determine the stereochemistry. The reaction could be further optimized to 82% NMR yield and 98:2 er by using L12 in the presence of TDG3 and Ag3PO4 (see the Supporting Information for optimization of reaction conditions).
Table 3.
Transient directing group evaluation for enantioselective β-C(sp3)‒H Arylation of cyclobutyl ketones[a,b]
 |
|---|
 |
Conditions: 1a (0.2 mmol, 2.0 equiv), methyl 4-iodobenzoate (1.0 equiv), Pd(OAc)2 (10 mol %), TDG (30 mol %), L9 (40 mol %), TFA (1.5 equiv), Ag3PO4 (1.0 equiv), HFIP (0.6 mL), 100 °C, under air, 24 h.
Yield determined by 1H NMR analysis of the crude product using CH2Br2 as internal standard.
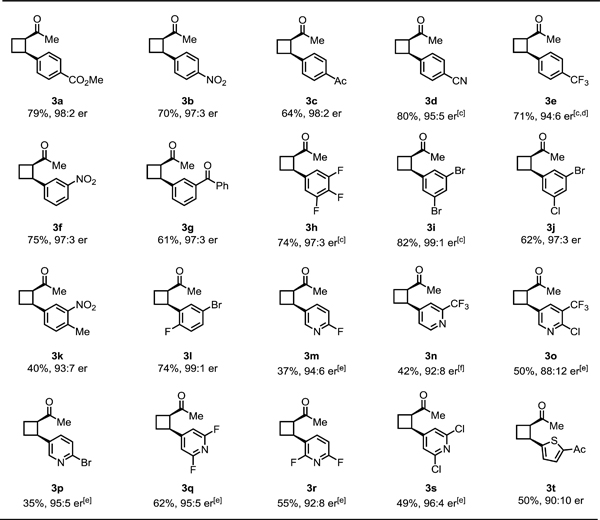
With the optimal reaction conditions established, we next investigated the scope of the aryl iodides and heteroaryl iodides using cyclobutyl ketone (1a) as the substrate (Table 4). The reaction of 1a with various aryl iodides furnished mono-arylated products in good yields and high enantioselectivities, exhibiting excellent functional group compatibility (3a−l). Various electron-withdrawing groups and coordinative cyano, nitro, acetyl, and benzoyl groups were well-tolerated. Ortho-fluorine substituted aryl iodide showed comparable reactivity and high enantioselectivity (3l). More importantly, to install diverse pyridines at β-position through asymmetric β-C(sp3)–H activation remains challenging, due to strong binding of iodopyridine to palladium. We optimized the reaction conditions for heteroarylation of 1a by increasing the pyridone ligand loading to one equivalent. Heteroarylation of 1a with a diverse range of 2-substituted iodopyridines proceeded smoothly in moderate yields and high enantioselectivities (3m–s). However, electron-neutral and electron-rich aryl iodides, such as iodobenzene and 4-iodoanisole, showed low activity affording around 5–10% yield of the desired products under our standard conditions.
Table 4.
 |
|---|
 |
Conditions: 1a (0.2 mmol, 2.0 equiv), aryl iodide 2 (1.0 equiv), Pd(OAc)2 (10 mol %), TDG3 (30 mol %), L12 (40 mol %), TFA (1.5 equiv), Ag3PO4 (1.0 equiv), HFIP (0.6 mL), 100 °C, under air, 24 h.
Isolated yields.
1a (0.1 mmol, 1.0 equiv), aryl iodide 2 (2.0 equiv).
TDG6.
L12 (1.0 equiv), 48 h.
TFA (0.5 equiv).
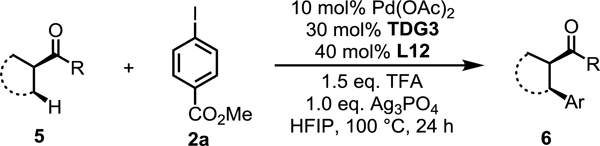
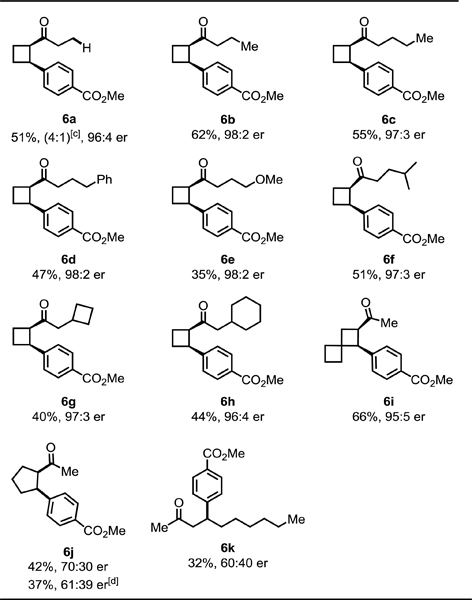
Next, we examined the scope of ketones (Table 5). Cyclobutyl alkyl ketones 5a−h were functionalized at the α-position in moderate yields and with good to excellent enantioselectivities (6a−h, 96:4–98:2 er). Notably, an array of functional groups, including phenyl, methoxy, cyclobutyl, and cyclohexyl groups were well tolerated. Interestingly, the C–H activation reaction also worked well with the spiro cyclobutyl ketone, affording a good yield and high enantioselectivity (6i). Although this catalytic system offers high enantioselectivity for C–H arylation of numerous cyclobutyl ketones where the enantiomer of product can be controlled by the choice of silver salt, it is not without limitations. For instance, cyclopentyl ketones and open chain ketones are reactive, but the enantioselectivities are lower (6j, 6k). For the cyclopentyl ketone, using AgTFA and Ag3PO4 did not generate inverse enantioselectivities.
Table 5.
 |
|---|
 |
Conditions: 5 (0.2 mmol, 2.0 equiv), methyl 4-iodobenzoate (1.0 equiv), Pd(OAc)2 (10 mol %), TDG3 (30 mol %), L12 (40 mol %), TFA (1.5 equiv), Ag3PO4 (1.0 equiv), HFIP (0.6 mL), 100 °C, under air, 24 h.
Isolated yields.
Numbers in parentheses indicate the ratio of the arylation of the cyclobutane and terminal methyl group.
AgTFA (2.0 equiv).
To demonstrate the robustness and the utility of this reaction, a gram-scale reaction and a sequential C−H activation were performed (Scheme 3). Arylation of 1a under the standard conditions with 3-iodo-4-fluorobromobenzene as the coupling partner afforded 1.2 g of enantioenriched product 3l in 75% isolated yield and 99:1 er. Cyclobutane 3l was then subjected to a second C−H arylation reaction employing simple glycine as the transient directing group and methyl 4-iodobenzoate as the coupling partner. This reaction afforded two enantiopure cyclobutanes bearing three contiguous stereogenic centers. Interestingly, the cis-mono-arylated ketone intermediate was epimerized to give the trans-isomer which readily form imine with TDG to direct subsequent diarylation.[15] The absolute configuration of these two products (7a, 7b) was confirmed by X-ray crystallographic analysis. This method allows rapid access to diverse chiral cyclobutanes which cannot be readily accessed by the previously reported asymmetric approaches.[12]
Scheme 3.

Diverse chiral cyclobutanes via sequential C–H arylation
To obtain some mechanistic insight into the reversed enantioselectivity observed when two different silver salts were employed in the reaction, we conducted deuterium incorporation experiments with 1a under the standard conditions with different silver salts in the presence of TFA-D and HFIP-ol-D (Scheme 4). No deuterium incorporation with AgTFA at the C-4 position on the cyclobutyl ring of the arylated product 3a suggests that the AgTFA conditions give rise to the irreversible C–H cleavage as the rate-limiting step (Scheme 4a). The distinct presence of 88% deuterium incorporation with Ag3PO4 at the C-4 position on the cyclobutyl ring of the arylated product 3a suggests that in these conditions C–H bond cleavage is a rapid, reversible step and the rate-limiting step might involve the Pd(IV) intermediate after C–H bond cleavage (Scheme 4b). Additionally, the lack of deuterium incorporation at the C-5 position on the cyclopentyl ring of the arylated product 6j with both AgTFA (Scheme 4c) and Ag3PO4 (Scheme 4d) additives suggests that the irreversible C–H cleavage is always the rate-limiting step for the arylation reaction of cyclopentyl ketones, regardless of the identity of the silver salt. These results could rationalize the lack of reverse enantioselectivity in C–H arylation reactions of cyclopentyl ketones with different silver salts (Table 5, 6j).
Scheme 4.

Deuterium-labeling experiments
On the basis of previous works from our laboratory[9,13,14] and the above-described experimental results, we propose a possible mechanism for the reversed enantioselectivity observed when two different silver salts, AgTFA and Ag3PO4, are employed as additives (Scheme 5). First, the pyridone ligand accelerates the C−H bond cleavage step and facilitates the generation of cyclometalated Pd(II)-complexes B and B’. Next, oxidative addition of the aryl iodide occurs to afford Pd(IV) complexes C and C’. A previous computational study suggests that abstraction of the iodide of this Pd(IV) intermediate by the silver additive triggers the reductive elimination to give the final arylation product.[16] Since AgTFA is an efficient additive that rapidly abstracts the iodide from Pd(IV) intermediates to promote C−C reductive elimination, the initial C–H bond cleavage will be the rate-limiting step, therefore, the chiral TDG-induced asymmetric C–H palladation controls the enantioselectivity. In contrast, when reductive elimination becomes the rate-limiting step with Ag3PO4 as the additive, the product formation from the chiral Pd(IV) intermediates is responsible for the chiral induction.
Scheme 5.

Proposed mechanism: combinations of pyridone ligands with silver salts to switch the rate-limiting steps (RLS)
Conclusion
We have developed the first example of a Pd(II)-catalyzed enantioselective C(sp3)−H arylation of ketones using an α-amino acid as a chiral transient directing group. A chiral trisubstituted cyclobutane is efficiently synthesized from a mono-substituted cyclobutane via sequential C–H arylation reactions, demonstrating the ability of this method to access structurally complex products from simple starting materials. The 3-nitro-5-trifluoromethyl-2-pyridone was identified as an effective ligand, serving as an acetate surrogate to accelerate C–H bond cleavage in the transient directing group strategy. The combination of an electron-deficient 2-pyridone ligand with different silver salts offers an effective method for controlling the rate-limiting steps which enables the high enantioselectivity and yield of this reaction. Our laboratory is currently applying these fundamental principles to achieve Pd-catalyzed enantioselective arylation of other alkyl C−H bonds using chiral transient directing groups.
Supplementary Material
Acknowledgements
We gratefully acknowledge The Scripps Research Institute, NIH (NIGMS, R01 GM084019), and Bristol-Myers Squibb for financial support. We thank Jiangsu Industrial Technology Research Institute (JITRI) for a postdoctoral fellowship (L.J.X). We thank Dr. Jason Chen, Ms. Brittany Sanchez, and Ms. Emily Sturgell from Automated Synthesis Facility (TSRI) for their assistance with HRMS analysis. We thank Arnie Rheingold, Jake Bailey and Milan Gembicky from X-ray Crystallography Facility (UCSD) for X-ray crystallography.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) For selected reviews of enantioselective C−H functionalization, see: Giri R, Shi B-F, Engle KM, Maugel N, Yu J-Q, Chem. Soc. Rev 2009, 38, 3242. [DOI] [PubMed] [Google Scholar]; b) Newton CG, Wang S-G, Oliveira CC, Cramer N, Chem. Rev 2017, 117, 8908. [DOI] [PubMed] [Google Scholar]; c) Saint-Denis TG, Zhu R-Y, Chen G, Wu Q-F, Yu J-Q, Science 2018, 359, eaao4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Shi B-F, Maugel N, Zhang Y-H, Yu J-Q, Angew. Chem. Int. Ed 2008, 47, 4882; Angew. Chem. 2018, 120, 4960. [DOI] [PubMed] [Google Scholar]; b) Wasa M, Engle KM, Lin DW, Yoo EJ, Yu J-Q, J. Am. Chem. Soc 2011, 133, 19598. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xiao K-J, Lin DW, Miura M, Zhu R-Y, Gong W, Wasa M, Yu J-Q, J. Am. Chem. Soc 2014, 136, 8138. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chan KSL, Fu H-Y, Yu J-Q, J. Am. Chem. Soc 2015, 137, 2042. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hu L, Shen P-X, Shao Q, Hong K, Qiao JX, Yu J-Q, Angew. Chem. Int. Ed 2019, 11, 2134; Angew. Chem. 2019, 131, 2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chen G, Gong W, Zhuang Z,; Andra MS,; Chen Y-Q, Hong X, Yang Y-F, Liu T, Houk KN,; Yu J-Q, Science 2016, 353, 1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Wu Q-F, Shen P-X, He J, Wang X-B, Zhang F, Shao Q, Zhu R-Y, Mapelli C, Qiao JX, Poss MA, Yu J-Q, Science 2017, 355, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shao Q, Wu Q-F, He J, Yu J-Q, J. Am. Chem. Soc 2018, 140, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wu Q-F, Wang X-B, Shen P-X, Yu J-Q, ACS Catal. 2018, 8, 2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shen P-X; Hu L; Shao Q; Hong K; Yu J-Q J. Am. Chem. Soc 2018, 140, 6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) For Pd (II)-catalyzed enantioselective C−H functionalization using phosphoric acid/amide and BINOL ligands, see: Yan S-B, Zhang S, Duan W-L, Org. Lett 2015, 17, 2458. [DOI] [PubMed] [Google Scholar]; b) Wang H, Tong H-R, He G, Chen G, Angew. Chem. Int. Ed 2016, 55, 15387; Angew. Chem. 2019, 128, 15613. [DOI] [PubMed] [Google Scholar]; c) Jain P, Verma P, Xia G, Yu J-Q, Nat. Chem 2017, 9, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Han Y-Q, Ding Y, Zhou T, Yan S-Y, Song H, Shi B-F, J. Am. Chem. Soc 2019, 141, 4558. [DOI] [PubMed] [Google Scholar]
- [7].a) For selected Pd(0)-catalyzed intramolecular enantioselective C−H functionalization, see: Nakanishi M, Katayev D, Besnard C, Kündig EP, Angew. Chem. Int. Ed 2011, 50, 7438; Angew. Chem. 2011, 123, 7576. [DOI] [PubMed] [Google Scholar]; b) Anas S, Cordi A, Kagan HB, Chem. Commun 2011, 47, 11483. [DOI] [PubMed] [Google Scholar]; c) Saget T, Lémouzy N, Cramer N, Angew. Chem. Int. Ed 2012, 51, 2238; Angew. Chem. 2012, 124, 2281. [DOI] [PubMed] [Google Scholar]; d) Martin N, Pierre C, Davi M, Jazzar R, Baudoin O, Chem. Eur. J 2012, 18, 4480. [DOI] [PubMed] [Google Scholar]; e) Zhu C, Wang D, Zhao Y, Sun W-Y, Shi Z, J. Am. Chem. Soc 2017, 139, 16486. [DOI] [PubMed] [Google Scholar]
- [8].a) For non-enantioselective examples of C(sp3)–H functionalization using a transient directing group, see: Wu Y, Chen Y-Q, Liu T, Eastgate MD, Yu J-Q, J. Am. Chem. Soc 2016, 138, 14554. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ma F, Lei M, Hu L, Org. Lett 2016, 18, 2708. [DOI] [PubMed] [Google Scholar]; c) Yang K, Li Q, Liu Y, Li G, G H, J. Am. Chem. Soc 2016, 138, 12775. [DOI] [PubMed] [Google Scholar]; d) Xu Y, Young MC, Wang C, Magness DM, Dong G, Angew. Chem. Int. Ed 2016, 55, 9084; Angew. Chem. 2016, 128, 9230. [DOI] [PubMed] [Google Scholar]; e) Hong K, Park H, Yu J-Q, ACS Catal. 2017, 7, 6938. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Liu Y, Ge H, Nat. Chem 2017, 9, 26. [Google Scholar]; g) Yada A, Liao W, Sato Y, Murakami M, Angew. Chem. Int. Ed 2017, 56, 1073; Angew. Chem. 2017, 129, 1093. [DOI] [PubMed] [Google Scholar]; h) St John-Campbell S, White AJP, Bull JA, Chem. Sci 2017, 8, 4840. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Chen Y-Q, Wang Z, Wu Y, Wisniewski SR, Qiao JX, Ewing WR, Eastgate MD, Yu J-Q, J. Am. Chem. Soc 2018, 140, 17884. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Pan L, Yang K, Li G, Ge H, Chem. Commun 2018, 54, 2759. [DOI] [PubMed] [Google Scholar]; k) Dong C, Wu L, Yao J, Wei K, Org. Lett 2019, 21, 2085. [DOI] [PubMed] [Google Scholar]
- [9].a) For enantioselective examples of C(sp3)–H functionalization using a transient directing group, see: Zhang F-L, Hong K, Li T-J, Park H, Yu J-Q, Science 2016, 351, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Park H, Verma P, Hong K, Yu J-Q, Nat. Chem 2018, 10, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) For selected enantioselective examples of C(sp2)–H functionalization using a transient directing group, see: Yao Q-J, Zhang S, Zhan B-B, Shi B-F, Angew. Chem. Int. Ed 2017, 56, 6617; Angew. Chem. 2017, 129, 6717. [DOI] [PubMed] [Google Scholar]; b) Xu J, Liu Y, Zhang J, Xu X, Jin Z, Chem. Commun 2018, 54, 689. [DOI] [PubMed] [Google Scholar]
- [11].a) Hansen TV, Stenstrøm Y, Naturally Occurring Cyclobutanes In Organic Synthesis: Theory and Applications; Hudlicky T, Ed.; Elsevier Science: Oxford, U.K., 2001; Vol. 5, p 1. [Google Scholar]; b) Dembitsky VM, J. Nat. Med 2008, 62, 1. [DOI] [PubMed] [Google Scholar]; c) Chi YM, Nakamura M, Zhao XY, Yoshizawa T, Yan WM, Hashimoto F, Kinjo J, Nohara T, Chem. Pharm. Bull 2005, 53, 1178. [DOI] [PubMed] [Google Scholar]; d) Kurosawa K, Takahashi K, Tsuda E, J. Antibiot 2001, 54, 541. [DOI] [PubMed] [Google Scholar]; e) Gutekunst WR, Baran PS, J. Am. Chem. Soc 2011, 133, 19076. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Gutekunst WR, Gianatassio R, Baran PS, Angew. Chem. Int. Ed 2012, 51, 7507; Angew. Chem. 2012, 124, 7625. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Gutekunst WR, Baran PS, J. Org. Chem 2014, 79, 2430. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Panish RA, Chintala SR, Fox JM, Angew. Chem. Int. Ed 2016, 55, 4983; Angew. Chem. 2016, 128, 5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) For selected recent examples of enantioselective synthesis of cyclobutane derivatives, see: Canales E, Corey EJ, J. Am. Chem. Soc 2007, 129, 12686. [DOI] [PubMed] [Google Scholar]; b) Kleinbeck F, Toste FD, J. Am. Chem. Soc 2009, 131, 9178. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Müller C, Bauer A, Maturi MM, Cuquerella MC, Miranda MA, Bach T, J. Am. Chem. Soc 2011, 133, 16689. [DOI] [PubMed] [Google Scholar]; d) Albrecht Ł, Dickmeiss G, Acosta FC, Rodriguez-Escrich C, Davis RL, Jørgensen KA, J. Am. Chem. Soc 2012, 134, 2543. [DOI] [PubMed] [Google Scholar]; e) Reeves M, Eidamshaus C, Kim J, Stoltz BM, Angew. Chem. Int. Ed 2013, 52, 6718; Angew. Chem. 2013, 125, 6850. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Panish R, Chintala SR, Boruta DT, Fang Y, Taylor MT, Fox JM, J. Am. Chem. Soc 2013, 135, 9283. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Du JN, Skubi KL, Schultz DM, Yoon TP, Science 2014, 344, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Conner ML, Xu Y, Brown MK, J. Am. Chem. Soc 2015, 137, 3482. [DOI] [PubMed] [Google Scholar]; i) Guisán-Ceinos M, Parra A, Martín-Heras V, Tortosa M, Angew. Chem. Int. Ed 2016, 55, 6969; Angew. Chem. 2016, 128, 7083. [DOI] [PubMed] [Google Scholar]; j) Hu J-L, Feng L-W, Wang L, Xie Z, Tang Y, Li X, J. Am. Chem. Soc 2016, 138, 13151. [DOI] [PubMed] [Google Scholar]
- [13].a) For selected examples of 2-pyridone-accelerated C(sp2)–H activation reactions: Wang P, Farmer ME, Huo X, Jain P, Shen P-X, Ishoey M, Bradner JE, Wisniewski SR, Eastgate ME, Yu J-Q, J. Am. Chem. Soc 2016, 138, 9269. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang P, Verma P, Xia G, Shi J, Qiao JX, Tao S, Cheng PTW, Poss MA, Farmer ME, Yeung KS, Yu J-Q, Nature 2017, 551, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li GC, Wang P, Farmer ME, Yu J-Q, Angew. Chem. Int. Ed 2017, 56, 6874; Angew. Chem. 2017, 129, 6978. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Farmer ME, Wang P, Shi H, Yu J-Q, ACS Catal. 2018, 8, 7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) For selected examples of 2-pyridone-accelerated C(sp3)–H activation reactions: Zhu R-Y, Li Z-Q, Park HS, Senanayake CH, Yu J-Q, J. Am. Chem. Soc 2018, 140, 3564. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) ref 8i. [Google Scholar]
- [15]. For proposed mechanism of the sequential C−H activation see SI, Scheme S3.
- [16].Feng W, Wang T, Liu D, Wang X, Dang Y, ACS Catal. 2019, 9, 6672. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.