

Abstract
Esophageal cancer (EC) causes hundreds of thousands of deaths a year worldwide, especially the major subtype esophageal squamous cell carcinoma (ESCC). With the advent of next-generation sequencing and the availability of commercial microarrays, abnormities in genetic levels have been revealed in various independent researches. High frequencies of structure variations (SVs), single nucleotide variations (SNVs) and copy-number alterations (CNAs) in ESCCs are uncovered, and ESCC shows high levels of inter- and intratumor heterogeneity, implying diverse evolutionary trajectories. This review tries to explain the pathogenesis of ESCC on the scope of most often mutated genes based on prior studies, hopes to offer some hints for diagnosis and therapy in clinic.
Keywords: ESCC, genomic alterations, heterogeneity, evolution, pathogenesis
Introduction
The esophagus is a hollow, muscular tube consisting of several layers. Esophageal cancer (EC) usually starts on the inner side of the mucosa and spreads outward through the other layers as it grows. According to the Global Cancer Statics 2018, about 572,000 people worldwide are diagnosed with EC a year, which ranked eighth among all new cancer cases, and approximately 509,000 patients die from it, ranking as sixth for cancer mortality [1].
The two most common forms of EC are esophageal squamous cell carcinoma (ESCC), most often found in the upper and middle parts of the esophagus, and esophageal adenocarcinoma (EAC), that usually forms in the lower part of the esophagus, near the stomach. ESCC is the most common subtype, accounting for 90% of all esophageal cancers worldwide [2]. It is different from EAC in terms of its pathogenesis, risk factors, genomic landscape and cellular transcriptome profile [3-5]. There is great variability in the incidence of EC with regard to geography, ethnicity, age, and gender (Figure 1) [6]. For example, In Europe and the United States the predominant histological subtype is EAC, while EAC remains rare in Asia. In China, EC is the third most frequently diagnosed cancer and the fourth leading cause of death from cancer [7]. The incidence in rural areas is more than that in urban areas [7,8], especially in regions such as Henan, Hebei, and Shanxi [9].
Figure 1.
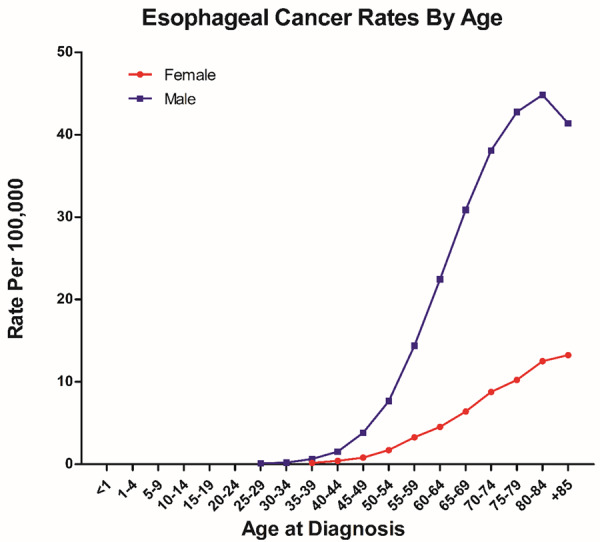
Based on SEER (Surveillance, Epidemiology, and End Results) website, esophagus cancer incidence rates by age at diagnosis in USA from 2012 to 2016.
EC is associated with a poor prognosis, despite advances in diagnosis and treatment, the overall 5-year survival rate is less than 20% worldwide [10]. As shown in Table 1, metastasis to lymph nodes and other organs markedly lowers the survival rate.
Table 1.
According to the SEER database, the 5-year survival rates of 21583 cases of esophageal cancer between 2009 and 2015 in the United States, based on how far the cancer has spread
| Stage | 5-Year Relative Survival Rate of EC | 5-Year Relative Survival Rate of ESCC |
|---|---|---|
| Localized | 46.7% | 31.2% |
| Regional | 25.1% | 23.8% |
| Distant | 4.8% | 6.3% |
| All stages combined | 19.9% | 17.6% |
Genomic alterations in ESCC
With the development of sequencing technology, whole-genome sequencing (WGS), whole-exome sequencing (WES), and transcriptome sequencing (RNA-seq) have been performed in many studies. In addition, various commercial microarrays have been developed and applied to ESCC. As shown in Tables 2 and 3, abnormalities in the molecular levels have been uncovered [11-18], providing comprehensive insights into the mutational signature of ESCCs and facilitating identification of biomarkers for early diagnosis and potential therapeutic targets.
Table 2.
Recent studies of ESCC using high-throughput sequencing
| Study | ESCC cases | District | Main Methods | Chromosome change | |
|---|---|---|---|---|---|
|
| |||||
| Amplification | Deletion | ||||
| [11] | 158 | Chaoshan, Guangdong, China | WGS (17), WES (71), array-CGH (123) | Large scale amplification of 3q, 5p, 8q, 12p, 20p and 20q; 43 focal CNAs, like 8q24.3, 11q13.3-13.4, 14q32.33 | Large scale deletion of 3p, 4q, 9p, 13q, 18q, 19p and 21q; 15 focal CNAs, such as 9p21.3, 3q26.1 |
| [12] | 220, (103)1 | North-central China | WES (139), RNA-seq (4), SNP-array (22), array-CGH (59) | 8p11.23, 8q24.21, 7p11.2, 11q13.2-13.3, 12p12.1, 12q15, 3q26, 3q26.32-33, 14q13.3 | Homozygous deletion of 2q22.1-22.2, 9p21.3, 5q12.1, 9p24.1, 3p14.2 |
| [13] | 113 | China | WES (113) | Recurrent CNAs like 11q13.3, 11q13.2, 11q13.4, 11q13.1, 7p11.2, 7q31.1 | Recurrent CNAs like 9p21, Yp11.3, Yq11.222 |
| [14] | 104, (88)2 | North-central China | WGS (14), WES (90), FISH, qPCR | Recurrent CNAs like 3q26.1-q29, 3q29, 3q25.1, 11q13.3-q13.4, 11q13.3, 8p12-p11.23, 7p12.1-p11.2 | Recurrent CNAs like 9p21.3, 19p11, 5p15.33, 21p11.2, 3p11.1, 4p11, Xq11.1 |
| [15] | 388, (70)3 | Guangdong, China | WGS (10), WES (60), IHC (120) | Recurrent CNAs like 3q26.33, 11q13, 20q13.12 | Recurrent CNAs like 2q22.1-22.2, 9p21.3, 13q13.3-q14.11 |
| [16] | 144 | Japan | WES SNP-array | 3q, 8q, 5p, 7p, 20q; recurrent focal CNVs, like 11q13.3, 5p15.33, 7p11.2, 11q22.1, 3q26.33 | 3p, 4p, 9p, 5q; recurrent focal CNVs, like 9p21.3, 2q22.1-22.2, 4q22, 3p14.2 |
| [17] | 90 | 10 counties, such as USA, Korea, Brazil, Germany | WES, SNP-array | CNAs, like 3q26.33, 3q28, 5p15.33, 7p11.2, 8p11.23, 11q13.3, 12p12.1, 12q14.3, 14q13.2 | CNAs, like 13q14.2, 3p25.2, 9p21.3, 2q22.1, 8p23.2, Xp11.3, 10p11.21, 3p14.3, 4p15.2, 4q22.1, 3p14.2 |
| [18] | 94 (704, 94)4 | China | WGS | 3q, 5p, 7p, 8q, 12p, 16p, 20p and 20q; 23 recurrent focal CNVs, like 11q13.3, 3q26.32, 5p15.33, 8q24.21, 7q22.1, 7p11.2, 19p13.3, 14q13.3 | 3p, 4p, 4q, 5q, 9p, 13q, 18q and 21p; 34 focal CNVs, like 9p21.3, 22q11.21, 10p11.22, 5q13.2, 6p24.3, 10q21.1, 12q23.1, 13q31.3 |
WGS: Whole-genome sequencing; WES: Whole-exome sequencing; TS: Targeted sequencing; CGH: Comparative genomic hybridization; RNA-seq: RNA sequence; SNP: Single nucleotide polymorphism; IHC: immunohistochemistry.
88 Chaoshan sequencing data from [10].
Table 3.
Recurrent affected genes in recent studies of ESCC
| Pathway | Gene | Location | Origin function on tumor | [11] | [12] | [13] | [14] | [15] | [16] | [17] | [18] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell cycle | TP53 | 17p13.1 | - | S | S | S | S | S | S | S | S |
| CDKN2A/B | 9p21.3 | - | S, L | S, L | S, L | S, L | S | S, L | S | S, L | |
| RB1 | 13q14.2 | - | S, L | S | S | S | L | F | |||
| CCND1 | 11q13.3 | + | G | G | G | G | G | G | G | ||
| CCND2 | 12p13.32 | + | G | ||||||||
| CDK4 | 12q14.1 | + | G | ||||||||
| CDK6 | 7q21.2 | + | G | G | |||||||
| E2F1 | 20q11.22 | - | G | ||||||||
| MDM2 | 12q15 | + | G | G | G | G | |||||
| TP63 | 3q28 | + | G | G | |||||||
| TP73 | 1p36.32 | + | G | G | |||||||
| MYBL2 | 20q13.12 | + | G | ||||||||
| Apoptosis | BCL2L1 | 20q11.21 | G | G | |||||||
| FADD | 11q13.3 | G | G | G | G | G | G | G | |||
| TRAF3 | 14q32.33 | G | |||||||||
| RTK-MAPK-PI3K signaling | FGFs | 11q13 | + | G | G | G | G | G | G | ||
| TGFBR2 | 3p24.1 | - | S | S | F | ||||||
| EGFR | 7p11.2 | + | G | G | G | G | G | G | |||
| FGFR1 | 8p11.23 | + | G | G | G | G | |||||
| ERBB2 | 17q12 | + | O | M | |||||||
| KRAS | 12p12.1 | + | G | G | F | G | |||||
| MYC | 8q24.21 | + | G | G | G | G | G | G | |||
| PIK3CA | 3q26.32 | + | S, G | S, G | F | S | S | G | F, G | ||
| AKT1 | 14q32.33 | + | G | G | |||||||
| PTEN | 10q23.31 | - | S, L | M | F | ||||||
| JAK-STAT signaling | IL7R | 5p13.2 | + | G | G | ||||||
| JAK1 | 1p31.3 | L | M | ||||||||
| STATs | G | O | |||||||||
| SOX2 | 3q26.33 | + | G | G | G | G | G | G | G | G | |
| Notch signaling | NOTCH1 | 9q34.3 | - | S, G | S | S | S | S | S | S | |
| NOTCH3 | 19p13.12 | - | S | F | |||||||
| RBPJ | 4p15.2 | - | S | F | |||||||
| Hedgehog signaling | PTCH1 | 9p22.32 | F | F | |||||||
| SUFU | 10q24.32 | - | M | ||||||||
| Hippo/Wnt signaling | FAT1 | 4q35.2 | - | S, D | F | S | S | F | |||
| FAT2 | 5q33.1 | - | S | M | |||||||
| FAT3 | 11q14.3 | - | F | ||||||||
| YAP1 | 11q22.1 | + | G | G | G | G | G | ||||
| AJUBA | 14q11.2 | - | S | S | S | S | |||||
| DVL3 | 3q27.1 | + | G | G | G | ||||||
| LRP5/6 | 12p13.2 | + | G | G | |||||||
| Chromatin regulator | MLL2 (KMT2D) | 12q13.12 | - | F | S | S | S | S | F | ||
| MLL3 (KMT2C) | 7q36.1 | - | F | F | F | ||||||
| ASH1L | 1q22 | F | |||||||||
| SETD1B | 12q24.31 | F | |||||||||
| CREBBP | 16p13.3 | - | F | F | S | S | F | ||||
| EP300 | 22q13.2 | - | F | S | F | S | |||||
| ZNF750 | 17q25.3 | - | S | S | S | S, L | F | ||||
| ARID2 | 12q12 | - | F | ||||||||
| PBRM1 | 3p21.1 | - | F | ||||||||
| KDM6A | Xp11.3 | - | S | F | F, L | ||||||
| BAP1 | 3p21.1 | - | S | ||||||||
| Oxidative response | NFE2L2 | 2q31.2 | + | S | S | S | S | S | S | S | S |
| KEAP1 | 19p13.2 | M | |||||||||
| CUL3 | 2q36.2 | - | M | F | |||||||
| Other | PTPRD | 9p24.1-p23 | - | L | |||||||
| PDE4D | 5q11.2-q12.1 | - | L | ||||||||
| LRP1B | 2q22.1-22.2 | L | L | L | S | ||||||
| FAM135B | 8q24.23 | + | S | ||||||||
| ADAM29 | 4q4.1 | S | |||||||||
| FBXW7 | 4q31.3 | - | F, D | S | S | S | S, L | ||||
| CBX4/8 | 17q25.3 | G | G | ||||||||
| TERT | 5p15.33 | + | G | G | G | G | |||||
| NKX2-1 | 14q13.3 | + | G | G | G | G | |||||
| FHIT | 3p14.2 | - | L | L | L | L | |||||
| TET2 | 4q24 | - | S | ||||||||
| TTN | 2q31.2 | G | S | ||||||||
| MIR548K | 11q13.3 | + | G | G | G | G | G | G | G | ||
| SHANK2 | 11q13.3-q13.4 | G | G | G | G | G | G | G | |||
| CTTN | 11q13.3 | + | G | G | G | G | G | G | G | ||
| ANO1 | 11q13.3 | + | G | G | G | G | G | G | G |
+: tumor promotion. -: tumor suppression. S: SMG, significantly mutated gene; F: frequently changed gene; G: gain of gene; L: loss of gene; O: overexpression; D: down-regulation.
Despite the fact that the presence and frequencies of gene alterations in ESCC vary greatly within studies because of geographic or ethnic differences of patients, stages and grades of tumors, the number of tumor samples, inter and intra-tumor heterogeneity, and depth of sequencing [19], some of the findings are consistent.
Numbers of structure variations (SVs) such as large-scale chromosome changes, gene copy number alterations (CNAs), small insertions and deletions (indels), and single-nucleotide variants (SNVs) have been revealed. Among all SNVs, there are high frequencies of C>G, C>T mutations at TpCpX, which are mostly attributed to the hyperactivity of APOBEC family of DNA deaminases [20], and C>T mutation at XpCpG, which may be the result of higher methylation levels and DNA repair inefficiency in tumors [21].
As a result, many genes are affected in ESCCs. The notable recurrent alterations are mutations of TP53, CDKN2A, NFE2L2, NOTCH1, PIK3CA, RB1, FAT1, MLL2, FBXW7 and ZNF750, amplification of CCND1 (11q13.3), MYC (8q24.21), and SOX2 (3q26.33), and deletion of CDKN2A (9p21.3). These aberrations are also involved in the progression of head and neck squamous cell carcinoma (HNSCC) [22] and lung squamous cell carcinoma (LSCC) [23]. In addition, as an amplicon at 13q13.3, MIR548K, ANO1, FADD, CTTN and SHANK2 are also amplified frequently but talked less in previous studies, they all play roles in the progression of ESCC. All these genes orchestrate many other genes to regulate cellular activities and maintain homeostasis. Thus, the disruption of the balance contributes to many cancers through various mechanisms like the discussion below.
Cell-cycle regulation
TP53 is a tumor suppressor gene, its protein product, p53, plays very important roles in cell cycle regulation [24], DNA repair [25], genomic stability maintenance [26], senescence [27], and apoptosis [28]. Various kinds of TP53 mutations have been found in numbers of human cancers, and it is the most frequently mutated gene in ESCC, with frequent protein loss mutations such as frameshift indels, nonsense mutations, and mutations at splice site. The majority of missense mutations are distributed in the DNA binding domain (amino acids 102-292) [29]. The classical G1/S phase checkpoint in the cell cycle is mediated through p16 (CDKN2A)-cyclin D (CCND1,2)-Rb and MDM2-p53-p21 (CDKN1A) pathways [30]. Functional loss of TP53, CDKN2A or RB1, and amplification of CCND1 lead to dysregulation of the cell cycle. In addition, other genes contributing to G1/S transition are also amplified in ESCCs, such as CDK4, CDK6, E2F1, and MDM2, although at much lower frequencies. Apart from the loss of the DNA-binding capability, such as mutations at residues R175, R248 or R273 [29], accumulating evidence has shown gain-of-function missense mutations of TP53, which drive tumor progression through different mechanisms [31]. Among them, p.R175H and p.R273H mutants not only lose tumor suppressor activity but also promote transformation and metastasis through the gained function of promoting integrin recycling [32], and both mutations have been found in ESCC by WGS and WES.
RTK-MAPK-PI3K signaling pathway
The binding of growth factors to their receptor tyrosine kinases (RTKs) could stimulates both PI3K-AKT and MAPK signaling pathways [33]. Activated Akt controls important cellular processes by phosphorylating substrates involved in apoptosis, protein synthesis, metabolism, migration, and cell cycle to promote cell survival and progression [34]. Activated ERK phosphorylates other factors such as c-Myc to enhance cell proliferation and differentiation [35]. Also, the aberrant activation of the RTK-MAPK-PI3K pathway is quite often detected in cancers. For Example, PIK3CA has been reported to be mutated quite frequently in various cancers, and the missense substitutions show a non-random distribution with hotspots at the helical domain (exon 9, commonly E542 and E545) and kinase domain (exon 20, commonly H1047), implying increased kinase activity of these mutants [36]. The three explicit oncogenic mutants p.E545K, p.E542K and p.H1047R [37] all have high frequencies in the current ESCC sequencing analyses. Apart from gain-of-function mutations of PIK3A, amplification or overexpression of MYC, FGFs, EGFR, FGFR1 and KRAS, and loss-of-function mutations of PTEN contribute to the survival and progression of ESCC.
NFE2L2
Reactive oxygen species (ROS) are highly cytotoxic, one of the cell’s defending strategies is to detoxicate ROS by encoding more than 20 phase II detoxifying enzymes [38]. The expression of the enzymes is regulated by the antioxidant response element (ARE)-binding transcriptional complex, and NF-E2-related factor 2 (Nrf2), the protein product of NFE2L2, is an essential component of the complex [39]. Besides the cytoprotective role, Nrf2 participates in many cellular activities like cell differentiation and metabolism, it can suppress carcinogenesis in early stages while promote tumor growth and metastasis in later stages [40]. A research has confirmed that oncogenic Nrf2 mutations is a feature of SCC and promote the development of various kinds of SCCs [41], which maintains high protein levels by inhibiting Keap1-Cul3-Roc1-dependent proteasomal degradation of Nrf2, such as mutations at DLG motif (23LxxQDxDLG31) or ETGE motif (77DxETGE82) [42]. In a recent EC cancer research, Nrf2 was confirmed to strongly promote glutathione metabolism, offering essential fuels for cancer progression [43]. Overall, gain-of-function mutation or higher expression of NFE2L2 is associated with tumor recurrence, poor prognosis, and resistance against chemoradiation therapy in ESCC [44].
FBXW7
FBXW7 is one of the most frequently mutated tumor suppressor genes in human cancers, which encodes the ubiquitin ligase FBW7 that targets many oncoproteins for ubiquitin-mediated protein degradation. The well-studied substrates involved in tumor progression are cyclin E, c-Myc, and c-Jun [45-48], thus, the aberrant function of FBW7 lead to the accumulation of numbers of oncoproteins to promote tumorigenesis in synergetic ways.
NOTCH1
Notch signaling is a highly evolutionarily conserved pathway implicated in cell fate determination, proliferation, and apoptosis [49]. It can play both tumor promotive and suppressive roles depending on the cancer cell type [50]. Quite a large fraction of NOTCH1 mutations in head and neck squamous cell carcinoma (HNSCC) are predicted to truncate the gene product or occur in the TAD domain required for transactivation of target genes, suggesting that Notch1 functions as a tumor suppressor in this type of tumor [51]. Similarly, the mutations of NOTCH1 in ESCCs are found to be stop-gain SNVs, frameshift indels, at splice sites, or harmful missense SNVs, implying a tumor suppressor role the functional protein plays in esophageal squamous cell. Activated Notch1 was shown to engage in cell cycle arrest by up-regulation of p21 and p27 in small cell lung cancer cells [52]. In human ESCC cell line EC9706, ectopic expression of Notch1 showed up-regulation of GSK3β and down-regulation of β-catenin, implying that Notch signaling inhibited proliferation and induces apoptosis through Wnt signaling pathway [53], though the mechanism remains to be elucidated.
SOX2
SOX2 is an indispensable factor for the induction of pluripotent stem cells in vitro [54]. It plays different and even opposite roles in various cancer types [55,56], in SCC, it has been proven to be essential for tumor initiation and cancer stem cell [57], and be an amplified lineage survival oncogene in lung squamous cell carcinoma (LSCC) and ESCC, but alone, it was insufficient to transform epithelial cell lines in vitro, implying co-operation with other signals to promote proliferation and metastasis [58]. As a transcription factor, SOX2 promotes tumorigenesis and tumor progression through diverse mechanisms. One was proved to be linked to the activation of the PI3K/AKT/mTOR1 signaling pathway [59], one was owed to epithelial-mesenchymal transition (EMT) through activation of STAT3/HIF-α signaling [60], and another was to promote the expression of SOX2 overlapping transcript (SOX2OT), an important lncRNA that in turn promoted the growth of ESCC [61]. Notably, ESCC patients with high expression levels of SOX2 had favorable prognosis, suggesting that it could serve as a prognostic factor and therapy target in clinic [62,63].
FAT1
FAT1 belongs to the cadherin superfamily, whose ortholog in Drosophila encodes a tumor suppressor essential to control cell proliferation during development [64]. FAT1 plays an essential role in cellular polarization, directs cell migration, and modulates cell-cell contact [65]. It was found to suppress multiple cancer cell growth in vitro and in vivo through quenching Wnt signaling though binding β-catenin to antagonize its nuclear localization [66]. In studies of HNSCC, functional loss of FAT1 resulted in YAP1 activation [67], promoted invasion of HNSCC cell lines, and was associated with nodal involvement, lymphovascular permeation and tumor recurrence [68]. In ESCC studies, FAT1 mutation disrupted the MAPK/ERK pathway to contribute to EMT [69], and affected cytoskeletal proteins to alter cellular mechanical properties, leading to deregulation of cell migration and invasion of ESCC cells [70].
AJUBA
AJUBA encodes a LIM domain-containing protein that acts as a scaffold protein [71], and shuttles between cytoplasm and nucleus to engage in many cellular activities. Generally, it is considered as a tumor suppressor gene. Ajuba is highly expressed in epithelial cells and interacts directly with E-cadherin and F-actin to mediate cell-cell junction formation [72]. In addition, it does not interact with F-actin but acts with Grb2 to augment MAPK signaling in fibroblasts that have much lower Ajuba expression level [73]. Within the nucleus, Ajuba can work as a co-repressor with transcriptional repressor protein Gfi1 to auto-regulate Gfi1 expression in 293T and myeloid cell lines [74]. Ajuba specifically associate with LATS/Wts and WW45/Sav in insect and mammalian cells to antagonize the phosphorylation of YAP1 [75], a downstream nuclear effector of the highly conserved Hippo signaling pathway. Moreover, Ajuba acts as a negative mediator of Wnt signaling by promoting GSK3β-mediated phosphorylation of β-catenin, that subsequently undergoes proteasome-dependent degradation [76]. Overall, loss-of-function mutations of AJUBA promote tumorigenesis.
Genes at 13q13.3
MIR548K encodes a microRNA miR-548k that regulates the expression of many proteins, the role it plays in ESCC has been revealed recently. It has been reported to reduce lncRNA-LET expression and increase NF90 expression in a feedback loop, which results in decreased p53 expression and increased HIF-1α and VEGF to enhance ESCC proliferation [77,78]. Besides, MiR-548k down-regulates ADAMTS1 (a disintegrin and metalloproteinase with thrombospondin motifs 1), resulting in increased secretion of VEGFC and tyrosine phosphorylation (p-Tyr) of VEGFR3 to promote lymphangiogenesis and lymph node metastasis. Additionally, miR-548k represses KLF10, an EGFR transcriptional repressor, to promote metastasis of ESCC cells [79]. Thus, serum abundance of miR-548k and VEGFC may be promising biomarkers for diagnosis of ESCC [79].
ANO1 encodes a Ca2+-activated Cl- channel, that is widely expressed and plays diverse functions in various tissues [80]. It is amplified or highly expressed in a variety of carcinomas, indicating an oncogenic role [81]. Overexpression of ANO1 enhances EGFR signaling and CAMKII (calmodulin-dependent protein kinase II) signaling in breast cancer [82], and inhibits apoptosis via downregulation of Bim in HNSCC [83]. In esophageal epithelial cell, the expression level of ANO1 was confirmed to highly corelate with the level of p63 and promote cell cycle, being rational to serve as a therapeutic target [84].
CTTN encodes an actin-associated scaffolding protein, that binds and activates the Arp (actin-related protein) 2/3 complex to regulate the formation of dynamic cortical actin-associated structures [85]. Ectopic expression of CTTN increases motility and invasion of tumor cells and correlates with metastasis of HNSCC [86]. Moreover, amplification and overexpression of CTTN correlates with lymph node metastasis by activating the PI3K/Akt pathway to inhibit anoikis in ESCC [87]. Thus, CTTN may serve as a candidate prognostic indicator and therapeutic target in ESCC [88].
SHANK2 also encodes a scaffolding protein, that organizes the postsynaptic density region in neurons and is related to many neuron disorders like autism [89]. In addition, Shank2 interacts with and activate NHE3 (Na+/H+ exchanger 3), involved in the fine regulation of transepithelial salt and water transport [90]. Furthermore, a truncated isoform, Shank2E, is highly expressed and associates with cortactin and actin at the apical membrane of liver epithelial cells [91]. However, the role of SHANK2 amplification in the development of tumors remains elusive.
FADD, the protein product of FADD, is a well-known key adaptor protein that transmits apoptotic signals mediated by death receptors [92]. In line with other scaffolds, it plays multifunctional roles in cells [93], such as regulating glucose and fat metabolisms [94]. In tumors, FADD was demonstrated to be required for Kras-mediated tumorigenesis in a murine model of lung cancer [95], as well as pancreatic cancer cell proliferation and drug resistance [96]. Amplification of FADD together with other genes was believed to contribute to the progression of HNSCC [97], but the role it plays in ESCC remains obscure.
Epigenetic regulation
Epigenetic regulation determines cell fate and dysregulation causes pathologies and diseases [98]. Modifications of histone proteins and changes of DNA methylations are the two major ways to regulate gene expression.
MLL2 (KMT2D) and MLL3 (KMT2C) belong to the KMT2 family, which methylate lysine 4 on the histone H3 tail to promote transcription [99]. Downregulation of MLL3 promotes tumorigenicity of ESCC cells, indicating a tumor suppressor role in ESCC [100]. MLL2 orchestrates H3K27 acetyltransferase p300 to promote differentiation [101], therefore, both MLL2 and p300 are tumor suppressors and are frequently mutated in many types of human cancers [102], which is consistent with the majority of their loss-of-function mutations in ESCCs.
ZNF750 encodes an important transcription factor that acts downstream of p63 to induce KLF4 and drive terminal epidermal differentiation [103]; Moreover, ZNF750 interacts directly with many chromatin regulators, such as RCOR1, KDM1A and CTBP1/2, to induce differentiation [104], it is regarded as a crucial SCC-specific tumor suppressors for its differentiation-induction and migration-reduction functions [105]. In oral squamous cell carcinoma, ZNF750 was shown to modulate the vascular microenvironment by regulating expression of many genes to inhibit malignant progression of tumor cells [106]. Overall, ZNF750 is regarded as a tumor suppressor gene in ESCC, and its low expression may be a biomarker for lymph node metastasis and be linked to poor prognosis [107].
Despite infrequent inactivation in many cancers and ESCCs, H3K27me2/3 demethylase KDM6A is thought to be a tumor suppressor [108], and the function greatly relies on its association with MLL3/MLL4 [109].
CREBBP, the ortholog of p300, is also inactivated in a range of cancer types, supporting it functions as a tumor suppressor [110]. The two acetyl transferases as cofactors participate in regulating the expression of thousands of genes, they are involved in many cellular activities like cell cycle regulation and DNA damage repair [111,112].
In ESCCs, large-scale chromosome deletion always occurs in 3p, and the 3p21 locus harbors three tumor suppressor genes: PBRM1, BAP1 and SETD2; In particular, SETD2 is responsible for H3K36 trimethylation, which directs DNA mismatch repair, and its loss triggers microsatellite instability and can increase genome-wide mutation rates [113]. Therefore, deletion of this region is catastrophic and greatly enhances tumor progression.
High heterogeneity and distinct evolution
Based on many valuable sequencing studies in recent years, the onset and development of ESCC has become clearer. As early as 40 years ago, esophageal dysplasia was believed to be the precursor lesion of ESCC [114], and the risk of developing ESCC was in parallel with the grade of dysplasia [115]. Until recently, this phenomenon was given a reasonable explanation. That was mutational burden, including gene mutations and CNAs, was already present in the physiologically normal esophageal epithelia (PNE) [116], and increased with dysplasia progression, reaching the top in high-grade dysplasia and ESCC [117,118]. Besides, high heterogeneity of gene mutations and CNAs existed among dysplasia and tumors from the same patient, showing highly distinct evolutionary trajectories [117,118]. In addition, the study revealed that inflammation promoted mutation and aggravated both dysplasia and carcinoma [118]. All these works highlighted the two genetic hits of TP53, which should play pivotal roles in tumorigenesis of ESCC. Nevertheless, the studies showed that the mechanisms of positive selection in PNE versus ESCC differed greatly, while it showed more similarities between high-grade dysplasia and ESCC.
Apart from the separate sites of abnormal esophageal tissues harboring high heterogeneity, a high level of intratumor heterogeneity (ITH) was also proved to be present in ESCC like many other tumors, showing divergent evolution in many aspects [119]. As high as 90% of all recurrent CNAs from different regions of a tumor were unraveled to be spatially heterogeneous [120], and a wide range of heterogeneously somatic variants were revealed in all researches about multi-region ESCC [119-121]. In addition, ITH of DNA methylation and T cell immune repertoire were also identified in two of the above studies, contributing to the heterogeneity and evolution of ESCC.
Research about clonal evolution of ESCC from normal mucosa to primary tumor and metastasis was conducted in 10 patients recently [122], the results also showed complexity in clones within each patient, both linear and parallel metastatic models were shown to be present in ESCC.
Discussion
Esophageal cancer, especially the esophageal squamous cell carcinoma subtype has a worldwide poor prognosis and the high death rate is largely attributed to late diagnosis and a lack of effective therapies. With the convenience of sequencing, molecular mechanisms underlying ESCC have been revealed, although the key step for transformation is yet to be determined. Not only the presence of the above recurrent mutated genes is important, but also the order of the occurrence plays critical roles in the development of ESCC. Every mitosis brings the chance of mutation to the genome, as a result, mutation density increases with age. Among the mutations, quite a number of them are passenger mutations that do not offer growth advantage to the host, or some of them offer limited growth promotion function to maintain the physiologically normal esophageal epithelia. With the accumulation of driver mutations, low-grade dysplasia manifests and it progresses to high-grade dysplasia, ESCC and eventually metastasizes to other organs. For its high frequency of mutations both in ESCC and high-grade dysplasia, and as the various phylogenetic trees show, the two-hits event of TP53 plays pivotal roles in tumorigenesis. Though diverse evolution patterns of ESCC coexist based on phylogenetic tree models, which is in accordance with studies of other cancer types, commonalities can still be inferred from the highly heterogeneous phylogenetic trees of ESCC. Driver mutations of tumor suppressor genes are more likely to be located at the trunk of phylogenetic trees than those of oncogenes, such as TP53 and CDKN2A. Amplification of 3q (SOX2) and 11q (CCND1), and the loss of 9p (CDKN2A) also tend to be early driver events in carcinogenesis.
Despite of the many known genomic alterations in ESCC through sequencing technology, we are far from the understanding of the dynamic process of tumorigenesis. Along with continuous evolution, both spatial and temporal heterogeneities exist in ESCC, contributing to the complexity and difficulty of researches in the development of ESCC. Besides, the huge number of tumor cells in a single tumor are not present as isolated ones, they communicate and even cooperate with each other and thus behave as a community to support and promote tumorigenesis [123]. Moreover, tumor cells co-evolve with their micro-environments; extracellular matrix, tumor vasculature and immune cells participate in this process and play multiple roles in carcinogenesis [124].
Many mechanisms such as carcinogens, APOBEC functions, and DNA repair deficiency contribute to genomic instability, which in turn greatly promote to the development and high heterogeneity of ESCC. To get better outcomes in clinic, Both early detection and personalized multi-targeted therapies are in need.
Acknowledgements
This work is supported by the Natural Science Foundation of Top Talent of SZTU (No: 1814309011180006). We thank Mitchell Arico from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Disclosure of conflict of interest
None.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Rustgi AK, El-Serag HB. Esophageal carcinoma. N Engl J Med. 2014;371:2499–2509. doi: 10.1056/NEJMra1314530. [DOI] [PubMed] [Google Scholar]
- 3.Bandla S, Pennathur A, Luketich JD, Beer DG, Lin L, Bass AJ, Godfrey TE, Litle VR. Comparative genomics of esophageal adenocarcinoma and squamous cell carcinoma. Ann Thorac Surg. 2012;93:1101–1106. doi: 10.1016/j.athoracsur.2012.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abnet CC, Arnold M, Wei WQ. Epidemiology of esophageal squamous cell carcinoma. Gastroenterology. 2018;154:360–373. doi: 10.1053/j.gastro.2017.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu H, Yu J, Li Y, Hou Q, Zhou R, Zhang N, Jing Z, Jiang M, Li Z, Hua Y, Brunicardi FC, Wu S. Single-cell RNA sequencing reveals diverse intratumoral heterogeneities and gene signatures of two types of esophageal cancers. Cancer Lett. 2018;438:133–143. doi: 10.1016/j.canlet.2018.09.017. [DOI] [PubMed] [Google Scholar]
- 6.Kollarova H, Machova L, Horakova D, Janoutova G, Janout V. Epidemiology of esophageal cancer - an overview article. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2007;151:17–20. doi: 10.5507/bp.2007.003. [DOI] [PubMed] [Google Scholar]
- 7.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 8.Chen W, He Y, Zheng R, Zhang S, Zeng H, Zou X, He J. Esophageal cancer incidence and mortality in China, 2009. J Thorac Dis. 2013;5:19–26. doi: 10.3978/j.issn.2072-1439.2013.01.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin Y, Totsuka Y, He Y, Kikuchi S, Qiao Y, Ueda J, Wei W, Inoue M, Tanaka H. Epidemiology of esophageal cancer in Japan and China. J Epidemiol. 2013;23:233–242. doi: 10.2188/jea.JE20120162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y. Epidemiology of esophageal cancer. World J Gastroenterol. 2013;19:5598–5606. doi: 10.3748/wjg.v19.i34.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song Y, Li L, Ou Y, Gao Z, Li E, Li X, Zhang W, Wang J, Xu L, Zhou Y, Ma X, Liu L, Zhao Z, Huang X, Fan J, Dong L, Chen G, Ma L, Yang J, Chen L, He M, Li M, Zhuang X, Huang K, Qiu K, Yin G, Guo G, Feng Q, Chen P, Wu Z, Wu J, Ma L, Zhao J, Luo L, Fu M, Xu B, Chen B, Li Y, Tong T, Wang M, Liu Z, Lin D, Zhang X, Yang H, Wang J, Zhan Q. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91–95. doi: 10.1038/nature13176. [DOI] [PubMed] [Google Scholar]
- 12.Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, Meng X, Sato Y, Okuno Y, Varela AM, Ding LW, Garg M, Liu LZ, Yang H, Yin D, Shi ZZ, Jiang YY, Gu WY, Gong T, Zhang Y, Xu X, Kalid O, Shacham S, Ogawa S, Wang MR, Koeffler HP. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet. 2014;46:467–473. doi: 10.1038/ng.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM, Zhang F, Zhao ZR, Li ZT, Liu ZY, Zhao YD, Sun J, Zhou CC, Yao R, Wang SY, Wang P, Sun N, Zhang BH, Dong JS, Yu Y, Luo M, Feng XL, Shi SS, Zhou F, Tan FW, Qiu B, Li N, Shao K, Zhang LJ, Zhang LJ, Xue Q, Gao SG, He J. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46:1097–1102. doi: 10.1038/ng.3076. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L, Zhou Y, Cheng C, Cui H, Cheng L, Kong P, Wang J, Li Y, Chen W, Song B, Wang F, Jia Z, Li L, Li Y, Yang B, Liu J, Shi R, Bi Y, Zhang Y, Wang J, Zhao Z, Hu X, Yang J, Li H, Gao Z, Chen G, Huang X, Yang X, Wan S, Chen C, Li B, Tan Y, Chen L, He M, Xie S, Li X, Zhuang X, Wang M, Xia Z, Luo L, Ma J, Dong B, Zhao J, Song Y, Ou Y, Li E, Xu L, Wang J, Xi Y, Li G, Xu E, Liang J, Yang X, Guo J, Chen X, Zhang Y, Li Q, Liu L, Li Y, Zhang X, Yang H, Lin D, Cheng X, Guo Y, Wang J, Zhan Q, Cui Y. Genomic analyses reveal mutational signatures and frequently altered genes in esophageal squamous cell carcinoma. Am J Hum Genet. 2015;96:597–611. doi: 10.1016/j.ajhg.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin HD, Liao XY, Chen YB, Huang SY, Xue WQ, Li FF, Ge XS, Liu DQ, Cai Q, Long J, Li XZ, Hu YZ, Zhang SD, Zhang LJ, Lehrman B, Scott AF, Lin D, Zeng YX, Shugart YY, Jia WH. Genomic characterization of esophageal squamous cell carcinoma reveals critical genes underlying tumorigenesis and poor prognosis. Am J Hum Genet. 2016;98:709–727. doi: 10.1016/j.ajhg.2016.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawada G, Niida A, Uchi R, Hirata H, Shimamura T, Suzuki Y, Shiraishi Y, Chiba K, Imoto S, Takahashi Y, Iwaya T, Sudo T, Hayashi T, Takai H, Kawasaki Y, Matsukawa T, Eguchi H, Sugimachi K, Tanaka F, Suzuki H, Yamamoto K, Ishii H, Shimizu M, Yamazaki H, Yamazaki M, Tachimori Y, Kajiyama Y, Natsugoe S, Fujita H, Mafune K, Tanaka Y, Kelsell DP, Scott CA, Tsuji S, Yachida S, Shibata T, Sugano S, Doki Y, Akiyama T, Aburatani H, Ogawa S, Miyano S, Mori M, Mimori K. Genomic landscape of esophageal squamous cell carcinoma in a Japanese population. Gastroenterology. 2016;150:1171–1182. doi: 10.1053/j.gastro.2016.01.035. [DOI] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research Network; Analysis Working Group: Asan University; BC Cancer Agency; Brigham and Women’s Hospital; Broad Institute; Brown University; Case Western Reserve University; Dana-Farber Cancer Institute; Duke University; Greater Poland Cancer Centre; Harvard Medical School; Institute for Systems Biology; KU Leuven; Mayo Clinic; Memorial Sloan Kettering Cancer Center; National Cancer Institute; Nationwide Children’s Hospital; Stanford University; University of Alabama; University of Michigan; University of North Carolina; University of Pittsburgh; University of Rochester; University of Southern California; University of Texas MD Anderson Cancer Center; University of Washington; Van Andel Research Institute; Vanderbilt University; Washington University; Genome Sequencing Center: Broad Institute; Washington University in St. Louis; Genome Characterization Centers: BC Cancer Agency; Broad Institute; Harvard Medical School; Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University; University of North Carolina; University of Southern California Epigenome Center; University of Texas MD Anderson Cancer Center; Van Andel Research Institute; Genome Data Analysis Centers: Broad Institute; Brown University:; Harvard Medical School; Institute for Systems Biology; Memorial Sloan Kettering Cancer Center; University of California Santa Cruz; University of Texas MD Anderson Cancer Center; Biospecimen Core Resource: International Genomics Consortium; Research Institute at Nationwide Children’s Hospital; Tissue Source Sites: Analytic Biologic Services; Asan Medical Center; Asterand Bioscience; Barretos Cancer Hospital; BioreclamationIVT; Botkin Municipal Clinic; Chonnam National University Medical School; Christiana Care Health System; Cureline; Duke University; Emory University; Erasmus University; Indiana University School of Medicine; Institute of Oncology of Moldova; International Genomics Consortium; Invidumed; Israelitisches Krankenhaus Hamburg; Keimyung University School of Medicine; Memorial Sloan Kettering Cancer Center; National Cancer Center Goyang; Ontario Tumour Bank; Peter MacCallum Cancer Centre; Pusan National University Medical School; Ribeirão Preto Medical School; St. Joseph’s Hospital &Medical Center; St. Petersburg Academic University; Tayside Tissue Bank; University of Dundee; University of Kansas Medical Center; University of Michigan; University of North Carolina at Chapel Hill; University of Pittsburgh School of Medicine; University of Texas MD Anderson Cancer Center; Disease Working Group: Duke University; Memorial Sloan Kettering Cancer Center; National Cancer Institute; University of Texas MD Anderson Cancer Center; Yonsei University College of Medicine; Data Coordination Center: CSRA Inc.; Project Team: National Institutes of Health. Integrated genomic characterization of oesophageal carcinoma. Nature. 2017;541:169–175. doi: 10.1038/nature20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang J, Tan W, Ling Z, Xi R, Shao M, Chen M, Luo Y, Zhao Y, Liu Y, Huang X, Xia Y, Hu J, Parker JS, Marron D, Cui Q, Peng L, Chu J, Li H, Du Z, Han Y, Tan W, Liu Z, Zhan Q, Li Y, Mao W, Wu C, Lin D. Genomic analysis of oesophageal squamous-cell carcinoma identifies alcohol drinking-related mutation signature and genomic alterations. Nat Commun. 2017;8:15290. doi: 10.1038/ncomms15290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dotto GP, Rustgi AK. Squamous cell cancers: a unified perspective on biology and genetics. Cancer Cell. 2016;29:622–637. doi: 10.1016/j.ccell.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo J, Huang J, Zhou Y, Zhou Y, Yu L, Li H, Hou L, Zhu L, Ge D, Zeng Y, Guleng B, Li Q. Germline and somatic variations influence the somatic mutational signatures of esophageal squamous cell carcinomas in a Chinese population. BMC Genomics. 2018;19:538. doi: 10.1186/s12864-018-4906-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, Pugh TJ, Stransky N, Helman E, Kim J, Sougnez C, Ambrogio L, Nickerson E, Shefler E, Cortés ML, Auclair D, Saksena G, Voet D, Noble M, DiCara D, Lin P, Lichtenstein L, Heiman DI, Fennell T, Imielinski M, Hernandez B, Hodis E, Baca S, Dulak AM, Lohr J, Landau DA, Wu CJ, Melendez-Zajgla J, Hidalgo-Miranda A, Koren A, McCarroll SA, Mora J, Crompton B, Onofrio R, Parkin M, Winckler W, Ardlie K, Gabriel SB, Roberts CWM, Biegel JA, Stegmaier K, Bass AJ, Garraway LA, Meyerson M, Golub TR, Gordenin DA, Sunyaev S, Lander ES, Getz G. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jäger N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdés-Mas R, van Buuren MM, van’t Veer L, Vincent-Salomon A, Waddell N, Yates LR Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain. Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim Y, Hammerman PS, Kim J, Yoon JA, Lee Y, Sun JM, Wilkerson MD, Pedamallu CS, Cibulskis K, Yoo YK, Lawrence MS, Stojanov P, Carter SL, McKenna A, Stewart C, Sivachenko AY, Oh IJ, Kim HK, Choi YS, Kim K, Shim YM, Kim KS, Song SY, Na KJ, Choi YL, Hayes DN, Kim J, Cho S, Kim YC, Ahn JS, Ahn MJ, Getz G, Meyerson M, Park K. Integrative and comparative genomic analysis of lung squamous cell carcinomas in east asian patients. J. Clin. Oncol. 2013;32:121–128. doi: 10.1200/JCO.2013.50.8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin Y, Tainsky MA, Bischoff FZ, Strong LC, Wahl GM. Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell. 1992;70:937–948. doi: 10.1016/0092-8674(92)90244-7. [DOI] [PubMed] [Google Scholar]
- 25.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–11. [PubMed] [Google Scholar]
- 26.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 27.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 28.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 29.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 30.Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB, Alon U. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36:147–150. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- 31.Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304–317. doi: 10.1016/j.ccr.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, Cromer A, Brugge JS, Sansom OJ, Norman JC, Vousden KH. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327–1341. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 33.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coulombe P, Rodier G, Pelletier S, Pellerin J, Meloche S. Rapid turnover of extracellular signal-regulated kinase 3 by the ubiquitin-proteasome pathway defines a novel paradigm of mitogen-activated protein kinase regulation during cellular differentiation. Mol Cell Biol. 2003;23:4542–4558. doi: 10.1128/MCB.23.13.4542-4558.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 37.Bader AG, Kang S, Vogt PK. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci U S A. 2006;103:1475–1479. doi: 10.1073/pnas.0510857103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- 39.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 40.Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564–571. doi: 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim YR, Oh JE, Kim MS, Kang MR, Park SW, Han JY, Eom HS, Yoo NJ, Lee SH. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J Pathol. 2010;220:446–451. doi: 10.1002/path.2653. [DOI] [PubMed] [Google Scholar]
- 42.Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M, Hirohashi S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A. 2008;105:13568–13573. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kitano Y, Baba Y, Nakagawa S, Miyake K, Iwatsuki M, Ishimoto T, Yamashita YI, Yoshida N, Watanabe M, Nakao M, Baba H. Nrf2 promotes oesophageal cancer cell proliferation via metabolic reprogramming and detoxification of reactive oxygen species. J Pathol. 2018;244:346–357. doi: 10.1002/path.5021. [DOI] [PubMed] [Google Scholar]
- 44.Shibata T, Kokubu A, Saito S, Narisawa-Saito M, Sasaki H, Aoyagi K, Yoshimatsu Y, Tachimori Y, Kushima R, Kiyono T, Yamamoto M. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia. 2011;13:864–873. doi: 10.1593/neo.11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004;303:1374–8. doi: 10.1126/science.1092880. [DOI] [PubMed] [Google Scholar]
- 46.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 47.Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nat Rev Cancer. 2014;14:233–247. doi: 10.1038/nrc3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer. 2018;17:115. doi: 10.1186/s12943-018-0857-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 50.Rangarajan A, Talora C, Okuyama R, Nicolas M, Mammucari C, Oh H, Aster JC, Krishna S, Metzger D, Chambon P, Miele L, Aguet M, Radtke F, Dotto GP. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001;20:3427–3436. doi: 10.1093/emboj/20.13.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, Carter SL, Voet D, Cortes ML, Auclair D, Berger MF, Saksena G, Guiducci C, Onofrio RC, Parkin M, Romkes M, Weissfeld JL, Seethala RR, Wang L, Rangel-Escareno C, Fernandez-Lopez JC, Hidalgo-Miranda A, Melendez-Zajgla J, Winckler W, Ardlie K, Gabriel SB, Meyerson M, Lander ES, Getz G, Golub TR, Garraway LA, Grandis JR. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sriuranpong V, Borges MW, Ravi RK, Arnold DR, Nelkin BD, Baylin SB, Ball DW. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001;61:3200–3205. [PubMed] [Google Scholar]
- 53.Lu Z, Liu H, Xue L, Xu P, Gong T, Hou G. An activated Notch1 signaling pathway inhibits cell proliferation and induces apoptosis in human esophageal squamous cell carcinoma cell line EC9706. Int J Oncol. 2008;32:643–51. [PubMed] [Google Scholar]
- 54.Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- 55.Weina K, Utikal J. SOX2 and cancer: current research and its implications in the clinic. Clin Transl Med. 2014;3:19. doi: 10.1186/2001-1326-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luo J, Yan R, He X, He J. SOX2 inhibits cell proliferation and metastasis, promotes apoptotic by downregulating CCND1 and PARP in gastric cancer. Am J Transl Res. 2018;10:639–647. [PMC free article] [PubMed] [Google Scholar]
- 57.Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, Delatte B, Caauwe A, Lenglez S, Nkusi E, Brohee S, Salmon I, Dubois C, del Marmol V, Fuks F, Beck B, Blanpain C. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014;511:246–250. doi: 10.1038/nature13305. [DOI] [PubMed] [Google Scholar]
- 58.Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, Ramos AH, Woo MS, Weir BA, Getz G, Beroukhim R, O’Kelly M, Dutt A, Rozenblatt-Rosen O, Dziunycz P, Komisarof J, Chirieac LR, Lafargue CJ, Scheble V, Wilbertz T, Ma C, Rao S, Nakagawa H, Stairs DB, Lin L, Giordano TJ, Wagner P, Minna JD, Gazdar AF, Zhu CQ, Brose MS, Cecconello I, Ribeiro U Jr, Marie SK, Dahl O, Shivdasani RA, Tsao MS, Rubin MA, Wong KK, Regev A, Hahn WC, Beer DG, Rustgi AK, Meyerson M. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41:1238–1242. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gen Y, Yasui K, Nishikawa T, Yoshikawa T. SOX2 promotes tumor growth of esophageal squamous cell carcinoma through the AKT/mammalian target of rapamycin complex 1 signaling pathway. Cancer Sci. 2013;104:810–816. doi: 10.1111/cas.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao H, Teng C, Huang W, Peng J, Wang C. SOX2 promotes the epithelial to mesenchymal transition of esophageal squamous cells by modulating slug expression through the activation of STAT3/HIF-alpha signaling. Int J Mol Sci. 2015;16:21643–21657. doi: 10.3390/ijms160921643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu Y, Chen X, Liang Y, Li J, Zhang K, Dai L, Guan X, Wang K, Bai Y. Overexpression of long non-coding RNA SOX2OT promotes esophageal squamous cell carcinoma growth. Cancer Cell Int. 2018;18:76. doi: 10.1186/s12935-018-0570-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maehara R, Fujikura K, Takeuchi K, Akita M, Abe-Suzuki S, Karbanova J, Corbeil D, Itoh T, Kakeji Y, Zen Y. SOX2-silenced squamous cell carcinoma: a highly malignant form of esophageal cancer with SOX2 promoter hypermethylation. Mod Pathol. 2018;31:83–92. doi: 10.1038/modpathol.2017.112. [DOI] [PubMed] [Google Scholar]
- 63.Liu Y, Xiong Z, Beasley A, D’Amico T, Chen XL. Personalized and targeted therapy of esophageal squamous cell carcinoma: an update. Ann N Y Acad Sci. 2016;1381:66–73. doi: 10.1111/nyas.13144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bennett FC, Harvey KF. Fat cadherin modulates organ size in Drosophila via the Salvador/Warts/Hippo signaling pathway. Curr Biol. 2006;16:2101–2110. doi: 10.1016/j.cub.2006.09.045. [DOI] [PubMed] [Google Scholar]
- 65.Tanoue T, Takeichi M. Mammalian Fat1 cadherin regulates actin dynamics and cell-cell contact. J Cell Biol. 2004;165:517–528. doi: 10.1083/jcb.200403006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morris LG, Kaufman AM, Gong Y, Ramaswami D, Walsh LA, Turcan S, Eng S, Kannan K, Zou Y, Peng L, Banuchi VE, Paty P, Zeng Z, Vakiani E, Solit D, Singh B, Ganly I, Liau L, Cloughesy TC, Mischel PS, Mellinghoff IK, Chan TA. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat Genet. 2013;45:253–261. doi: 10.1038/ng.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martin D, Degese MS, Vitale-Cross L, Iglesias-Bartolome R, Valera JLC, Wang Z, Feng X, Yeerna H, Vadmal V, Moroishi T, Thorne RF, Zaida M, Siegele B, Cheong SC, Molinolo AA, Samuels Y, Tamayo P, Guan KL, Lippman SM, Lyons JG, Gutkind JS. Assembly and activation of the Hippo signalome by FAT1 tumor suppressor. Nat Commun. 2018;9:2372. doi: 10.1038/s41467-018-04590-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin SC, Lin LH, Yu SY, Kao SY, Chang KW, Cheng HW, Liu CJ. FAT1 somatic mutations in head and neck carcinoma are associated with tumor progression and survival. Carcinogenesis. 2018;39:1320–1330. doi: 10.1093/carcin/bgy107. [DOI] [PubMed] [Google Scholar]
- 69.Hu X, Zhai Y, Kong P, Cui H, Yan T, Yang J, Qian Y, Ma Y, Wang F, Li H, Cheng C, Zhang L, Jia Z, Li Y, Yang B, Xu E, Wang J, Yang J, Bi Y, Chang L, Wang Y, Zhang Y, Song B, Li G, Shi R, Liu J, Zhang M, Cheng X, Cui Y. FAT1 prevents epithelial mesenchymal transition (EMT) via MAPK/ERK signaling pathway in esophageal squamous cell cancer. Cancer Lett. 2017;397:83–93. doi: 10.1016/j.canlet.2017.03.033. [DOI] [PubMed] [Google Scholar]
- 70.Hu X, Zhai Y, Shi R, Qian Y, Cui H, Yang J, Bi Y, Yan T, Yang J, Ma Y, Zhang L, Liu Y, Li G, Zhang M, Cui Y, Kong P, Cheng X. FAT1 inhibits cell migration and invasion by affecting cellular mechanical properties in esophageal squamous cell carcinoma. Oncol Rep. 2018;39:2136–2146. doi: 10.3892/or.2018.6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dawid IB, Breen JJ, Toyama R. LIM domains: multiple roles as adapters and functional modifiers in protein interactions. Trends Genet. 1998;14:156–162. doi: 10.1016/s0168-9525(98)01424-3. [DOI] [PubMed] [Google Scholar]
- 72.Marie H, Pratt SJ, Betson M, Epple H, Kittler JT, Meek L, Moss SJ, Troyanovsky S, Attwell D, Longmore GD, Braga VM. The LIM protein Ajuba is recruited to cadherin-dependent cell junctions through an association with alpha-catenin. J Biol Chem. 2003;278:1220–1228. doi: 10.1074/jbc.M205391200. [DOI] [PubMed] [Google Scholar]
- 73.Goyal RK, Lin P, Kanungo J, Payne AS, Muslin AJ, Longmore GD. Ajuba, a novel LIM protein, interacts with Grb2, augments mitogen-activated protein kinase activity in fibroblasts, and promotes meiotic maturation of Xenopus oocytes in a Grb2- and Ras-dependent manner. Mol Cell Biol. 1999;19:4379–4389. doi: 10.1128/mcb.19.6.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Montoya-Durango DE, Velu CS, Kazanjian A, Rojas ME, Jay CM, Longmore GD, Grimes HL. Ajuba functions as a histone deacetylase-dependent co-repressor for autoregulation of the growth factor-independent-1 transcription factor. J Biol Chem. 2008;283:32056–32065. doi: 10.1074/jbc.M802320200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Das Thakur M, Feng Y, Jagannathan R, Seppa MJ, Skeath JB, Longmore GD. Ajuba LIM proteins are negative regulators of the Hippo signaling pathway. Curr Biol. 2010;20:657–662. doi: 10.1016/j.cub.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haraguchi K, Ohsugi M, Abe Y, Semba K, Akiyama T, Yamamoto T. Ajuba negatively regulates the Wnt signaling pathway by promoting GSK-3β-mediated phosphorylation of β-catenin. Oncogene. 2007;27:274–84. doi: 10.1038/sj.onc.1210644. [DOI] [PubMed] [Google Scholar]
- 77.Chen Z, Lin J, Wu S, Xu C, Chen F, Huang Z. Up-regulated miR-548k promotes esophageal squamous cell carcinoma progression via targeting long noncoding RNA-LET. Exp Cell Res. 2018;362:90–101. doi: 10.1016/j.yexcr.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 78.Lin J, Chen Z, Wu S, Huang W, Chen F, Huang Z. An NF90/long noncoding RNA-LET/miR-548k feedback amplification loop controls esophageal squamous cell carcinoma progression. J Cancer. 2019;10:5139–5152. doi: 10.7150/jca.30816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang W, Hong R, Li L, Wang Y, Du P, Ou Y, Zhao Z, Liu X, Xiao W, Dong D, Wu Q, Chen J, Song Y, Zhan Q. The chromosome 11q13.3 amplification associated lymph node metastasis is driven by miR-548k through modulating tumor microenvironment. Mol Cancer. 2018;17:125. doi: 10.1186/s12943-018-0871-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang F, Rock JR, Harfe BD, Cheng T, Huang X, Jan YN, Jan LY. Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc Natl Acad Sci U S A. 2009;106:21413–21418. doi: 10.1073/pnas.0911935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bill A, Alex Gaither L. The mechanistic role of the calcium-activated chloride channel ANO1 in tumor growth and signaling. Adv Exp Med Biol. 2017;966:1–14. doi: 10.1007/5584_2016_201. [DOI] [PubMed] [Google Scholar]
- 82.Britschgi A, Bill A, Brinkhaus H, Rothwell C, Clay I, Duss S, Rebhan M, Raman P, Guy CT, Wetzel K, George E, Popa M, Lilley S, Choudhury H, Gosling M, Wang L, Fitzgerald S, Borawski J, Baffoe J, Bentires-Alj M. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci U S A. 2013;110:E1026–34. doi: 10.1073/pnas.1217072110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Godse NR, Khan N, Yochum ZA, Gomez-Casal R, Kemp C, Shiwarski DJ, Seethala RS, Kulich S, Seshadri M, Burns TF, Duvvuri U. TMEM16A/ANO1 inhibits apoptosis via downregulation of bim expression. Clin Cancer Res. 2017;23:7324–7332. doi: 10.1158/1078-0432.CCR-17-1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vanoni S, Zeng C, Marella S, Uddin J, Wu D, Arora K, Ptaschinski C, Que J, Noah T, Waggoner L, Barski A, Kartashov A, Rochman M, Wen T, Martin L, Spence J, Collins M, Mukkada V, Putnam P, Naren A, Chehade M, Rothenberg ME, Hogan SP. Identification of anoctamin 1 (ANO1) as a key driver of esophageal epithelial proliferation in eosinophilic esophagitis. J Allergy Clin Immunol. 2020;145:239–254. e232. doi: 10.1016/j.jaci.2019.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weed SA, Parsons JT. Cortactin: coupling membrane dynamics to cortical actin assembly. Oncogene. 2001;20:6418–6434. doi: 10.1038/sj.onc.1204783. [DOI] [PubMed] [Google Scholar]
- 86.Rothschild BL, Shim AH, Ammer AG, Kelley LC, Irby KB, Head JA, Chen L, Varella-Garcia M, Sacks PG, Frederick B, Raben D, Weed SA. Cortactin overexpression regulates actin-related protein 2/3 complex activity, motility, and invasion in carcinomas with chromosome 11q13 amplification. Cancer Res. 2006;66:8017–25. doi: 10.1158/0008-5472.CAN-05-4490. [DOI] [PubMed] [Google Scholar]
- 87.Luo ML, Shen XM, Zhang Y, Wei F, Xu X, Cai Y, Zhang X, Sun YT, Zhan QM, Wu M, Wang MR. Amplification and overexpression of CTTN (EMS1) contribute to the metastasis of esophageal squamous cell carcinoma by promoting cell migration and anoikis resistance. Cancer Res. 2006;66:11690–9. doi: 10.1158/0008-5472.CAN-06-1484. [DOI] [PubMed] [Google Scholar]
- 88.Lu P, Qiao J, He W, Wang J, Jia Y, Sun Y, Tang S, Fu L, Qin Y. Genome-wide gene expression profile analyses identify CTTN as a potential prognostic marker in esophageal cancer. PLoS One. 2014;9:e88918. doi: 10.1371/journal.pone.0088918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Leblond CS, Heinrich J, Delorme R, Proepper C, Betancur C, Huguet G, Konyukh M, Chaste P, Ey E, Rastam M, Anckarsater H, Nygren G, Gillberg IC, Melke J, Toro R, Regnault B, Fauchereau F, Mercati O, Lemiere N, Skuse D, Poot M, Holt R, Monaco AP, Jarvela I, Kantojarvi K, Vanhala R, Curran S, Collier DA, Bolton P, Chiocchetti A, Klauck SM, Poustka F, Freitag CM, Waltes R, Kopp M, Duketis E, Bacchelli E, Minopoli F, Ruta L, Battaglia A, Mazzone L, Maestrini E, Sequeira AF, Oliveira B, Vicente A, Oliveira G, Pinto D, Scherer SW, Zelenika D, Delepine M, Lathrop M, Bonneau D, Guinchat V, Devillard F, Assouline B, Mouren MC, Leboyer M, Gillberg C, Boeckers TM, Bourgeron T. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012;8:e1002521. doi: 10.1371/journal.pgen.1002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Han W, Kim KH, Jo MJ, Lee JH, Yang J, Doctor RB, Moe OW, Lee J, Kim E, Lee MG. Shank2 associates with and regulates Na+/H+ exchanger 3. J Biol Chem. 2006;281:1461–1469. doi: 10.1074/jbc.M509786200. [DOI] [PubMed] [Google Scholar]
- 91.McWilliams RR, Gidey E, Fouassier L, Weed SA, Doctor RB. Characterization of an ankyrin repeat-containing Shank2 isoform (Shank2E) in liver epithelial cells. Biochem J. 2004;380:181–191. doi: 10.1042/BJ20031577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 93.Tourneur L, Chiocchia G. FADD: a regulator of life and death. Trends Immunol. 2010;31:260–269. doi: 10.1016/j.it.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 94.Zhuang H, Wang X, Zha D, Gan Z, Cai F, Du P, Yang Y, Yang B, Zhang X, Yao C, Zhou Y, Jiang C, Guan S, Zhang X, Zhang J, Jiang W, Hu Q, Hua ZC. FADD is a key regulator of lipid metabolism. EMBO Mol Med. 2016;8:895–918. doi: 10.15252/emmm.201505924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bowman BM, Sebolt KA, Hoff BA, Boes JL, Daniels DL, Heist KA, Galbán CJ, Patel RM, Zhang J, Beer DG, Ross BD, Rehemtulla A, Galbán S. Phosphorylation of FADD by the kinase CK1α promotes KRASG12D-induced lung cancer. Sci Signal. 2015;8:ra9. doi: 10.1126/scisignal.2005607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang R, Liu Y, Hammache K, He L, Zhu B, Cheng W, Hua ZC. The role of FADD in pancreatic cancer cell proliferation and drug resistance. Oncol Lett. 2017;13:1899–1904. doi: 10.3892/ol.2017.5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Van Waes C, Musbahi O. Genomics and advances towards precision medicine for head and neck squamous cell carcinoma. Laryngoscope Investig Otolaryngol. 2017;2:310–319. doi: 10.1002/lio2.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Roy DM, Walsh LA, Chan TA. Driver mutations of cancer epigenomes. Protein Cell. 2014;5:265–296. doi: 10.1007/s13238-014-0031-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015;15:334–346. doi: 10.1038/nrc3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xia M, Xu L, Leng Y, Gao F, Xia H, Zhang D, Ding X. Downregulation of MLL3 in esophageal squamous cell carcinoma is required for the growth and metastasis of cancer cells. Tumour Biol. 2015;36:605–613. doi: 10.1007/s13277-014-2616-3. [DOI] [PubMed] [Google Scholar]
- 101.Wang C, Lee JE, Lai B, Macfarlan TS, Xu S, Zhuang L, Liu C, Peng W, Ge K. Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc Natl Acad Sci U S A. 2016;113:11871–11876. doi: 10.1073/pnas.1606857113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23:4225–4231. doi: 10.1038/sj.onc.1207118. [DOI] [PubMed] [Google Scholar]
- 103.Sen GL, Boxer LD, Webster DE, Bussat RT, Qu K, Zarnegar BJ, Johnston D, Siprashvili Z, Khavari PA. ZNF750 is a p63 target gene that induces KLF4 to drive terminal epidermal differentiation. Dev Cell. 2012;22:669–677. doi: 10.1016/j.devcel.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Boxer LD, Barajas B, Tao S, Zhang J, Khavari PA. ZNF750 interacts with KLF4 and RCOR1, KDM1A, and CTBP1/2 chromatin regulators to repress epidermal progenitor genes and induce differentiation genes. Genes Dev. 2014;28:2013–2026. doi: 10.1101/gad.246579.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hazawa M, Lin DC, Handral H, Xu L, Chen Y, Jiang YY, Mayakonda A, Ding LW, Meng X, Sharma A, Samuel S, Movahednia MM, Wong RW, Yang H, Tong C, Koeffler HP. ZNF750 is a lineage-specific tumour suppressor in squamous cell carcinoma. Oncogene. 2017;36:2243–2254. doi: 10.1038/onc.2016.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pan L, Yang H, Xu C, Chen S, Meng Z, Li K, Chen H. ZNF750 inhibited the malignant progression of oral squamous cell carcinoma by regulating tumor vascular microenvironment. Biomed Pharmacother. 2018;105:566–572. doi: 10.1016/j.biopha.2018.06.001. [DOI] [PubMed] [Google Scholar]
- 107.Otsuka R, Akutsu Y, Sakata H, Hanari N, Murakami K, Kano M, Toyozumi T, Takahashi M, Matsumoto Y, Sekino N, Yokoyama M, Okada K, Shiraishi T, Komatsu A, Iida K, Matsubara H. ZNF750 expression is a potential prognostic biomarker in esophageal squamous cell carcinoma. Oncology. 2018;94:142–148. doi: 10.1159/000484932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Herz HM, Madden LD, Chen Z, Bolduc C, Buff E, Gupta R, Davuluri R, Shilatifard A, Hariharan IK, Bergmann A. The H3K27me3 demethylase dUTX is a suppressor of Notch- and Rb-dependent tumors in Drosophila. Mol Cell Biol. 2010;30:2485–2497. doi: 10.1128/MCB.01633-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang L, Shilatifard A. UTX mutations in human cancer. Cancer Cell. 2019;35:168–176. doi: 10.1016/j.ccell.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 111.Dutto I, Scalera C, Prosperi E. CREBBP and p300 lysine acetyl transferases in the DNA damage response. Cell Mol Life Sci. 2018;75:1325–1338. doi: 10.1007/s00018-017-2717-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Attar N, Kurdistani SK. Exploitation of EP300 and CREBBP lysine acetyltransferases by cancer. Cold Spring Harb Perspect Med. 2017;7:a026534. doi: 10.1101/cshperspect.a026534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li F, Mao G, Tong D, Huang J, Gu L, Yang W, Li GM. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell. 2013;153:590–600. doi: 10.1016/j.cell.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Qiu S, Yang G. Precursor lesions of esophageal cancer in high-risk populations in Henan province, China. Cancer. 1988;62:551–557. doi: 10.1002/1097-0142(19880801)62:3<551::aid-cncr2820620319>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 115.Dawsey SM, Lewin KJ, Wang GQ, Liu FS, Nieberg RK, Yu Y, Li JY, Blot WJ, Li B, Taylor PR. Squamous esophageal histology and subsequent risk of squamous cell carcinoma of the esophagus. A prospective follow-up study from Linxian, China. Cancer. 1994;74:1686–1692. doi: 10.1002/1097-0142(19940915)74:6<1686::aid-cncr2820740608>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 116.Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, Shiozawa Y, Sato Y, Aoki K, Kim SK, Fujii Y, Yoshida K, Kataoka K, Nakagawa MM, Inoue Y, Hirano T, Shiraishi Y, Chiba K, Tanaka H, Sanada M, Nishikawa Y, Amanuma Y, Ohashi S, Aoyama I, Horimatsu T, Miyamoto Si, Tsunoda S, Sakai Y, Narahara M, Brown JB, Sato Y, Sawada G, Mimori K, Minamiguchi S, Haga H, Seno H, Miyano S, Makishima H, Muto M, Ogawa S. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. 2019;565:312–317. doi: 10.1038/s41586-018-0811-x. [DOI] [PubMed] [Google Scholar]
- 117.Chen XX, Zhong Q, Liu Y, Yan SM, Chen ZH, Jin SZ, Xia TL, Li RY, Zhou AJ, Su Z, Huang YH, Huang QT, Huang LY, Zhang X, Zhao YN, Yun JP, Wu QL, Lin DX, Bai F, Zeng MS. Genomic comparison of esophageal squamous cell carcinoma and its precursor lesions by multi-region whole-exome sequencing. Nat Commun. 2017;8:524. doi: 10.1038/s41467-017-00650-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu X, Zhang M, Ying S, Zhang C, Lin R, Zheng J, Zhang G, Tian D, Guo Y, Du C, Chen Y, Chen S, Su X, Ji J, Deng W, Li X, Qiu S, Yan R, Xu Z, Wang Y, Guo Y, Cui J, Zhuang S, Yu H, Zheng Q, Marom M, Sheng S, Zhang G, Hu S, Li R, Su M. Genetic alterations in esophageal tissues from squamous dysplasia to carcinoma. Gastroenterology. 2017;153:166–177. doi: 10.1053/j.gastro.2017.03.033. [DOI] [PubMed] [Google Scholar]
- 119.Cao W, Wu W, Yan M, Tian F, Ma C, Zhang Q, Li X, Han P, Liu Z, Gu J, Biddle FG. Multiple region whole-exome sequencing reveals dramatically evolving intratumor genomic heterogeneity in esophageal squamous cell carcinoma. Oncogenesis. 2015;4:e175. doi: 10.1038/oncsis.2015.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hao JJ, Lin DC, Dinh HQ, Mayakonda A, Jiang YY, Chang C, Jiang Y, Lu CC, Shi ZZ, Xu X, Zhang Y, Cai Y, Wang JW, Zhan QM, Wei WQ, Berman BP, Wang MR, Koeffler HP. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat Genet. 2016;48:1500–1507. doi: 10.1038/ng.3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yan T, Cui H, Zhou Y, Yang B, Kong P, Zhang Y, Liu Y, Wang B, Cheng Y, Li J, Guo S, Xu E, Liu H, Cheng C, Zhang L, Chen L, Zhuang X, Qian Y, Yang J, Ma Y, Li H, Wang F, Liu J, Liu X, Su D, Wang Y, Sun R, Guo S, Li Y, Cheng X, Liu Z, Zhan Q, Cui Y. Multi-region sequencing unveils novel actionable targets and spatial heterogeneity in esophageal squamous cell carcinoma. Nat Commun. 2019;10:1670. doi: 10.1038/s41467-019-09255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yuan W, Liu Z, Wang Y, Liu M, Pan Y, Lei W, Yang H, Xu R, Zhang L, Cai H, Li J, Ke Y. Clonal evolution of esophageal squamous cell carcinoma from normal mucosa to primary tumor and metastases. Carcinogenesis. 2019;40:1445–1451. doi: 10.1093/carcin/bgz162. [DOI] [PubMed] [Google Scholar]
- 123.Tabassum DP, Polyak K. Tumorigenesis: it takes a village. Nat Rev Cancer. 2015;15:473–483. doi: 10.1038/nrc3971. [DOI] [PubMed] [Google Scholar]
- 124.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]