

Abstract
Hepatocellular carcinoma (HCC) is a common cancer with high morbidity and mortality. Poorer differentiation status indicates worse prognosis of HCC patients. Regain of better differentiation status may improve the prognosis. Differentiation therapy for HCC is based on the fact that agents may reverse the dedifferentiation process from hepatocytes to HCC cells and thus improve tumor differentiation status. Reversal of progenitor-like property and restoration of hepatic characteristics are main objectives of HCC differentiation therapy. Comprehending the mechanisms of HCC dedifferentiation provides ideas for drug design. Diverse dysregulated molecules and signalings cooperatively cause HCC dedifferentiation. Dysregulation of liver enriched transcription factors, especially hepatocyte nuclear factor 4α, was a critical determinant of HCC dedifferentiation. Aberrant pivotal signaling molecules such as transforming factor-β, β-catenin and Yes-associated protein caused disordered signalings, which promoted HCC dedifferentiation. Loss of epithelial morphology during epithelial-mesenchymal transition (EMT) concurred with HCC dedifferentiation. Some EMT-related molecules exerted double-sided role in concurrently inducing EMT and HCC dedifferentiation. Besides, microRNAs (e.g. miR-122 and miR-148a) as well as some impressive proteins (i.e. KLF4, gankyrin and CHD1L) functioned in manipulating HCC differentiation status. Restoring normal expression levels of these molecules could induce HCC differentiation and inhibited malignant tumor behaviors. Based on the knowledge above, some agents have been found effective in lab, but need more data to support their reliability. Additionally, peretinoin as a potential drug is in progress of several phase III clinical trials. It’s promising that differentiation therapy for HCC may be a part of options in future HCC treatment.
Keywords: Hepatocellular carcinoma, dedifferentiation, molecular mechanism, differentiation therapy, liver cancer stem cell, epithelial-mesenchymal transition
Introduction
Liver cancer is the sixth most common cancer and the fourth leading cause of cancer-related death worldwide. Hepatocellular carcinoma (HCC) is the major subtype of liver cancer, and comprises 75%-85% of the cases [1]. Stepwise dedifferentiation is a certain event in the development of HCC. Regenerative nodules in cirrhotic liver incipiently breed dysplastic nodules (DNs). Then, DNs gradually develop into well, moderately and poorly differentiated HCCs [2]. Many clinical reports have highlighted that HCC patients with poorer tumor differentiation had worse prognosis [3-5]. It can be speculated that regain of better differentiation status may improve the prognosis of HCC patients. Therefore, differentiation therapy focusing on inducing tumor differentiation may be a promising strategy for HCC.
Recent convincing evidence from murine models suggested that HCCs originated from hepatocytes but never from the progenitor/biliary compartment [6]. During the dedifferentiation process, hepatocytes transform into neoplastic hepatocytes, then dedifferentiate into HCC cells, and partially become HCC cells with hepatic progenitor markers [7]. Several hepatic progenitor markers such as epithelial cell adhesion molecule (EpCAM), CD90 and CD133 are useful for identifying and isolating liver cancer stem cell (LCSCs) [8]. Expansion of these hepatic progenitor markers is a sign of HCC dedifferentiation. In addition, HCC dedifferentiation is accompanied by the loss of hepatic functions and morphology, whose alterations can be reflected by detecting hepatocyte markers including functional proteins and surface markers [9,10]. Therefore, reversal of progenitor-like property and restoration of hepatocyte differentiation are considered as the main objectives of HCC differentiation therapy (Figure 1). Diverse dysregulated molecules and signaling pathways cooperatively contribute to HCC dedifferentiation. Regulation of relevant molecules may contribute to better differentiation status. Knowledge of the underlying mechanisms of HCC dedifferentiation provides profound insights for drug design. In this review, we summarized potent molecules that manipulate HCC differentiation status and emerging agents for inducing HCC differentiation as well as depicted the prospect of differentiation therapy in HCC.
Figure 1.

HCC differentiation therapy aims to reverse HCC dedifferentiation process. HCC dedifferentiation is a stepwise process from hepatocytes to LCSCs exhibiting two obvious characteristics. These two characteristics serve as the objectives of HCC differentiation therapy to reverse the dedifferentiation process.
Potent molecules manipulate HCC differentiation status
Liver enriched transcription factors
Hepatocyte differentiation is results from the vitality of liver enriched transcription factors (LETFs) including hepatocyte nuclear factor 1 (HNF1), HNF3, HNF4, HNF6 and CCAAT/enhancer binding protein (C/EBP). The relative abundance of LETFs greatly impacts hepatocyte differentiation [11]. In hepatocarcinogenesis, the expression levels of LETFs are frequently changed. In a mouse model of tumor progression, cellular dedifferentiation that occurred during HCC progression was accompanied by decreased mRNA expression of LETFs [12]. HNF4α is a central regulator among LETFs. Hepatocyte-specific knockdown of HNF4α in a mouse model significantly increased the size and number of diethylnitrosamine (DEN)-induced hepatic tumors [13]. Reexpression of HNF4α induced differentiation of HCC cells in vitro and led to a less progressive phenotype in vivo [12]. Adenovirus-mediated gene delivery of HNF4α gene (Ad-HNF4α) into HCC cells induced cell differentiation by re-establishing expression profile of hepatocyte and decreasing hepatic progenitor markers. Besides, systemic injection and intratumoral injection of Ad-HNF4α inhibited tumor growth in mouse models [14]. HNF4γ2 is an isoform variant of HNF4α with similar binding affinity to the targets of HNF4α. HNF4γ2 induced expression of typical HNF4α target genes to a greater degree than did HNF4α. Evidence showed that HNF4γ2 could restore dedifferentiated hepatoma cells to a more differentiated state by improving expression of hepatocyte markers and critical hepatic functions [15]. HNF1α is implicated in hepatocyte differentiation and regulate most liver-specific genes at transcriptional level. Bi-allelic inactivation of HNF1α gene (also known as HNF1 or TCF1) was frequently found in DNs, indicating that the loss of HNF1α was an early event in hepatocarcinogenesis [16]. Decreased expression of HNF1α was observed in human HCC samples and HCC cells. Forced reexpression of HNF1α in HCC cells significantly suppressed their malignant behaviors in vitro and abolished their tumorigenicity in vivo [17]. An adenovirus vector expressing HNF1α gene (Ad-HNF1α) was transfected to hepatoma cells, which induced differentiation of hepatoma cells and abolished their tumorigenicity in vivo. Besides, injection of Ad-HNF1α could inhibit HCC xenograft growth in mice [17]. HNF1β as an isoform of HNF1α seems to play an opposite role in regulating HCC differentiation status since higher expression of HNF-1β suggested a worse prognosis of HCC patients. In addition, HNF-1β promoted the dedifferentiation of HCC cells into LCSCs via activation of the Notch signaling, and enhanced tumorigenicity of HCC cells in vivo [18]. HNF6 is a member of LETFs and is able to directly active HNF3α and HNF4 at the transcriptional level [11]. Our study found that the expression level of HNF6 in human HCCs was significantly lower than those in the paired nontumor samples, and HNF6 expression level was negatively correlated with HCC differentiation grade. Furthermore, forced expression of HNF6 in poor differentiated HCC cells elicited distinguishable morphology changes toward well-differentiated phenotype and increased expression of hepatocyte markers, whereas inhibiting HNF6 expression repressed expression of hepatocyte markers [10]. C/EBPα also functions as a tumor suppressor protein. Phosphorylation of C/EBPα at Ser193 (Ser190 in human protein) enhanced its tumor suppressor activities, whereas de-phosphorylation of C/EBPα at Ser193 caused dedifferentiation of hepatocytes into cancer cells with pre-neoplastic foci that gave rise to HCC [19]. Reebye et al. have designed an injectable short-activating RNA to enhance C/EBPα expression, which improved liver function and simultaneously reduced tumor burden in a rat model [20]. Collectively, normalization of LTEFs may acquire better differentiation status of HCC.
Pivotal signaling molecules
Dysregulation of Wnt signaling is a frequent event during hepatocarcinogenesis, and β-catenin is a pivotal signaling molecule representing canonical Wnt/β-catenin signaling. Normally, β-catenin is located at plasma membrane with E-cadherin. However, analysis of a series of patient samples revealed that 58.6% of human HCCs exhibited nuclear β-catenin accumulation. And a large percentage of poorly differentiated HCCs showed nuclear β-catenin at moderate to high levels, while most of well or moderately differentiated HCCs exhibited nuclear β-catenin at low to moderate levels [21]. Nevertheless, nuclear β-catenin accumulation doesn’t mean the direct activation of canonical Wnt/β-catenin signaling since the existence of corresponding antagonists. TCF reporter assay demonstrated more frequent activation of canonical Wnt/β-catenin signaling in well-differentiated HCC cell lines as compared to poorly differentiated HCC cell lines. It could be explained that canonical Wnt/β-catenin signaling was inhibited by Wnt5a (a noncanonical Wnt ligand) in poorly differentiated HCC cells, which was independent of the expression or cellular localization of β-catenin [22]. It could be speculated that canonical Wnt signaling might not play a predominant role in triggering the dedifferentiation process of well-differentiated HCC to poorly differentiated HCC. Thus, nuclear β-catenin may participate in other pathways promoting HCC dedifferentiation. An in vivo study identified that HCCs established by transforming factor (TGF)-β1-treated neoplastic hepatocytes exhibited a less differentiated phenotype than those without TGF-β1 treatment. TGF-β1 treatment caused nuclear β-catenin accumulation, whereas loss of nuclear β-catenin led to a more differentiated HCC [21]. It suggested that TGF-β1 might cause dedifferentiation of neoplastic hepatocytes via nuclear β-catenin. Further in vitro study observed that exogenous TGF-β1 stimulation promoted dedifferentiation of neoplastic hepatocytes [9]. Expression of Interleukin 6 (IL-6) mRNA and protein were found to be upregulated after TGF-β1 stimulation. Also, exogenous IL-6 inhibited expression of hepatocyte markers via the signal transducer and activator of transcription 3 (STAT3). Simultaneously adding exogenous TGF-β1 and a specific STAT3 inhibitor to neoplastic hepatocytes abolished the TGFβ1-induced reduction of hepatocyte markers, indicating that TGF-β1 might mediate neoplastic hepatocytes dedifferentiation via IL-6/STAT3 [9].
Yes-associated protein (YAP), a transcriptional regulator in cooperation with TEAD, is a central effector of the Hippo signaling. Overexpression of YAP was frequently observed in human HCC samples compared with the nontumor counterparts. Also, positive YAP expression was detected in more than half of the cases and was significantly associated with poorer tumor differentiation [23]. Fitamant et al. identified that knockout of YAP in genetically engineered mouse models inhibited a progenitor-like phenotype of HCC. And systemic injection of siRNA to silence YAP restored hepatic differentiation and caused pronounced tumor regression [24]. Besides, they verified that HNF4α was a prominently inhibitory target of YAP/TEAD. Inhibiting YAP expression showed pronounced activation of the HNF4α gene signature (expression of a set of hepatocyte functional genes) [24]. Consistent results have been found in human HCC samples that YAP1 expression increased while HNF4α expression decreased with higher grade HCC compared to lower grade HCC [25]. Further results showed that the HNF4α protein level, not the mRNA level, was downregulated by YAP1 via a ubiquitin proteasome pathway. Reciprocally, HNF4α did not affect the expression of YAP1 but directly competed with YAP for binding to TEAD4. The existence of a double-negative feedback loop between YAP/TEAD and HNF4α has also been evidenced in a DEN-induced rat model [25]. Therefore, YAP1 and YAP/TEAD complex may be promising targets for designing differentiation-inducing agents.
Epithelial-mesenchymal transition-related molecules
Hepatocytes are epithelial cells with highly specialized morphology. Epithelial-mesenchymal transition (EMT) is a multistep biological process where epithelial cells dedifferentiated into a mesenchymal phenotype. To some extent, EMT and HCC dedifferentiation describe the same thing, but with different emphasis. During EMT, loss of epithelial morphology is evidenced by decreased epithelial markers, which concurs with HCC dedifferentiation [26]. For example, one of the common changes is the loss of plasma membrane E-cadherin, an epithelial marker and also a marker of hepatocyte differentiation. In a set of human HCC samples, well-differentiated HCCs exhibited plasma membrane E-cadherin in 63% of cases, whereas poorly differentiated human HCCs frequently displayed loss or redistribution of plasma membrane localization. The loss of E-cadherin could cause disruption of normal plasma membrane E-cadherin/β-catenin complexes and further led to accumulation of nuclear β-catenin [21].
Snail, Twist or ZEB are key EMT-inducing transcription factors which are involved in the downregulation of E-cadherin, and TGF-β has been widely proposed as a major inducer of these transcription factors [26]. We have described in the previous section that TGF-β could promote HCC dedifferentiation, indicating its multifaceted roles. Wu et al. confirmed that TGF-β1 could induce dedifferentiation of HCC cells in vitro, and they found that canonical TGF-β1/Smad/Snail1 signaling pathway was involved in the dysregulation of HNF4α [27]. Besides, increased expression level of CD147 mRNA, a tumor-related glycoprotein, was found in the TGF-β1-induced dedifferentiation via the PI3K/AKT signaling. Interestingly, overexpression of CD147 could also induced dedifferentiation of HCC cells. Further results showed that CD147 could induce the expression, secretion and activation of TGF-β1 through upregulation of β-catenin and its nuclear translocation, and thus negatively regulated HNF4α expression via the TGF-β1 signaling [27]. Consistent results have been observed in DEN-treated mice and human HCC samples as well as paired serum samples [27]. Another study found that CD147 was normally located at basolateral membrane of hepatocyte. In human HCC samples, normal localization of CD147 significantly exhibited better differentiation status than disordered localization of CD147 [28]. Additionally, CD147 not only promoted the TGF-β1-mediated loss of morphology, but also directly induced endocytosis and ubiquitination degradation of E-cadherin, which contributed to the loss of hepatic morphology [28]. On the basis of these studies, potential agents designed to target TGF-β1 signaling or CD147 may concurrently induce HCC differentiation and reverse EMT.
MicroRNAs
MicroRNAs (miRNAs) are conserved small non-coding RNAs involved in post-transcriptional gene regulation and play an important role in physiological and pathological processes. Evidence has shown miRNA dysregulation during HCC dedifferentiation. The expression levels of miR-18, precursor miR-18, miR-20 and miR-92 were inversely correlated with the degree of HCC differentiation. In contrast, miR-99a expression exhibited a positive correlation between expression level and tumor differentiation status [29]. Another microarray analysis demonstrated that the expression of miR-18a, miR-18b, miR-221 and miR-423-5p were higher in poorly differentiated HCCs than in well-differentiated HCCs, whereas the expression of miR-22, miR-99a, miR-100, miR-122, miR-215, miR-455-3p, miR-1914 and let-7b were lower in poorly differentiated HCCs than in well-differentiated HCCs [30]. Altered expression of miRNAs may involve in the regulation of HCC differentiation status.
MiR-122 is the most abundant miRNA in the liver. However, miR-122 levels were highly variable among the clinical HCC samples and HCC cell lines. Low miR-122 expression has been shown to be associated with poor differentiation status in human HCCs [31]. In HCC cell lines, high expression level of miR-122 was correlated with high expression of hepatocyte-specific genes [31]. Forced expression of miR-122 to a high level in HepG2 cells could induce mRNA expression of liver functional genes [32]. Several forenamed LETFs (i.e. C/EBPα, HNF1α, HNF3β, and HNF4α) were able to directly bind to miR-122 as transactivators and upregulated miR-122 expression in HCC cells. It indicated that these LETFs may induce HCC differentiation with the help of miR-122 [32,33]. MiR-148a have been identified as a liver specific miRNA highly expressed in adult liver, whose expression was frequently downregulated in samples from HCC patients as well as in mouse and human HCC cell lines [34]. Furthermore, miR-148a expression was lower in poorly differentiated HCC tissues relative to well-differentiated HCC tissues [35]. Overexpression of miR-148a in HCC cell lines led to a significant increase of hepatocyte markers. Intraperitoneal injection of nanoliposomal miR-148a in a genetically engineered mouse model induced a better differentiated phenotype of HCC and also improved liver functions [34,36].
Other impressive proteins
Krüppel-like factor 4 (KLF4) is a transcription factor functioning as an imperative regulator of cell differentiation, and is physiologically expressed in terminally differentiated hepatocytes [10]. Our previous results demonstrated that KLF4 expression in human HCC samples was remarkably lower than that in paired nontumor specimens, and decreased KLF4 expression was closely correlated with poor tumor differentiation [10]. KLF4 was found to induce differentiation of poorly differentiated HCC cells via its transcriptional activity and synchronously upregulated HNF6 expression. HNF6 knockdown evidently reversed the KLF4-induced differentiation. Further mechanistic study revealed that KLF4 could bind to the HNF6 promoter and transactivated HNF6 transcription [10].
Gankyrin, also known as a proteasome regulatory subunit, was overexpressed in human HCC, whose expression was inversely correlated with the differentiation status of human HCCs. Consistent results have been observed in a DEN-treated Wistar rat model [37]. Overexpressed gankyrin in primary rat hepatocytes facilitated the dedifferentiation of hepatocytes to LCSCs, while inhibition of gankyrin expression improved differentiation status of hepatoma cells and their subcutaneous xenograft [37]. A notably inverse correlation between gankyrin and HNF4α was found in human HCCs and the DEN-treated rats. A mechanistic study showed that gankyrin could bind to HNF4α and subsequently enhanced proteasome-dependent degradation of HNF4α, indicating that gankyrin might mediate dedifferentiation process by downregulating HNF4α [37].
Chromodomain-helicase-DNA-binding-protein 1-like (CHD1L) is a genome-wide chromatin remodeling protein found frequently amplified in HCC. CHD1L expression increased progressively from well-differentiated to moderately and poorly differentiated human HCCs [38]. Overexpression of CHD1L could promote HCC dedifferentiation both in vitro and in vivo, whereas knockdown of CHD1L significantly upregulated hepatocyte markers in HCC cells and sensitized HCC cells to sorafenib treatment [38]. Additionally, CHD1L could bind to the 5’-untranslated region of TCF4 gene and activated TCF4 transcription in HCC cells. In HCC cells, overexpressed TCF4 exhibited worse differentiation status, whereas knockdown of TCF4 partially abolished the effects of CHD1L in HCC dedifferentiation, suggesting that CHD1L facilitated HCC dedifferentiation via TCF4 and unknown downstream factors [38].
Emerging agents for inducing HCC differentiation
ATRA and arsenic trioxide (As2O3) are clinically promising drugs for the treatment of leukemia. ATRA-treated HCC cell lines showed better differentiation features reflected by elevated expression of differentiation marker E-cadherin and HNF4α, and reduced expression of LCSC marker CD90 and CD133 [27]. A mechanistic study demonstrated that binding of ATRA to retinoic acid receptor (RAR) induced inactivation of the PI3K/AKT pathway, thus enhancing GSK-3β-dependent phosphorylation of β-catenin, which attenuated the function of β-catenin in maintaining the quiescent status of LCSCs [39]. Moreover, ATRA could induce differentiation of EpCAM+ LCSCs both in vitro and in vivo. The combined administration of ATRA with cisplatin or sorafenib potentiated the therapeutic effect of a single chemotherapeutic agent [40,41]. Likewise, As2O3 could induce differentiation of LCSCs by downregulating expression of GLI, a pivotal transcription factor in Hedgehog pathway, and prevented tumor recurrence after radical resection in mice [42].
Some molecule-targeting agents have been found effective in lab and exert functions both in vitro and in vivo. Oncostatin M (OSM) is an IL-6-related cytokine that induced hepatocyte differentiation of EpCAM+ HCC cells in an OSM receptor-specific manner [43]. Combination of OSM with 5-fluorouracil (5-FU) dramatically increased the number of apoptotic cells in vitro and suppressed tumor growth in vivo as compared to saline control, OSM, or 5-FU treatment alone [43]. Mouse chemokine C-X-C motif ligand 1 (mCXCL1) as the mouse homolog of human IL-8 (hCXCL8) could block differentiation of premalignant cells and activated quiescence in LCSCs by activating mTOR complex 1 (mTORC1) kinase [44]. Blocking of the mTORC1 kinase induced differentiation of LCSCs and facilitated their elimination using the chemotherapeutic drug doxorubicin [44]. Stearoyl-CoA desaturase-1 (SCD1) was one of the most upregulated enzymes in a subset of LCSCs. Suppression of SCD1 by a specific inhibitor forced LCSCs to differentiate, and augmented the efficacy of sorafenib in vivo [45]. A demethylating compound 5-azacytidine (5-AZA) could promote expression of characteristic hepatocyte markers in HCC cells via CpG demethylation of these genes. Injection of 5-AZA into mouse models inhibited tumor growth and restored hepatic differentiation, demonstrating the therapeutic significance of epigenetic reconditioning [46].
We have summarized the potential differentiation-inducing agents for HCC in Table 1. These agents have multifaceted effects that 1) induce cell differentiation and inhibit malignant behaviors of hepatoma cells; 2) activate quiescence and promote maturation of LCSCs; 3) reverse progenitor-like property as well as suppress HCC growth and metastasis in vivo. Currently, available systemic therapy is unable to completely destroy hepatoma cells, especially LCSCs. These agents may facilitate drug sensitivity of systemic therapy and compensate for its limitation.
Table 1.
Emerging agents for inducing HCC differentiation
| Compounds | Mechanisms | Functions | Cell lines and models | Ref. |
|---|---|---|---|---|
| ATRA | inactivation of PI3K/AKT pathway enhancing GSK-3β-dependent phosphorylation of β-catenin | (i) Induce differentiation | (i) Cell lines (SMMC-7721, BEL-7402 and HepG2) | [27,39] |
| (ii) Inhibit proliferation | ||||
| (iii) Enhance the drug sensitivity to docetaxel | ||||
| ATRA + cisplatin | Inhibition of AKT phosphorylation in Thr308 | (i) Induce differentiation and apoptosis | (i) Cell lines (Huh7 and CSQT-2) | [40] |
| (ii) Inhibit proliferation and migration | (ii) Cell-derived xenograft (HuH7 and CSQT-2) | |||
| (iii) Enhance the drug sensitivity to cisplatin | ||||
| (iiii) Inhibit HCC metastasis | ||||
| ATRA + sorafenib | Inhibition of AKT phosphorylation | (i) Induce differentiation | (i) Cell lines (Huh7) | [41] |
| (ii) Inhibit proliferation | (ii) Patient-derived xenograft | |||
| (iii) Enhance the drug sensitivity to sorafenib | ||||
| (iiii) Inhibit HCC growth | ||||
| As2O3 | Downregulation of the GLI1 expression | (i) Induce differentiation | (i) Cell lines (Huh7 and Hep3B) | [42] |
| (ii) Inhibit self-renewal ability | (ii) Cell-derived xenograft (Huh7 and Hep3B) | |||
| (iii) Inhibit tumorigenic capacity and HCC recurrence | ||||
| OSM | Unknown | (i) Induce differentiation | (i) Cell lines (Huh1 and Huh7) | [43] |
| (ii) Enhance the drug sensitivity to 5-FU | (ii) Cell-derived xenograft (Huh1 and Huh7) | |||
| (iii) Inhibit HCC growth | (iii) Patient-derived xenograft | |||
| Rad001 | Inhibition of mTORC1 | (i) Induce differentiation | (i) Cell lines (HepT1, HepG2, Huh6 and Huh7) | [44] |
| (ii) Enhance the drug sensitivity to doxorubicin | (ii) Cell-derived xenograft (Huh7) | |||
| (iii) Inhibit HCC growth | ||||
| A939572 | Inhibition of SCD1 | (i) Induce differentiation | (i) Cell lines (PLC/PRF/5 and Huh7) | [45] |
| (ii) Inhibit self-renewal ability, migration and invasion | (ii) Patient-derived xenograft | |||
| (iii) Enhance the drug sensitivity to sorafenib | ||||
| (iiii) Inhibit HCC growth | ||||
| 5-AZA | CpG demethylation | (i) Induce differentiation | (i) Cell lines (HepG2, Hep3B and Huh7) | [46] |
| (ii) Inhibit tumorigenicity | (ii) Cell-derived xenograft (HepG2 and Hep3B) | |||
| (iii) Enhance the drug sensitivity to sorafenib | ||||
| (iiii) Inhibit HCC growth |
Abbreviations: ATRA, all-trans retinoic acid; As2O3, arsenic trioxide; OSM, oncostatin M; SCD1, stearoyl-CoA desaturase-1; 5-AZA, 5-azacytidine.
Conclusions and perspectives
HCC is a global medical problem due to its high morbidity and mortality. Surgical treatments (e.g. liver resection and transplantation) and local regional strategies (e.g. ablation and chemoembolization) remain the mainstay to maximally increase survival. But for the patients whose tumors progress to advanced stage or those with newly diagnosed advanced HCC, systemic therapy is their only choice. In recent years, several systemic drugs have substantially changed the field of palliative strategies in patients with advanced HCC. Sorafenib and lenvatinib are currently the first line treatment for patients with advanced HCC. Regorafenib, cabozantinib and ramucirumab are approved second line medication [47]. Although current sequential approaches have evolved to benefit HCC patients irrespective of the stage of disease at diagnosis, the treatment decision pivots around what can be done rather than what is worth being done. Oncologists and surgeons have focused on adjuvant therapy of HCC for a long time. Excellent results have been achieved in other solid tumors such as colorectal cancer and breast cancer. To date, there is no widely accepted adjuvant option to reliably improve survival of HCC patients. Novel therapeutic options are urgently required to prevent recurrence after curative therapy or progression after effective chemoembolisation. To fill the gap, HCC differentiation therapy is proposed. The concept of differentiation therapy is based on the fact that agents may induce differentiation of cancer cells into benign or normal cells. Its landmark success is the treatment of acute promyelocytic leukaemia [48]. HCC differentiation therapy might be designed as either the single use of differentiation-inducing agents or the combined administration of differentiation inducers and other therapeutic drugs. It might act as a neoadjuvant therapy for HCC downstaging or could serve as an adjuvant strategy to reduce postoperative recurrence, which would increase tumor resectability and improve prognosis.
Application of HCC differentiation therapy is based on the development of differentiation-inducing drugs. Discovery of the molecular mechanisms of HCC dedifferentiation contributes to the development of new agents. Accumulated evidence reveals that dysregulated molecules and aberrant signaling pathways cooperatively cause HCC dedifferentiation. We drew Figure 2 to show predominant molecular mechanisms of HCC dedifferentiation, among which the PI3K/AKT signaling has been widely confirmed. Peretinoin is an orally synthetic retinoid with a RAR agonist activity (that could inhibit the PI3K/AKT signaling) as the only potential drug currently for secondary chemoprevention of HCC after curative therapy. It is worth mentioning that peretinoin has been shown to reduce the postoperative recurrence of hepatitis C virus-related HCC in a phase II/III randomized controlled study [49,50]. The significant reduction in recurrence after 2 years (600 mg group vs. the placebo group) was clinically meaningful (hazard ratio (HR) 0.27, 95% confidence cinterval (CI) 0.07-0.96). In the subset of patients with Child-Pugh A, the 5-year survival of the 600 mg group was significantly longer than the placebo group (HR 0.575, 95% CI 0.341-0.967, P=0.0347). Although achieving positive results, the study indicated that only a specific group could reliably benefit from peretinoin. Several ongoing phase III clinical trials expand recruitment criteria and patient groups, which are promising to further identify the targeted groups of peretinoin.
Figure 2.
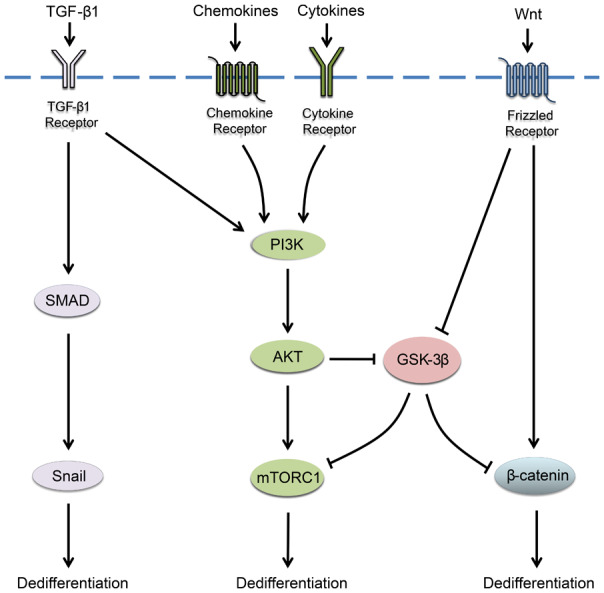
Predominant molecular mechanisms of HCC dedifferentiation. Oncogenic TGF-β/SMAD signaling, PI3K/AKT signaling and Wnt/β-catenin signaling are widely activated in HCC and cooperatively promote HCC dedifferentiation.
The need for effective therapeutic agents is continuous. Although a growing number of agents are shown to induce HCC differentiation, most of the agents are only indentified to be effective in lab, and they need more data to support their reliability. Elaborative preclinical studies are necessary to ensure their safety, tolerability and efficiency. Besides, in vivo assessment of tumor differentiation status is more difficult in HCC than in leukaemia. It’s hard to directly confirm whether a differentiation inducer functions at a tissue level, since repeated liver biopsy has high risk. Therefore, developing a non-invasive method to stratify HCC differentiation status by detecting reliable biomarkers (i.e. liquid biopsy) is of great value. In addition, the characteristics of patients and their tumors as well as potential biomarkers may guide the patients who will benefit from the differentiation therapy. Existing treatment modalities for HCC are evolving with emerging agents. Differentiation therapy for HCC holds great promise for being part of options in future HCC treatment.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant number 81672846).
Disclosure of conflict of interest
None.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Kojiro M. Histopathology of liver cancers. Best Pract Res Clin Gastroenterol. 2005;19:39–62. doi: 10.1016/j.bpg.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Eguchi S, Kanematsu T, Arii S, Omata M, Kudo M, Sakamoto M, Takayasu K, Makuuchi M, Matsuyama Y, Monden M Liver Cancer Study Group of Japan. Recurrence-free survival more than 10 years after liver resection for hepatocellular carcinoma. Br J Surg. 2011;98:552–7. doi: 10.1002/bjs.7393. [DOI] [PubMed] [Google Scholar]
- 4.Sasaki K, Matsuda M, Ohkura Y, Kawamura Y, Inoue M, Hashimoto M, Ikeda K, Kumada H, Watanabe G. The influence of histological differentiation grade on the outcome of liver resection for hepatocellular carcinomas 2 cm or smaller in size. World J Surg. 2015;39:1134–41. doi: 10.1007/s00268-014-2806-6. [DOI] [PubMed] [Google Scholar]
- 5.Guerrini GP, Pinelli D, Di Benedetto F, Marini E, Corno V, Guizzetti M, Aluffi A, Zambelli M, Fagiuoli S, Luca MG, Lucianetti A, Colledan M. Predictive value of nodule size and differentiation in HCC recurrence after liver transplantation. Surg Oncol. 2016;25:419–428. doi: 10.1016/j.suronc.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Mu X, Espanol-Suner R, Mederacke I, Affo S, Manco R, Sempoux C, Lemaigre FP, Adili A, Yuan D, Weber A, Unger K, Heikenwälder M, Leclercq IA, Schwabe RF. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J Clin Invest. 2015;125:3891–903. doi: 10.1172/JCI77995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sia D, Villanueva A, Friedman SL, Llovet JM. Liver Cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology. 2017;152:745–761. doi: 10.1053/j.gastro.2016.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest. 2013;123:1911–8. doi: 10.1172/JCI66024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubois-Pot-Schneider H, Fekir K, Coulouarn C, Glaise D, Aninat C, Jarnouen K, Le Guevel R, Kubo T, Ishida S, Morel F, Corlu A. Inflammatory cytokines promote the retrodifferentiation of tumor-derived hepatocyte-like cells to progenitor cells. Hepatology. 2014;60:2077–90. doi: 10.1002/hep.27353. [DOI] [PubMed] [Google Scholar]
- 10.Sun H, Tang H, Xie D, Jia Z, Ma Z, Wei D, Mishra L, Gao Y, Zheng S, Xie K, Peng Z. Kruppel-like Factor 4 Blocks Hepatocellular Carcinoma dedifferentiation and progression through activation of hepatocyte nuclear factor-6. Clin Cancer Res. 2016;22:502–12. doi: 10.1158/1078-0432.CCR-15-0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayashi Y, Wang W, Ninomiya T, Nagano H, Ohta K, Itoh H. Liver enriched transcription factors and differentiation of hepatocellular carcinoma. Mol Pathol. 1999;52:19–24. doi: 10.1136/mp.52.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazarevich NL, Cheremnova OA, Varga EV, Ovchinnikov DA, Kudrjavtseva EI, Morozova OV, Fleishman DI, Engelhardt NV, Duncan SA. Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology. 2004;39:1038–47. doi: 10.1002/hep.20155. [DOI] [PubMed] [Google Scholar]
- 13.Walesky C, Edwards G, Borude P, Gunewardena S, O’Neil M, Yoo B, Apte U. Hepatocyte nuclear factor 4 alpha deletion promotes diethylnitrosamine-induced hepatocellular carcinoma in rodents. Hepatology. 2013;57:2480–90. doi: 10.1002/hep.26251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yin C, Lin Y, Zhang X, Chen YX, Zeng X, Yue HY, Hou JL, Deng X, Zhang JP, Han ZG, Xie WF. Differentiation therapy of hepatocellular carcinoma in mice with recombinant adenovirus carrying hepatocyte nuclear factor-4α gene. Hepatology. 2008;48:1528–39. doi: 10.1002/hep.22510. [DOI] [PubMed] [Google Scholar]
- 15.Sasaki S, Urabe M, Maeda T, Suzuki J, Irie R, Suzuki M, Tomaru Y, Sakaguchi M, Gonzalez FJ, Inoue Y. Induction of hepatic metabolic functions by a novel variant of hepatocyte nuclear factor 4γ. Mol Cell Biol. 2018;38 doi: 10.1128/MCB.00213-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bluteau O, Jeannot E, Bioulac-Sage P, Marques JM, Blanc JF, Bui H, Beaudoin JC, Franco D, Balabaud C, Laurent-Puig P, Zucman-Rossi J. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat Genet. 2002;32:312–5. doi: 10.1038/ng1001. [DOI] [PubMed] [Google Scholar]
- 17.Zeng X, Lin Y, Yin C, Zhang X, Ning BF, Zhang Q, Zhang JP, Qiu L, Qin XR, Chen YX, Xie WF. Recombinant adenovirus carrying the hepatocyte nuclear factor-1alpha gene inhibits hepatocellular carcinoma xenograft growth in mice. Hepatology. 2011;54:2036–47. doi: 10.1002/hep.24647. [DOI] [PubMed] [Google Scholar]
- 18.Zhu JN, Jiang L, Jiang JH, Yang X, Li XY, Zeng JX, Shi RY, Shi Y, Pan XR, Han ZP, Wei LX. Hepatocyte nuclear factor-1beta enhances the stemness of hepatocellular carcinoma cells through activation of the Notch pathway. Sci Rep. 2017;7:4793. doi: 10.1038/s41598-017-04116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cast A, Valanejad L, Wright M, Nguyen P, Gupta A, Zhu L, Shin S, Timchenko N. C/EBPα-dependent preneoplastic tumor foci are the origin of hepatocellular carcinoma and aggressive pediatric liver cancer. Hepatology. 2018;67:1857–1871. doi: 10.1002/hep.29677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reebye V, Saetrom P, Mintz PJ, Huang KW, Swiderski P, Peng L, Liu C, Liu X, Lindkaer-Jensen S, Zacharoulis D, Kostomitsopoulos N, Kasahara N, Nicholls JP, Jiao LR, Pai M, Spalding DR, Mizandari M, Chikovani T, Emara MM, Haoudi A, Tomalia DA, Rossi JJ, Habib NA. Novel RNA oligonucleotide improves liver function and inhibits liver carcinogenesis in vivo. Hepatology. 2014;59:216–27. doi: 10.1002/hep.26669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zulehner G, Mikula M, Schneller D, van Zijl F, Huber H, Sieghart W, Grasl-Kraupp B, Waldhör T, Peck-Radosavljevic M, Beug H, Mikulits W. Nuclear β-catenin induces an early liver progenitor phenotype in hepatocellular carcinoma and promotes tumor recurrence. Am J Pathol. 2010;176:472–81. doi: 10.2353/ajpath.2010.090300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuzugullu H, Benhaj K, Ozturk N, Senturk S, Celik E, Toylu A, Tasdemir N, Yilmaz M, Erdal E, Akcali KC, Atabey N, Ozturk M. Canonical Wnt signaling is antagonized by noncanonical Wnt5a in hepatocellular carcinoma cells. Mol Cancer. 2009;8:90. doi: 10.1186/1476-4598-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu MZ, Yao TJ, Lee NP, Ng IO, Chan YT, Zender L, Lowe SW, Poon RT, Luk JM. Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer. 2009;115:4576–85. doi: 10.1002/cncr.24495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fitamant J, Kottakis F, Benhamouche S, Tian HS, Chuvin N, Parachoniak CA, Nagle JM, Perera RM, Lapouge M, Deshpande V, Zhu AX, Lai A, Min B, Hoshida Y, Avruch J, Sia D, Campreciós G, McClatchey AI, Llovet JM, Morrissey D, Raj L, Bardeesy N. YAP inhibition restores hepatocyte differentiation in advanced hcc, leading to tumor regression. Cell Rep. 2015;10:1692–1707. doi: 10.1016/j.celrep.2015.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cai WY, Lin LY, Hao H, Zhang SM, Ma F, Hong XX, Zhang H, Liu QF, Ye GD, Sun GB, Liu YJ, Li SN, Xie YY, Cai JC, Li BA. Yes-associated protein/TEA domain family member and hepatocyte nuclear factor 4-alpha (HNF4α) repress reciprocally to regulate hepatocarcinogenesis in rats and mice. Hepatology. 2017;65:1206–1221. doi: 10.1002/hep.28911. [DOI] [PubMed] [Google Scholar]
- 26.Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. 2016;65:798–808. doi: 10.1016/j.jhep.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 27.Wu J, Lu M, Li Y, Shang YK, Wang SJ, Meng Y, Wang Z, Li ZS, Chen H, Chen ZN, Bian H. Regulation of a TGF-β1-CD147 self-sustaining network in the differentiation plasticity of hepatocellular carcinoma cells. Oncogene. 2016;35:5468–5479. doi: 10.1038/onc.2016.89. [DOI] [PubMed] [Google Scholar]
- 28.Lu M, Wu J, Hao ZW, Shang YK, Xu J, Nan G, Li X, Chen ZN, Bian H. Basolateral CD147 induces hepatocyte polarity loss by E-cadherin ubiquitination and degradation in hepatocellular carcinoma progress. Hepatology. 2018;68:317–332. doi: 10.1002/hep.29798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murakami Y, Yasuda T, Saigo K, Urashima T, Toyoda H, Okanoue T, Shimotohno K. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene. 2006;25:2537–45. doi: 10.1038/sj.onc.1209283. [DOI] [PubMed] [Google Scholar]
- 30.Murakami Y, Tamori A, Itami S, Tanahashi T, Toyoda H, Tanaka M, Wu W, Brojigin N, Kaneoka Y, Maeda A, Kumada T, Kawada N, Kubo S, Kuroda M. The expression level of miR-18b in hepatocellular carcinoma is associated with the grade of malignancy and prognosis. BMC Cancer. 2013;13:99. doi: 10.1186/1471-2407-13-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coulouarn C, Factor VM, Andersen JB, Durkin ME, Thorgeirsson SS. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene. 2009;28:3526–36. doi: 10.1038/onc.2009.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu H, He JH, Xiao ZD, Zhang QQ, Chen YQ, Zhou H, Qu LH. Liver-enriched transcription factors regulate microRNA-122 that targets CUTL1 during liver development. Hepatology. 2010;52:1431–42. doi: 10.1002/hep.23818. [DOI] [PubMed] [Google Scholar]
- 33.Zeng C, Wang R, Li D, Lin XJ, Wei QK, Yuan Y, Wang Q, Chen W, Zhuang SM. A novel GSK-3 beta-C/EBP alpha-miR-122-insulin-like growth factor 1 receptor regulatory circuitry in human hepatocellular carcinoma. Hepatology. 2010;52:1702–12. doi: 10.1002/hep.23875. [DOI] [PubMed] [Google Scholar]
- 34.Gailhouste L, Gomez-Santos L, Hagiwara K, Hatada I, Kitagawa N, Kawaharada K, Thirion M, Kosaka N, Takahashi RU, Shibata T, Miyajima A, Ochiya T. miR-148a plays a pivotal role in the liver by promoting the hepatospecific phenotype and suppressing the invasiveness of transformed cells. Hepatology. 2013;58:1153–65. doi: 10.1002/hep.26422. [DOI] [PubMed] [Google Scholar]
- 35.Yan H, Dong X, Zhong X, Ye J, Zhou Y, Yang X, Shen J, Zhang J. Inhibitions of epithelial to mesenchymal transition and cancer stem cells-like properties are involved in miR-148a-mediated anti-metastasis of hepatocellular carcinoma. Mol Carcinog. 2014;53:960–9. doi: 10.1002/mc.22064. [DOI] [PubMed] [Google Scholar]
- 36.Jung KH, Zhang J, Zhou C, Shen H, Gagea M, Rodriguez-Aguayo C, Lopez-Berestein G, Sood AK, Beretta L. Differentiation therapy for hepatocellular carcinoma: multifaceted effects of miR-148a on tumor growth and phenotype and liver fibrosis. Hepatology. 2016;63:864–79. doi: 10.1002/hep.28367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun W, Ding J, Wu K, Ning BF, Wen W, Sun HY, Han T, Huang L, Dong LW, Yang W, Deng X, Li Z, Wu MC, Feng GS, Xie WF, Wang HY. Gankyrin-mediated dedifferentiation facilitates the tumorigenicity of rat hepatocytes and hepatoma cells. Hepatology. 2011;54:1259–72. doi: 10.1002/hep.24530. [DOI] [PubMed] [Google Scholar]
- 38.Liu M, Chen L, Ma NF, Chow RK, Li Y, Song Y, Chan TH, Fang S, Yang X, Xi S, Jiang L, Li Y, Zeng TT, Li Y, Yuan YF, Guan XY. CHD1L promotes lineage reversion of hepatocellular carcinoma through opening chromatin for key developmental transcription factors. Hepatology. 2016;63:1544–59. doi: 10.1002/hep.28437. [DOI] [PubMed] [Google Scholar]
- 39.Zhu X, Wang W, Zhang X, Bai J, Chen G, Li L, Li M. All-trans retinoic acid-induced deficiency of the Wnt/β-catenin pathway enhances hepatic carcinoma stem cell differentiation. PLoS One. 2015;10:e0143255. doi: 10.1371/journal.pone.0143255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Guan DX, Shi J, Gao H, Li JJ, Zhao JS, Qiu L, Liu J, Li N, Guo WX, Xue J, Zhou FG, Wu MC, Wang HY, Xie D, Cheng SQ. All-trans retinoic acid potentiates the chemotherapeutic effect of cisplatin by inducing differentiation of tumor initiating cells in liver cancer. J Hepatol. 2013;59:1255–63. doi: 10.1016/j.jhep.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 41.Guan DX, Shi J, Zhang Y, Zhao JS, Long LY, Chen TW, Zhang EB, Feng YY, Bao WD, Deng YZ, Qiu L, Zhang XL, Koeffler HP, Cheng SQ, Li JJ, Xie D. Sorafenib enriches epithelial cell adhesion molecule-positive tumor initiating cells and exacerbates a subtype of hepatocellular carcinoma through TSC2-AKT cascade. Hepatology. 2015;62:1791–803. doi: 10.1002/hep.28117. [DOI] [PubMed] [Google Scholar]
- 42.Zhang KZ, Zhang QB, Zhang QB, Sun HC, Ao JY, Chai ZT, Zhu XD, Lu L, Zhang YY, Bu Y, Kong LQ, Tang ZY. Arsenic trioxide induces differentiation of CD133+ hepatocellular carcinoma cells and prolongs posthepatectomy survival by targeting GLI1 expression in a mouse model. J Hematol Oncol. 2014;7:28. doi: 10.1186/1756-8722-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamashita T, Honda M, Nio K, Nakamoto Y, Yamashita T, Takamura H, Tani T, Zen Y, Kaneko S. Oncostatin m renders epithelial cell adhesion molecule-positive liver cancer stem cells sensitive to 5-Fluorouracil by inducing hepatocytic differentiation. Cancer Res. 2010;70:4687–97. doi: 10.1158/0008-5472.CAN-09-4210. [DOI] [PubMed] [Google Scholar]
- 44.Wolf B, Krieg K, Falk C, Breuhahn K, Keppeler H, Biedermann T, Schmid E, Warmann S, Fuchs J, Vetter S, Thiele D, Nieser M, Avci-Adali M, Skokowa Y, Schöls L, Hauser S, Ringelhan M, Yevsa T, Heikenwalder M, Kossatz-Boehlert U. Inducing differentiation of premalignant hepatic cells as a novel therapeutic strategy in hepatocarcinoma. Cancer Res. 2016;76:5550–61. doi: 10.1158/0008-5472.CAN-15-3453. [DOI] [PubMed] [Google Scholar]
- 45.Ma MKF, Lau EYT, Leung DHW, Lo J, Ho NPY, Cheng LKW, Ma S, Lin CH, Copland JA, Ding J, Lo RCL, Ng IOL, Lee TKW. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J Hepatol. 2017;67:979–990. doi: 10.1016/j.jhep.2017.06.015. [DOI] [PubMed] [Google Scholar]
- 46.Gailhouste L, Liew LC, Yasukawa K, Hatada I, Tanaka Y, Nakagama H, Ochiya T. Differentiation therapy by epigenetic reconditioning exerts antitumor effects on liver cancer cells. Mol Ther. 2018;26:1840–1854. doi: 10.1016/j.ymthe.2018.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301–1314. doi: 10.1016/S0140-6736(18)30010-2. [DOI] [PubMed] [Google Scholar]
- 48.de The H. Differentiation therapy revisited. Nat Rev Cancer. 2018;18:117–127. doi: 10.1038/nrc.2017.103. [DOI] [PubMed] [Google Scholar]
- 49.Okita K, Izumi N, Matsui O, Tanaka K, Kaneko S, Moriwaki H, Ikeda K, Osaki Y, Numata K, Nakachi K, Kokudo N, Imanaka K, Nishiguchi S, Okusaka T, Nishigaki Y, Shiomi S, Kudo M, Ido K, Karino Y, Hayashi N, Ohashi Y, Makuuchi M, Kumada H Peretinoin Study Group. Peretinoin after curative therapy of hepatitis C-related hepatocellular carcinoma: a randomized double-blind placebo-controlled study. J Gastroenterol. 2015;50:191–202. doi: 10.1007/s00535-014-0956-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Okita K, Izumi N, Ikeda K, Osaki Y, Numata K, Ikeda M, Kokudo N, Imanaka K, Nishiguchi S, Kondo S, Nishigaki Y, Shiomi S, Ueshima K, Isoda N, Karino Y, Kudo M, Tanaka K, Kaneko S, Moriwaki H, Makuuchi M, Okusaka T, Hayashi N, Ohashi Y, Kumada H Peretinoin Study Group. Survey of survival among patients with hepatitis C virus-related hepatocellular carcinoma treated with peretinoin, an acyclic retinoid, after the completion of a randomized, placebo-controlled trial. J Gastroenterol. 2015;50:667–74. doi: 10.1007/s00535-014-0996-1. [DOI] [PMC free article] [PubMed] [Google Scholar]