

Abstract
Pulmonary arterial hypertension (PAH), a chronic and progressive disease of the lung vascular system, is characterized by vasculopathy in the pulmonary arterioles, especially in endothelial cells and pulmonary vascular smooth cells. Several mechanisms are involved in PAH occurrence and development, and all are characterized by excessive pulmonary vasoconstriction and abnormal vascular remodeling, which leads to a progressive resistance to blood flow and an increase in pulmonary artery pressure. Recent studies have shown that endoplasmic reticulum (ER) stress is implicated in the pathophysiology of PAH. In this review, we highlight the effect of ER stress on the proliferation and apoptosis of endothelial cells and pulmonary vascular smooth muscle cells, and discuss the feasibility of targeting unfolded protein response components as a strategy to reverse or alleviate the progression of PAH.
Keywords: Pulmonary arterial hypertension (PAH), endoplasmic reticulum (ER) stress, unfolded protein response (UPR), endothelial cells (ECs), pulmonary artery smooth muscle cells (PASMCs)
Introduction
Pulmonary arterial hypertension (PAH) is a serious lung disease that results in right heart failure. The main symptoms of PAH are dyspnea, chest pain, dizziness and syncope; some patients may have hemoptysis. The World Health Organization (WHO) classifies PAH into four groups: primary PAH formerly encompassed idiopathic, familial, and anoxia-induced PAH as group I; other factors induce PAH as group II; PAH with left heart disease is classified as group III; PAH associated with lung diseases and/or hypoxemia comprises group IV; and PAH due to chronic thrombotic and/or embolic disease is classified as group V. Several mechanisms are involved in PAH occurrence and development, and all are characterized by excessive pulmonary vasoconstriction and abnormal vascular remodeling, which leads to progressive resistance to blood flow and an increase in pulmonary artery (PA) pressure [1]. Molecular biology techniques have revealed three signal transduction pathways involved in PAH: the nitric oxide (NO) pathway [2,3], the prostacyclin (PGI2) pathway [4], and the endothelin-1 (ET-1) pathway [5]. The discovery of those key signaling pathways has deepened our understanding of PAH and promoted the treatment of PAH from the era of symptomatic supportive therapy to the era of targeted therapy. Pharmacological interventions, including NO, PGI2, and ET-1 receptor agonists and phosphodiesterase-5 inhibitors, are available, but the therapeutic effect of these drugs is still limited. These diverse pathogenic mechanisms increase the difficulty of treatment and lead to an unsatisfactory prognosis. Studies have shown that the mortality of PAH is 15% within 1 year of modern therapy [6].
The endoplasmic reticulum (ER), an essential cellular organelle, produces diverse molecules ranging from hormones to signaling molecules and regulates systemic metabolic, inflammatory, and endocrine processes and other activities in the body [7]. The ER can be influenced by hypoxia, oxidative injury, toxins, aberrant Ca2+ regulation, viral infection and other physiological and pathological insults during protein folding processes [8], leading to an increase in unfolded proteins. Recent studies have suggested that ER stress is involved in the pathogenesis of PAH through unfolded and/or misfolded proteins [9,10]. To help elucidate the pathomechanism of PAH, in this review, we focus on the relationship between ER stress and PAH at the cellular level and hope to provide a new strategy against PAH progression in clinical settings.
The occurrence and development of PAH
PAH is characterized by excessive pulmonary vasoconstriction and abnormal vascular remodeling [11]. Although the etiology and categories of PAH are diverse and complex, the pathological processes of various types of PAH are similar. Several studies have suggested that inflammation plays a vital role in the initiation and progression of PAH. Cytokines such as tumor necrosis factor, IL-6, and IL-1β were increased in IPAH [12-14], and animal models also support this view [14]. Some chemokines lead to inflammatory cell recruitment [15]. Previous research investigated the levels of vasoactive mediators among patients with PAH, and the results suggested that some mediators (ET-1, thromboxane, 5-hydroxytryptamine) increase while others (PGI2) decrease [16]. These changes may induce or inhibit signaling pathways that regulate the proliferation, apoptosis and remodeling of endothelial cells (ECs) and pulmonary artery smooth muscle cells (PASMCs). Recent studies have reported that the presence of perivascular inflammatory cells and factors might influence the microenvironment, leading to disordered angiogenesis [17,18]. Moreover, increased plasma levels of tissue factor, fibrinopeptide A, and plasminogen activator inhibitor 1 result in a hypercoagulable state in patients [19,20]. In summary, the imbalance in vasoactive mediators, dysfunction of ECs and PASMCs, vascular remodeling, and thrombogenesis mediate the pathogenesis of PAH [11] (Figure 1).
Figure 1.
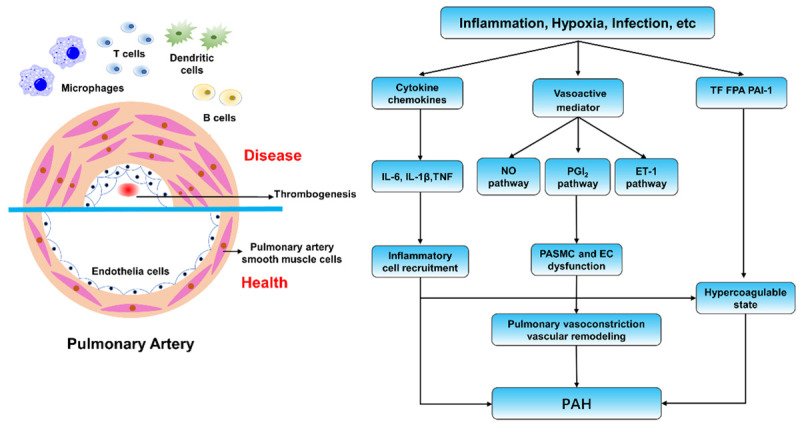
Overview of the mechanism of pulmonary hypertension. Multiple stimuli activate three signal transduction pathways: the nitric oxide (NO) pathway, the prostacyclin (PGI2) pathway, and the endothelin-1 (ET-1) pathway and eventually cause various pathological processes. The imbalance in vasoactive mediators, dysfunction of endothelial and smooth muscle cells, vascular remodeling, and thrombogenesis mediate the pathogenesis of PAH.
ER stress and signal transduction pathways
The ER is one of the most important organelles in eukaryotes and participates in the synthesis, folding, modification and transportation of proteins. Notably, protein folding is the most error-prone step in gene expression [21] (Figure 2). Under normal conditions, the ER chaperone BiP/GRP78 is combined with three canonical unfolded protein response (UPR) sensors, PKR-like eukaryotic initiating factor α kinase (PERK), activating transcription factor-6 (ATF6), and inositol-requiring enzyme 1 (IRE1) [7], and the related downstream pathway is blocked. Oxidative stress, hypoxia, Ca+ disorder or other pathologic conditions will disturb protein folding, leading to an accumulation of unfolded proteins, which results in the separation of BiP/GRP78 and UPR sensors and finally activates the signal transduction pathways.
Figure 2.
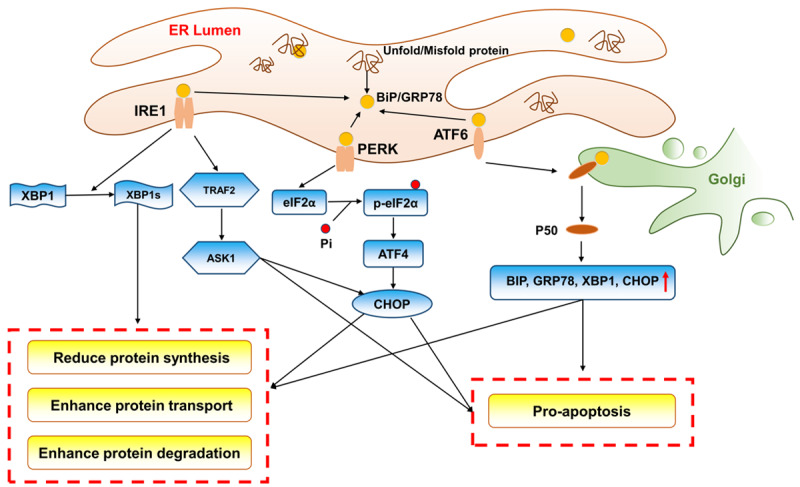
Schematic illustration of ER stress and the activation of the three UPR pathways. Under stressed conditions, the separation of BiP/GRP78 and UPR sensors activates the signal transduction pathways. PERK is activated to phosphorylate eIF2α at Ser51, and this process increases the ATF4 level. DDIT/CHOP is the target of ATF4 and can help to upregulate apoptosis genes and downregulate antiapoptosis genes. IRE1 cleaves a 26-nucleotide segment from XBP1 mRNA to produce a transcriptionally active XBP1 (XBP1s). XBP1 can accelerate protein folding and transport as well as degeneration, and IRE1 can also react directly with TRAF2, leading to activation of the apoptosis signaling pathway and inhibition of the antiapoptosis signaling pathway. ATF6 transits to the Golgi and is processed by the proteases S1P and S2P to become an activated fragment called PSOATF6. PSOATF6 moves into the nucleus and subsequently activates the expression of BiP, ER protein 57, and Grp 94. These proteins enhance the capacity to alleviate protein loading and maintain ER homeostasis through accelerating protein folding, transduction and degradation.
The PERK signaling pathway
PERK, a type-I transmembrane protein, phosphorylates eIF2α at Ser51. On the one hand, eIF2α-P can inhibit the formation of a translation initiation complex to reduce global protein synthesis [22]. On the other hand, this process increases the levels of ATF4, which can enhance protein transportation in the ER [23]. Additionally, DDIT/CHOP is a target of ATF4 and can help upregulate apoptosis genes and downregulate anti-apoptosis genes [24]. When ER stress protein folding homeostasis is restored, ATF4 and CHOP can induce the upregulation of DNA-damage-inducible protein 34 (GADD34) and the dephosphorylation of eIF2α-P, restoring the protein synthesis [25].
The IRE1 signaling pathway
IRE1 is the most evolutionarily conserved bifunctional type-I transmembrane protein of the UPR [8]. This protein cleaves a 26-nucleotide segment from XBP1 mRNA to produce a transcriptionally active XBP1 (XBP1s). XBP1 can accelerate protein folding and transport as well as degeneration, which alleviates protein loading in the ER and reverses the UPR [21]. Additionally, IRE1 can react directly with tumor necrosis-associated receptor 2 (TRAF2) through a series of cascade reactions, leading to activation of the apoptosis signaling pathway and inhibition of the anti-apoptosis signaling pathway [26,27].
The ATF6 signaling pathway
ATF6 is a type-II transmembrane-activating transcription factor. Under ER stress conditions, ATF6 transitions to the Golgi and is processed by the proteases S1P and S2P, yielding an activated fragment called p50. P50 moves to the nucleus and subsequently increases the expression of protein chaperones (binding immunoglobulin protein (BiP), ER protein 57), glucose-regulated proteins (glucose-regulated protein 94 (Grp 94)), and ER-associated proteins [21]. These proteins enhance the capacity to alleviate protein loading and maintain ER homeostasis by accelerating protein folding, transduction and degradation.
Pathological mechanisms of ER stress promoting PAH
PAH is related to excessive pulmonary vasoconstriction and abnormal vascular remodeling, and past research has identified a potential link between ER stress and PAH. Therefore, we hypothesized that ER stress causes vascular pathological changes and thus participates in the occurrence and development of PAH. In this review, we examined the relevant research and literature to explore how ER stress affects the function of vascular ECs and PASMCs.
ECs
Persistent ER stress and the UPR are important pathogenic mechanisms for many chronic diseases, such as neurodegenerative disease, atherosclerosis, type 2 diabetes, liver disease, and cancer [28,29]. Prolonged perturbation of ER stress and UPR activation can cause increased oxidative stress and inflammation in ECs, which often leads to dysfunction and disease [30]. On the one hand, studies have shown that EC injury or dysfunction may be a pivotal pathophysiological mechanism that increases the susceptibility to PAH [31,32]. On the other hand, various studies have proven that damage to ECs can result from the excessive activation of ER stress and the UPR [33-37]. On this basis, Guignabert [38] explored the correlation between PAH and ER stress in ECs using a dasatinib-treated experimental group and a control group treated with imatinib. Dasatinib is a drug used to treat chronic myeloid leukemia and can also induce PAH in humans and rodents, while imatinib cannot. This study examined the levels of endothelial and vascular dysfunction and damage markers, both of which increased significantly in the experimental group compared with the control group. Furthermore, the author examined markers of ER stress and found an increase in the molecular chaperone GRP78, ATF6 and XBP1 in rat lung homogenates and a significant decrease in eIF2α in the dasatinib-treated group but not the control group. These results indicated that EC damage and ER stress caused by dasatinib treatment of chronic myeloid leukemia may be one of the pathological mechanisms. The results of other studies are similar. In another review, the author explored whether the UPR pathways are activated in ECs [39]. They examined the expression of UPR markers in the lung vasculature of patients with systemic sclerosis-associated PAH (SSc-PAH). The researchers found that the BiP and CHOP levels were markedly increased in the vasculature and microphages of SSc-PAH lungs compared with those of the control group. Notably, the increase in CHOP was primarily observed in ECs. These studies investigated the relationship between PAH and ECs through testing the expression of BiP and CHOP as well as other ER stress/UPR markers, and the results indicated that the pathogenesis of PAH is correlated with ER stress and the UPR in ECs.
Interestingly, ER stress does not only influence the function of ECs through these three signal transduction pathways. Past studies have shown that increased levels of ET-1 can be a risk factor in the development of SSc patients with iPHT [40-43]. Further research indicated that the gene HLA-B35 can induce ER stress in ECs, and consequently, the ER stress will increase the expression of ET-1. However, the molecular mechanism of the HLA-B35-ER stress-ET-1 axis requires further research [44].
PASMCs
Pulmonary vascular remodeling is a key component in the pathological process of PAH; therefore, analysis of the proliferation and apoptosis of PAMCs is needed. Under anoxic or other conditions, PAMCs develop a synthetic phenotype, resulting in proliferation, hypertrophy, and migration; activating a series of signal transduction pathways; and eventually participating in the progression of PAH [45]. In the above section, we mainly discuss the role of ECs. In this section, we focus on the relationship between SMCs and PAH.
Therapeutic studies have explored whether drugs that can inhibit ER stress as chemical chaperones can attenuate PAH. In Wu’s research [46], exogenous H2S was verified to inhibit hypoxia-induced PASMC proliferation and reverse proliferation and migration, which correlated with the suppressed expression of UPR markers. These results suggested that using chemical factors to prevent proliferation and migration in PASMCs via inhibition of ER stress and the UPR is effective and significant. From another perspective, Mao. [47] established a PAH model in rats through hypoxia to examine the effect of intermedin (IMD) on hypoxic pulmonary vascular remodeling. IMD is a member of the calcitonin gene-related peptide family that can dilate systemic and pulmonary vessels [48-50]. In this study, the author found that IMD not only reduced hypoxic PAH and right ventricular hypertrophy but also alleviated pulmonary vascular remodeling through inhibiting PASMC proliferation and promoting apoptosis. Notably, the GRP78, GRP94 and caspase-12 protein levels increased in the IMD treatment group, indicating that IMD treatment exacerbates the ER stress apoptosis pathway in PASMCs. These results suggest that drugs that can improve PAMSC function can be used to treat PAH and that the activation of the ER stress apoptosis pathway in PASMCs can be used as a mechanism for antivascular remodeling. Other therapeutic studies yielded similar results; drugs that restrain ER stress could normalize proliferation and induce apoptosis [37,51,52]. During the few past years, our laboratory has focused on the effect of ER stress on PAH. In our research, we demonstrated that 4u8c, an inhibitor of the IRE1α/XBP1 pathway, could restrain hypoxia-induced cell proliferation and migration and reverse hypoxia-induced apoptosis arrest [53]. Thus, suppressing ER stress by altering UPR signaling pathways can normalize PASMC function, which is of therapeutic significance for PAH.
Since the above studies have shown that ER stress and the UPR signaling pathways are associated with the onset of PAH, an abnormal ER structure may also have a negative effect. Nogo, a member of the reticulon family of proteins that regulates the tubular structure of the ER, is implicated in vascular remodeling and in the tissue response to hypoxia [54-56]. Sutendra [57] found that the lack of Nogo-B in PASMCs from Nogo-A/B-deficient mice (missing both the Nogo-A and Nogo-B isoforms) prevented hypoxia-induced changes in vitro and in vivo, resulting in complete resistance to PAH. The researchers also found that the increased levels of Nogo-B during ER stress may lead to a restructuring of the ER and disruption of the mitochondria-ER unit, resulting in mitochondrial hyperpolarization, closure of the mitochondrial transition pore, and suppression of apoptosis. These findings imply that the absence of Nogo, especially Nogo-B, can disrupt PASMC hyperproliferation. The normal structure of the ER is the basis of its normal function. Structural abnormalities can not only affect the function of the ER itself but also cause dysfunction of mitochondria or other related organelles and eventually participate in the development of PAH. Remarkably, the rising phenomenon of Nogo-B does not appear similarly in carotid arteries, which suggested that this kind of artery soomth muscle cells response is vascular selective [58,59].
In addition, multiple studies have identified genes associated with PAH, such as bone morphogenetic protein receptor 2 (BMPR2) [60-63], TGF-β superfamily genes [64], activin receptor-like kinase 1 (ALK1) [65], SMAD1/4/8 [66], and BMPR1B [67]. The guidelines of the European Cardiology Society recommend analyses for genetic alterations in BMPR2, BMPR1B, EIF2AK4, CAV1, KCNK3 and ENG, as BMPR2 has a key role in the etiology of the disease [68]. These genetic studies have substantially increased our understanding of the molecular mechanisms of pulmonary hypertension. Research has shown that PAH can be identified by overexpression of NOTCH3 in PASMCs [69]. On this basis, Chida analyzed two novel NOTCH3 mutants in PAH patients and subsequently established stable cell lines expressing wild-type and mutant NOTCH3. The researchers found that the majority of the protein-folding chaperone GRP78/BiP was translocated into nuclei in the mutant group, while in the wild-type group, GRP78/BiP was located in the ER. These changes might influence unfolded protein degradation or transduction. NOTCH3 mutants impaired NOTCH3-HES5 signaling and affected the proliferation and viability of PASMCs. These results, which assessed the occurrence and development of PAH at a genetic level, may help to illuminate the pathogenesis of PAH. Currently, personalized treatment is the developmental direction of many refractory diseases, such as tumors. For PAH, research on the mechanism at the genetic level is forward-looking, and genes related to ER structure and function can be potential targets.
Summary and perspectives
In this review, we summarized studies of ER stress in PAH, focusing on how ER stress and the UPR interact with ECs and PASMCs (Figure 3). From these studies, we found that ER stress is part of the complex process of the onset and progression of PAH. Many triggers of PAH, including hypoxia, gene mutation, and NOTCH induction, can affect the degree of ER stress. Excessive ER stress and the UPR affect the state and function of ECs and PASMCs, thus influencing the formation and aggravation of pulmonary hypertension. Therefore, targeting ER stress might be a broad-spectrum and effective treatment strategy in PAH. From these studies, we can conclude that the dysfunction of ER stress in ECs and PASMCs exists during the occurrence and development of PAH, but the specific pathway of how ER stress and PAH interact remains unclear. In the future, with the advancement of molecular mechanisms and signaling pathways, there will be an increasing number of new drugs or treatments targeting PAH.
Figure 3.
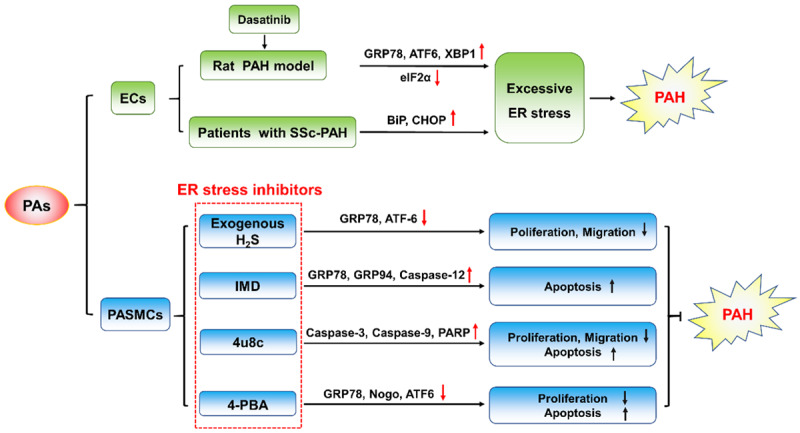
The relationship between ER stress and PAH. Excessive ER stress can cause dysfunction of endothelial cells and smooth muscle cells, leading to excessive pulmonary vasoconstriction and abnormal vascular remodeling and eventually participating in the onset of pulmonary hypertension.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81800068).
Disclosure of conflict of interest
None.
References
- 1.Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res. 2014;115:115–130. doi: 10.1161/CIRCRESAHA.115.301146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roldan T, Landzberg MJ, Deicicchi DJ, Atay JK, Waxman AB. Anticoagulation in patients with pulmonary arterial hypertension: an update on current knowledge. J Heart Lung Transplant. 2016;35:151–164. doi: 10.1016/j.healun.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Lan NSH, Massam BD, Kulkarni SS, Lang CC. Pulmonary arterial hypertension: pathophysiology and treatment. Diseases. 2018;6:38. doi: 10.3390/diseases6020038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med. 2010;104:9–21. doi: 10.1016/j.rmed.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 5.Fulton DJR, Li X, Bordan Z, Haigh S, Bentley A, Chen F, Barman SA. Reactive oxygen and nitrogen species in the development of pulmonary hypertension. Antioxidants (Basel) 2017;6:54. doi: 10.3390/antiox6030054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006. Eur Respir J. 2007;30:1103–1110. doi: 10.1183/09031936.00042107. [DOI] [PubMed] [Google Scholar]
- 7.Ochoa CD, Wu RF, Terada LS. ROS signaling and ER stress in cardiovascular disease. Mol Aspects Med. 2018;63:18–29. doi: 10.1016/j.mam.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: autophagy induction, inhibition and selection. Autophagy. 2015;11:1956–1977. doi: 10.1080/15548627.2015.1091141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakao S, Tatsumi K, Voelkel NF. Endothelial cells and pulmonary arterial hypertension: apoptosis, proliferation, interaction and transdifferentiation. Respir Res. 2009;10:95. doi: 10.1186/1465-9921-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, Krishnamachary B, Zhen L, Mao T, Richter A, Erzurum SC, Tuder RM. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol. 2010;176:1130–1138. doi: 10.2353/ajpath.2010.090832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8:443–455. doi: 10.1038/nrcardio.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151:1628–1631. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 13.Itoh T, Nagaya N, Ishibashi-Ueda H, Kyotani S, Oya H, Sakamaki F, Kimura H, Nakanishi N. Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology. 2006;11:158–163. doi: 10.1111/j.1440-1843.2006.00821.x. [DOI] [PubMed] [Google Scholar]
- 14.Bhargava A, Kumar A, Yuan N, Gewitz MH, Mathew R. Monocrotaline induces interleukin-6 mRNA expression in rat lungs. Heart Dis. 1999;1:126–132. [PubMed] [Google Scholar]
- 15.Dorfmuller P, Zarka V, Durand-Gasselin I, Monti G, Balabanian K, Garcia G, Capron F, Coulomb-Lhermine A, Marfaing-Koka A, Simonneau G, Emilie D, Humbert M. Chemokine RANTES in severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002;165:534–539. doi: 10.1164/ajrccm.165.4.2012112. [DOI] [PubMed] [Google Scholar]
- 16.Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, Loyd JE. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327:70–75. doi: 10.1056/NEJM199207093270202. [DOI] [PubMed] [Google Scholar]
- 17.Partovian C, Adnot S, Eddahibi S, Teiger E, Levame M, Dreyfus P, Raffestin B, Frelin C. Heart and lung VEGF mRNA expression in rats with monocrotaline- or hypoxia-induced pulmonary hypertension. Am J Physiol. 1998;275:H1948–1956. doi: 10.1152/ajpheart.1998.275.6.H1948. [DOI] [PubMed] [Google Scholar]
- 18.Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144:275–285. [PMC free article] [PubMed] [Google Scholar]
- 19.White RJ, Meoli DF, Swarthout RF, Kallop DY, Galaria II, Harvey JL, Miller CM, Blaxall BC, Hall CM, Pierce RA, Cool CD, Taubman MB. Plexiform-like lesions and increased tissue factor expression in a rat model of severe pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L583–590. doi: 10.1152/ajplung.00321.2006. [DOI] [PubMed] [Google Scholar]
- 20.Johnson SR, Granton JT, Mehta S. Thrombotic arteriopathy and anticoagulation in pulmonary hypertension. Chest. 2006;130:545–552. doi: 10.1378/chest.130.2.545. [DOI] [PubMed] [Google Scholar]
- 21.Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529:326–335. doi: 10.1038/nature17041. [DOI] [PubMed] [Google Scholar]
- 22.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 23.Di Prisco GV, Huang W, Buffington SA, Hsu CC, Bonnen PE, Placzek AN, Sidrauski C, Krnjevic K, Kaufman RJ, Walter P, Costa-Mattioli M. Translational control of mGluR-dependent long-term depression and object-place learning by eIF2alpha. Nat Neurosci. 2014;17:1073–1082. doi: 10.1038/nn.3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith M, Wilkinson S. ER homeostasis and autophagy. Essays Biochem. 2017;61:625–635. doi: 10.1042/EBC20170092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harding HP, Zhang Y, Scheuner D, Chen JJ, Kaufman RJ, Ron D. Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc Natl Acad Sci U S A. 2009;106:1832–1837. doi: 10.1073/pnas.0809632106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Q, Lan T, Lu J, Zhang H, Zhang D, Lou T, Xu P, Ren J, Zhao D, Sun L, Li X, Wang J. DiDang tang inhibits endoplasmic reticulum stress-mediated apoptosis induced by oxygen glucose deprivation and intracerebral hemorrhage through blockade of the GRP78-IRE1/PERK pathways. Front Pharmacol. 2018;9:1423. doi: 10.3389/fphar.2018.01423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carew NT, Nelson AM, Liang Z, Smith SM, Milcarek C. Linking endoplasmic reticular stress and alternative splicing. Int J Mol Sci. 2018;19:3919. doi: 10.3390/ijms19123919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ozcan L, Tabas I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu Rev Med. 2012;63:317–328. doi: 10.1146/annurev-med-043010-144749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839–850. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guignabert C, Tu L, Girerd B, Ricard N, Huertas A, Montani D, Humbert M. New molecular targets of pulmonary vascular remodeling in pulmonary arterial hypertension: importance of endothelial communication. Chest. 2015;147:529–537. doi: 10.1378/chest.14-0862. [DOI] [PubMed] [Google Scholar]
- 32.Huertas A, Perros F, Tu L, Cohen-Kaminsky S, Montani D, Dorfmuller P, Guignabert C, Humbert M. Immune dysregulation and endothelial dysfunction in pulmonary arterial hypertension: a complex interplay. Circulation. 2014;129:1332–1340. doi: 10.1161/CIRCULATIONAHA.113.004555. [DOI] [PubMed] [Google Scholar]
- 33.Witte I, Horke S. Assessment of endoplasmic reticulum stress and the unfolded protein response in endothelial cells. Methods Enzymol. 2011;489:127–146. doi: 10.1016/B978-0-12-385116-1.00008-X. [DOI] [PubMed] [Google Scholar]
- 34.McAlpine CS, Werstuck GH. The development and progression of atherosclerosis: evidence supporting a role for endoplasmic reticulum (ER) stress signaling. Cardiovasc Hematol Disord Drug Targets. 2013;13:158–164. doi: 10.2174/1871529x11313020009. [DOI] [PubMed] [Google Scholar]
- 35.Lenna S, Han R, Trojanowska M. Endoplasmic reticulum stress and endothelial dysfunction. IUBMB Life. 2014;66:530–537. doi: 10.1002/iub.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dromparis P, Paulin R, Sutendra G, Qi AC, Bonnet S, Michelakis ED. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ Res. 2013;113:126–136. doi: 10.1161/CIRCRESAHA.112.300699. [DOI] [PubMed] [Google Scholar]
- 37.Dromparis P, Paulin R, Stenson TH, Haromy A, Sutendra G, Michelakis ED. Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation. 2013;127:115–125. doi: 10.1161/CIRCULATIONAHA.112.133413. [DOI] [PubMed] [Google Scholar]
- 38.Guignabert C, Phan C, Seferian A, Huertas A, Tu L, Thuillet R, Sattler C, Le Hiress M, Tamura Y, Jutant EM, Chaumais MC, Bouchet S, Maneglier B, Molimard M, Rousselot P, Sitbon O, Simonneau G, Montani D, Humbert M. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J Clin Invest. 2016;126:3207–3218. doi: 10.1172/JCI86249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Battson ML, Lee DM, Gentile CL. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2017;312:H355–H367. doi: 10.1152/ajpheart.00437.2016. [DOI] [PubMed] [Google Scholar]
- 40.Gobin SJ, van Zutphen M, Woltman AM, van den Elsen PJ. Transactivation of classical and nonclassical HLA class I genes through the IFN-stimulated response element. J Immunol. 1999;163:1428–1434. [PubMed] [Google Scholar]
- 41.Braun-Moscovici Y, Nahir AM, Balbir-Gurman A. Endothelin and pulmonary arterial hypertension. Semin Arthritis Rheum. 2004;34:442–453. doi: 10.1016/j.semarthrit.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 42.Ramirez A, Varga J. Pulmonary arterial hypertension in systemic sclerosis: clinical manifestations, pathophysiology, evaluation, and management. Treat Respir Med. 2004;3:339–352. doi: 10.2165/00151829-200403060-00002. [DOI] [PubMed] [Google Scholar]
- 43.Yoshibayashi M, Nishioka K, Nakao K, Saito Y, Matsumura M, Ueda T, Temma S, Shirakami G, Imura H, Mikawa H. Plasma endothelin concentrations in patients with pulmonary hypertension associated with congenital heart defects. Evidence for increased production of endothelin in pulmonary circulation. Circulation. 1991;84:2280–2285. doi: 10.1161/01.cir.84.6.2280. [DOI] [PubMed] [Google Scholar]
- 44.Lenna S, Townsend DM, Tan FK, Kapanadze B, Markiewicz M, Trojanowska M, Scorza R. HLA-B35 upregulates endothelin-1 and downregulates endothelial nitric oxide synthase via endoplasmic reticulum stress response in endothelial cells. J Immunol. 2010;184:4654–4661. doi: 10.4049/jimmunol.0903188. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Ghosh S, Gupta M, Xu W, Mavrakis DA, Janocha AJ, Comhair SA, Haque MM, Stuehr DJ, Yu J, Polgar P, Naga Prasad SV, Erzurum SC. Phosphorylation inactivation of endothelial nitric oxide synthesis in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2016;310:L1199–1205. doi: 10.1152/ajplung.00092.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu J, Pan W, Wang C, Dong H, Xing L, Hou J, Fang S, Li H, Yang F, Yu B. H2S attenuates endoplasmic reticulum stress in hypoxia-induced pulmonary artery hypertension. Biosci Rep. 2019;39:BSR20190304. doi: 10.1042/BSR20190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mao SZ, Fan XF, Xue F, Chen R, Chen XY, Yuan GS, Hu LG, Liu SF, Gong YS. Intermedin modulates hypoxic pulmonary vascular remodeling by inhibiting pulmonary artery smooth muscle cell proliferation. Pulm Pharmacol Ther. 2014;27:1–9. doi: 10.1016/j.pupt.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 48.Bell D, McDermott BJ. Intermedin (adrenomedullin-2): a novel counter-regulatory peptide in the cardiovascular and renal systems. Br J Pharmacol. 2008;153(Suppl 1):S247–262. doi: 10.1038/sj.bjp.0707494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grossini E, Molinari C, Mary DA, Uberti F, Caimmi PP, Vacca G. Intracoronary intermedin 1-47 augments cardiac perfusion and function in anesthetized pigs: role of calcitonin receptors and beta-adrenoreceptor-mediated nitric oxide release. J Appl Physiol (1985) 2009;107:1037–1050. doi: 10.1152/japplphysiol.00569.2009. [DOI] [PubMed] [Google Scholar]
- 50.Burak Kandilci H, Gumusel B, Wasserman A, Witriol N, Lippton H. Intermedin/adrenomedullin-2 dilates the rat pulmonary vascular bed: dependence on CGRP receptors and nitric oxide release. Peptides. 2006;27:1390–1396. doi: 10.1016/j.peptides.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 51.Koyama M, Furuhashi M, Ishimura S, Mita T, Fuseya T, Okazaki Y, Yoshida H, Tsuchihashi K, Miura T. Reduction of endoplasmic reticulum stress by 4-phenylbutyric acid prevents the development of hypoxia-induced pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2014;306:H1314–1323. doi: 10.1152/ajpheart.00869.2013. [DOI] [PubMed] [Google Scholar]
- 52.Wu Y, Adi D, Long M, Wang J, Liu F, Gai MT, Aierken A, Li MY, Li Q, Wu LQ, Ma YT, Hujiaaihemaiti M. 4-phenylbutyric acid induces protection against pulmonary arterial hypertension in rats. PLoS One. 2016;11:e0157538. doi: 10.1371/journal.pone.0157538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cao X, He Y, Li X, Xu Y, Liu X. The IRE1alpha-XBP1 pathway function in hypoxia-induced pulmonary vascular remodeling, is upregulated by quercetin, inhibits apoptosis and partially reverses the effect of quercetin in PASMCs. Am J Transl Res. 2019;11:641–654. [PMC free article] [PubMed] [Google Scholar]
- 54.Acevedo L, Yu J, Erdjument-Bromage H, Miao RQ, Kim JE, Fulton D, Tempst P, Strittmatter SM, Sessa WC. A new role for Nogo as a regulator of vascular remodeling. Nat Med. 2004;10:382–388. doi: 10.1038/nm1020. [DOI] [PubMed] [Google Scholar]
- 55.Wright PL, Yu J, Di YP, Homer RJ, Chupp G, Elias JA, Cohn L, Sessa WC. Epithelial reticulon 4B (Nogo-B) is an endogenous regulator of Th2-driven lung inflammation. J Exp Med. 2010;207:2595–2607. doi: 10.1084/jem.20100786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- 57.Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, McMurtry MS, Michalak M, Vance JE, Sessa WC, Michelakis ED. The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med. 2011;3:88ra55. doi: 10.1126/scitranslmed.3002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Munoz JP, Zorzano A. Endoplasmic reticulum stress enters a Nogo zone. Sci Transl Med. 2011;3:88ps26. doi: 10.1126/scitranslmed.3002708. [DOI] [PubMed] [Google Scholar]
- 59.Shanahan CM, Furmanik M. Endoplasmic reticulum stress in arterial smooth muscle cells: a novel regulator of vascular disease. Curr Cardiol Rev. 2017;13:94–105. doi: 10.2174/1573403X12666161014094738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–744. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soubrier F, Chung WK, Machado R, Grunig E, Aldred M, Geraci M, Loyd JE, Elliott CG, Trembath RC, Newman JH, Humbert M. Genetics and genomics of pulmonary arterial hypertension. Turk Kardiyol Dern Ars. 2014;42(Suppl 1):17–28. [PubMed] [Google Scholar]
- 62.Machado RD, Southgate L, Eichstaedt CA, Aldred MA, Austin ED, Best DH, Chung WK, Benjamin N, Elliott CG, Eyries M, Fischer C, Graf S, Hinderhofer K, Humbert M, Keiles SB, Loyd JE, Morrell NW, Newman JH, Soubrier F, Trembath RC, Viales RR, Grunig E. Pulmonary arterial hypertension: a current perspective on established and emerging molecular genetic defects. Hum Mutat. 2015;36:1113–1127. doi: 10.1002/humu.22904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Girerd B, Coulet F, Jais X, Eyries M, Van Der Bruggen C, De Man F, Houweling A, Dorfmuller P, Savale L, Sitbon O, Vonk-Noordegraaf A, Soubrier F, Simonneau G, Humbert M, Montani D. Characteristics of pulmonary arterial hypertension in affected carriers of a mutation located in the cytoplasmic tail of bone morphogenetic protein receptor type 2. Chest. 2015;147:1385–1394. doi: 10.1378/chest.14-0880. [DOI] [PubMed] [Google Scholar]
- 64.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA 3rd, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 65.Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, Simonneau G, Galie N, Loyd JE, Humbert M, Nichols WC, Morrell NW, Berg J, Manes A, McGaughran J, Pauciulo M, Wheeler L. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2001;345:325–334. doi: 10.1056/NEJM200108023450503. [DOI] [PubMed] [Google Scholar]
- 66.Shintani M, Yagi H, Nakayama T, Saji T, Matsuoka R. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J Med Genet. 2009;46:331–337. doi: 10.1136/jmg.2008.062703. [DOI] [PubMed] [Google Scholar]
- 67.Chida A, Shintani M, Nakayama T, Furutani Y, Hayama E, Inai K, Saji T, Nonoyama S, Nakanishi T. Missense mutations of the BMPR1B (ALK6) gene in childhood idiopathic pulmonary arterial hypertension. Circ J. 2012;76:1501–1508. doi: 10.1253/circj.cj-11-1281. [DOI] [PubMed] [Google Scholar]
- 68.Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT) Eur Heart J. 2016;37:67–119. doi: 10.1093/eurheartj/ehv317. [DOI] [PubMed] [Google Scholar]
- 69.Li X, Zhang X, Leathers R, Makino A, Huang C, Parsa P, Macias J, Yuan JX, Jamieson SW, Thistlethwaite PA. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med. 2009;15:1289–1297. doi: 10.1038/nm.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]