

Abstract
Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) have been used as first-line recommended therapy for EGFR mutant non-small cell lung cancer patients. However, epithelial-mesenchymal transition (EMT) can reduce EGFR-TKI sensitivity and lead to resistance. This study was designed to investigate the reversal effect of astragalus polysaccharides (APS) on gefitinib resistance (GR) and to elucidate the underlying mechanisms. PC9 and HCC827 lung cancer cells were stimulated by TGF-β1 to develop EMT-associated GR cells. Cell proliferation, migration and apoptosis assays were used to confirm the effect of gefitinib on GR cells and the therapeutic effect of APS on GR cells after knockdown and over-expression of related signaling pathways. Reverse transcription polymerase chain reaction, western blotting, and immunofluorescent staining assays were used to evaluate the expression levels of E-cadherin, N-cadherin, vimentin, PD-L1, and SREBP-1. Furthermore, proliferation and migration abilities were enhanced, while apoptosis ability was weakened in EMT-associated GR cells. After over-expression of PD-L1, expression levels of N-cadherin, vimentin and SREBP-1 increased, while expression of E-cadherin decreased. After knockdown of PD-L1 or SREBP-1, E-cadherin expression increased, while expression of N-cadherin and vimentin decreased. Further studies revealed that APS promoted apoptosis and reduced proliferation and migration abilities in GR cells. Moreover, APS increased expression of E-cadherin and decreased expression of N-cadherin and vimentin, indicating that it may be related to inhibition of the PD-L1/SREBP-1/EMT signaling pathway. Based on these findings, it can be concluded that APS can reverse acquired resistance to gefitinib in lung cancer cells by inhibiting the PD-L1/SREBP-1/EMT signaling pathway.
Keywords: Gefitinib, resistance, astragalus polysaccharides, lung adenocarcinoma, PD-L1, epithelial-mesenchymal transition (EMT)
Introduction
Lung cancer is a common malignant tumor and its morbidity and mortality rank first in the world. Non-small cell lung cancer (NSCLC) accounts for ~80-90% of lung cancers. Lung adenocarcinoma is the main pathological type of NSCLC, accounting for ~50-60% of NSCLC types. NSCLC five-year survival rate is only 15% [1]. In terms of treatment, epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) have a significant effect on EGFR mutant NSCLC and have been used as first-line recommended treatment for these patients [2]. However, most patients may develop resistance 9-13 months after the initial treatment with EGFR-TKIs [3]. Research has shown that approximately half of the patients developed epithelial-mesenchymal transition (EMT) after using EGFR-TKIs [4].
EMT refers to transformation of cells from the epithelial to mesenchymal phenotype, which is closely related to occurrence, in-situ invasion, and distant metastasis of tumors [5,6]. It is also closely related to NSCLC prognosis and its sensitivity and resistance to EGFR-TKIs [7,8]. Therefore, EMT may be closely related to the generation of acquired EGFR-TKI resistance in NSCLC patients. Current studies have confirmed that EMT in cancer cells is closely related to up-regulation of programmed death ligand 1 (PD-L1) [9]. PD-L1 is an important regulatory molecule of the immune system [10]. Cancer cells can up-regulate PD-L1 expression, thus inhibiting the function of T cells and antigen-presenting cells, thereby causing immune escape of cancer cells. It has been reported that EGFR-TKIs can down-regulate the expression of PD-L1 in lung cancer cells [11]. Studies have shown that PD-L1 induces EMT in cells by activating sterol regulatory element-binding protein 1 (SREBP-1) and is involved in promoting invasion and metastasis of skin and kidney cancer cells [12,13]. SREBP-1 is a major transcription factor regulating expression of lipid synthesis genes and is involved in the occurrence and development of cancers. Abnormal expression of SREBP-1 exists in many kinds of cancers, including lung adenocarcinoma, prostate cancer, and breast cancer [14]. It has been reported that inhibition of SREBP-1 increases lung adenocarcinoma sensitivity to gefitinib [15].
Some studies [16,17] have shown astragalus polysaccharides (APS) inhibits metastasis in non-small cell lung carcinoma cell lines and clinical feasibility of APS for maintenance therapy in patients with lung cancer. Moreover, the combined treatment of APS significantly improved clinical symptoms [17,18]. Traditional Chinese medicine can act on multiple targets, participating in overall regulation and having the advantage of improving or reversing drug resistance. This study was designed to explore whether APS could reverse the acquired resistance of lung adenocarcinoma cells to gefitinib by inhibiting the PD-L1/SREBP-1/EMT signaling pathway.
Materials and methods
Cell culture and treatment
Human lung adenocarcinoma cell lines (PC9, HCC827, Cell Resource Center of the Chinese Academy of Medical Sciences, Beijing, China) were cultured in 5% CO2 at 37°C in RPMI 1640 (Hyclone, USA) supplemented with 10% fetal bovine serum (FBS, Excell, Australia), 100 U/mL penicillin, and 100 U/mL streptomycin. Cells treated with 10 ng/mL transforming growth factor-β1 (TGF-β1, Peprotech, USA) for six days were used in the following experiments as an example of morphological and EMT phenomena. The culture medium was replaced every two days. TGF-β1 was dissolved in citric acid (pH 3.0) to a concentration of 10 µg/mL, stored at -20°C, and diluted in culture medium to the required concentration of 10 ng/mL. Gefitinib (AstraZeneca Medicine, UK) was dissolved in DMSO to a concentration of 20 mM, stored at -20°C, and diluted in culture medium to the required concentration (0.1-20 µM). APS (Pujingkangli Technology, China) were dissolved in DMSO to a concentration of 28 mg/mL, stored at -20°C, and diluted in culture medium to the required concentration of 200 mg/L.
Small interfering RNA (siRNA) transfection
PD-L1-siRNA (STB0010934A), SREBP-1-siRNA (STB0007997A), and negative control siRNA were purchased from RiboBio (Guangzhou, China). Transfection was performed using jetPRIME transfection reagent (Polyplus Transfection, France) following the manufacturer’s instructions. The culture medium was changed 12 h after transfection. Cells were harvested after 24 h for use in various assays.
Construction of expression plasmids and transfection
The entire coding sequence of human PD-L1 was inserted into the mammalian expression plasmid pcDNA3.0 along with the N-terminal Flag tag. Cells were transfected with the construct plasmid.
RNA isolation and reverse transcriptase-polymerase chain reaction (RT-PCR)
Total RNA was isolated from cell lines and tissues using Trizol reagent (Thermo, USA). A total of 2 µg RNA were reverse transcribed into single-strand cDNA in 20 µL of reaction buffer using a reverse transcriptase kit (TIANGEN, China). For RT-PCR, each experiment was performed in 20 µL of reaction volume according to the manufacturer’s protocol. Thermal cycle conditions were as follows: 95°C for 10 min, followed by 40 cycles of 95°C for 30 s and 60°C for 1 min. The primers used for real-time PCR are listed in Table 1. GAPDH was chosen as an internal control. All reactions were carried out in triplicate to evaluate their reproducibility.
Table 1.
Primers used for RT-PCR
| Name | Accession No. | Sequences (5’-3’) | Product size (bp) | |
|---|---|---|---|---|
| E-cadherin for human | NM_004360 | Forward | CGAGAGCTACACGTTCACGG | 119 |
| Reverse | GGGTGTCGAGGGAAAAATAGG | |||
| vimentin for human | NM_003380 | Forward | AGTCCACTGAGTACCGGAGAC | 98 |
| Reverse | CATTTCACGCATCTGGCGTTC | |||
| N-cadherin for human | NM_001792 | Forward | TGCGGTACAGTGTAACTGGG | 123 |
| Reverse | GAAACCGGGCTATCTGCTCG | |||
| PD-L1 for human | NM_014143 | Forward | TGGCATTTGCTGAACGCATTT | 120 |
| Reverse | TGCAGCCAGGTCTAATTGTTTT | |||
| SREBP-1 for human | NM_001005291 | Forward | CGGAACCATCTTGGCAACAGT | 141 |
| Reverse | CGCTTCTCAATGGCGTTGT | |||
| GAPDH for rat | NM_001256799 | Forward | ACAACTTTGGTATCGTGGAAGG | 101 |
| Reverse | GCCATCACGCCACAGTTTC | |||
Protein quantification and western blot analysis
Protein was extracted and lysed in the RIPA buffer. After quantification using the BCA method, 30 µg of protein were re-suspended in Laemmli buffer, separated by 12% SDS-PAGE, and then transferred to a membrane and subjected to immunoblotting. Membranes were incubated with primary antibodies (anti-E-cadherin: #3195, Cell Signaling; anti-vimentin: #5741, Cell Signaling; anti-N-cadherin: #14215, Cell Signaling; anti-PD-L1: ab213524, Abcam; anti-SREBP-1: ab28481, Abcam; and anti-GAPDH; #ab6276, Abcam) at 4°C overnight, followed by incubation with an HRP-conjugated secondary antibody. GAPDH was used as an internal control. All antibodies were diluted with a TBST buffer solution (primary antibodies: 1:1000, secondary antibodies: 1:5000). The ECL reagent was used for protein detection. Membrane immunocomplexes were visualized using Image Lab Software (BIO-RAD) with Immobilon Western Chemiluminescent HRP Substrate (Millipore, USA).
Cell proliferation assay
CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Dojindo, Japan) was used to determine the viability of cells with different treatments according to the manufacturer’s protocol. Cells with or without transfection for the duration of 24 h were seeded in 96-well plates. After 4 h, cells were exposed to gefitinib and/or APS (vehicle was used as a control) in fresh medium for additional 72 h. After washing with PBS, cells in each well were incubated with 10 μL of CellTiter 96 AQueous One Solution reagent mixed with 100 μL of culture medium at 37°C for 2 h in a humidified atmosphere with 5% CO2. The supernatant culture medium was collected and absorbance at 450 nm was recorded using a 96-well plate reader.
Cell apoptosis assay
Annexin V-FITC/propidium iodide (PI, 70-AP101-30, MultiSciences, China) double-staining was used to measure apoptosis of human lung adenocarcinoma cells according to the manufacturer’s protocol. Human lung adenocarcinoma cells were seeded into 12-well plates and then harvested after different treatments. The cells were then fluorescently labeled for detection of apoptotic and necrotic cells by adding 5 µL of annexin V-FITC and 5 µL of PI to each sample. Samples were mixed gently and incubated at room temperature in the dark for 15 min. Just before flow cytometry analysis, 300 µL of binding buffer were added to each sample (BD Bioscience, USA). Acquired data were analyzed using FlowJo software (Treestar Inc, USA).
Transwell migration assay
Transwell migratory apparatus (Costar, Corning Costar, MD, USA) was used for the migration assay. Approximately 1×105 cells with different treatments in 200 µL of RPMI 1640 medium were placed in the upper chamber and 1 mL of complete RPMI 1640 medium was placed in the lower chamber. After 24-48 h in culture, cells were fixed in 4% paraformaldehyde methanol for 30 min and then stained with 0.1% crystal violet in PBS for 30 min. Cells on the upper side of the filters were removed with cotton-tipped swabs and the filters were washed with PBS. Cells on the underside of the filters were viewed and counted under a microscope (Leica, Germany).
Immunofluorescent staining
Slides with cells undergoing different treatments were washed in PBS three times, fixed with 4% paraformaldehyde in PBS for 15 min, and permeabilized with 0.3% Triton X-100 in PBS for 10 min. Non-specific binding sites were blocked by a 15-min incubation in 20% normal donkey serum (Solarbio, Beijing) in PBS. The cells were then incubated with primary antibodies at 4°C overnight. After three 5-min washes with PBS, cells were incubated with anti-rabbit Alexa Fluor 488-conjugated secondary antibodies (1:200, Thermo, USA) for 2 h at room temperature. Following three 5-min washes with PBS, cells were mounted on slides with mounting medium with DAPI (Molecular Probes, USA). Cells were examined and photographed with a confocal microscope (FV300, Olympus).
Statistical analysis
All experiments were carried out at least three times. Data were represented as means ± SEM. Differences between groups were calculated using Student’s t-test. Statistical significance was defined as P<0.05 for a two-tailed test. All statistical analyses were carried out with Graphpad Prism 7.0 software.
Results
Establishment of EMT-associated lung adenocarcinoma cells
EMT-associated lung adenocarcinoma cells PC9 and HCC827 were established as described previously [19]. Cells stimulated with TGF-β1 (10 ng/mL) showed remarkable morphological changes into spindle-shaped cells with loose cell connections (Figure 1A, 1B). EMT markers were examined to evaluate whether EMT had occurred in lung adenocarcinoma cells. It was found that vimentin and N-cadherin protein expression was elevated, while E-cadherin expression was lower in both cell types compared to their respective parental cells (Figure 1C, 1D). EMT marker mRNA expression was also examined using RT-PCR. It was shown that mRNA expression of vimentin and N-cadherin was significantly increased, while E-cadherin mRNA expression was decreased after TGF-β1 stimulation. These results were consistent with those of western blot assays (Figure 1E, 1F).
Figure 1.
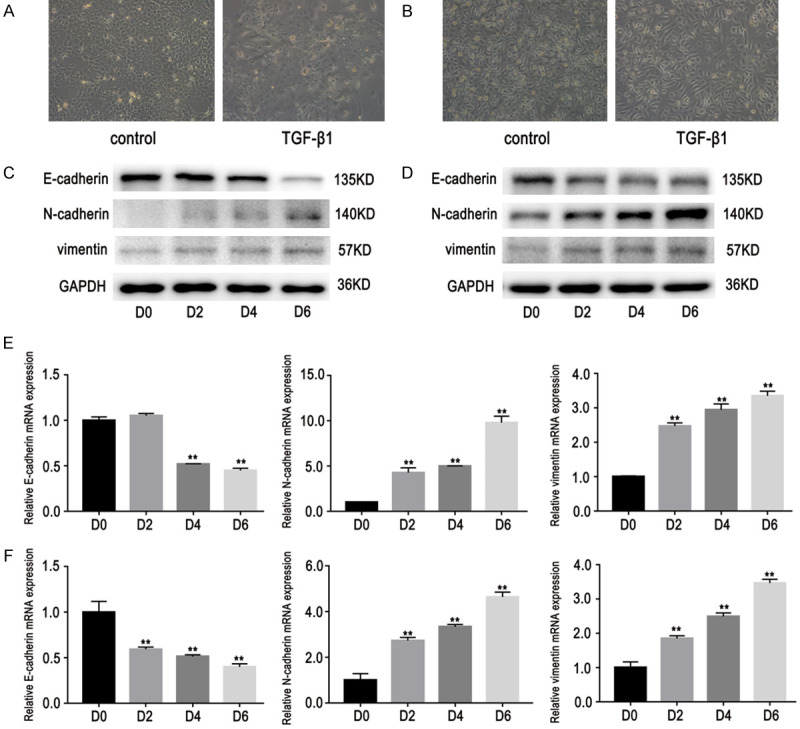
Effects of TGF-β1 on expression of E-cadherin, N-cadherin and vimentin. Cells were stimulated by TGF-β1 (10 ng/mL) at different times (D0, 2, 4, and 6). A, B. Cells stimulated with TGF-β1 exhibited morphological changes into spindle-shaped cells with loose cell connections. Observations were made using a light microscope (×200). C, D. Western blot results showed that vimentin and N-cadherin expression levels increased significantly, while E-cadherin expression decreased after TGF-β1 stimulation. E, F. RT-PCR results showed that vimentin and N-cadherin expression levels increased significantly, while E-cadherin expression decreased after TGF-β1 stimulation. Data are represented as mean ± SEM (n=3). All experiments were repeated at least three times and representative data are shown. **P<0.01, compared to D0 group. A, C, E. HCC827 cells; B, D, F. PC9 cells.
Relationship between EMT-associated lung adenocarcinoma cells and gefitinib resistance (GR)
This study sought to determine the relationship between EMT-associated lung adenocarcinoma cells and GR. It was found that gefitinib-induced inhibition of proliferation and promotion of apoptosis were weakened in EMT-associated lung adenocarcinoma cells (Figure 2A-D). In addition, the inhibition effect of gefitinib on lung adenocarcinoma cell migration was also significantly weakened. This indicated that EMT-associated lung adenocarcinoma cells were resistant to gefitinib (Figure 2E, 2F).
Figure 2.

Effects of gefitinib on lung adenocarcinoma cells with EMT (cells were stimulated with TGF-β1 [10 ng/mL] for six days). Cells with EMT treated with or without gefitinib (G, 1 µM, D1). A-D. Cell proliferation and apoptosis assays showed gefitinib effects on proliferation inhibition and apoptosis promotion were weakened in EMT-associated lung adenocarcinoma cells. Data represent percentage of annexin V single positive (early apoptosis) and annexin V/PI double positive (late apoptosis) cells. Lower left quadrant contains the vital (double negative) population, upper left quadrant contains the damaged (annexin V-/PI+) cells, upper right quadrant contains the late apoptotic (annexin V+/PI+) cells, and lower right quadrant contains the early apoptotic (annexin V+/PI-) population. E, F. Cell migration assays showed the effect of gefitinib on migration inhibition in lung adenocarcinoma cells was also significantly weakened. Data are represented as mean ± SEM (n=3). All experiments were repeated at least three times and representative data are shown. *P<0.05, **P<0.01, compared to control group. A, C, E. HCC827/EMT cells; B, D, F. PC9/EMT cells.
Expression of PD-L1 and SREBP-1 in EMT-associated lung adenocarcinoma cells
To investigate potential roles of PD-L1 and SREBP-1 in EMT-associated lung adenocarcinoma cells, their expression levels were detected. It was found that PD-L1 and SREBP-1 expression levels were significantly increased in these cells (Figure 3A, 3B).
Figure 3.
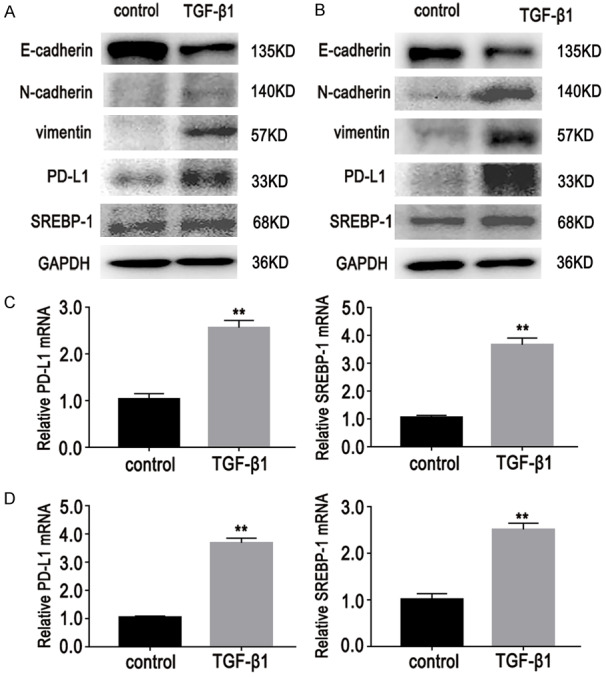
Effects of TGF-β1 (10 ng/mL, D6) on expression of PD-L1 and SREBP-1 in lung adenocarcinoma cells. As mentioned above, TGF-β1 induced EMT and resistance to gefitinib in cells. A-D. Western blot and RT-PCR results showed that expression levels of PD-L1 and SREBP-1 were significantly increased in lung adenocarcinoma cells after TGF-β1 stimulation. Data are represented as mean ± SEM (n=3). All experiments were repeated at least three times and representative data are shown. A, C. HCC827/GR/EMT cells; B, D. PC9/GR/EMT cells.
EMT is regulated in lung adenocarcinoma cells via the PD-L1/ SREBP-1 signaling pathway
The PD-L1/SREBP-1 signaling pathway role in promoting EMT in lung adenocarcinoma cells was further explored. E-cadherin, N-cadherin, and vimentin are well-established EMT biomarkers. In this study, western blot, RT-PCR, and immunofluorescence (IF) data showed that over-expression of PD-L1 promoted expression of SREBP-1, induced a decrease in E-cadherin expression, and increased vimentin and N-cadherin expression in NSCLC cells (Figure 4). PD-L1 and SREBP-1 gene knockdown with specific siRNAs decreased expression of vimentin or N-cadherin and increased expression of E-cadherin (Figure 5A-D). In addition, IF staining was also used to examine the expression of E-cadherin and vimentin (Figure 5E, 5F). Results showed that EMT was regulated by the PD-L1/SREBP-1 signaling pathway.
Figure 4.
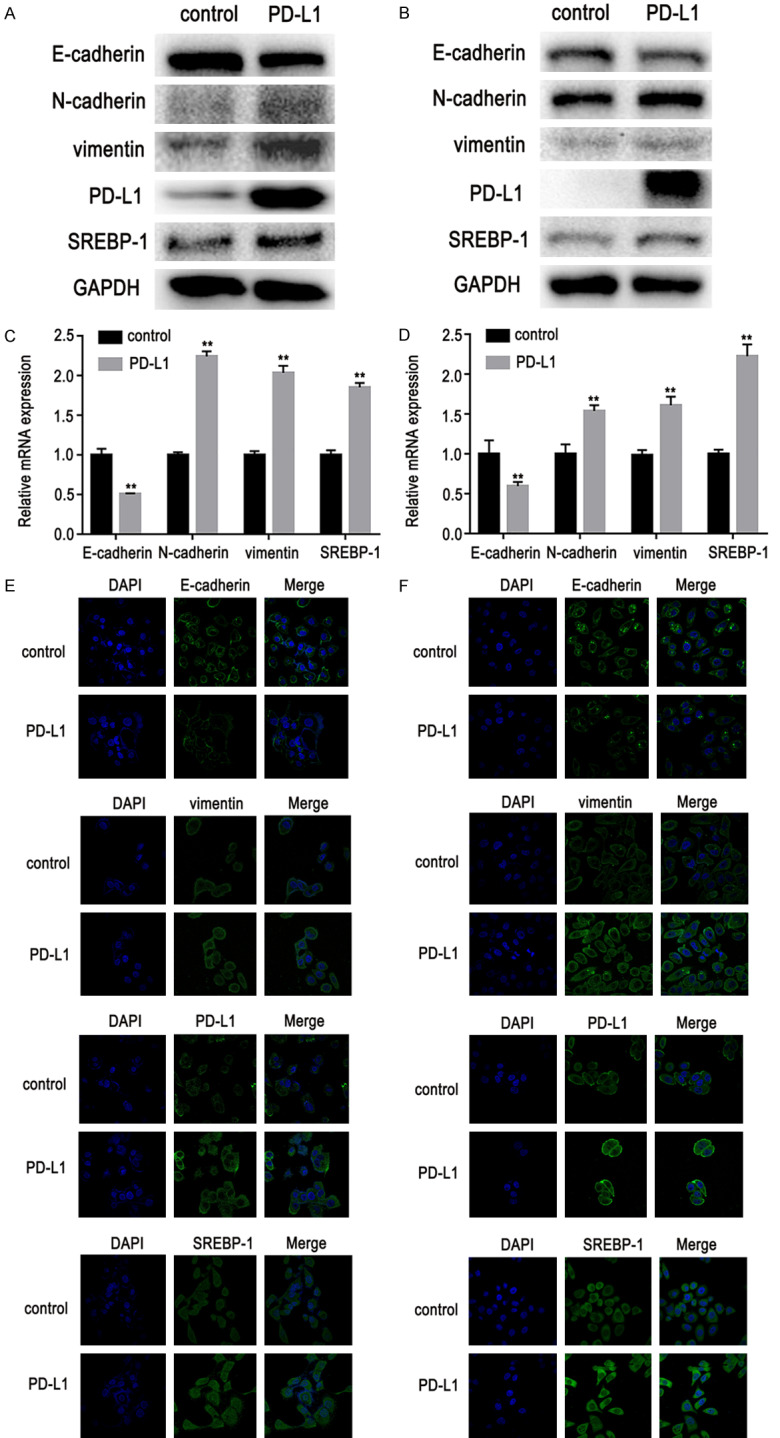
Effects of PD-L1 over-expression on expression of SREBP-1, E-cadherin, N-cadherin, and vimentin in lung adenocarcinoma cells. A, B. Western blot results showed that over-expression of PD-L1 promoted expression of SREBP-1, induced a decrease in E-cadherin expression, and increased vimentin and N-cadherin expression in lung adenocarcinoma cells. C, D. RT-PCR also showed that SREBP-1, vimentin, and N-cadherin expression was increased and E-cadherin expression was decreased. E, F. Immunofluorescent staining showed that SREBP-1 and vimentin expression was increased and E-cadherin expression was decreased. Data are represented as mean ± SEM (n=3). All experiments were repeated at least three times and representative data are shown. **P<0.01, compared to control group. A, C, E. HCC827 cells; B, D, F. PC9 cells.
Figure 5.

Effects of PD-L1 or SREBP-1 knockdown on expression of E-cadherin, N-cadherin, and vimentin in GR cells with EMT. A, B. Western blot results showed that PD-L1 or SREBP-1 gene knockdown decreased expression of vimentin and N-cadherin and increased expression of E-cadherin. C, D. RT-PCR showed that expression of E-cadherin was increased. E, F. Immunofluorescent staining showed that expression of vimentin was decreased and expression of E-cadherin was increased. Data are represented as mean ± SEM (n=3). All experiments were repeated at least three times and representative data are shown. *P<0.05, **P<0.01, compared to control group. A, C, E. HCC827/GR/EMT cells; B, D, F. PC9/GR/EMT cells.
APS reversed GR in EMT-associated lung adenocarcinoma cells
In our present study, the inhibitory concentration of APS in GR cells was 187.7 mg/L. An APS concentration of 200 mg/L was selected for this study according to experimental results and relevant literature [20]. The proliferation inhibition rate of GR cells treated with gefitinib and APS was higher than that of GR cells treated with gefitinib (Figure 6A, 6B). Cell apoptosis assay showed that APS could further promote apoptosis of GR cells (Figure 6C, 6D). After treatment with APS combined with gefitinib, GR cells’ migration ability was further reduced (Figure 6E, 6F).
Figure 6.

Effects of APS on GR cells with EMT. Cells were treated with gefitinib (G, 1 µM, D1) and/or APS (200 mg/L, D1). A, B. Cell proliferation assays showed that proliferation inhibition rate of GR cells treated with gefitinib and APS was higher than that of GR cells treated with gefitinib. C, D. Cell apoptosis assays showed that after treatment with APS combined with gefitinib, GR cell apoptosis ability was enhanced. Data represented percentage of annexin V single positive (early apoptosis) and annexin V/PI double positive (late apoptosis) cells. Lower left quadrant contains the vital (double negative) population, upper left quadrant contains the damaged (annexin V-/PI+) cells, upper right quadrant contains the late apoptotic (annexin V+/PI+) cells, and lower right quadrant contains the early apoptotic (annexin V+/PI-) population. E, F. Cell migration assays showed that GR cell migration ability was weakened. Data are represented as mean ± SEM (n=3). All data are representative of at least three independent experiments. *P<0.05, **P<0.01, compared to control group. A, C, E. HCC827/GR/EMT cells; B, D, F. PC9/GR/EMT cells.
APS reversed GR by regulating the PD-L1/SREBP-1/EMT signaling pathway
Lung adenocarcinoma cells were treated with APS for 48 h after TGF-β1 stimulation for six days. Western blot and RT-PCR data showed that APS can reduce expression of PD-L1 and SREBP-1 and increase expression of E-cadherin, thus enhancing sensitivity of GR cells (Figure 7). In order to further verify whether APS can reverse GR cell resistance via the PD-L1/SREBP-1 pathway, genes in the PD-L1/SREBP-1 pathway were knocked down. Results showed that the ability of APS to inhibit proliferation of GR cells was decreased (Figure 8A, 8B, 8E, 8F). At the same time, the ability of APS to inhibit cell migration was also weakened (Figure 8C, 8D, 8G, 8H), indicating that APS reversed GR cell resistance via the PD-L1/SREBP-1 signaling pathway.
Figure 7.
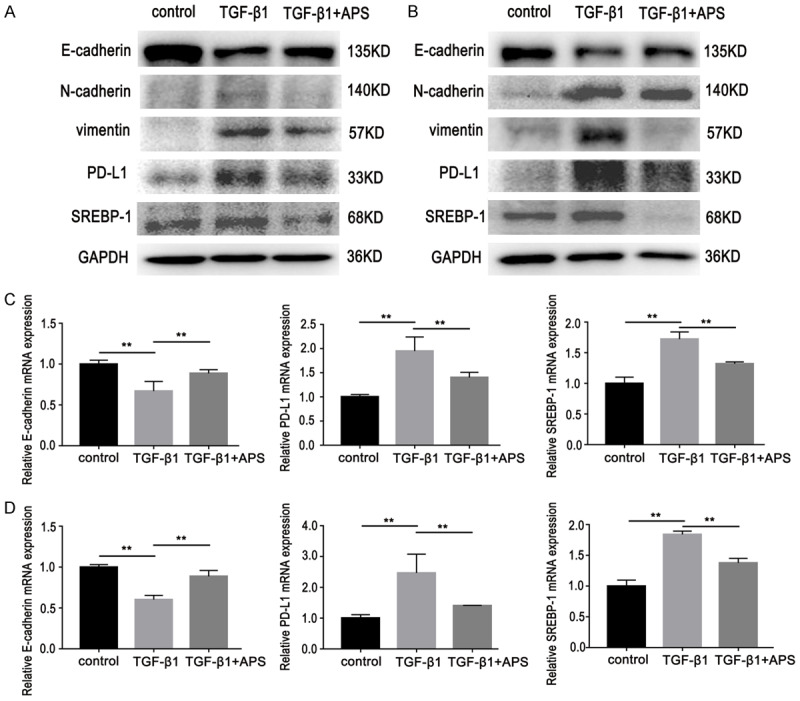
Effects of APS (200 mg/L, D2) on expression of E-cadherin, N-cadherin, vimentin, PD-L1 and SREBP-1 in GR cells with EMT. A, B. Western blot showed that APS can reduce expression of PD-L1, SREBP-1, N-cadherin, and vimentin and concurrently increase expression of E-cadherin. C, D. RT-PCR results showed the expression of E-cadherin was increased and PD-L1 and SREBP-1 expression levels were decreased. Data are represented as mean ± SEM (n=3). All data are representative of at least three independent experiments. **P<0.01, compared to control group. A, C. HCC827/GR/EMT cells; B, D. PC9/GR/EMT cells.
Figure 8.

Effects of gefitinib (G, 1 µM, D1) and/or APS (200 mg/L, D1) on GR cells with EMT after knockdown of PD-L1 or SREBP-1. A, B, E, F. Cell proliferation assays showed that the ability of APS to inhibit proliferation of GR cells was decreased. C, D, G, H. Cell migration assays showed that the ability of APS to inhibit cell migration was also weakened. Data are represented as mean ± SEM (n=3). All experiments were repeated at least three times and representative data are shown. **P<0.01, compared to control group. A, C, E, G. HCC827/GR/EMT cells; B, D, F, H. PC9/GR/EMT cells.
Discussion
The main active ingredients of astragalus are APS, astragalus methyl glycoside, kaempferol, and quercetin, which have a variety of pharmacological effects [21]. It has been showed that APS can improve the quality of life in patients with digestive system cancer [22,23]. Some studies have also shown that APS have unique advantages in regulating the immune response, inhibiting tumor proliferation, promoting apoptosis, and improving cancer resistance [24-31]. It has been shown that APS can exert an anti-tumor effect in mouse lung cancer [32,33]. This study demonstrated that APS can effectively inhibit proliferation and migration of GR lung cancer cells in vitro, suggesting that APS can reverse GR. This effect may be due to the inhibition of the PD-L1/SREBP-1/EMT signaling pathway.
In recent years, EGFR-TKIs have become important molecular target drugs for treatment of NSCLC. However, acquired resistance inevitably occurs during the application process, seriously affecting patient quality of life and survival. In acquired resistance, secondary mutation at the EGFR T790M site and c-met gene amplification accounted for ~50-60% of changes, while 40% of the resistance mechanism remain unclear [34]. Currently, third-generation EGFR-TKIs and c-met inhibitors can be selected for acquired drug resistance caused by secondary mutation at the T790M site of EGFR and c-met gene amplification. However, a considerable proportion of drug resistance still exists without corresponding treatment strategies. EMT is a common mechanism underlying the resistance to EGFR-TKIs, beyond secondary EGFR mutation. EMT accompanied by a loss of E-cadherin and increase in vimentin expression, is a process in which epithelial cells lose their polarity and adhesion, gain migratory abilities, and adopt a mesenchymal phenotype [35]. This mesenchymal phenotype has been shown to be associated with aggressive biological behavior, such as increased migration and invasion [36-38]. So far, no available drugs specifically kill cancer cells via EMT and novel treatment strategies that overcome or prevent EMT are needed. Studies have shown that APS can enhance sensitivity of NSCLC, ovarian cancer, and liver cancer cells to chemotherapeutic drugs [16,39,40]. Therefore, the effect of APS on EMT was also investigated. It was found that APS significantly improved expression of epithelial marker E-cadherin and reduced expression of mesenchymal markers N-cadherin and vimentin. APS also inhibited proliferation and migration and promoted apoptosis of EMT-associated GR cells. Therefore, these data indicate that APS can effectively reverse EMT in GR lung adenocarcinoma cells, which may lead to further inhibition of migration in lung cancer cells. However, the underlying mechanism remains unknown.
The underlying EMT mechanisms have been extensively studied in recent years. It has been reported that PD-L1 over-expression promotes EMT and cancerization of skin epithelial cells, suggesting that EMT is closely related to the immune escape signaling pathway [12]. PD-L1 is closely related to EMT, invasion, and metastasis of breast, lung, and kidney cancers and other malignant tumors [41,42]. Recent studies have shown that PD-L1 is one of the main mechanisms for tumor resistance induced by the interaction between tumor cells and the tumor microenvironment. The up-regulation of PD-L1 expression in tumor cells is often closely related to the occurrence of EMT [9,43]. Studies have shown that PD-L1 can induce EMT in cells by activating SREBP-1, thereby promoting invasion and metastasis of skin and renal cancer cells [12,13]. SREBP-1 is one of the key transcriptional regulators of lipid metabolism and cellular growth. The process of lipogenesis has been shown to be involved in the progression of cancer and maintenance of cancer stem cells [44]. SREBP-1 activity has been found to maintain cancer cell proliferation and tumor growth. Studies on the PD-L1/SREBP-1 signaling pathway are rare. It has been reported that SREBP-1 is regulated by PD-L1 in renal cell carcinoma (RCC) cells [45]. The PD-L1/SREBP-1 signaling pathway has been shown to be involved in RCC cell metastasis [13]. SREBP-1 has been found to mediate the initial stages of EMT [46]. In the present study, PD-L1 over-expression promoted SREBP-1 expression, increased E-cadherin expression, and decreased expression of vimentin and N-cadherin in lung cancer cells, suggesting that it promoted EMT. PD-L1 over-expression also enhanced lung cancer cells proliferation and migration and weakened their apoptotic ability. This effect may be attributed to EMT occurrence. All of the aforementioned results were reversed when PD-L1 and SREBP-1 were knocked down, suggesting that the PD-L1/SREBP-1/EMT pathway may be involved in acquired resistance to gefitinib. APS did not only restore GR cell sensitivity to gefitinib, but also inhibited the EMT process by blocking the PD-L1/SREBP-1 pathway.
In conclusion, the present study demonstrates that APS can reverse EMT and significantly inhibit the proliferation and migration of GR lung cancer cells and promote their apoptosis. The underlying mechanism may depend on inhibition of the PD-L1/SREBP-1/EMT signaling pathway.
Acknowledgements
This study was supported by The Capital Health Research and Development of Special (No. 2018-2-1113), National Natural Science Foundation of China (No. 81774221), Basic-Clinical Cooperation Program from Capital Medical University (No. 17JL14), Research Foundation of Beijing Friendship Hospital, Capital Medical University (No. yyqdkt2016-4). Beijing Municipal 215 High-level Health Person Foundation Project (No. 2014-3-004). New Teacher Launch Fund Project, Beijing University of Chinese Medicine (No. 2020-JYB-XJSJJ-055).
Disclosure of conflict of interest
None.
References
- 1.Kumarakulasinghe NB, van Zanwijk N, Soo RA. Molecular targeted therapy in the treatment of advanced stage non-small cell lung cancer (NSCLC) Respirology. 2015;20:370–378. doi: 10.1111/resp.12490. [DOI] [PubMed] [Google Scholar]
- 2.Mitsudomi T, Yatabe Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci. 2007;98:1817–1824. doi: 10.1111/j.1349-7006.2007.00607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu HA, Riely GJ, Lovly CM. Therapeutic strategies utilized in the setting of acquired resistance to EGFR tyrosine kinase inhibitors. Clin Cancer Res. 2014;20:5898–5907. doi: 10.1158/1078-0432.CCR-13-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uramoto H, Iwata T, Onitsuka T, Shimokawa H, Hanagiri T, Oyama T. Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res. 2010;30:2513–2517. [PubMed] [Google Scholar]
- 5.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 7.Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II, Yang SH, Kim CH, Lee JC. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer. 2009;63:219–226. doi: 10.1016/j.lungcan.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 8.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, Nilsson MB, Gudikote J, Tran H, Cardnell RJ, Bearss DJ, Warner SL, Foulks JM, Kanner SB, Gandhi V, Krett N, Rosen ST, Kim ES, Herbst RS, Blumenschein GR, Lee JJ, Lippman SM, Ang KK, Mills GB, Hong WK, Weinstein JN, Wistuba II, Coombes KR, Minna JD, Heymach JV. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279–290. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lou Y, Diao L, Cuentas ER, Denning WL, Chen L, Fan YH, Byers LA, Wang J, Papadimitrakopoulou VA, Behrens C, Rodriguez JC, Hwu P, Wistuba II, Heymach JV, Gibbons DL. Epithelial-mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clin Cancer Res. 2016;22:3630–3642. doi: 10.1158/1078-0432.CCR-15-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Li F, Jiang F, Lv X, Zhang R, Lu A, Zhang G. A mini-review for cancer immunotherapy: molecular understanding of PD-1/PD-L1 pathway & translational blockade of immune checkpoints. Int J Mol Sci. 2016;17:1151. doi: 10.3390/ijms17071151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin K, Cheng J, Yang T, Li Y, Zhu B. EGFR-TKI down-regulates PD-L1 in EGFR mutant NSCLC through inhibiting NF-kappaB. Biochem Biophys Res Commun. 2015;463:95–101. doi: 10.1016/j.bbrc.2015.05.030. [DOI] [PubMed] [Google Scholar]
- 12.Cao Y, Zhang L, Kamimura Y, Ritprajak P, Hashiguchi M, Hirose S, Azuma M. B7-H1 overexpression regulates epithelial-mesenchymal transition and accelerates carcinogenesis in skin. Cancer Res. 2011;71:1235–1243. doi: 10.1158/0008-5472.CAN-10-2217. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Wang H, Zhao Q, Xia Y, Hu X, Guo J. PD-L1 induces epithelial-to-mesenchymal transition via activating SREBP-1c in renal cell carcinoma. Med Oncol. 2015;32:212. doi: 10.1007/s12032-015-0655-2. [DOI] [PubMed] [Google Scholar]
- 14.Li W, Tai Y, Zhou J, Gu W, Bai Z, Zhou T, Zhong Z, McCue PA, Sang N, Ji JY, Kong B, Jiang J, Wang C. Repression of endometrial tumor growth by targeting SREBP1 and lipogenesis. Cell Cycle. 2012;11:2348–2358. doi: 10.4161/cc.20811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Yan H, Zhao L, Jia W, Yang H, Liu L, Zhou X, Miao P, Sun X, Song S, Zhao X, Liu J, Huang G. Inhibition of SREBP increases gefitinib sensitivity in non-small cell lung cancer cells. Oncotarget. 2016;7:52392–52403. doi: 10.18632/oncotarget.10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Y, Hong T, Tong L, Liu W, Yang X, Luo J, Wang F, Li J, Yan L. Astragalus polysaccharide combined with 10-hydroxycamptothecin inhibits metastasis in non-small cell lung carcinoma cell lines via the MAP4K3/mTOR signaling pathway. Int J Mol Med. 2018;42:3093–3104. doi: 10.3892/ijmm.2018.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bamodu OA, Kuo KT, Wang CH, Huang WC, Wu ATH, Tsai JT, Lee KY, Yeh CT, Wang LS. Astragalus polysaccharides (PG2) enhances the M1 polarization of macrophages, functional maturation of dendritic cells, and T cell-mediated anticancer immune responses in patients with lung cancer. Nutrients. 2019;11:2264. doi: 10.3390/nu11102264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo L, Bai SP, Zhao L, Wang XH. Astragalus polysaccharide injection integrated with vinorelbine and cisplatin for patients with advanced non-small cell lung cancer: effects on quality of life and survival. Med Oncol. 2012;29:1656–1662. doi: 10.1007/s12032-011-0068-9. [DOI] [PubMed] [Google Scholar]
- 19.Soucheray M, Capelletti M, Pulido I, Kuang Y, Paweletz CP, Becker JH, Kikuchi E, Xu C, Patel TB, Al-Shahrour F, Carretero J, Wong KK, Jänne PA, Shapiro GI, Shimamura T. Intratumoral heterogeneity in EGFR-mutant NSCLC results in divergent resistance mechanisms in response to EGFR tyrosine kinase inhibition. Cancer Res. 2015;75:4372–4383. doi: 10.1158/0008-5472.CAN-15-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang YM, Liu YQ, Liu D, Zhang L, Qin J, Zhang Z, Su Y, Yan C, Luo YL, Li J, Xie X, Guan Q. The effects of astragalus polysaccharide on bone marrow-derived mesenchymal stem cell proliferation and morphology induced by A549 lung cancer cells. Med Sci Monit. 2019;25:4110–4121. doi: 10.12659/MSM.914219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang QZ, Wang ZH, Fu J, Liu DW, Huang LF. Correlation between chemical constituents and ecological factors of Astragalus membranaceus var. mongholicus. Ying Yong Sheng Tai Xue Bao. 2015;26:732–738. [PubMed] [Google Scholar]
- 22.Ge L, Mao L, Tian JH, Shi FY, Lou LL, Qiu X, Li JL, Yang KH. Network meta-analysis on selecting Chinese medical injections in radiotherapy for esophageal cancer. Zhongguo Zhong Yao Za Zhi. 2015;40:3674–3681. [PubMed] [Google Scholar]
- 23.Wang JC, Tian JH, Ge L, Gan YH, Yang KH. Which is the best Chinese herb injection based on the FOLFOX regimen for gastric cancer? A network meta-analysis of randomized controlled trials. Asian Pac J Cancer Prev. 2014;15:4795–4800. doi: 10.7314/apjcp.2014.15.12.4795. [DOI] [PubMed] [Google Scholar]
- 24.Liu L, Zhang J, Li M, Zhang X, Zhang J, Li Z, Wang L, Wu J, Luo C. Inhibition of HepG2 cell proliferation by ursolic acid and polysaccharides via the downregulation of cyclooxygenase-2. Mol Med Rep. 2014;9:2505–2511. doi: 10.3892/mmr.2014.2059. [DOI] [PubMed] [Google Scholar]
- 25.Luo C, Urgard E, Vooder T, Metspalu A. The role of COX-2 and Nrf2/ARE in anti-inflammation and antioxidative stress: aging and anti-aging. Med Hypotheses. 2011;77:174–178. doi: 10.1016/j.mehy.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 26.Huang WH, Liao WR, Sun RX. Astragalus polysaccharide induces the apoptosis of human hepatocellular carcinoma cells by decreasing the expression of Notch1. Int J Mol Med. 2016;38:551–557. doi: 10.3892/ijmm.2016.2632. [DOI] [PubMed] [Google Scholar]
- 27.Tian QE, Li HD, Yan M, Cai HL, Tan QY, Zhang WY. Astragalus polysaccharides can regulate cytokine and P-glycoprotein expression in H22 tumor-bearing mice. World J Gastroenterol. 2012;18:7079–7086. doi: 10.3748/wjg.v18.i47.7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lai X, Xia W, Wei J, Ding X. Therapeutic effect of astragalus polysaccharides on hepatocellular carcinoma H22-bearing mice. Dose Response. 2017;15:1559325816685182. doi: 10.1177/1559325816685182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang B, Xiao B, Sun T. Antitumor and immunomodulatory activity of Astragalus membranaceus polysaccharides in H22 tumor-bearing mice. Int J Biol Macromol. 2013;62:287–290. doi: 10.1016/j.ijbiomac.2013.09.016. [DOI] [PubMed] [Google Scholar]
- 30.Li Q, Bao JM, Li XL, Zhang T, Shen XH. Inhibiting effect of Astragalus polysaccharides on the functions of CD4+CD25 highTreg cells in the tumor microenvironment of human hepatocellular carcinoma. Chin Med J (Engl) 2012;125:786–793. [PubMed] [Google Scholar]
- 31.Sun S, Zheng K, Zhao H, Lu C, Liu B, Yu C, Zhang G, Bian Z, Lu A, He X. Regulatory effect of astragalus polysaccharides on intestinal intraepithelial γδT cells of tumor bearing mice. Molecules. 2014;19:15224–15236. doi: 10.3390/molecules190915224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ming H, Chen Y, Zhang F, Wang Q, Dong X, Gu J, Li Y. Astragalus polysaccharides combined with cisplatin decreases the serum levels of CD44 and collagen type IV and hyaluronic acid in mice bearing Lewis lung cancer. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2015;31:909–913. [PubMed] [Google Scholar]
- 33.Zhou X, Liu Z, Long T, Zhou L, Bao Y. Immunomodulatory effects of herbal formula of astragalus polysaccharide (APS) and polysaccharopeptide (PSP) in mice with lung cancer. Int J Biol Macromol. 2018;106:596–601. doi: 10.1016/j.ijbiomac.2017.08.054. [DOI] [PubMed] [Google Scholar]
- 34.Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11:473–481. doi: 10.1038/nrclinonc.2014.104. [DOI] [PubMed] [Google Scholar]
- 35.Raoof S, Mulford IJ, Frisco-Cabanos H, Nangia V, Timonina D, Labrot E, Hafeez N, Bilton SJ, Drier Y, Ji F, Greenberg M, Williams A, Kattermann K, Damon L, Sovath S, Rakiec DP, Korn JM, Ruddy DA, Benes CH, Hammerman PS, Piotrowska Z, Sequist LV, Niederst MJ, Barretina J, Engelman JA, Hata AN. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene. 2019;38:6399–6413. doi: 10.1038/s41388-019-0887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27:2192–2206. doi: 10.1101/gad.225334.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 38.Meng F, Wu G. The rejuvenated scenario of epithelial-mesenchymal transition (EMT) and cancer metastasis. Cancer Metastasis Rev. 2012;31:455–467. doi: 10.1007/s10555-012-9379-3. [DOI] [PubMed] [Google Scholar]
- 39.Li C, Hong L, Liu C, Min J, Hu M, Guo W. Astragalus polysaccharides increase the sensitivity of SKOV3 cells to cisplatin. Arch Gynecol Obstet. 2018;297:381–386. doi: 10.1007/s00404-017-4580-9. [DOI] [PubMed] [Google Scholar]
- 40.Boye A, Wu C, Jiang Y, Wang J, Wu J, Yang X, Yang Y. Compound Astragalus and Salvia miltiorrhiza extracts modulate MAPK-regulated TGF-beta/Smad signaling in hepatocellular carcinoma by multi-target mechanism. J Ethnopharmacol. 2015;169:219–228. doi: 10.1016/j.jep.2015.04.013. [DOI] [PubMed] [Google Scholar]
- 41.Wilcox RA, Feldman AL, Wada DA, Yang ZZ, Comfere NI, Dong H, Kwon ED, Novak AJ, Markovic SN, Pittelkow MR, Witzig TE, Ansell SM. B7-H1 (PD-L1, CD274) suppresses host immunity in T-cell lymphoproliferative disorders. Blood. 2009;114:2149–2158. doi: 10.1182/blood-2009-04-216671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother. 2007;56:739–745. doi: 10.1007/s00262-006-0272-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8:467–477. doi: 10.1038/nri2326. [DOI] [PubMed] [Google Scholar]
- 44.Pandey PR, Xing F, Sharma S, Watabe M, Pai SK, Iiizumi-Gairani M, Fukuda K, Hirota S, Mo YY, Watabe K. Elevated lipogenesis in epithelial stem-like cell confers survival advantage in ductal carcinoma in situ of breast cancer. Oncogene. 2013;32:5111–5122. doi: 10.1038/onc.2012.519. [DOI] [PubMed] [Google Scholar]
- 45.Raghow R, Yellaturu C, Deng X, Park EA, Elam MB. SREBPs: the crossroads of physiological and pathological lipid homeostasis. Trends Endocrinol Metab. 2008;19:65–73. doi: 10.1016/j.tem.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 46.Schelch K, Wagner C, Hager S, Pirker C, Siess K, Lang E, Lin R, Kirschner MB, Mohr T, Brcic L, Marian B, Holzmann K, Grasl-Kraupp B, Krupitza G, Laszlo V, Klikovits T, Dome B, Hegedus B, Garay T, Reid G, van Zandwijk N, Klepetko W, Berger W, Grusch M, Hoda MA. FGF2 and EGF induce epithelial-mesenchymal transition in malignant pleural mesothelioma cells via a MAPKinase/MMP1 signal. Carcinogenesis. 2018;39:534–545. doi: 10.1093/carcin/bgy018. [DOI] [PubMed] [Google Scholar]