

Abstract
HOE-642 has been shown to provide significant protection in a variety of models of cerebral and myocardial ischemia/reperfusion injury. In this study, we examined the impact of HOE-642, a selective Na+/H+ exchanger 1 inhibitor, with or without hypothermia on neuronal and neuronal mitochondrial function during resuscitation. Cardiac arrest was induced by 8 min of asphyxia in rats. Five groups were included in this study: sham; normothermia (N); HOE-642 (HOE, 1 mg/kg); hypothermia (Hypo, 33±0.5°C); and HOE-642 plus hypothermia (HOE+Hypo). Survival and neurological deficit scores (NDS) were evaluated after 24 h of resuscitation. ΔΨm, mitochondrial swelling, ROS production, mitochondrial complex I-IV activity, and ultrastructural changes of the hippocampal mitochondria were evaluated. Survival in the HOE+Hypo group (85.7%) was higher than in the N group (42.9%) and HOE group (31.8%), P<0.05. NDS in the Hypo and HOE+Hypo groups were lower than in the N and HOE groups, P<0.05. ΔΨm in the HOE group (2.7±0.9) were higher than in the N (1.3±0.3) and Hypo (1.4±0.4) groups, P<0.05. Mitochondrial swelling in the N group was severe than in the HOE and Hypo groups, P<0.05. The production of ROS in the HOE and HOE+Hypo groups were lower than in the N group, P<0.05. Complex I-IV activity in the HOE+Hypo group was higher than in the other groups. The ultrastructure of mitochondria in the N group was severely damaged. The mitochondria maintained structural integrity in the HOE, Hypo and HOE+Hypo groups. HOE-642 plus hypothermia during resuscitation was beneficial than HOE-642 or hypothermia alone.
Keywords: CPR, hypothermia, HOE642, ischemia/reperfusion, mitochondria, neuroprotection
Introduction
Cerebral recirculation disturbances and complex metabolic derangements usually lead to brain injury after successful cardiopulmonary resuscitation (CPR), which results in deteriorative sequelae and a high post-resuscitative mortality [1]. A lack of neuroprotective therapies to ameliorate ischemia/reperfusion (I/R) injury during the post-resuscitation period has caused poor outcomes [2]. Mitochondrial dysfunction is considered to be main determinant of the extent of cerebral injury [3]. Impairment of mitochondrial function leads to reduced ATP production, impaired Ca2+ buffering, and in particular, overproduction of reactive oxygen species (ROS) [4]. The mitochondrial death pathway features the sequential loss of the mitochondrial membrane potential (ΔΨm), which is accompanied by the irreversible opening of the mitochondrial permeability transition pore (mPTP), release of cytochrome c (Cyt c) into the cytosol, activation of the apoptotic cascade, and activation of the proteolytic activity of caspases [5].
The activation of the Na+/H+ exchanger 1 (NHE1) after ischemia may subsequently cause cerebral damage via Na+ and Ca2+ mediated toxic effects [6]. Concomitant Na+ accumulation leads to the reversal of the Na+/Ca2+ exchanger and Ca2+ overload, which ultimately contributes to ischemic cell death. Administration of HOE-642, a selective NHE1 inhibitor, has been shown to provide significant protection in a variety of models of cerebral and myocardial I/R injury [6-11]. Inhibition of NHE1 significantly reduced cerebral edema and the infarct volume [6,12,13]. Prevention of Ca2+ overload by NHE1 blockade has been proposed to be a potential mechanism of cardioprotection [14,15]. Inhibition of NHE1 reduces cardiac I/R injury by delaying mPTP opening and reducing mitochondrial-derived superoxide production [16]. Administration of HOE-642 at the start of chest compressions is helpful for the restoration of electrically and mechanically stable circulation, improves resuscitability, and ameliorates post resuscitation myocardial dysfunction after resuscitation from cardiac arrest [17,18]. No studies have examined the effects of HOE-642 when used in combination with therapeutic hypothermia and the effects of HOE-642 on mitochondria after cardiac arrest. In this study, we sought to characterize the effects of administration of HOE-642 with or without hypothermia at the initiation of resuscitation on neuronal outcome and neuronal mitochondrial function.
Materials and methods
All of the animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals of Harbin Medical University and were approved by the Institutional Animal Care and Use Committee of the Cancer Hospital, Harbin Medical University. Male Wistar rats (240-340 g) were from the Animal Center of Harbin Medical University. All rats were allowed to acclimate for 6 days before the experiments.
Experimental groups
Five groups were included in this study: sham (S group), in which rats received surgery and ventilation only; normothermia group (N group), in which rats were subjected to cardiac arrest induced by 8 min of asphyxia and the tympanic and rectal temperatures of rats were maintained at 37±0.5°C after the return of spontaneous circulation (ROSC); HOE-642 group (HOE group), in which HOE-642 (1 mg/kg) [19-21] was administered intravenously during the initiation of CPR in the setting of normothermia; mild hypothermia group (Hypo group), in which rats were cooled following ROSC, tympanic and rectal temperature was maintained at 33±0.5°C for 1 h, and animals were rewarmed for 1 h; HOE-642 plus mild hypothermia group (HOE+Hypo group), in which HOE-642 and mild hypothermia were administered as in the HOE group and Hypo group.
Experimental protocol
Rats were anesthetized with 4% isoflurane and then intubated. Mechanical ventilation was initiated with a volume rodent ventilator (Model 683, Harvard Apparatus, South Natick, MA, USA) at a rate of 40/min and positive end-expiratory pressure (PEEP) of 5 cmH2O, I:E = 1:1, FiO2 = 0.5. The tidal volume (8-12 ml/kg) was regulated by end-tidal CO2 maintained between 35 and 45 mmHg. During surgical preparation, the tympanic and rectal temperatures of rats were maintained at 37.0±0.5°C. Anesthesia was sustained with 2% isoflurane. After the mean arterial pressure (MAP), heart rate (HR), and blood gas values were maintained within a normal range, experimental animals were given vecuronium (2 mg/kg, intravenously) 5 min before asphyxia. Cardiac arrest was induced via asphyxia by turning off the ventilator and clamping the endotracheal tube. Cardiac arrest was confirmed by MAP <20 mmHg. The body temperatures were maintained at 37±0.5°C during the entire asphyxia period. Following 8 min of asphyxia, chest compressions were performed at a rate of 200-300/min in combination with 0.01 mg/kg epinephrine. Ventilation was resumed at the same time. Body temperature was maintained in different groups with either a heating lamp and carpet or surface cooling following ROSC. Experimental animals in all groups were mechanically ventilated for 1 h after ROSC. Epinephrine, 1 µg iv, was used after ROSC if MAP <50 mmHg in all groups. The neurological deficit scores (NDS, 0, normal, 500, brain death) were evaluated 24 h after resuscitation [22]. Animals in each group after 24 h of resuscitation were decapitated under deep anesthesia for hippocampal mitochondria isolation to determine the mitochondrial transmembrane potential (ΔΨm), mitochondrial swelling, ROS production, and complex I-IV activity as well as for electron micrograph inspection.
Electron microscopy inspection
Ultrastructural changes in isolated cerebral mitochondria were assessed by transmission electron microscopy. The cerebral tissue section acquired at 24 h after resuscitation was fixed with 4% glutaraldehyde in 0.1 mol/L cacodylate buffer (pH 7.4) and post fixed with 1% osmium tetroxide. The preparations were dehydrated through an ethanol gradient, processed for Epon 812 embedding and sectioned at a thickness of 80 nm on a rotary microtome. The ultrathin sections were stained with 4% uranyl acetate-lead citrate and examined with a HITACHI H-7650 Transmission Electron Microscope (HITACHI Ltd., Japan).
Isolation of hippocampal mitochondria
Hippocampal tissue was identified and rapidly separated from the brain. Hippocampal mitochondria were extracted with a tissue mitochondria isolation kit (Beyotime Institute of Biotechnology, China). The hippocampal tissue homogenate was centrifuged at 1,000 g for 5 min at 4°C to remove nuclei and any unbroken cells. The supernatant was collected and centrifuged at 35,000 g for 10 min at 4°C to obtain the pellet, which is the mitochondrial fraction. All of the isolation procedures were performed under ice-cold conditions.
Detection of ΔΨm
ΔΨm was monitored with a sensitive fluorescent JC-1 probe (ΔΨm detection kit, Beyotime Institute of Biotechnology, China) and inspected with an epifluorescence microscope. All of the steps were performed in the dark. Mitochondria (1 μg/10 μL) were suspended in incubation buffer. ΔΨm was determined using the ratio of JC-1 aggregates (red), which were expressed at an excitation and emission of 490 and 530 nm, to JC-1 monomers (green), which were expressed at an excitation and emission of 525 and 590 nm. Mitochondrial depolarization was expressed as the decrease in the intensity ratio of red/green fluorescence.
Determination of mitochondrial swelling
Mitochondrial swelling was measured with a swelling assay kit (GENMED Bioengineering Institute, Shanghai, China). Mitochondrial swelling was caused by the influx of solutes through the opened mPTP, which decreased absorbance. Mitochondria (200 μg/20 μL) were suspended in incubation buffer. After a 1-minute equilibration period, mitochondrial swelling was assessed in a fluorescence microplate reader via the decrease in absorbance at 520 nm. Mitochondrial swelling was triggered by the addition of CaCl2. Measurements were repeated every 60 s for 10 min. The decrease in absorbance was calculated for each sample as the difference between the values before and after the addition of CaCl2.
Measurement of ROS production
ROS production was measured with a ROS assay kit (GENMED Bioengineering Institute, Shanghai, China). The chromogenic reaction mixture was added to equal amounts of mitochondria (50 μg/50 μL) in each sample at 37°C for 15 min. All steps were performed in the dark. Production of ROS was observed at an excitation and emission of 490 and 530 nm with an epifluorescence microscope.
Spectrophotometric assays of electron transport chain complex activity
Complex I (NADH-ubiquinone reductase) activity
The electron transfer activity of complex I was determined by ubiquinone-stimulated NADH oxidation. Ten micrograms of mitochondrial protein was mixed with 1,000 μl of assay buffer (Mitochondrial respiratory chain Complex I activity testing kit, Nanjing Jiancheng Bioengineering Institute) in a quartz cuvette. The change in the absorbance of NADH was measured at 340 nm. The specificity of the assay was validated by rotenone-sensitive inhibition. Complex I activity was expressed as the oxidation rate of NADH (nmol NADH oxidized/min/mg protein).
Complex II (Succinate-ubiquinone reductase) activity
Complex II activity was analyzed by ubiquinone-2 stimulated dichlorophenolindophenol (DCPIP) reduction. Ten micrograms of mitochondrial protein was mixed with 1,000 μl of assay buffer (Mitochondrial respiratory chain Complex II activity testing kit, Nanjing Jiancheng Bioengineering Institute) in a quartz cuvette. The decrease in absorbance at 600 nm was measured. Complex II activity was expressed as the reduction rate of DCPIP (nmol DCPIP reduced/min/mg protein).
Complex III (Ubiquinol-Cyt c reductase) activity
Complex III activity was evaluated by ubiquinol-mediated ferricytochrome c reduction. Ten micrograms of mitochondrial protein was mixed with 1,000 μl of assay buffer (Mitochondrial respiratory chain Complex III activity testing kit, Nanjing Jiancheng Bioengineering Institute) in a quartz cuvette. The change of absorbance for ferricytochrome c was measured at 550 nm. Inhibition of the assay with Antimycin A was used to verify the specificity of the assay. Complex III activity was expressed as the reduction rate of ferricytochrome c (nmol ferricytochrome c reduced/min/mg protein).
Complex IV (Cyt c oxidase) activity
Two micrograms of mitochondrial protein was added to 1,000 μl of assay buffer (Mitochondrial respiratory chain Complex IV activity testing kit, Nanjing Jiancheng Bioengineering Institute) in a quartz cuvette. Complex IV activity was determined by the decrease in the rate of absorbance of ferrocytochrome c at 550 nm. Complex IV activity was expressed as the oxidation rate of ferrocytochrome c (nmol ferrocytochrome c oxidized/min/mg protein).
Statistical analyses
All data are presented as the means ± SD unless otherwise stated. Differences in physiologic parameters, animal weight, body temperature, the duration from asphyxia to cardiac arrest, CPR time, MAP, the dosages of epinephrine, ΔΨm, mitochondrial swelling, ROS production, and complex I-IV activities between groups were evaluated by one-way ANOVA, followed by the LSD multiple comparison test. A Chi-square was used to compare differences in the rate of ROSC and survival between groups. The Kruskal-Wallis test and Mann-Whitney U test were used to compare NDS between groups. Values of P<0.05 were considered statistically significant. All statistical assessments were performed with SPSS statistical software, version 22.0.
Results
The number of experimental animals used are indicated in the figures or figure legends.
Physiologic variables and hemodynamic measurements
Baseline measurements, including animal weight, body temperature, and hemodynamic parameters, did not vary between groups. The duration from asphyxia to cardiac arrest was not different among the N (214.3±9.3 s), HOE (205.2±7.7 s), Hypo (201.1±8.2 s) and HOE+Hypo (203.5±9.3 s) groups. The rate of ROSC was not significantly different between the no HOE-642 treatment groups (84.6%) and HOE-642 treated groups (81.3%). CPR time was not significantly different between the N (32.6±2.8 s), HOE (36.0±1.1 s), Hypo (36.5±1.0 s) and HOE+Hypo (35.4±2.4 s) groups. MAP is presented in Figure 1. MAP in the N, HOE, Hypo and HOE+Hypo groups was significantly higher than in the S group at 5 and 10 min after ROSC (P<0.05). MAP in the N and HOE groups was significantly lower than in the S, Hypo and HOE+Hypo groups from 20 min to 60 min after ROSC (P<0.05). During 1 h of mechanical ventilation after ROSC, the dosages of epinephrine used in the different groups were 3.78±3.58 µg (N group), 5.5±6.18 µg (HOE group), and 0 µg (S, Hypo, and HOE+Hypo groups). But, these values were not statistically significantly different.
Figure 1.
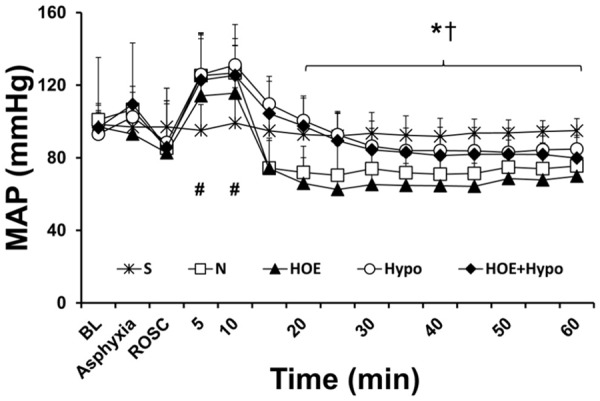
MAP during experiments. *P<0.05, S (n = 16), Hypo (n = 17), HOE+Hypo (n = 13) groups vs. N (n = 38) group. †P<0.05, S, Hypo, HOE+Hypo groups vs. HOE (n = 26) group; #P<0.05, N, HOE, Hypo and HOE+Hypo groups vs. S group. MAP, mean arterial pressure; BL, baseline; ROSC, return of spontaneous circulation. S, sham; N, normothermia; HOE, HOE-642; Hypo, hypothermia; HOE+Hypo, HOE-642 plus mild hypothermia.
Survival and neurological outcome
Animal survival is shown in Figure 2A. The survival in the HOE+Hypo group (85.7%) was significantly higher than in the N group (42.9%) and HOE group (31.8%), P<0.05. There was no significant difference between the N, HOE and Hypo groups (70%). NDS is shown in Figure 2B. NDS in the Hypo and HOE+Hypo groups was significantly lower than in the N and HOE groups at 24 h after ROSC (P<0.05). There was no significant difference between the N and HOE groups.
Figure 2.
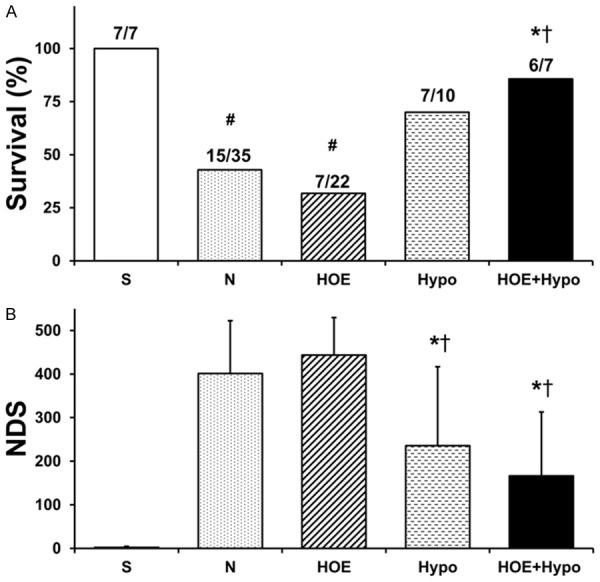
Survival and NDS at 24 h after resuscitation. A. Survival; B. NDS. *P<0.05, vs. N group; †P<0.05, vs. HOE group; #P<0.05, vs. S group. NDS, neurological deficit scores. S, sham; N, normothermia; HOE, HOE-642; Hypo, hypothermia; HOE+Hypo, HOE-642 plus mild hypothermia.
Electron microscopy inspection for ultrastructural changes of mitochondria
Ultrastructural changes of mitochondria are shown in Figure 3. Mitochondria in the sham group exhibited normal membrane integrity. The cristae showed no signs of swelling or injury. A typical homogeneous staining pattern of the matrix was observed without density changes. Mitochondria in the N group are severely damaged and exhibited swelling, decreased matrix density and cristae disintegration. The ultrastructure of the mitochondria maintained structural integrity, and the cristae showed no significant signs of swelling, injury, or density changes in the HOE, Hypo and HOE+Hypo groups.
Figure 3.
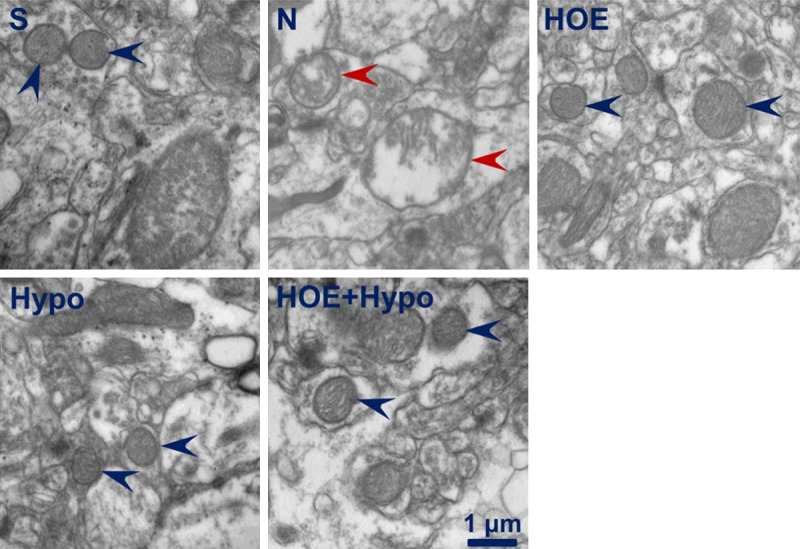
Representative isolated mitochondrial ultrastructural changes assessed by electron microscopy at 24 h after resuscitation. Blue arrowheads, mitochondrial ultrastructural exhibited normal membrane integrity and no signs of swelling or injury; red arrowheads, mitochondrial ultrastructural exhibited swelling, decreased matrix density, and cristae disintegration. S, sham; N, normothermia; HOE, HOE-642; Hypo, hypothermia; HOE+Hypo, HOE-642 plus mild hypothermia. Magnification, 30,000X. Scale bar, 1 µm.
ΔΨm
ΔΨm is presented in Figure 4A. ΔΨm in the HOE group was significantly higher than in the N group and Hypo group (P<0.05). There was no difference between the HOE group and HOE+Hypo group.
Figure 4.
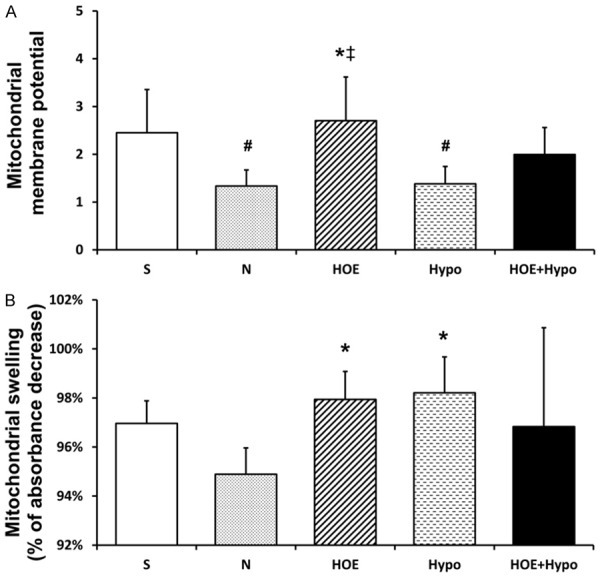
Mitochondrial membrane potential and swelling at 24 h after resuscitation. A. Mitochondrial membrane potential was expressed by the fluorescence intensity ratio of JC-1 aggregation and monomer, n = 4-5/group; B. Mitochondrial swelling, n = 3-5/group. *P<0.05, vs. N groups; ‡P<0.05, vs. Hypo group; #P<0.05, vs. S group. S, sham; N, normothermia; HOE, HOE-642; Hypo, hypothermia; HOE+Hypo, HOE-642 plus mild hypothermia.
Mitochondrial swelling
Mitochondrial swelling is shown in Figure 4B. Mitochondrial swelling in the N group was significantly severe than in the HOE and Hypo groups at 24 h after ROSC (P<0.05). There was no significant difference between the HOE, Hypo and HOE+Hypo groups.
ROS production
ROS production is shown in Figure 5. Production of ROS in the N group was significantly higher than in the HOE, and HOE+Hypo groups at 24 h after ROSC (P<0.05). There was no significant difference between the HOE, Hypo and HOE+Hypo groups.
Figure 5.
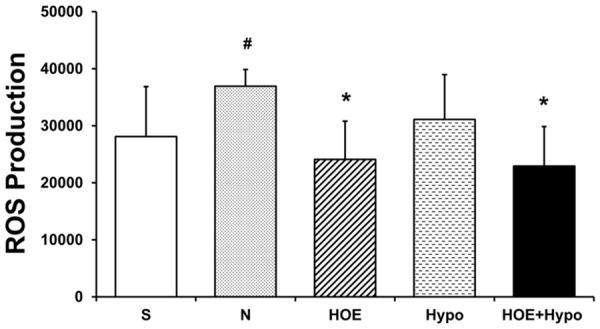
ROS production at 24 h after resuscitation. n = 4-6/group. *P<0.05, vs. N group; #P<0.05, vs. S group. ROS, reactive oxygen species. S, sham; N, normothermia; HOE, HOE-642; Hypo, hypothermia; HOE+Hypo, HOE-642 plus mild hypothermia.
Complex I-IV activities
The mitochondrial complex activities are shown in Figure 6. Complex I activity in the HOE+Hypo group was higher than in the HOE group (P<0.05, Figure 6A). There was no difference between the N, HOE, Hypo groups. Complex II activity in the HOE+Hypo group was higher than in the HOE and Hypo groups (P<0.05, Figure 6B). There was no difference between the N, HOE, and Hypo groups. Complex III activity in the N and HOE groups were lower than in the S group (P<0.05, Figure 6C). Complex III activity in the HOE+Hypo group was higher than in the N, HOE and Hypo groups (P<0.05). There was no difference between the N, HOE, and Hypo groups. Complex IV activity in the HOE+Hypo group was higher than in the N and HOE groups (P<0.05). Complex IV activity in the Hypo group was higher than in the HOE group (P<0.05).
Figure 6.
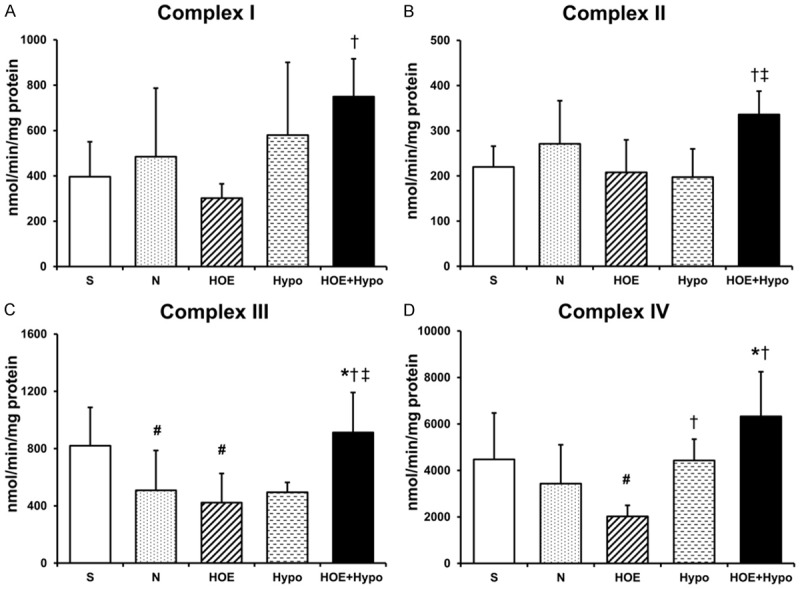
Electron transport chain complex activities in isolated mitochondria at 24 h after resuscitation. A. Complex I activity, n = 5-8/group; B. Complex II activity, n = 5-8/group; C. Complex III activity, n = 5-7/group; D. Complex IV activity at 24 h after ROSC, n = 5/group. *P<0.05, vs. N group; †P<0.05, vs. HOE group; ‡P<0.05, vs. Hypo group; #P<0.05, vs. S group. S, sham; N, normothermia; HOE, HOE-642; Hypo, hypothermia; HOE+Hypo, HOE-642 plus mild hypothermia.
Discussion
The present study was the first to demonstrate the effects of HOE-642 with or without therapeutic hypothermia on neuronal mitochondrial function and the opening of mPTP during the initiation of resuscitation after cardiac arrest. Our data demonstrated that HOE-642 in combination with hypothermia was significantly more beneficial than HOE-642 or hypothermia alone. HOE-642 plus hypothermia improved survival and neurological function. HOE-642 plus hypothermia alleviated microscopic and ultrastructural changes in the cerebral hippocampal mitochondria. HOE-642 plus hypothermia decreased the production of ROS and increased mitochondrial complex activity. HOE-642 alone did not improve survival, neuronal function recovery, or complex I-IV activity. However, HOE-642 alleviated microscopic and ultrastructural changes in the cerebral hippocampal mitochondria, enhanced ΔΨm, prevented mitochondrial swelling, and decreased production of ROS. HOE-642 induced hypotension, which could be prevented by hypothermia. HOE-642 had no influence on the rate of ROSC. Hypothermia improved survival and neurological function. Hypothermia preserved the mitochondrial ultrastructure and prevented mitochondrial swelling. Hypothermia alone did not decrease ROS production or improve complex activity.
HOE-642 has beneficial myocardial effects for resuscitation from cardiac arrest, presumably by protecting mitochondrial integrity and function [20,23]. HOE-642 leads to a hemodynamically more effective chest resuscitation during closed-chest resuscitation and attenuates post-resuscitation myocardial dysfunction [24]. However, it was reported that intravenous injection of HOE-642 reduced myocardial infarction but increased the rate of cerebrovascular occlusive acute events and severe acute events and decreased survival. HOE-642 induced hypotension according to the current study, our preliminary data (Circulation, 2015; 132: A13895.), and other report [21]. HOE-642 used in cardiopulmonary bypass (CPB) resuscitation resulted in prolonged CPB and higher pump flows of CPB were required, which caused severe vasodilation and increased the need for vasopressor administration [21]. A potential culprit is the adverse effect of HOE-642 on the vascular tone. One of the reasons why HOE-642 plus hypothermia was more effective than HOE-642 or hypothermia alone might be because hypothermia prevented the post resuscitation hypotension caused by HOE-642. The hypotension caused by HOE-642 explained why HOE-642 had protective effects on mitochondria but did not improve survival and neurological function. In addition, importantly, a previous study showed that the NHE mechanism remains functional even under hypothermic conditions and is a potential target for inhibition when hypothermic conditions are present [25]. Therefore, the conclusion of this study that HOE-642 plus hypothermia improved recovery compared to hypothermia alone during resuscitation could be explained by which the NHE mechanism remained functional under hypothermic conditions was inhibited by HOE-642.
HOE-642 improves post-ischemic recovery due to the attenuation of Ca2+ overload and prolonged acidosis during reperfusion [26]. Evidence from studies about I/R other than cardiac arrest indicates that NHE1 is activated by the profound intracellular acidosis that accompanies ischemia [14,27]. NHE1 activation prompts Na+ entry into cardiomyocytes in exchange for H+. Na+ accumulates in the cytosol and drives Ca2+ entry through the sarcolemmal Na+/Ca2+ exchanger, which operates in reverse mode [28,29]. Cytosolic and mitochondrial Ca2+ overload in conjunction with ATP depletion and oxidative stress open the mPTP and lead to mitochondrial swelling, depolarization, uncoupling of oxidative phosphorylation, and release of proapoptotic factors [30]. The beneficial effect of HOE-642 during myocardial I/R is thought to primarily result from inhibition of NHE1, thereby limiting Na+-induced cytosolic Ca2+ overload and subsequent mitochondrial Ca2+ overload [5,26,31]. The main mitochondrial damage after resuscitation is the opening of mPTP, which is deemed to be the common pathway for cell apoptosis and death after injury [32]. Many factors, such as intra-cellular calcium overload, ROS bursts, and reduced energy generation, induce mPTP opening, which results in decreased ΔΨm during I/R injury [33]. In vitro, silencing of cardiac mitochondrial NHE1 prevented mPTP opening [16]. In addition, NHE1 inhibitors may have direct effects on mitochondrial ion-exchange mechanisms, increasing the threshold for opening mPTP [34]. No studies have shown the effects of HOE-642 on mPTP after cardiac arrest. Our data showed that both HOE-642 and mild hypothermia prevented the opening of mPTP and increased ΔΨm. These results confirmed that mild hypothermia prevented the opening of mPTP after cardiac arrest [35]. In this study, HOE-642 prevented the swelling of mitochondria and enhanced ΔΨm, while hypothermia prevented the swelling of mitochondria without preventing the decrease in ΔΨm. This finding likely indicates that hypothermia partially inhibited the opening of mPTP, while HOE-642 prevented the opening of mPTP to a larger extent. We propose that when hypothermia was used, opening of mPTP was partially inhibited and larger molecules and water could not enter the mitochondrial matrix, resulting in the prevention of mitochondrial swelling. However, ions could pass through mPTP freely, resulting in a loss of ΔΨm and ion balance disruption. The fact HOE-642 enhanced the inhibition on the opening of mPTP by hypothermia might be the other reason why HOE-642 plus hypothermia was significantly more beneficial than hypothermia only.
Mitochondrial dysfunction is characterized by faulty electron transport through the respiratory complexes. Slow electron transfer is directly related to an abnormal increase in the generation of reactive ROS by electron “leakage” at the partially or totally blocked respiratory complex, principally at complex I (NADH dehydrogenase) or complex III [36]. In this study, a significant increase in complex I-IV activity was found when HOE-642 plus hypothermia was used after cardiac arrest compared to HOE-642 or hypothermia alone. This finding was consistent with the protective effects of HOE-642 in combination with hypothermia on improving survival and neurological function compared to HOE-642 or hypothermia alone.
This study had some limitations. First, there was no mitochondrial respiratory function evaluation. Although the mitochondrial complex activities were investigated in this study, mitochondrial respiration was an appropriate method to determine mitochondria function. Second, the effects of NHE1 inhibition were not confirmed by measuring calcium, and sodium and no HOE-642 dose-effective investigation was performed. The dosage of HOE-642, 1 mg/kg, chosen in this study was based on previous reports [7,20,21]. The optimal dosage and the treatment of HOE-642 for the protection of neuronal mitochondria after cardiac arrest should be investigated. Third, animals were not randomly allocated to treatment groups before each study and investigators were not blinded to drug treatment, so an inherent risk of bias is possible.
In conclusion, our results demonstrated that HOE-642 plus hypothermia used after cardiac arrest improved survival, neurological recovery, and hippocampal mitochondria function compared with the normothermic group. HOE-642 in combination with hypothermia showed significantly more beneficial than HOE-642 or hypothermia treatment alone. HOE-642 inhibited the opening of mPTP when used after cardiac arrest. Hypothermia did not adequately inhibit the opening of mPTP.
Acknowledgements
The authors greatly appreciate the technical assistance of the staff in the Lab of Anesthesiology and Critical Care Medicine, the Second Affiliated Hospital, Harbin Medical University. This study was supported by the National Natural Science Foundation of China (grant number 81871515), the Nature Science Foundation, Department of Science and Technology, Heilongjiang, China (grant number LC2012C40), the fund of the Third Affiliated Hospital, Harbin Medical University (grant number JJZD2014-02), and the Nn10 program of the Third Affiliated Hospital, Harbin Medical University, China (grant number Nn10py2017-05).
Disclosure of conflict of interest
None.
References
- 1.Madl C, Holzer M. Brain function after resuscitation from cardiac arrest. Curr Opin Crit Care. 2004;10:213–217. doi: 10.1097/01.ccx.0000127542.32890.fa. [DOI] [PubMed] [Google Scholar]
- 2.Gong P, Li CS, Hua R, Zhao H, Tang ZR, Mei X, Zhang MY, Cui J. Mild hypothermia attenuates mitochondrial oxidative stress by protecting respiratory enzymes and upregulating MnSOD in a pig model of cardiac arrest. PLoS One. 2012;7:e35313. doi: 10.1371/journal.pone.0035313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan PH. Mitochondrial dysfunction and oxidative stress as determinants of cell death/survival in stroke. Ann N Y Acad Sci. 2005;1042:203–209. doi: 10.1196/annals.1338.022. [DOI] [PubMed] [Google Scholar]
- 4.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 5.Teshima Y, Akao M, Jones SP, Marban E. Cariporide (HOE642), a selective Na+-H+ exchange inhibitor, inhibits the mitochondrial death pathway. Circulation. 2003;108:2275–2281. doi: 10.1161/01.CIR.0000093277.20968.C7. [DOI] [PubMed] [Google Scholar]
- 6.Luo J, Chen H, Kintner DB, Shull GE, Sun D. Decreased neuronal death in Na+/H+ exchanger isoform 1-null mice after in vitro and in vivo ischemia. J Neurosci. 2005;25:11256–11268. doi: 10.1523/JNEUROSCI.3271-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Luo J, Chen X, Chen H, Cramer SW, Sun D. Gene inactivation of Na+/H+ exchanger isoform 1 attenuates apoptosis and mitochondrial damage following transient focal cerebral ischemia. Eur J Neurosci. 2008;28:51–61. doi: 10.1111/j.1460-9568.2008.06304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Myers ML, Farhangkhoee P, Karmazyn M. Hydrogen peroxide induced impairment of post-ischemic ventricular function is prevented by the sodium-hydrogen exchange inhibitor HOE 642 (cariporide) Cardiovasc Res. 1998;40:290–296. doi: 10.1016/s0008-6363(98)00183-7. [DOI] [PubMed] [Google Scholar]
- 9.Rupprecht HJ, vom Dahl J, Terres W, Seyfarth KM, Richardt G, Schultheibeta HP, Buerke M, Sheehan FH, Drexler H. Cardioprotective effects of the Na(+)/H(+) exchange inhibitor cariporide in patients with acute anterior myocardial infarction undergoing direct PTCA. Circulation. 2000;101:2902–2908. doi: 10.1161/01.cir.101.25.2902. [DOI] [PubMed] [Google Scholar]
- 10.Sakurai S, Kuroko Y, Shimizu S, Kawada T, Akiyama T, Yamazaki T, Sugimachi M, Sano S. Effects of intravenous cariporide on release of norepinephrine and myoglobin during myocardial ischemia/reperfusion in rabbits. Life Sci. 2014;114:102–106. doi: 10.1016/j.lfs.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 11.Bourahla V, Dubouchaud H, Mourmoura E, Vitiello D, Faure P, Migne C, Pujos-Guillot E, Richardson M, Demaison L. Disruption of chronic cariporide treatment abrogates myocardial ion homeostasis during acute ischemia reperfusion. J Cardiovasc Pharmacol. 2011;58:284–294. doi: 10.1097/FJC.0b013e318223ebb2. [DOI] [PubMed] [Google Scholar]
- 12.O’Donnell ME, Chen YJ, Lam TI, Taylor KC, Walton JH, Anderson SE. Intravenous HOE-642 reduces brain edema and Na uptake in the rat permanent middle cerebral artery occlusion model of stroke: evidence for participation of the blood-brain barrier Na/H exchanger. J Cereb Blood Flow Metab. 2013;33:225–234. doi: 10.1038/jcbfm.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrazzano P, Shi Y, Manhas N, Wang Y, Hutchinson B, Chen X, Chanana V, Gerdts J, Meyerand ME, Sun D. Inhibiting the Na+/H+ exchanger reduces reperfusion injury: a small animal MRI study. Front Biosci (Elite Ed) 2011;3:81–88. doi: 10.2741/e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avkiran M. Rational basis for use of sodium-hydrogen exchange inhibitors in myocardial ischemia. Am J Cardiol. 1999;83:10G–17G. doi: 10.1016/s0002-9149(99)00215-5. [DOI] [PubMed] [Google Scholar]
- 15.Keul P, van Borren MM, Ghanem A, Muller FU, Baartscheer A, Verkerk AO, Stumpel F, Schulte JS, Hamdani N, Linke WA, van Loenen P, Matus M, Schmitz W, Stypmann J, Tiemann K, Ravesloot JH, Alewijnse AE, Hermann S, Spijkers LJ, Hiller KH, Herr D, Heusch G, Schafers M, Peters SL, Chun J, Levkau B. Sphingosine-1-phosphate receptor 1 regulates cardiac function by modulating Ca2+ sensitivity and Na+/H+ exchange and mediates protection by ischemic preconditioning. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.116.003393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villa-Abrille MC, Cingolani E, Cingolani HE, Alvarez BV. Silencing of cardiac mitochondrial NHE1 prevents mitochondrial permeability transition pore opening. Am J Physiol Heart Circ Physiol. 2011;300:H1237–1251. doi: 10.1152/ajpheart.00840.2010. [DOI] [PubMed] [Google Scholar]
- 17.Ayoub IM, Kolarova J, Gazmuri RJ. Cariporide given during resuscitation promotes return of electrically stable and mechanically competent cardiac activity. Resuscitation. 2010;81:106–110. doi: 10.1016/j.resuscitation.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gazmuri RJ, Ayoub IM, Hoffner E, Kolarova JD. Successful ventricular defibrillation by the selective sodium-hydrogen exchanger isoform-1 inhibitor cariporide. Circulation. 2001;104:234–239. doi: 10.1161/01.cir.104.2.234. [DOI] [PubMed] [Google Scholar]
- 19.Scholz W, Albus U, Counillon L, Gogelein H, Lang HJ, Linz W, Weichert A, Scholkens BA. Protective effects of HOE642, a selective sodium-hydrogen exchange subtype 1 inhibitor, on cardiac ischaemia and reperfusion. Cardiovasc Res. 1995;29:260–268. [PubMed] [Google Scholar]
- 20.Radhakrishnan J, Kolarova JD, Ayoub IM, Gazmuri RJ. AVE4454B--a novel sodium-hydrogen exchanger isoform-1 inhibitor--compared less effective than cariporide for resuscitation from cardiac arrest. Transl Res. 2011;157:71–80. doi: 10.1016/j.trsl.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liakopoulos OJ, Hristov N, Buckberg GD, Triana J, Trummer G, Allen BS. Resuscitation after prolonged cardiac arrest: effects of cardiopulmonary bypass and sodium-hydrogen exchange inhibition on myocardial and neurological recovery. Eur J Cardiothorac Surg. 2011;40:978–984. doi: 10.1016/j.ejcts.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liakopoulos OJ, Allen BS, Buckberg GD, Hristov N, Tan Z, Villablanca JP, Trummer G. Resuscitation after prolonged cardiac arrest: role of cardiopulmonary bypass and systemic hyperkalemia. Ann Thorac Surg. 2010;89:1972–1979. doi: 10.1016/j.athoracsur.2010.02.052. [DOI] [PubMed] [Google Scholar]
- 23.Lin X, Kraut JA, Wu D. Coadministration of a Na+-H+ exchange inhibitor and sodium bicarbonate for the treatment of asphyxia-induced cardiac arrest in piglets. Pediatr Res. 2014;76:118–126. doi: 10.1038/pr.2014.65. [DOI] [PubMed] [Google Scholar]
- 24.Kolarova JD, Ayoub IM, Gazmuri RJ. Cariporide enables hemodynamically more effective chest compression by leftward shift of its flow-depth relationship. Am J Physiol Heart Circ Physiol. 2005;288:H2904–2911. doi: 10.1152/ajpheart.01181.2004. [DOI] [PubMed] [Google Scholar]
- 25.Yamauchi T, Ichikawa H, Sawa Y, Fukushima N, Kagisaki K, Maeda K, Matsuda H, Shirakura R. The contribution of Na+/H+ exchange to ischemia-reperfusion injury after hypothermic cardioplegic arrest. Ann Thorac Surg. 1997;63:1107–1112. doi: 10.1016/s0003-4975(96)01390-2. [DOI] [PubMed] [Google Scholar]
- 26.Stromer H, de Groot MC, Horn M, Faul C, Leupold A, Morgan JP, Scholz W, Neubauer S. Na(+)/H(+) exchange inhibition with HOE642 improves postischemic recovery due to attenuation of Ca(2+) overload and prolonged acidosis on reperfusion. Circulation. 2000;101:2749–2755. doi: 10.1161/01.cir.101.23.2749. [DOI] [PubMed] [Google Scholar]
- 27.Yuen N, Lam TI, Wallace BK, Klug NR, Anderson SE, O’Donnell ME. Ischemic factor-induced increases in cerebral microvascular endothelial cell Na/H exchange activity and abundance: evidence for involvement of ERK1/2 MAP kinase. Am J Physiol Cell Physiol. 2014;306:C931–942. doi: 10.1152/ajpcell.00021.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imahashi K, Kusuoka H, Hashimoto K, Yoshioka J, Yamaguchi H, Nishimura T. Intracellular sodium accumulation during ischemia as the substrate for reperfusion injury. Circ Res. 1999;84:1401–1406. doi: 10.1161/01.res.84.12.1401. [DOI] [PubMed] [Google Scholar]
- 29.Karmazyn M, Sostaric JV, Gan XT. The myocardial Na+/H+ exchanger: a potential therapeutic target for the prevention of myocardial ischaemic and reperfusion injury and attenuation of postinfarction heart failure. Drugs. 2001;61:375–389. doi: 10.2165/00003495-200161030-00006. [DOI] [PubMed] [Google Scholar]
- 30.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res. 2004;61:372–385. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 31.Sun HY, Wang NP, Halkos ME, Kerendi F, Kin H, Wang RX, Guyton RA, Zhao ZQ. Involvement of Na+/H+ exchanger in hypoxia/re-oxygenation-induced neonatal rat cardiomyocyte apoptosis. Eur J Pharmacol. 2004;486:121–131. doi: 10.1016/j.ejphar.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 32.Schriewer JM, Peek CB, Bass J, Schumacker PT. ROS-mediated PARP activity undermines mitochondrial function after permeability transition pore opening during myocardial ischemia-reperfusion. J Am Heart Assoc. 2013;2:e000159. doi: 10.1161/JAHA.113.000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woodfield K, Ruck A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J. 1998;336:287–290. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukuhiro Y, Wowk M, Ou R, Rosenfeldt F, Pepe S. Cardioplegic strategies for calcium control: low Ca(2+), high Mg(2+), citrate, or Na(+)/H(+) exchange inhibitor HOE-642. Circulation. 2000;102:III319–325. [PubMed] [Google Scholar]
- 35.Gong P, Hua R, Zhang Y, Zhao H, Tang Z, Mei X, Zhang M, Cui J, Li C. Hypothermia-induced neuroprotection is associated with reduced mitochondrial membrane permeability in a swine model of cardiac arrest. J Cereb Blood Flow Metab. 2013;33:928–934. doi: 10.1038/jcbfm.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borutaite V, Mildaziene V, Brown GC, Brand MD. Control and kinetic analysis of ischemia-damaged heart mitochondria: which parts of the oxidative phosphorylation system are affected by ischemia? Biochim Biophys Acta. 1995;1272:154–158. doi: 10.1016/0925-4439(95)00080-1. [DOI] [PubMed] [Google Scholar]