

Abstract
Recent preclinical evidence has indicated that both androgen receptor (AR) inactivation and glucocorticoid receptor (GR) transrepression are associated with suppression of urothelial carcinogenesis. We therefore assessed the effect of a unique compound, 2-(4-acetoxyphenyl)-2-chloro-N-methylethylammonium chloride (Compound A; CpdA), which could function as an AR antagonist as well as a GR ligand, on urothelial tumorigenesis. Using the in vitro system with GR-positive non-neoplastic urothelial SVHUC cells stably expressing AR (SVHUC-AR), neoplastic transformation induced by a chemical carcinogen 3-methylcholanthrene (MCA) was inhibited similarly by an anti-androgen hydroxyflutamide and a glucocorticoid prednisone, and more strongly by CpdA. CpdA also prevented the neoplastic transformation of AR-negative MCA-SVHUC cells, which was diminished by a GR antagonist RU486, but failed to prevent that of GR knockdown MCA-SVHUC cells. In MCA-SVHUC-AR cells, CpdA significantly reduced the expression levels of oncogenes (c-Fos/c-Jun/c-Myc) and induced those of tumor suppressors (UGT1A/p21/p27/p53/PTEN). Additionally, a potent carcinogen N-butyl-N-(4-hydroxybutyl)nitrosamine induced bladder cancer in all of 8 mock-treated mice versus 4 (50%) of flutamide-treated (P = 0.021), 4 (50%) of prednisone-treated (P = 0.021), or 2 (25%) of CpdA-treated (P = 0.002) animals. Finally, CpdA was found to reduce AR transactivation and selectively induce GR transrepression (i.e. suppression of NF-κB transactivation and expression of its regulated genes), but not GR transactivation (i.e. activation of glucocorticoid-response element-mediated transcription and expression of its targets) in SVHUC cells. These findings suggest that CpdA suppresses urothelial tumorigenesis via both the AR and GR pathways, which may consequently provide an effective option of chemoprevention for bladder cancer, especially in patients with superficial disease following transurethral surgery.
Keywords: Anti-androgen, dexamethasone, malignant transformation, prednisone, urothelial cancer
Introduction
Urinary bladder cancer, which is mostly a urothelial carcinoma, has been one of the most frequently diagnosed neoplasms predominantly affecting men [1]. Up to 80% of patients with bladder tumor present with non-muscle-invasive disease in which tumor recurrence is common even after transurethral surgery and currently available intravesical pharmacotherapy with bacillus Calmette-Guèrin or cytotoxic agents [2]. In spite of therapeutic advances, the prognosis for non-muscle-invasive bladder tumor has not significantly improved during the past several decades. Therefore, identification of molecules or pathways that play a key role in urothelial tumorigenesis is urgently required, which may successively offer new targeted therapy that more effectively prevents the recurrence of superficial bladder tumor.
Emerging evidence has indicated the involvement of androgen-mediated androgen receptor (AR) signaling in modulating both of two distinct events/processes, urothelial tumorigenesis and tumor progression, while precise mechanisms for the functions of AR and related signals in urothelial cells remain far from being fully understood (reviewed in [3]). In particular, we have demonstrated preclinical findings suggesting that AR activation is associated with the induction of urothelial tumorigenesis [4-11], which may explain the sex-related disparity in bladder cancer incidence. Retrospective cohort studies by us [12,13] and another group [14] also indicated that androgen deprivation therapy in men with prostate cancer could considerably reduce the risk of recurrence of their superficial bladder cancer.
In addition to AR, glucocorticoid receptor (GR), another steroid hormone receptor, has been implicated in the progression of bladder cancer (reviewed in [15]). Specifically, we have demonstrated that several glucocorticoids, including corticosterone, dexamethasone (DEX), and prednisone (PRED), inhibit the invasion of GR-positive tumor cells, while DEX contradictorily induces tumor cell proliferation and reduces apoptosis in the presence or absence of a cytotoxic agent cisplatin [16,17]. Recently, we have shown preclinical data suggesting that the neoplastic transformation of urothelial cells or the development of bladder cancer is prevented by PRED which selectively induces GR transrepression, but not by DEX which induces both transactivation and transrepression of GR [18].
It is thus likely that AR inactivation and GR transrepression are associated with the suppression of urothelial carcinogenesis. Meanwhile, we have demonstrated that a unique steroid hormone receptor modulator, 2-(4-acetoxyphenyl)-2-chloro-N-methylethylammonium chloride (Compound A; CpdA), which is known to function as an AR antagonist as well as a GR ligand [19,20], inhibits not only the migration/invasion but also the proliferation of AR-positive/GR-positive bladder cancer cells [21]. Accordingly, the present study aimed to assess the effect of CpdA on urothelial tumorigenesis.
Materials and methods
Cell lines and chemicals
An immortalized human normal urothelial cell line, SVHUC, was originally obtained from the American Type Culture Collection and recently authenticated, using GenePrint 10 System (Promega), by the institutional core facility. Stable sublines, including SVHUC-AR expressing a full-length wild-type human AR, as well as SVHUC-control-short hairpin RNA (shRNA) and SVHUC-GR-shRNA, were established in our previous studies [6,16,18] (also see Figure 1). The parental cells and SVHUC-derived sublines were maintained in Ham’s F-12K (Kaighn’s) medium (Mediatech) supplemented with 10% fetal bovine serum (FBS) in a humidified atmosphere of 5% CO2 at 37°C and routinely tested for Mycoplasma contamination, using PCR Mycoplasma Detection Kit (Applied Biological Materials). Phenol red-free medium supplemented with either 5% regular FBS or 5% charcoal-stripped FBS was then used during actual assays. We obtained DEX, PRED, CpdA, hydroxyflutamide (HF), mifepristone (RU486), and flutamide from Sigma-Aldrich.
Figure 1.
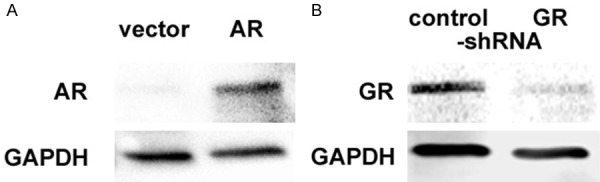
Expression of AR and GR in SVHUC-derived sublines. Western blotting of AR (A) and GR (B), using protein extracts from SVHUC-vector vs. SVHUC-AR and SVHUC-control-shRNA vs. SVHUC-GR-shRNA. GAPDH served as an internal control.
Western blot
Proteins (30 µg) obtained from cell extracts were separated in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membrane electronically, blocked, and incubated with an appropriate dilution of each specific antibody [an anti-AR antibody (clone N20; Santa Cruz Biotechnology), an anti-GR antibody (clone H-300; Santa Cruz Biotechnology), or an anti-GAPDH antibody (clone 6c5; Santa Cruz Biotechnology)] and then a secondary antibody (anti-mouse IgG HRP-linked antibody or anti-rabbit IgG HRP-linked antibody; Cell Signaling Technology), which was followed by scanning with an imaging system (ChemiDOC™ MP, Bio-Rad).
In vitro transformation
An in vitro neoplastic/malignant transformation system was employed, using the SVHUC line upon exposure to a carcinogen 3-methylcholanthrene (MCA), as established in a previous study [22], with minor modifications. In brief, cells (2×106/10-cm culture dish incubated for 24 hours) were cultured in FBS-free F-12K containing 5 µg/ml MCA (Sigma-Aldrich). After the first 24 hours of MCA exposure, 1% FBS was added to the medium. After additional 24 hours of MCA exposure, the cells were cultured in medium containing 5% FBS (without MCA) until near confluence. Subcultured cells (1:3 split ratio) were again incubated with MCA for two 48-hour exposure periods, using the above protocol. These MCA-exposed cells were subcultured for 6 weeks in the presence or absence of AR/GR ligands (without MCA) and then utilized for subsequent assays.
Cell proliferation assay
We used the methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay to assess cell viability. Cells (500-1000/well) seeded in 96-well tissue culture plates were cultured for up to 72 hours, and then incubated with 0.5 mg/mL of MTT (Sigma-Aldrich) in 100 μL of medium for 3 hours at 37°C. MTT was dissolved by DMSO, and the absorbance was measured at a wavelength of 570 nm with background subtraction at 630 nm.
Plate colony formation assay
Cells (500/well) seeded in 12-well tissue culture plates were allowed to grow until colonies in the control well were certainly detectable. The cells were then fixed with methanol and stained with 0.1% crystal violet. The number of colonies in photographed pictures was quantitated, using ImageJ software (National Institutes of Health).
Reverse transcription (RT) and real-time polymerase chain reaction (PCR)
Total RNA isolated from cultured cells by TRIzol (Invitrogen) was subject to RT, using oligo-dT primers and Ominiscript reverse transcriptase (Qiagen). Real-time PCR was then conducted, using RT2 SYBR Green FAST Mastermix (Qiagen). The primer sequences are given in Table 1.
Table 1.
Sequences of PCR primers
| Gene | Sense | Anti-sense |
|---|---|---|
| c-Fos | 5’-CGAGATGGAGATCGGTATGGT-3’ | 5’-GGGTCTTCTTACCCGGCTTG-3’ |
| c-Jun | 5’-TGTACCGACTGAGAGTTCTTGA-3’ | 5’-ACAGAGCGAGTGAAAATGTGTAT-3’ |
| c-Myc | 5’-ACCAGATCCCGGAGTTGGAA-3’ | 5’-CGTCGTTTCCGCAACAAGTC-3’ |
| UGT1A | 5’-TGGGTGGAGTTTGTGATGAGGC-3’ | 5’-CAATGGGTCTTGGATTTGTGGG-3’ |
| p21 | 5’-AAGACCATGTGGACCTGTCACTGT-3’ | 5’-GAAGATCAGCCGGCGTTTG-3’ |
| p27 | 5’-CGAGTGGCAAGAGGTGGAGA-3’ | 5’-GGAGCCCCAATTAAAGGCG-3’ |
| p53 | 5’-GGAGGGGCGATAAATACC-3’ | 5’-AACTGTAACTCCTCAGGCAGGC-3’ |
| PTEN | 5’-GTTTACCGGCAGCATCAAAT-3’ | 5’-CCCCCACTTTAGTGCACAGT-3’ |
| FKBP51 | 5’-CTCCCTAAAATTCCCTCGAATGC-3’ | 5’-CCCTCTCCTTTCCGTTTGGTT-3’ |
| GILZ | 5’-AACACCGAAATGTATCAGACCC-3’ | 5’-TGTCCAGCTTAACGGAAACCA-3’ |
| IL-6 | 5’-AAATTCGGTACATCCTCGACGG-3’ | 5’-GGAAGGTTCAGGTTGTTTTCTGC-3’ |
| VEGF | 5’-CTGTACCTCCACCATGCCAAG-3’ | 5’-GGTACTCCTGGAAGATGTCCACC-3’ |
| GAPDH | 5’-AAGGTGAAGGTCGGAGTCAAC-3’ | 5’-GGGGTCATTGATGGCAACAATA-3’ |
Reporter gene assay
Cells at a density of 50-70% confluence in 24-well tissue culture plates were co-transfected with 250 ng of a luciferase reporter plasmid DNA, androgen-response element (ARE)- and glucocorticoid-response element (GRE)-driven MMTV-Luc [4,23] or NF-κB-Luc (Signosis), and 2.5 ng of a control reporter plasmid (pRL-CMV), using Lipofectamine® 3000 transfection reagent (Life Technologies). After transfection, the cells were cultured in the presence or absence of AR/GR ligands for 24 hours. Cell lysates were then assayed for luciferase activity measured using a Dual-Luciferase Reporter Assay kit (Promega).
Mouse models
The animal protocol in accordance with the National Institutes of Health Guidelines for the Care and Use of Experimental Animals was approved by the Institutional Animal Care and Use Committee. The male C57BL/6 mice (Johns Hopkins University Research Animal Resources) at age of 6 weeks were supplied ad libitum with tap water containing 0.1% N-butyl-N-(4-hydroxybutyl)nitrosamine (BBN) (Sigma-Aldrich) for 12 weeks, as we described previously [4,10,18]. These mice also received daily subcutaneous injections of vehicle (1/2000 ethanol in 0.2 mL sterile distilled water), flutamide (500 µg), DEX (10 µg), PRED (10 µg), or CpdA (10 µg). At 18 weeks of their age, all the animals were euthanized for macroscopic and microscopic analyses of the bladder and other major organs.
Statistical analysis
Chi-square test and Student’s t-test were used to assess statistical significance for categorized variables and those with ordered distribution, respectively. P values less than 0.05 were considered statistically significant.
Results
The efficacy of CpdA for neoplastic transformation of urothelial cells
Human normal urothelial SVHUC cells have been shown to express GR [18], but not AR [6] (see Figure 1). We first investigated the effects of an anti-androgen, HF, and glucocorticoids, DEX and PRED, as well as CpdA, for their inhibitory activity in the neoplastic/malignant transformation of AR-positive/GR-positive SVHUC-AR urothelial cells, using an in vitro system. Following exposure to chemical carcinogens, such as MCA, non-neoplastic SVHUC cells are known to undergo stepwise transformation during subsequent 6-week culture [22] (see Figure 2A). In this period of the neoplastic transformation, each ligand was treated for 6 weeks. Carcinogen-mediated oncogenic activity (i.e. degree of neoplastic transformation) was then monitored by the viability (via MTT assay; Figure 2B) and colony formation (via clonogenic assay; Figure 2C) of resultant cells without further drug treatment that could directly affect their growth during the assays. In accordance with our previous observations [8,18], the 6-week culture with HF or PRED resulted in a significant delay in cell growth, indicating their preventive effects on urothelial tumor initiation, while DEX showed an even marginal stimulatory activity. Additionally, as expected, treatment with CpdA more strongly inhibited the neoplastic transformation of MCA-SVHUC-AR cells.
Figure 2.
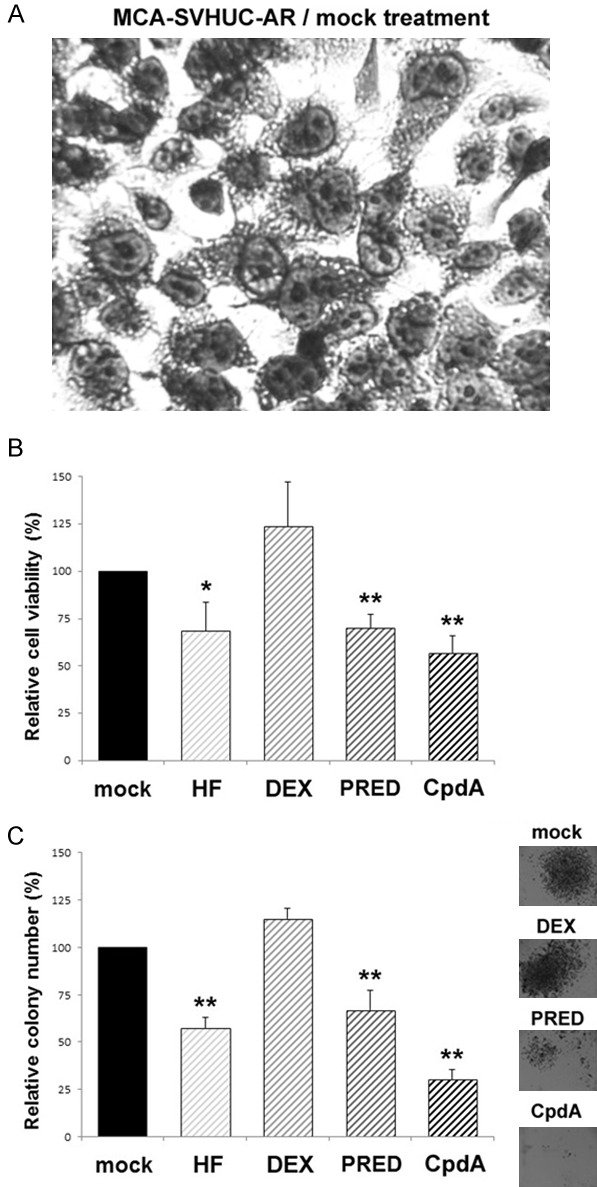
Effects of CpdA on the neoplastic transformation of AR-positive/GR-positive urothelial cells. Morphology of SVHUC-AR cells exposed to MCA and subsequently cultured for 6 weeks (A; original magnification: ×400). MCA-exposed SVHUC-AR cells cultured with ethanol (mock), HF (1 µM), PRED (10 nM), or CpdA (10 nM) for 6 weeks were seeded for MTT assay (B; additional 72-hour culture without drug treatment) or clonogenic assay (C; additional 2-week culture without drug treatment). Cell viability or colony number (≥20 cells) presented relative to that of mock-treated cells represents the mean (+SD) from three independent experiments. *P<0.05 (vs. mock treatment). **P<0.01 (vs. mock treatment).
We further assessed the effects of CpdA on the neoplastic transformation of AR-negative/GR-positive SVHUC cells with the carcinogen challenge (see Figure 3A). PRED or CpdA treatment during the process of neoplastic transformation resulted in a striking delay in cell viability (Figure 3B) or colony formation (Figure 3C), which was diminished by a GR antagonist RU486, suggesting the suppressive effects of PRED/CpdA via the GR pathway. Correspondingly, in GR knockdown MCA-SVHUC where the neoplastic transformation was significantly induced, PRED and CpdA failed to considerably prevent it (P>0.1).
Figure 3.
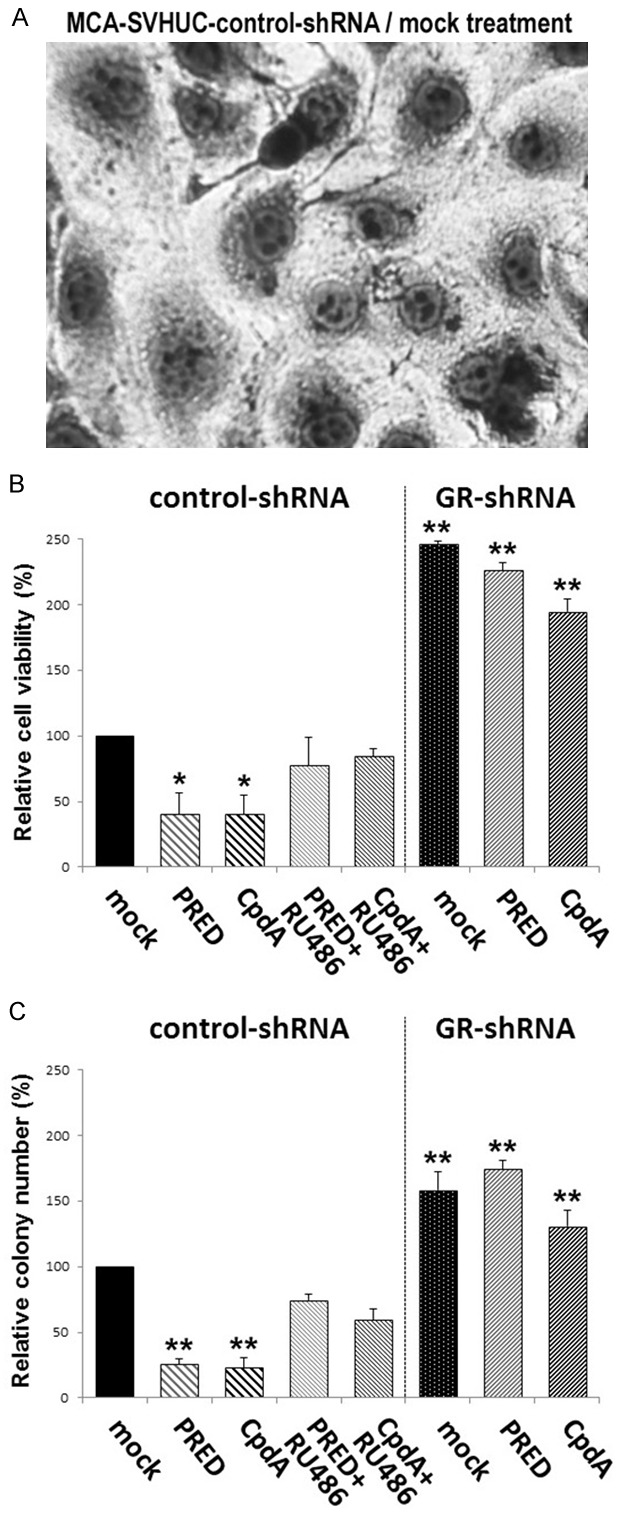
Effects of CpdA on the neoplastic transformation of AR-negative/GR-positive urothelial cells. Morphology of SVHUC-control-shRNA cells exposed to MCA and subsequently cultured for 6 weeks (A; original magnification: ×400). MCA-exposed SVHUC-control-shRNA/SVHUC-GR-shRNA cells cultured with ethanol (mock), PRED (10 nM), CpdA (10 nM), and/or RU486 (1 µM) for 6 weeks were seeded for MTT assay (B; additional 72-hour culture without drug treatment) or clonogenic assay (C; additional 2-week culture without drug treatment). Cell viability or colony number (≥20 cells) presented relative to that of mock-treated SVHUC-control-shRNA cells represents the mean (+SD) from three independent experiments. *P<0.05 (vs. mock-treated control-shRNA subline). **P<0.01 (vs. mock-treated control-shRNA subline).
To support the preventive effects of the AR/GR ligands, we compared the expression levels of oncogenic molecules, as well as other molecules having suppressive functions in bladder tumorigenesis, in SVHUC-AR cells undergoing the neoplastic transformation, using a quantitative RT-PCR method. In MCA-SVHUC-AR cells, HF and PRED (except c-Myc) significantly reduced the expression levels of oncogenes, including c-Fos (Figure 4A), c-Jun (Figure 4B), and c-Myc (Figure 4C), and induced those of tumor suppressors, including UGT1A (Figure 5A), p21 (Figure 5B), p27 (Figure 5C), p53 (Figure 5D), and PTEN (Figure 5E), while CpdA exhibited stronger effects.
Figure 4.
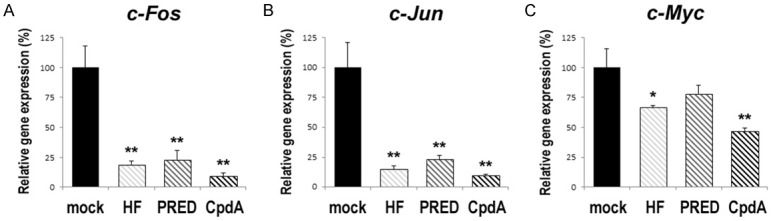
Effects of CpdA on the expression of oncogenes in urothelial cells undergoing the neoplastic transformation. SVHUC-AR cells exposed to MCA and subsequently cultured with ethanol (mock), HF (1 µM), PRED (10 nM), or CpdA (10 nM) for 6 weeks were subjected to RNA extraction, RT, and real-time PCR. GAPDH was used to normalize the expression of c-Fos (A), c-Jun (B), or c-Myc (C), which is presented relative to that of mock-treated cells (mean + SD of triplicate experiments). *P<0.05 (vs. mock treatment). **P<0.01 (vs. mock treatment).
Figure 5.
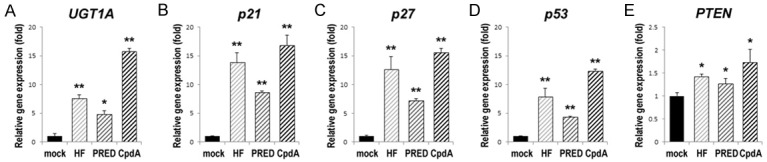
Effects of CpdA on the expression of tumor suppressor genes in urothelial cells undergoing the neoplastic transformation. SVHUC-AR cells exposed to MCA and subsequently cultured with ethanol (mock), HF (1 µM), PRED (10 nM), or CpdA (10 nM) for 6 weeks were subjected to RNA extraction, RT, and real-time PCR. GAPDH was used to normalize the expression of UGT1A (A), p21 (B), p27 (C), p53 (D), or PTEN (E), which is presented relative to that of mock-treated cells (mean + SD of triplicate experiments). *P<0.05 (vs. mock treatment). **P<0.01 (vs. mock treatment).
The efficacy of CpdA for bladder cancer development
As recently reported [18], we utilized an animal carcinogenesis model where a chemical carcinogen BBN could reliably induce the development of bladder tumor especially in male rodents to further assess the effects of CpdA on urothelial tumorigenesis. Male C57BL/6 mice were treated with BBN as well as an AR/GR ligand for 12 weeks and euthanized at 18 weeks of age to detect urothelial tumors macroscopically and microscopically (Table 2). Bladder tumors grossly identified were histologically confirmed as high-grade carcinomas (see Figure 6A), while in situ carcinoma was also found in some of the animals (see Figure 6B). Overall, all 8 (100%) of 8 mock-treated mice, as well as 4 (50%) of flutamide-treated, 7 (87.5%) of DEX-treated, 4 (50%) of PRED-treated, and 2 (25%) of CpdA-treated mice, developed bladder cancer. Thus, there was a significant difference in the incidence of bladder cancer between mock versus flutamide (P = 0.021), PRED (P = 0.021), or CpdA (P = 0.002) treatment, but not DEX treatment (P = 0.302). None of the mice in any group developed upper urinary tract or metastatic tumors.
Table 2.
Bladder tumor development in BBN-treated mice
| Group | N | Gross tumor (%) | In situ tumor (%) | All cancer (%) | P value (vs. mock) |
|---|---|---|---|---|---|
| Mock | 8 | 5 (62.5) | 3 (37.5) | 8 (100) | |
| Flutamide | 8 | 3 (37.5) | 1 (12.5) | 4 (50) | 0.021 |
| DEX | 8 | 2 (25) | 5 (62.5) | 7 (87.5) | 0.302 |
| PRED | 8 | 2 (25) | 2 (25) | 4 (50) | 0.021 |
| CpdA | 8 | 0 (0) | 2 (25) | 2 (25) | 0.002 |
Figure 6.
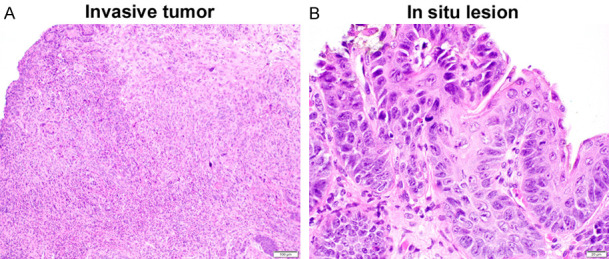
Histology of bladder tumors in BBN-treated mice. Representative H&E sections of invasive tumor (A; original magnification: ×100) and in situ lesion (B; original magnification: ×400).
The efficacy of CpdA for AR transactivation and GR transactivation/transrepression
In our previous study, CpdA was shown to antagonize androgen-enhanced AR transactivation, as well as induce GR transrepression, in bladder cancer cells [21]. To assess the impact of CpdA treatment on AR transactivation, GR transactivation, and GR transrepression in non-neoplastic urothelial cells, we performed luciferase assay and RT-PCR in SVHUC-derived cells for determining ARE-mediated transcriptional activity (Figure 7A), GRE-mediated transcriptional activity (Figure 7B) or the expression of canonical targets of GR transcription, FKBP51 and GILZ (Figure 7C), and NF-κB transcriptional activity (Figure 7D) or the expression of NF-κB-regulated genes, IL-6 and VEGF (Figure 7E), respectively. In SVHUC-AR cells, HF and CpdA similarly reduced the activity of AR transcription. In AR-negative/GR-positive SVHUC cells, CpdA did not induce GR transactivation but considerably induced GR transrepression.
Figure 7.
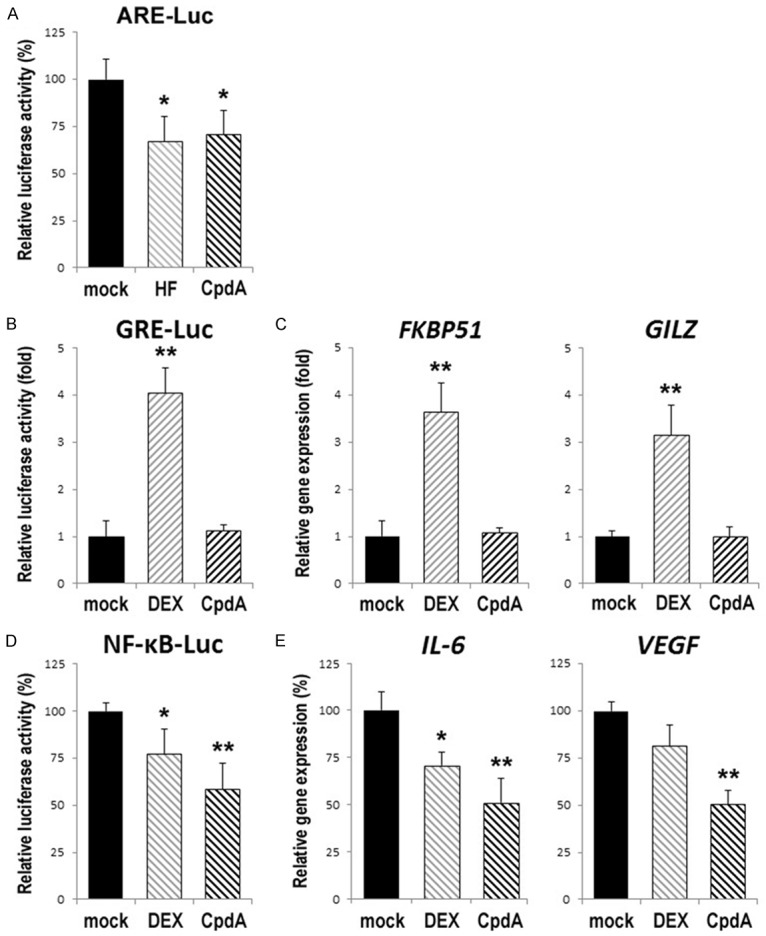
Effects of CpdA on AR transactivation (A), GR transactivation (B, C), and GR transrepression (D, E) in urothelial cells. The MMTV-Luc (A, B) or NF-κB-Luc (D) reporter activity, as well as the level of FKBP51/GILZ (C) or IL-6/VEGF (E) expression, was determined in SVHUC-AR (A) or SVHUC (B-E) cells cultured in medium supplemented with normal FBS (A) or charcoal-stripped FBS (B-E) plus ethanol (mock), HF (1 µM), DEX (100 nM), or CpdA (1 µM) for 24 hours. The luciferase activity (normalized by pRL-CMV activity) or the expression of each gene (normalized by GAPDH expression) is presented relative to that of mock treatment (mean + SD of triplicate experiments). *P<0.05 (vs. mock treatment). **P<0.01 (vs. mock treatment).
Discussion
As aforementioned, the AR and GR signaling pathways have been shown to be involved in urothelial cancer initiation which is a pathological event/process distinct from cancer progression. Specifically, our preclinical [4-11] and clinical [12,13] studies have indicated that AR activation is associated with the promotion of urothelial tumorigenesis and that androgen deprivation therapy effectively prevents the development of recurrent superficial bladder tumors, respectively. Moreover, our recent study, using preclinical models that are similar to those in the current study, has suggested that knockdown or transrepression of GR leads to the promotion or retardation, respectively, of urothelial tumorigenesis [18]. Down-regulation of GR expression in bladder [24] and upper urinary tract [25] cancers, as well as its association with a significantly higher risk of intravesical recurrence [24], in our immunohistochemical studies has further suggested the role of GR as a tumor suppressor.
In our current assays using an in vitro system with the carcinogen challenge, the suppressive effects of a GR modulator CpdA on the neoplastic transformation of GR-positive/AR-positive urothelial cells were even stronger than those of a glucocorticoid PRED or an anti-androgen HF. An in vivo experiment demonstrated similar findings in the incidence of BBN-induced bladder cancer in mock/PRED/flutamide/CpdA-treated male mice. Six-week treatment with CpdA during the process of the neoplastic transformation was also found to result in significant decreases and increases in the expression levels of oncogenic and tumor suppressive molecules, respectively, which were known to involve modulating urothelial tumorigenesis. However, the effects of CpdA versus PRED on the neoplastic transformation of GR-positive/AR-negative cells, both of which could be antagonized by a GR antagonist RU486, were similar, and no further suppression by CpdA was seen in GR-knockdown/AR-negative cells. Because CpdA is known to possess anti-androgenic property via mechanisms similar to those for classic anti-androgens [19-21], as also shown in our ARE reporter assay in SVHUC-AR cells, these observations suggest the inhibition of urothelial tumorigenesis by CpdA through not only GR but also AR signals.
The action of glucocorticoids appears to be complex and has been suggested to be dependent on a balance between transactivation and transrepression of GR [26,27]. Importantly, GR transactivation is thought to be associated with adverse effects of glucocorticoids, whereas GR transrepression usually yields their therapeutic effects. In our previous studies in non-neoplastic urothelial [18] or bladder cancer [16] cells, DEX, which could substantially induce both transactivation and transrepression of GR [18,21], failed to inhibit their neoplastic transformation or growth, respectively. By contrast, PRED, which was found to preferentially induce GR transrepression in non-neoplastic urothelial cells, was able to considerably prevent their neoplastic transformation [18]. These findings, along with data in GR knockdown cells, suggest that GR signals, particularly those mediated via GR transrepression, contribute to modulating urothelial tumorigenesis. In addition, CpdA originally identified as a “dissociated” GR ligand, which could alter GR structure favorable for its transrepression over transactivation [28], was shown to induce only GR transrepression in bladder cancer cells [21]. In the present study, we confirmed this in non-neoplastic urothelial cells by demonstrating that the selective activity of CpdA for GR transactivation was virtually absent. Apart from its anti-androgenic property, CpdA may thus be superior to other glucocorticoids for preventing urothelial tumorigenesis as to minimizing associated side effects/immunosuppression by shifting GR functions toward transrepression.
We have indicated that NF-κB whose activity represents GR transrepression plays a critical role in modulating both the development and progression of bladder cancer via cooperation with AR signaling [10]. Indeed, the functional interplay between AR and GR signals has been documented in endocrine-related tumors, such as prostate cancer [29]. Although our data suggest the inhibition of urothelial tumorigenesis by CpdA via both the AR and GR pathways, interaction of these two needs to be further investigated in urothelial cells.
In conclusion, the present study provides evidence suggesting that a dual AR/GR modulator CpdA more effectively inhibits urothelial tumorigenesis, compared with other glucocorticoids/pure GR ligands or classic AR antagonists. These findings may thus offer the application of CpdA therapy to chemoprevention of bladder cancer in patients with superficial disease following transurethral surgery or otherwise in high-risk populations. For this purpose, however, long-term treatment is unavoidable. Further preclinical studies followed by clinical trials are therefore required to determine its optimal dose that strongly prevents the development of urothelial cancer yet is unlikely to induce unfavorable effects.
Disclosure of conflict of interest
None.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:397–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Chang SS, Boorjian SA, Chou R, Clark PE, Daneshmand S, Konety BR, Pruthi R, Quale DZ, Ritch CR, Seigne JD, Skinner EC, Smith ND, McKiernan JM. Diagnosis and treatment of non-muscle-invasive bladder cancer: AUA/SUO guideline. J Urol. 2016;196:1021–1029. doi: 10.1016/j.juro.2016.06.049. [DOI] [PubMed] [Google Scholar]
- 3.Inoue S, Mizushima T, Miyamoto H. Role of the androgen receptor in urothelial cancer. Mol Cell Endocrinol. 2018;465:73–81. doi: 10.1016/j.mce.2017.06.021. [DOI] [PubMed] [Google Scholar]
- 4.Miyamoto H, Yang Z, Chen YT, Ishiguro H, Uemura H, Kubota Y, Nagashima Y, Chang YJ, Hu YC, Tsai MY, Yeh S, Messing EM, Chang C. Promotion of bladder cancer development and progression by androgen receptor signals. J Natl Cancer Inst. 2007;99:558–568. doi: 10.1093/jnci/djk113. [DOI] [PubMed] [Google Scholar]
- 5.Hsu JW, Hsu IW, Xu D, Miyamoto H, Liang L, Wu XR, Shyr CR, Chang C. Decreased tumorigenesis and mortality from bladder cancer in mice lacking urothelial androgen receptor. Am J Pathol. 2013;182:1811–1820. doi: 10.1016/j.ajpath.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Izumi K, Zheng Y, Hsu JW, Chang C, Miyamoto H. Androgen receptor signals regulate UDP-glucuronosyltransferases in the urinary bladder: a potential mechanism of androgen-induced bladder carcinogenesis. Mol Carcinog. 2013;52:94–102. doi: 10.1002/mc.21833. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Ishiguro H, Kawahara T, Miyamoto Y, Izumi K, Miyamoto H. GATA3 in the urinary bladder: suppression of neoplastic transformation and down-regulation by androgens. Am J Cancer Res. 2014;4:461–473. [PMC free article] [PubMed] [Google Scholar]
- 8.Kawahara T, Inoue S, Kashiwagi E, Chen J, Ide H, Mizushima T, Li Y, Zheng Y, Miyamoto H. Enzalutamide as an androgen receptor inhibitor prevents urothelial tumorigenesis. Am J Cancer Res. 2017;7:2041–2050. [PMC free article] [PubMed] [Google Scholar]
- 9.Inoue S, Ide H, Mizushima T, Jiang G, Kawahara T, Miyamoto H. ELK1 promotes urothelial tumorigenesis in the presence of an activated androgen receptor. Am J Cancer Res. 2018;8:2325–2336. [PMC free article] [PubMed] [Google Scholar]
- 10.Inoue S, Ide H, Mizushima T, Jiang G, Netto GJ, Gotoh M, Miyamoto H. Nuclear factor-κB promotes urothelial tumorigenesis and cancer progression via cooperation with androgen receptor signaling. Mol Cancer Ther. 2018;17:1303–1314. doi: 10.1158/1535-7163.MCT-17-0786. [DOI] [PubMed] [Google Scholar]
- 11.Inoue S, Mizushima T, Ide H, Jiang G, Goto T, Nagata Y, Netto GJ, Miyamoto H. ATF2 promotes urothelial cancer outgrowth via cooperation with androgen receptor signaling. Endocr Connect. 2018;7:1397–1408. doi: 10.1530/EC-18-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Izumi K, Taguri M, Miyamoto H, Hara Y, Kishida T, Chiba K, Murai T, Hirai K, Suzuki K, Fujinami K, Ueki T, Udagawa K, Kitami K, Moriyama M, Miyoshi Y, Tsuchiya F, Ikeda I, Kobayashi K, Sato M, Morita M, Noguchi K, Uemura H. Androgen deprivation therapy prevents bladder cancer recurrence. Oncotarget. 2014;5:12665–12674. doi: 10.18632/oncotarget.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izumi K, Ito Y, Miyamoto H, Miyoshi Y, Ota J, Moriyama M, Murai T, Hayashi H, Inayama Y, Ohashi K, Yao M, Uemura H. Expression of androgen receptor in non-muscle-invasive bladder cancer predicts the preventive effect of androgen deprivation therapy on tumor recurrence. Oncotarget. 2016;7:14153–14160. doi: 10.18632/oncotarget.7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shiota M, Kiyoshima K, Yokomizo A, Takeuchi A, Kashiwagi E, Dejima T, Takahashi R, Inokuchi J, Tatsugami K, Eto M. Suppressed recurrent bladder cancer after androgen suppression with androgen deprivation therapy or 5α-reductase inhibitor. J Urol. 2017;197:308–313. doi: 10.1016/j.juro.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Ide H, Inoue S, Miyamoto H. The role of glucocorticoid receptor signaling in bladder cancer progression. Cancers. 2018;10:484. doi: 10.3390/cancers10120484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng Y, Izumi K, Li Y, Ishiguro H, Miyamoto H. Contrary regulation of bladder cancer cell proliferation and invasion by dexamethasone-mediated glucocorticoid receptor signals. Mol Cancer Ther. 2012;11:2621–2632. doi: 10.1158/1535-7163.MCT-12-0621. [DOI] [PubMed] [Google Scholar]
- 17.Ishiguro H, Kawahara T, Zheng Y, Kashiwagi E, Li Y, Miyamoto H. Differential regulation of bladder cancer growth by various glucocorticoids: corticosterone and prednisone inhibit cell invasion without promoting cell proliferation or reducing cisplatin cytotoxicity. Cancer Chemother Pharmacol. 2014;74:249–255. doi: 10.1007/s00280-014-2496-7. [DOI] [PubMed] [Google Scholar]
- 18.Ide H, Inoue S, Mizushima T, Kashiwagi E, Zheng Y, Miyamoto H. Role of glucocorticoid signaling: inhibition by prednisone presumably through inducing glucocorticoid receptor transrepression. Mol Carcinog. 2019;58:2297–2305. doi: 10.1002/mc.23118. [DOI] [PubMed] [Google Scholar]
- 19.Tanner TM, Verrijdt G, Rombauts W, Louw A, Hapgood JP, Claessens F. Anti-androgenic properties of compound A, an analog of a non-steroidal plant compound. Mol Cell Endocrinol. 2003;201:155–164. doi: 10.1016/s0303-7207(02)00411-2. [DOI] [PubMed] [Google Scholar]
- 20.De Bosscher K, Vanden Berghe W, Beck IM, Van Molle W, Hennuyer N, Hapgood J, Libert C, Staels B, Louw A, Haegeman G. A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci U S A. 2005;102:15827–15832. doi: 10.1073/pnas.0505554102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng Y, Ishiguro H, Ide H, Inoue S, Kashiwagi E, Kawahara T, Jalalizadeh M, Reis LO, Miyamoto H. Compound A inhibits bladder cancer growth predominantly via glucocorticoid receptor transrepression. Mol Endocrinol. 2015;29:1486–1497. doi: 10.1210/me.2015-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reznikoff CA, Loretz LJ, Christian BJ, Wu SQ, Meisner LF. Neoplastic transformation of SV40-immortalized human urinary tract epithelial cells by in vitro exposure to 3-methylcholanthrene. Carcinogenesis. 1988;9:1427–1436. doi: 10.1093/carcin/9.8.1427. [DOI] [PubMed] [Google Scholar]
- 23.Miyamoto H, Marwah P, Marwah A, Yang Z, Chung CY, Altuwaijri S, Chang C, Lardy H. Identification of steroid derivatives that function as potent antiandrogens. Int J Cancer. 2005;117:866–872. doi: 10.1002/ijc.21217. [DOI] [PubMed] [Google Scholar]
- 24.Ishiguro H, Kawahara T, Zheng Y, Netto GJ, Miyamoto H. Reduced glucocorticoid receptor expression predicts bladder tumor recurrence and progression. Am J Clin Pathol. 2014;142:157–164. doi: 10.1309/AJCPU8UCEZYG4WTV. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kashiwagi E, Fujita K, Yamaguchi S, Fushimi H, Ide H, Inoue S, Mizushima T, Reis LO, Sharma R, Netto GJ, Nonomura N, Miyamoto H. Expression of steroid hormone receptors and its prognostic significance in urothelial carcinoma of the upper urinary tract. Cancer Biol Ther. 2016;17:1188–1196. doi: 10.1080/15384047.2016.1235667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, De Bosscher K. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol. 2013;380:41–51. doi: 10.1016/j.mce.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 27.Patel R, Williams-Dautovich J, Cummins CL. Minireview: new molecular mediators of glucocorticoid receptor activity in metabolic tissues. Mol Endocrinol. 2014;28:999–1011. doi: 10.1210/me.2014-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schäcke H, Berger M, Rehwinkel H, Asadullah K. Selective glucocorticoid receptor agonists (SEGRAs): novel ligands with an improved therapeutic index. Mol Cell Endocrinol. 2007;275:109–117. doi: 10.1016/j.mce.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 29.Xie N, Cheng H, Lin D, Liu L, Yang O, Jia L, Gleave ME, Wang Y, Rennie P, Dong X. The expression of glucocorticoid receptor is negatively regulated by active androgen receptor signaling in prostate tumors. Int J Cancer. 2015;136:E27–E38. doi: 10.1002/ijc.29147. [DOI] [PubMed] [Google Scholar]