

Abstract
New treatments for Ewing’s sarcoma (ES) are urgently needed. Magnolol, an active ingredient in Magnolia officinalis, shows anti-oxidative, anti-microbial, and anti-tumor effects, but its effect on ES is unknown. We examined the effect of magnolol on ES cell proliferation and apoptosis in vitro as well as the mechanism of its anticancer effect. The results demonstrated that magnolol inhibited the proliferation of ES and induced ES cell apoptosis through the mitochondrial and death receptor pathways. Magnolol reduced MEK1/2, ERK1/2, and STAT3 phosphorylation in ES cells, suggesting that the MAPK/ERK and JAK/STAT3 signal transduction pathways are involved in the inhibition of ES cell growth by magnolol. In short, magnolol is a potential anti-ES drug.
Keywords: Anticancer botanical drug, apoptosis, human Ewing’s sarcoma, invasion, migration, magnolol
Introduction
Ewing’s sarcoma (ES) is a malignant bone tumor commonly found in children and adolescents. The long-term survival rate of patients with ES was less than 10% before the application of multiple chemotherapeutic drugs [1]. In recent years, with further improvements in imaging techniques, surgical techniques, and chemotherapy regimens, the 5-year survival rate of ES has reached 60-70%, but the prognosis of patients with metastasis and recurrence remains poor [2]. Treatments for improving the survival rate and quality of life are currently being examined. As a natural source with few side effects, traditional Chinese medicine plays a unique role in the comprehensive treatment of malignant tumors [3]. Therefore, using modern scientific theory to interpret the roles and molecular mechanisms of traditional Chinese medicines in the adjuvant treatment of ES will more effectively improve the survival rate and quality of life of patients with ES.
Magnolia officinalis is a widely used drug in traditional Chinese medicine and Japanese Kampo medicine and is administered clinically to treat bacterial infections, inflammation, and gastrointestinal diseases [4]. Magnolol is one of the main active ingredients in M.officinalis. Previous studies showed that magnolol exerts anti-inflammatory [5], anti-oxidation [6], neuroprotection [7], and antibacterial effects [8] through various molecular mechanisms. Recent studies confirmed that magnolol has good anti-tumor effects in vitro and can inhibit human bladder cancer cells [9], prostate cancer cells [10], ovarian cancer cells [11], glioma cells [12], thyroid cancer cells [13], histiocytic lymphoma [14], and other malignant tumor cells, and its anti-tumor effect has multiple targets in multiple pathways. However, no studies have examined the effect of magnolol on ES.
Apoptosis is a cell suicide phenomenon that occurs in a specific time and space and is tightly regulated by the body. It plays an important role in the occurrence of many normal physiological functions [15]. It can be triggered by two different pathways: an intrinsic pathway (mitochondrial pathway) and extrinsic pathway (death receptor pathway) [16]. Currently, a variety of anti-tumor Chinese medicines have been shown to exert pharmacological effects by inducing the apoptosis of tumor cells [17]. Studies have shown that magnolol induces apoptosis of human malignant melanocytes A375-S2 via the mitochondrial and death receptor pathways [18]. We predicted that magnolol promotes apoptosis in ES cells by activating the mitochondrial pathway and/or death receptor pathway. Additionally, the Janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3) and mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signal transduction pathways are closely related to cell proliferation and apoptosis. We also hypothesized that magnolol can inhibit ES cell proliferation by affecting the phosphorylation levels of ERK and STAT3.
In this study, we investigated the effects of magnolol on the activity and apoptosis of ES SK-ES-1 cells and further examined the molecular mechanisms involved.
Materials and methods
Cell culture
The human ES cell line SK-ES-1 was obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in RPMI-1640 medium supplemented with 10% (v/v) fetal bovine serum, 100 U/ml penicillin, and 100 µg/ml streptomycin. The cells were grown at 37°C in a humidified atmosphere containing 5% CO2. The cells used in this study were passaged fewer than 20 times and all cells examined were exponentially growing.
Material
Magnolol (Source: stem bark of M.officinalis; MF: C18H18O2; MW: 266.33; purity: 99.72%, HPLC) was purchased from Shanghai Wildlife Technology (Shanghai, China). A stock solution of magnolol was prepared in dimethyl sulfoxide (DMSO; Sigma, St. Louis, MO, USA) and an equal volume of DMSO was added to the control. The final concentration of DMSO in the medium was <0.5%. All other chemicals were purchased from Sigma-Aldrich unless otherwise stated.
Cell viability was determined by CCK-8 assay
SK-ES-1 cells were cultured in 96-well plates (5×103 cells/well), and cell viability was determined by the CCK-8 colorimetric assay. Briefly, the cells were treated with magnolol (10, 20, 30, 40 and 50 μM) for 24, 48 and 72 h, while control cells were treated with <0.5% (v/v) DMSO. After the indicated incubation times, 10 μl of CCK-8 was added to the plates, which were incubated for an additional 1-4 h at 37°C. Thereafter, the absorbance was measured at 450 nm using an ELISA plate reader (Model EXL800; BioTek Instruments, Inc., Winooski, VT, USA).
Hoechst 33258 staining for observation of nuclei
SK-ES-1 cells were incubated with magnolol (0, 20, 30 and 40 μM) in 6-well plates for 24 h. The cells were then fixed with 4% paraformaldehyde for 30 min. Thereafter, the cells were washed three times with pre-cooled PBS and stained with 10 mg/l Hoechst 33258 solution for 10 min at 25°C in the dark. Subsequently, the stained nuclei were observed under a fluorescence microscope (Olympus Corp., Tokyo, Japan) at 350 nm excitation and 460 nm emission wavelengths (original magnification, 100×).
Annexin V-FITC/PI apoptosis assay
SK-ES-1 cells were cultured for 24 h with magnolol (0, 20, 30 and 40 μM), washed twice with ice-cold PBS, and resuspended at a concentration of 1×106 cells/ml in 1x binding buffer. The cell suspension (100 μl) was incubated with 1 μl annexin V-FITC and 2 μl PI solution for 15 min at 25°C in the dark. After adding 150 μl of 1x binding buffer, the samples were analyzed using a BD Accuri C6 flow cytometer (BD Biosciences, San Jose, CA). Apoptosis rates were analyzed using the BD Accuri C6 Plus software (Version 1.0.23.1, BD Biosciences, San Jose, CA). The Annexin V-FITC/PI Apoptosis Assay Kit was obtained from Becton-Dickinson (San Jose, CA).
Cell migration and invasion assay
Migration and invasion of SK-ES-1 cells were examined using a Transwell cell culture chamber (8 μm pore size). When detecting cell invasion, the membrane in each chamber was first coated with 100 μg/mL Matrigel, and then 1×105 cells were seeded into the upper chamber and treated with magnolol (0, 20, 30, 40 μM). When the cells migrated, 1×105 cells were directly inoculated into the upper chamber and treated with magnolol (0, 20, 30, 40 μM). Subsequently, the lower wells were filled with RPMI-1640 medium supplemented with 15% (v/v) fetal bovine serumin 24-well plates. After 24 h of incubation, the non-invasive cells in the upper chamber were removed with a cotton swab, and the cells attached to the lower surface were fixed with pre-cooled methanol and stained with a 0.1% crystal violet solution. Five areas of each chamber were randomly selected, and the number of cells was counted under the microscope (original magnification, 40×). The number of migrated or invading cells was analyzed by ImageJ software (NIH, Bethesda, MD, USA).
Western blot analysis
For STAT3 (RabMAbs, 1:2000, ab68153, Abcam, Cambridge, UK), p-STAT3 (RabMAbs, 1:2000, ab76315, Abcam), MEK1/2 (RabMAbs, 1:1000, ab131517, Abcam), p-MEK1/2 (RabMAbs, 1:1000, ab194754, Abcam), ERK1/2 (RabMAbs, 1:10000, ab184699, Abcam), p-ERK1/2 (RabMAbs, 1:2000, ab201015, Abcam), B-cell lymphoma 2 (Bcl-2, RabMAbs, 1:2000, ab182858, Abcam), Bcl-2-associated X protein (Bax, RabMAbs, 1:1000, ab32503, Abcam), cytochrome C (RabMAbs, 1:5000, ab133504, Abcam), caspase-3 (RabMAbs, 1:5000, ab32351, Abcam), caspase-8 (RabMAbs, 1:1000, ab108333, Abcam), caspase-9 (RabMAbs, 1:1000, ab32539, Abcam), poly (ADP-ribose) polymerase (PARP, RabMAbs, 1:1000, ab32138, Abcam), and GAPDH (rabbit polyclonal antibody, 1:2500, ab9485, Abcam) were used. The goat anti-rabbit secondary antibody (1:2000, 10285-1-AP) was purchased from Proteintech Group, Inc. of Wuhan Sanying (Wuhan, China).
SK-ES-1 cells were cultured in a 6-well plate at a density of 2×105 cells/well. After treatment with magnolol (0, 20, 30 and 40 μM) for 24 hours, cells were harvested andlysed in RIPA buffer containing a protease inhibitor cocktail (Sigma-Aldrich). The lysate was centrifuged at 12,000×g for 10 minutes at 4°C. The supernatant was then collected, and the protein concentration was determined by a BCA protein assay kit (Thermo Fisher scientific). Proteins (10 μl) were loaded and separated using SDS-PAGE (10% gel), followed by transfer onto polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% (w/v) fat-free milk in Tris-buffered saline containing 0.05% Tween-20 (TBS-T) and then incubated with primary antibodies at 4°C overnight. The next day, the PVDF membranes were washed three times in TBS-T. Secondary HRP-conjugated antibodies were incubated for 2 h at room temperature. Immunoreactive proteins were detected using an enhanced chemiluminescence system (Amersham LifeScience, Inc., Pittsburg, PA, USA) according to the manufacturer’s instructions. The gray values of the bands were analyzed using the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All data were calculated as the mean ± standard deviation (SD) and analyzed with Graphpad Prism 7.0 software (La Jolla, CA, USA). One-way analysis of variance (ANOVA) was conducted with the Newman-Keuls method to determine the significance of the differences between the experimental conditions. All experiments were repeated at least three times. P<0.05 was considered significant and represented by *, P<0.01 was represented by ** and P<0.001 by ***.
Results
Effect of magnolol on viability of SK-ES-1 cells
Relative absorbance was measured in the CCK-8 assay, and SK-ES-1 cells were exposed to different concentrations of magnolol. Control cells (0 μM magnolol) were considered as the baseline (100% viable) for the analysis. Figure 1 shows that magnolol appeared to be an effective inhibitor of SK-ES-1 cell viability, which was inhibited in a dose-dependent manner. The IC50 values of SK-ES-1 cells treated with magnolol were 35.85, 28.63 and 25.40 μM at 24, 48 and 72 h, respectively. Subsequently, SK-ES-1 cells were treated with magnolol at concentrations of 0, 20, 30 and 40 μM for 24 has described below.
Figure 1.
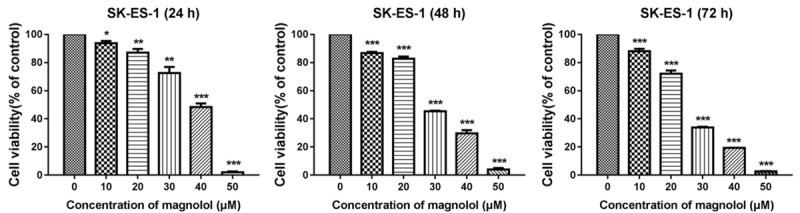
Magnolol inhibits SK-ES-1 cells growth. SK-ES-1 cells were treated with different concentrations of magnolol for 24, 48 and 72 h separately, and a CCK-8 assay was performed to detect cell viability. It was observed that magnolol can inhibit the viability of SK-ES-1 cells in a dose-dependent manner at 24, 48 and 72 h. *P<0.05, **P<0.01, ***P<0.001 vs 0 μM concentration magnolol treated group. Statistically significant difference was determined using one-way ANOVA.
Magnolol causes morphological changes in SK-ES-1 cells
After treatment with magnolol (0, 20, 30 and 40 μM) for 24 h, SK-ES-1 cells were stained with the fluorescent DNA-binding dye Hoechst 33258, revealing concentrated and broken nuclei compared to the control group, which is a typical morphological feature of apoptotic cells (Figure 2). These findings indicate that cell death occurs through apoptosis.
Figure 2.

Magnolol leads to changes in cell morphology. Apoptotic nuclei stained with Hoechst 33258 (24 h) showed nuclear chromosome condensation and fragmentation (magnification, 100×). Apoptosis is a gene-regulated phenomenon and characterized by cell membrane blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, DNA fragmentation, and formation of apoptotic bodies.
Magnolol induces apoptosis
Apoptosis was measured by flow cytometry with Annexin V-FITC/PI double labeling. The apoptosis rate of the control group (sum of early and late apoptosis) was 13.2±2.25%. After 24 h of treatment with magnolol (20, 30 and 40 μM), the apoptotic rate increased to 20.94±4.09%, 28.84±2.05% and 41.81±3.56%, respectively (Figure 3A). Thus, magnolol-induced apoptosis is dose-dependent (Figure 3B).
Figure 3.
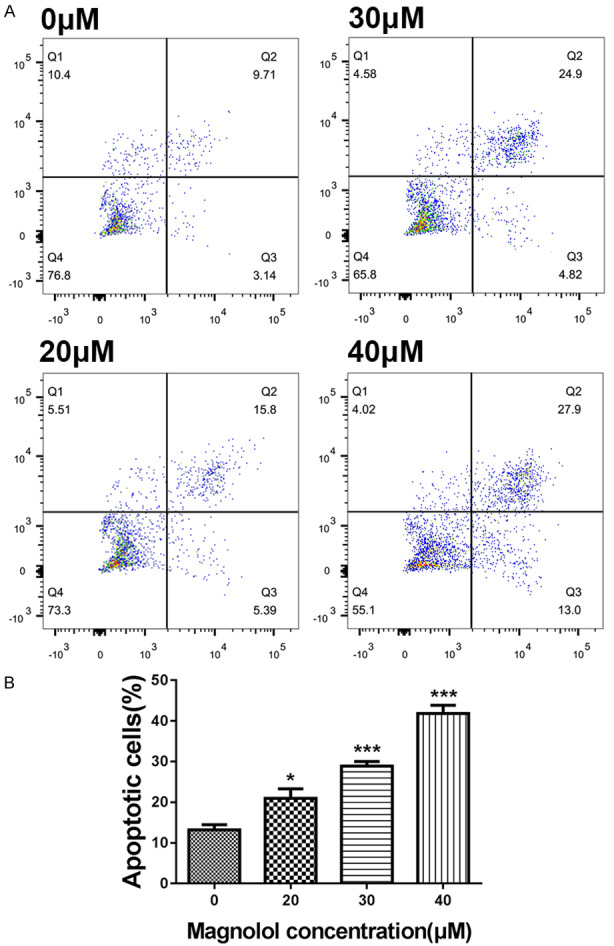
Magnolol induces SK-ES-1 cells apoptosis. A. Representative images obtained by flow cytometry after double staining with Annexin V-FITC/PI and apoptosis rate of SK-ES-1 cells in the control and magnolol treatment groups (24 h). B. *P<0.05, ***P<0.001 vs 0 μM concentration magnolol treated group. Statistically significant difference was determined using one-way ANOVA.
Effect of magnolol on migration and invasion of SK-ES-1 cells
We measured the effects of magnolol on the migration and invasion of SK-ES-1 cells in Transwell chamber experiments. After treatment with magnolol (0, 20, 30, and 40 μM) for 24 h, the Transwell chamber test showed that the number of cells migrating or invading into the transwell ventricular membrane was decreased by magnolol treatment in a dose-dependent manner (Figures 4 and 5).
Figure 4.
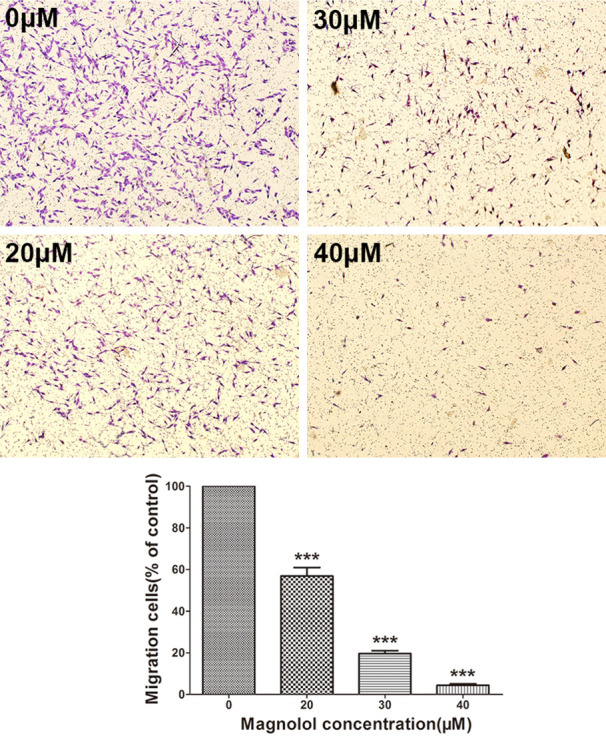
Magnolol inhibits SK-ES-1 cells migration. Transwell cell culture chamber was used to test SK-ES-1 cell migration after treatment with magnolol (0, 20, 30, 40 μM) (magnification, 40×). SK-ES-1 migrating cells were quantified with ImageJ software. Data are expressed as the mean ± SD of three independent experiments. ***P<0.001 vs control.
Figure 5.
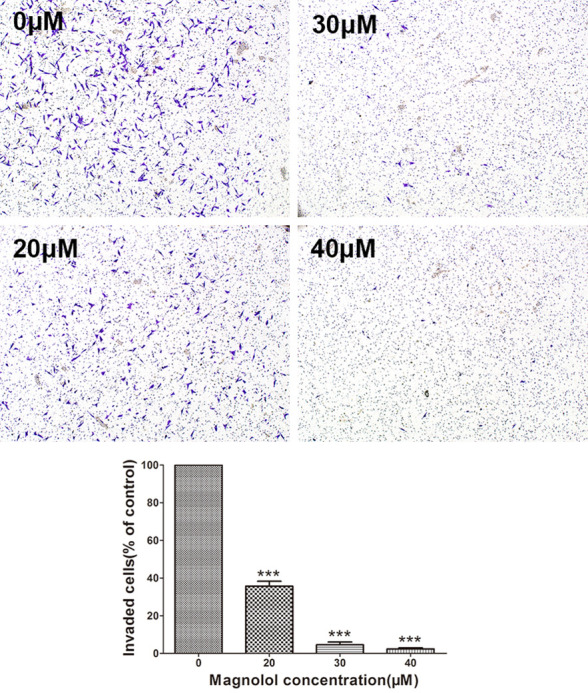
Magnolol inhibits SK-ES-1 cells invasion. Transwell cell culture chamber coated with Matrigel to test SK-ES-1 cell invasion after treatment with magnolol (0, 20, 30, 40 μM) (magnification, 40×). SK-ES-1 invading cells were quantified by ImageJ software. Data are expressed as the mean ± SD of three independent experiments. ***P<0.001 vs control.
Western blot
Western blotting was used to detect the expression of apoptosis-related protein Bcl-2, pro-apoptotic Bax, cytochrome C, caspase-3, caspase-8, caspase-9 and poly ADP ribose polymerase (PARP) to determine the apoptosis of SK-ES-1 cells induced by magnolol. Magnolol caused an increase in Bax protein (Figure 6) and cytochrome C release (Figure 7), and a decrease in Bcl-2 protein (Figure 6). This demonstrates that magnolol activates the mitochondrial apoptotic pathway in SK-ES-1 cells by modulating the expression of Bcl-2 family proteins. We also evaluated the protein expression levels of caspase-3, -8 and -9. A decrease in pro caspase-3 was observed and the expression level of pro caspase-9 was down-regulated, both of which occurred in a concentration-dependent manner (Figure 7), while the key cellular substrate PARP was decreased (Figure 6). Additionally, the expression level of caspase-8 was decreased in a concentration-dependent manner (Figure 6). These results indicate that magnolol-induced apoptosis involves the caspase cascade and is triggered by the mitochondrial and death receptor pathways.
Figure 6.
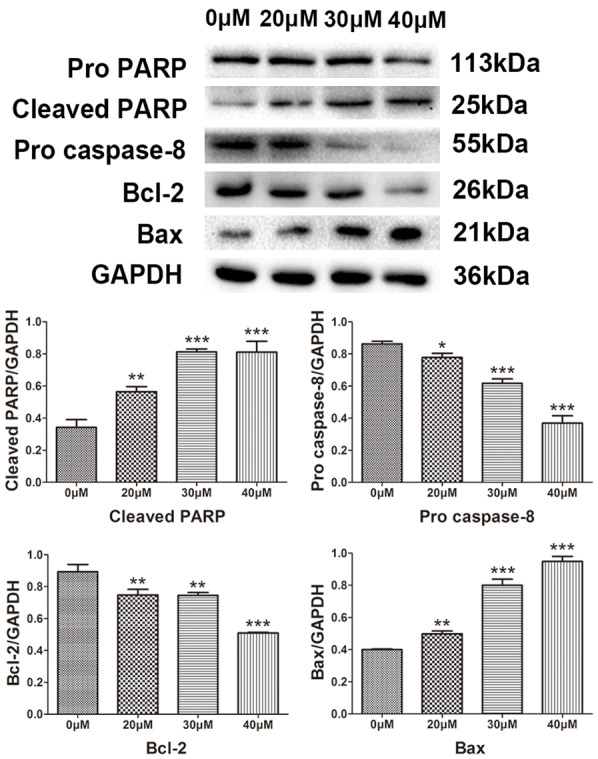
Protein expression levels of pro PARP, cleaved PARP, pro caspase-8, Bcl-2 and Bax by western blot analysis. Effect of magnolol on the protein expression levels of PARP, cleaved PARP, pro caspase-8, Bcl-2 and Bax in human SK-ES-1 cells. GAPDH was used as aninternal loading control. *P<0.05, **P<0.01, ***P<0.001 vs 0 μM concentration magnolol treated group. Statistically significant difference was determined using one-way ANOVA.
Figure 7.
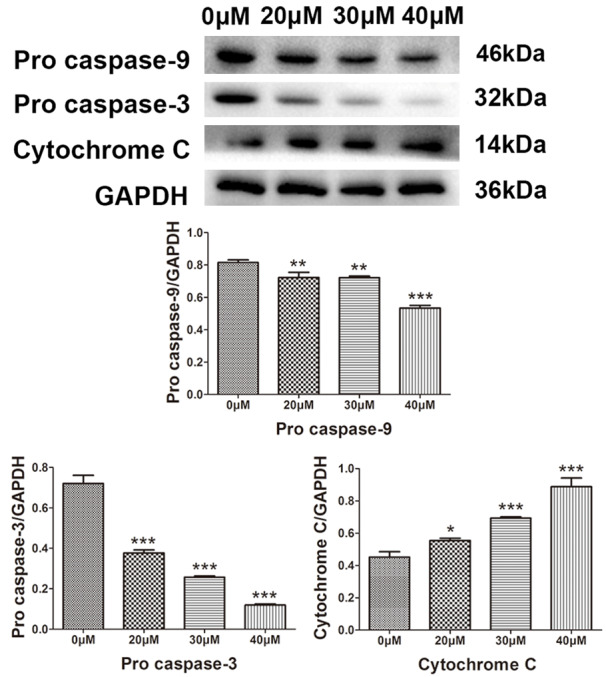
Protein expression levels of pro caspase-9, pro caspase-3 and cytochrome C determined by western blot analysis. Effect of magnolol on the protein expression levels of pro caspase-9, pro caspase-3 and cytochrome C in human SK-ES-1 cells. GAPDH was used as aninternal loading control. *P<0.05, **P<0.01, ***P<0.001 vs 0 μM concentration magnolol treated group. Statistically significant difference was determined using one-way ANOVA.
Additionally, magnolol reduced the expression levels of p-STAT3, p-MEK and p-ERK. The JAK/STAT3 and MAPK/ERK signaling pathways are closely related to cell proliferation and apoptosis. We found that magnolol significantly reduced the expression levels of p-STAT3, p-MEK and p-ERK (P<0.05) in a dose-dependent manner (Figure 8).
Figure 8.
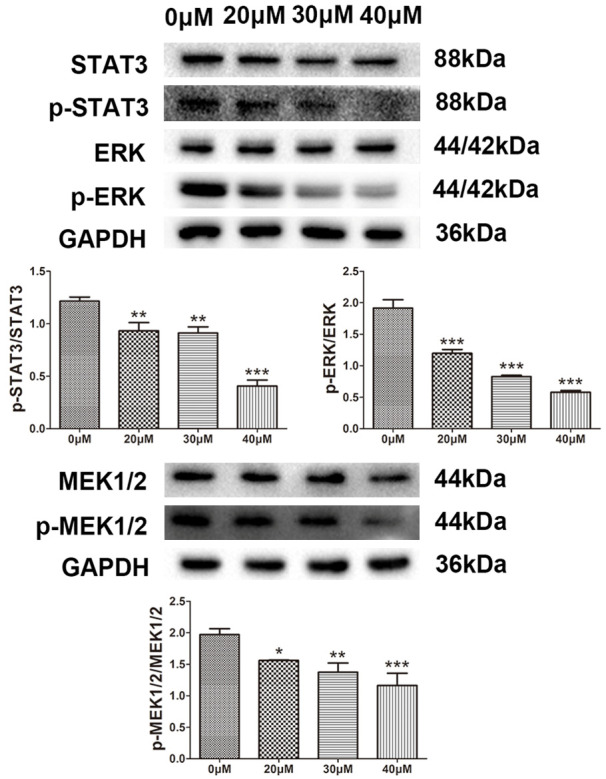
Protein expression levels of STAT3, p-STAT3, MEK1/2, p-MEK1/2, ERK1/2 and p-ERK1/2 by western blot analysis. Effect of magnolol on the protein expression levels of STAT3, p-STAT3, MEK1/2, p-MEK1/2, ERK1/2 and p-ERK1/2 in human SK-ES-1 cells. GAPDH was used as aninternal loading control. *P<0.05, **P<0.01, ***P<0.001 vs 0 μM concentration magnolol treated group. Statistically significant difference was determined using one-way ANOVA.
Discussion
The results of this study indicate that magnolol significantly inhibits the activity of SK-ES-1 cells in human ES and induces apoptosis in SK-ES-1 cells in time- and dose-dependent manners. Additionally, magnolol inhibited the migration and invasion of SK-ES-1 cells in a dose-dependent manner. These findings are consistent with those reported for other types of tumors [9-14], confirming that magnolol has good in vitro antitumor effects. Subsequently, we examined the precise role of magnolol in ES and the related molecular mechanisms. Caspases are semi-proline-aspartate-specific proteases. Caspase-3 is a member of the caspase family and is located downstream of the apoptotic signal and is also the executor of apoptosis. After activation, caspase-3 cleaves its substrate, resulting in apoptosis. Thus, during apoptosis, particularly in the early stage, the detection of caspase-3 activity can indicate whether cells undergo apoptosis. PARP repairs DNA and is the first caspase-3 substrate to be detected. In the early stage of apoptosis, PARP can be cleaved into two fragments with molecular masses of 89 and 25 kDa, and thus is a marker for the early detection of apoptosis [19,20]. Our results indicate that the expression level of procaspase-3 in SK-ES-1 cells treated with magnolol decreased in a dose-dependent manner, while the cleavage of PARP increased significantly in a dose-dependent manner, indicating that magnolol induces apoptosis in SK-ES-1 cells. The intrinsic pathway (mitochondrial pathway) and extrinsic pathway (death receptor pathway) are regulated by caspase-9 and caspase-8, respectively [21,22]. In the intrinsic pathway, apoptosis triggers the mitochondrial membrane PT pore (regular transition pore) regulated by the proapoptotic protein Bax in the Bcl-2 family, leading to a decrease in the mitochondrial membrane potential and release of cytochrome C, thereby activating caspase-9. Caspase-9 activates the downstream molecule, caspase-3, which re-cleaves its substrate PARP [23]. The extrinsic apoptotic pathway triggered by the binding of cell surface-specific death receptors and their corresponding ligands first activates caspase-8 and then activates its downstream molecule caspase-3, which in turn cleaves PARP and leads to apoptosis [24]. Our results indicate that magnolol-induced apoptosis is accompanied by cytochrome C release, an increase in Bax/Bcl-2 ratio, and activation of caspase-9 and caspase-8, further demonstrating that magnolol induces apoptosis in ES cells through the mitochondrial and death receptor pathways.
MAPKs are important signal transduction proteinsineukaryotic cells that mediate intracellular responses to extracellular signals, regulating physiological processes such as cell growth, differentiation, and apoptosis. MEK/ERK is one of the many pathways of “MAPK”, and the abnormal activation of this pathway is inextricably linked to the production and malignant evolution of many cancers [25,26]. Under the stimulation of various potential carcinogenic factors, cytoplasmic Raf is activated and then it activates its downstream molecule MEK, which also activates ERK through phosphorylation, and then activated ERK enters the nucleus. ERKs are members of the MAPK family and are divided into two isoforms, ERK1 (P44) and ERK2 (P42). ERKs are primarily involved in cell growth, differentiation, and anti-apoptosis [27-29]. Studies have shown that ERKs can promote the anti-apoptotic proteins Bcl-2 and Bcl-xl as well as other activities to increase the survival of tumor cells [30]. STATs are a class of deoxyribonucleic acid binding proteins. The STAT family has seven members: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B and STAT6. STAT3 is widely expressed in different types of cells and tissues, and is involved in physiological processes such as cell growth, central nervous system diseases, and myocardial ischemic preconditioning [31]. Recent studies showed that activation of STAT3 protein is strictly controlled under normal physiological conditions. In many tumors (such as breast cancer, ovarian cancer, prostate cancer, malignant melanoma, and non-small cell lung cancer), the STAT3 protein is over-activated and expressed at a high level. To further explore the mechanism by which magnolol induces apoptosis, we examined the phosphorylation levels of MEK, ERK and STAT3. The results showed that magnolol significantly reduced the expression levels of p-MEK, p-ERK and p-STAT3 (P<0.05) in a dose-dependent manner, indicating that magnolol induced SK-ES-1 cell apoptosis and is closely related to the MAPK/ERK and JAK/STAT3 signal transduction pathways, further demonstrating that the anti-tumor effect of magnolol involves multiple targets and multiple pathways.
Notably, STAT3 and ERK, as key molecules in the JAK/STAT3 and MAPK/ERK signal transduction pathways, playa common role in regulating cell biological behavior and malignant transformation [32], may be closely related, and interact to affect signaling pathway activity. Studies have shown that under the influence of certain stimulating factors, both the MARK pathway and STAT3 pathway may be activated, and the regulation of MAPK-STAT signaling may occur in cells [33]. This study showed that magnolol inhibits both STAT3 and ERK activation. Whether the JAK/STAT3 and MAPK/ERK signaling pathways are involved in the induction of SK-ES-1 cell apoptosis by magnolol remains unclear.
Recent studies have confirmed that magnolol has a wide range of pharmacological effects and great potential for clinical application. However, it also has several limitations. First, safety must be guaranteed before a “natural medicine” can be applied in the clinic. Magnolol has been shown to cause severe cytotoxicity in human mesenchymal stem cells at a concentration of 100 μM [34], and thus cytotoxicity must be assessed before use. Next, the oxidative properties and poor water solubility of magnolol are physical properties that prevent its clinical utility, which must be overcome in the future. Finally, pharmacological studies of magnolol are based on preclinical research. It is difficult to reproduce the complex in vivo environment under relatively single conditions in vitro, making it difficult to determine the specific role and mechanism of the drug in organisms, particularly in humans. Therefore, its many pharmacological effects remain to be verified and must be further explored in large randomized, controlled clinical trials.
In summary, magnolol inhibited the proliferation, invasion and metastasis of ES cells in vitro and induced apoptosis of SK-ES-1 cells through the mitochondrial and death receptor pathways. Additionally, the MAPK/ERK and JAK/STAT3 signal transduction pathways are closely related to the apoptosis of SK-ES-1 cells induced by magnolol, further demonstrating that the targets and pathways of magnolol are diverse. However, the present study has certain limitations. First, we only employed in vitro experiments and, thus, further research is needed to elucidate the in vivo effects of magnolol on SK-ES-1 xenograft tumors in nude mice. Second, the present study was more observational than an in-depth investigation of the mechanism. There was no detailed examination of the molecular mechanism concerning the findings obtained. Our in vitro results support that magnolol is an effective candidate for the chemoprevention and/or treatment of ES.
Disclosure of conflict of interest
None.
References
- 1.Iwamoto Y. Diagnosis and treatment of Ewing’s sarcoma. Jpn J Clin Oncol. 2007;37:79–89. doi: 10.1093/jjco/hyl142. [DOI] [PubMed] [Google Scholar]
- 2.Leavey PJ, Collier AB. Ewing sarcoma: prognostic criteria, outcomes and future treatment. Expert Rev Anticancer Ther. 2008;8:617–624. doi: 10.1586/14737140.8.4.617. [DOI] [PubMed] [Google Scholar]
- 3.Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981-2002. J Nat Prod. 2003;66:1022–37. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- 4.Weng TI, Wu HY, Kuo CW, Liu SH. Honokiol rescues sepsis-associated acute lung injury and lethality via the inhibition of oxidative stress and inflammation. Intensive Care Med. 2011;37:533–41. doi: 10.1007/s00134-010-2104-1. [DOI] [PubMed] [Google Scholar]
- 5.Fu Y, Liu B, Zhang N, Liu Z, Liang D, Li F, Cao Y, Feng X, Zhang X, Yang Z. Magnolol inhibits lipopolysaccharide-induced inflammatory response by interfering with TLR4 mediated NF-κB and MAPKs signaling pathways. J Ethnopharmacol. 2013;145:193–199. doi: 10.1016/j.jep.2012.10.051. [DOI] [PubMed] [Google Scholar]
- 6.Amorati R, Zotova J, Baschieri A, Valgimigli L. Antioxidant activity of magnolol and honokiol: kinetic and mechanistic investigations of their reaction with peroxyl radicals. J Org Chem. 2015;80:10651–10659. doi: 10.1021/acs.joc.5b01772. [DOI] [PubMed] [Google Scholar]
- 7.Dong L, Zhou S, Yang X, Chen Q, He Y, Huang W. Magnolol protects against oxidative stress-mediated neural cell damage; by modulating mitochondrial dysfunction and PI3K/Akt signaling. J Mol Neurosci. 2013;50:469–81. doi: 10.1007/s12031-013-9964-0. [DOI] [PubMed] [Google Scholar]
- 8.Liu T, Pan Y, Lai R. New mechanism of magnolol and honokiol from Magnolia officinalis against Staphylococcus aureus. Nat Prod Commun. 2014;9:1307–9. [PubMed] [Google Scholar]
- 9.Lee SJ, Park SS, Lee US, Kim WJ, Moon SK. Signaling pathway for TNF-alpha-induced MMP-9 expression: mediation through p38 MAP kinase, and inhibition by anti-cancer molecule magnolol in human urinary bladder cancer 5637 cells. Int Immunopharmacol. 2008;8:1821–1826. doi: 10.1016/j.intimp.2008.08.018. [DOI] [PubMed] [Google Scholar]
- 10.Lee DH, Lee YJ. Magnolol induces apoptosis via inhibiting the EGFR/PI3K/Akt signaling pathway in human prostate cancer cells. J Cell Biochem. 2010;106:1113–1122. doi: 10.1002/jcb.22098. [DOI] [PubMed] [Google Scholar]
- 11.Chuang TC, Hsu SC, Cheng YT, Shao WS, Wu K, Fang GS, Ou CC, Wang V. Magnolol down-regulates HER2 gene expression, leading to inhibition of HER2-mediated metastatic potential in ovarian cancer cells. Cancer Lett. 2011;311:11–19. doi: 10.1016/j.canlet.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 12.Chen LC, Liu YC, Liang YC, Ho YS, Lee WS. Magnolol inhibits human glioblastoma cell proliferation through upregulation of p21/Cip1. J Agric Food Chem. 2009;57:7331–7. doi: 10.1021/jf901477g. [DOI] [PubMed] [Google Scholar]
- 13.Huang S, Chen Y, Tung P, Wu J, Chen K, Wu J, Wang S. Mechanisms for the magnolol-induced cell death of CGTH W-2 thyroid carcinoma cells. J Cell Biochem. 2007;101:1011–22. doi: 10.1002/jcb.21100. [DOI] [PubMed] [Google Scholar]
- 14.Ikai T, Akao Y, Nakagawa Y, Ohguchi K, Sakai Y, Nozawa Y. Magnolol-induced apoptosis is mediated via the intrinsic pathway with release of AIF from mitochondria in U937 cells. Biol Pharm Bull. 2006;29:2498–501. doi: 10.1248/bpb.29.2498. [DOI] [PubMed] [Google Scholar]
- 15.Kasibhatla S, Tseng B. Why target apoptosis in cancer treatment? Molecular Cancer Therapeutics. 2003;2:573–580. [PubMed] [Google Scholar]
- 16.Schulzebergkamen H, Krammer PH. Apoptosis in cancer--implications for therapy. Semin Oncol. 2004;31:90–119. doi: 10.1053/j.seminoncol.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Piao MJ, Kim KC, Zheng J, Yao CW, Cha JW, Boo SJ, Yoon WJ, Kang HK, Yoo ES, Koh YS. The ethyl acetate fraction of Sargassum muticum attenuates ultraviolet B radiation-induced apoptotic cell death via regulation of MAPK- and caspase-dependent signaling pathways in human HaCaT keratinocytes. Pharm Biol. 2014;52:1110–1118. doi: 10.3109/13880209.2013.879186. [DOI] [PubMed] [Google Scholar]
- 18.You Q, Li M, Jiao G. Magnolol induces apoptosis via activation of both mitochondrial and death receptor pathways in A375-S2 cells. Arch Pharm Res. 2009;32:1789–1794. doi: 10.1007/s12272-009-2218-6. [DOI] [PubMed] [Google Scholar]
- 19.Po-ki Ho, Hawkins CJ. Mammalian initiator apoptotic caspases. FEBS J. 2005;272:5436–5453. doi: 10.1111/j.1742-4658.2005.04966.x. [DOI] [PubMed] [Google Scholar]
- 20.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 21.Lorenzo HK, Susin SA. Therapeutic potential of AIF-mediated caspase-independent programmed cell death. Drug Resist Updat. 2007;10:235–255. doi: 10.1016/j.drup.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 22.Chang HY, Yang X. Proteases for cell suicide: functions and regulation of caspases. Microbiol Mol Biol Rev. 2000;64:821–846. doi: 10.1128/mmbr.64.4.821-846.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia L, Srinivasula SM, Liu FT, Newland AC, Fernandesalnemri T, Alnemri ES, Kelsey SM. Apaf-1 protein deficiency confers resistance to cytochrome c-dependent apoptosis in human leukemic cells. Blood. 2001;98:414. doi: 10.1182/blood.v98.2.414. [DOI] [PubMed] [Google Scholar]
- 24.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D’Angiolella V, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, DeBerardinis RJ, Deshmukh M, Di Daniele N, Di Virgilio F, Dixit VM, Dixon SJ, Duckett CS, Dynlacht BD, El-Deiry WS, Elrod JW, Fimia GM, Fulda S, García-Sáez AJ, Garg AD, Garrido C, Gavathiotis E, Golstein P, Gottlieb E, Green DR, Greene LA, Gronemeyer H, Gross A, Hajnoczky G, Hardwick JM, Harris IS, Hengartner MO, Hetz C, Ichijo H, Jäättelä M, Joseph B, Jost PJ, Juin PP, Kaiser WJ, Karin M, Kaufmann T, Kepp O, Kimchi A, Kitsis RN, Klionsky DJ, Knight RA, Kumar S, Lee SW, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, López-Otín C, Lowe SW, Luedde T, Lugli E, MacFarlane M, Madeo F, Malewicz M, Malorni W, Manic G, Marine JC, Martin SJ, Martinou JC, Medema JP, Mehlen P, Meier P, Melino S, Miao EA, Molkentin JD, Moll UM, Muñoz-Pinedo C, Nagata S, Nuñez G, Oberst A, Oren M, Overholtzer M, Pagano M, Panaretakis T, Pasparakis M, Penninger JM, Pereira DM, Pervaiz S, Peter ME, Piacentini M, Pinton P, Prehn JHM, Puthalakath H, Rabinovich GA, Rehm M, Rizzuto R, Rodrigues CMP, Rubinsztein DC, Rudel T, Ryan KM, Sayan E, Scorrano L, Shao F, Shi Y, Silke J, Simon HU, Sistigu A, Stockwell BR, Strasser A, Szabadkai G, Tait SWG, Tang D, Tavernarakis N, Thorburn A, Tsujimoto Y, Turk B, Vanden Berghe T, Vandenabeele P, Vander Heiden MG, Villunger A, Virgin HW, Vousden KH, Vucic D, Wagner EF, Walczak H, Wallach D, Wang Y, Wells JA, Wood W, Yuan J, Zakeri Z, Zhivotovsky B, Zitvogel L, Melino G, Kroemer G. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on cell death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monica Rosaria M, Amelia DA, Simona B, Marianna G, Nicola N, Antonella DL. EGFR and MEK blockade in triple negative breast cancer cells. J Cell Biochem. 2015;116:2778–2785. doi: 10.1002/jcb.25220. [DOI] [PubMed] [Google Scholar]
- 26.Jiang L, Yan Q, Fang S, Liu M, Li Y, Yuan YF, Li Y, Zhu Y, Qi J, Yang X, Kwong DLW, Guan XY. Calcium binding protein 39 promotes hepatocellular carcinoma growth and metastasis by activating ERK signaling pathway. Hepatology. 2017;66:1529–1545. doi: 10.1002/hep.29312. [DOI] [PubMed] [Google Scholar]
- 27.Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by HO ROLE IN CELL SURVIVAL FOLLOWING OXIDANT INJURY. J Biol Chem. 1996;271:4138. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- 28.Troadec JD, Marien M, Mourlevat S, Debeir T, Ruberg M, Colpaert F, Michel PP. Activation of the mitogen-activated protein kinase (ERK(1/2)) signaling pathway by cyclic AMP potentiates the neuroprotective effect of the neurotransmitter noradrenaline on dopaminergic neurons. Molecular Pharmacology. 2002;62:1043–1052. doi: 10.1124/mol.62.5.1043. [DOI] [PubMed] [Google Scholar]
- 29.Nielsen FC. KCl potentiates forskolin-induced PC12 cell neurite outgrowth via protein kinase A and extracellular signal-regulated kinase signaling pathways. Neurosci Lett. 2003;347:57–61. doi: 10.1016/s0304-3940(03)00581-0. [DOI] [PubMed] [Google Scholar]
- 30.Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009;16:368. doi: 10.1038/cdd.2008.148. [DOI] [PubMed] [Google Scholar]
- 31.Xuan YT, Guo Y, Han H, Zhu Y, Bolli R. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc Natl Acad Sci U S A. 2001;98:9050–9055. doi: 10.1073/pnas.161283798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haura EB, Turkson J, Jove R. Mechanisms of disease: insights into the emerging role of signal transducers and activators of transcription in cancer. Nat Clin Pract Oncol. 2005;2:315–24. doi: 10.1038/ncponc0195. [DOI] [PubMed] [Google Scholar]
- 33.Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, Franklin RA, McCubrey JA. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17:1263. doi: 10.1038/sj.leu.2402945. [DOI] [PubMed] [Google Scholar]
- 34.Wei YJ, Tsai KS, Lin LC, Lee YT, Chi CW, Chang MC, Tsai TH, Hung SC. Catechin stimulates osteogenesis by enhancing PP2A activity in human mesenchymal stem cells. Osteoporos Int. 2011;22:1469–1479. doi: 10.1007/s00198-010-1352-9. [DOI] [PubMed] [Google Scholar]