

Abstract
Acute lymphoblastic leukaemia (ALL) is the most common form of paediatric cancer and epigenetic aberrations are determinants of leukaemogenesis. The aim of this study was to investigate the methylation degree of a distinct phospholipase A2 receptor 1 (PLA2R1) promoter region in paediatric ALL patients and to evaluate its relevance as new biomarker for monitoring treatment response and burden of residual disease. The impact of PLA2R1 re-expression on proliferative parameters was assessed in vitro in Jurkat cells with PLA2R1 naturally silenced by DNA methylation. Genomic DNA was isolated from bone marrow (BM) and peripheral blood (PB) of 44 paediatric ALL patients. PLA2R1 methylation was analysed using digital PCR and compared to 20 healthy controls. Transfected Jurkat cells were investigated using cell growth curve analysis and flow cytometry. PLA2R1 was found hypermethylated in BM and PB from pre-B and common ALL patients, and in patients with the disease relapse. PLA2R1 methylation decreased along with leukaemic blast cell reduction during ALL induction treatment. In vitro analysis revealed an anti-proliferative phenotype associated with PLA2R1 re-expression, suggesting a tumour-suppressive function of PLA2R1. Collected data indicates that PLA2R1 promoter methylation quantitation can be used as biomarker for ALL induction treatment control, risk stratification, and early detection of ALL relapse.
Subject terms: Tumour biomarkers, Haematological cancer
Introduction
Acute Lymphoblastic Leukaemia (ALL) is the most frequent form of childhood cancer worldwide1,2. More than 7000 new cases of ALL were diagnosed in the European Union in 2018, with a peak incidence between two and five years of age3,4. ALL is characterized by an altered proliferation of lymphoid progenitor cells and a subsequent accumulation of leukaemic blast cells in the bone marrow, blood, and other organs5–7. Primary ALL diagnosis is based on the microscopic analysis of bone marrow aspirates7. Subtypes of childhood ALL are defined by immunohistological identification of the affected cell lineage (B- or T-cell), chromosomal and genetic alterations, and the cell differentiation status8. In addition to germline and somatic mutations, epigenetic alterations (especially DNA promoter hypermethylation) contribute to ALL subtype classification9 and may have prognostic value as an indicator of the likelihood of relapse under standardized treatment10–12.
The phospholipase A2 receptor 1 (PLA2R1) is a type I transmembrane receptor13 that is suggested to play a crucial role in the regulation of cellular senescence/apoptosis in primary human fibroblasts14 and different breast cancer cell lines15–18. Our previous study was the first to establish that epigenetic mechanisms are involved in the regulation of PLA2R1 expression in leukaemic cells19. According to the PLA2R1 bisulfite sequencing analysis, 77 CpG sites at −473 bp to +586 bp from exon 1 were found to be hypermethylated in blood leukocytes of adult patients with acute myeloid leukaemia compared to healthy individuals. PLA2R1 methylation quantification by methylation-sensitive high-resolution melting analysis demonstrated a significantly higher methylation degree in adult leukaemia patients. Additionally, the PLA2R1 methylation degree was found to increase with disease stage progression in a group of myelodysplastic syndrome (MDS) patients19. The analysed values correlated with the International Prognostic Scoring System (IPSS) classification, suggesting that PLA2R1 methylation measurement can be used as an additional biomarker for risk stratification. Preliminary analysis of the PLA2R1 methylation degrees of high-risk MDS and AML patients during azacitidine treatment indicated that the response to treatment also correlated with the PLA2R1 methylation degrees, and measuring quantitatively the receptor methylation was considered a useful early indicator for the requirement of follow-up therapy19. Furthermore, our study provided evidence that PLA2R1 promoter methylation is inversely correlated with PLA2R1 expression in the human T lymphocyte acute leukaemia (Jurkat) cell line19, which is extensively used to investigate ALL20–22.
Based on these previous findings, the aim of the present study was to investigate the following: (i) whether the PLA2R1 promoter is also hypermethylated in patients with childhood ALL at diagnosis in comparison to healthy individuals; (ii) whether the PLA2R1 promoter methylation in blood leukocyte DNA can be used as a biomarker for treatment response and control of residual disease. Additionally, the effect of PLA2R1 expression on cell proliferation and apoptosis/necrosis of Jurkat cells as a cell model for childhood ALL was assessed.
Results
Differential PLA2R1 promoter methylation in healthy and childhood ALL samples at diagnosis
To investigate the effect of PLA2R1 in childhood ALL, the PLA2R1 promoter methylation status was analysed by droplet digital polymerase chain reaction (ddPCR) in PB samples and BM aspirates of children with ALL and AML. The samples were then compared to a healthy, age-matched control group (Fig. 1).
Figure 1.
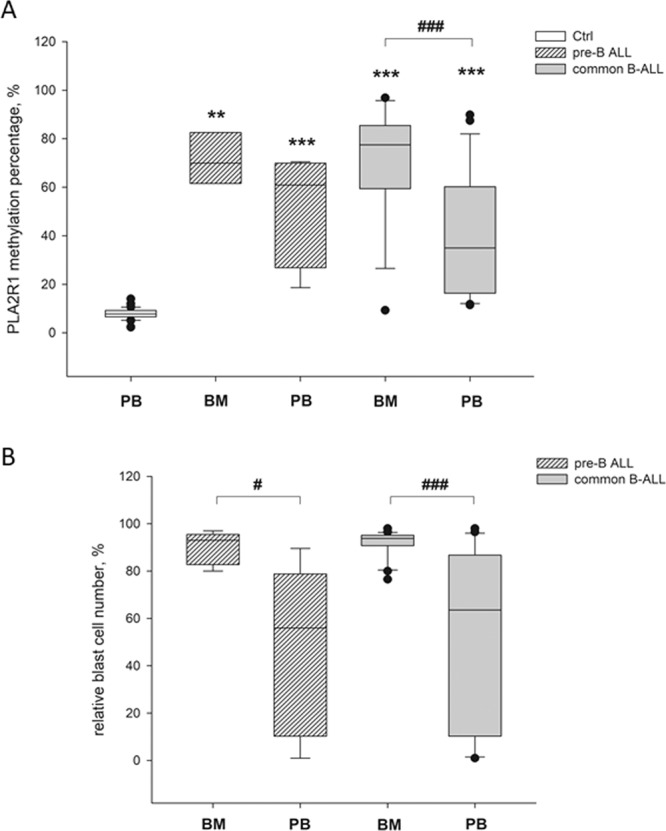
Differential PLA2R1 promoter methylation and blast cell occurrence in healthy and childhood ALL samples. Box plots consist of the median as ‘center value’, the 25th and 75th percentiles as box edges, and the 10th and 90th percentiles as whisker boundaries. (A) Percentage of PLA2R1 promoter methylation at diagnosis was determined in PB from healthy children (Ctrl, n = 20) and in BM aspirates or PB from children with pre-B cell (nBM = 3, nPB = 5) or common ALL (nBM = 17, nPB = 19) using droplet digital PCR. (B) The relative blast cell number (number of blast cells in relation to the number of total leukocytes in %) in BM aspirates and PB of childhood pre-B cell (nBM = 5, nPB = 5) or common ALL samples (nBM = 21, nPB = 22) were determined at diagnosis using light microscopic and flow cytometric analysis. The symbols * and # indicate significant differences compared to the healthy control group or between marked cohorts, respectively. Levels of significance are defined as p < 0.05 (#), p < 0.01 (**), and p < 0.001 (***, ###).
The mean PLA2R1 promoter methylation percentage of the healthy, age-matched control group was 7.8% ± 2.3%. The 97.5% percentile of the control group was estimated 12.05% and was defined as cutoff. In comparison to the control group, PLA2R1 methylation was approximately nine times higher in the BM of patients at diagnosis of pre-B (71.3% ± 8.6%, p = 0.005) and common ALL (70.7% ± 22.1%, p < 0.001) (Fig. 1A). In PB samples, the mean PLA2R1 methylation percentage was 6.5 or 5.1 times higher in pre-B (50.9% ± 20.6%, p < 0.001) and common ALL samples (40.2% ± 25.7%, p < 0.001) in comparison with the control group, respectively (Fig. 1A). In the common ALL cohort, significant differences between PLA2R1 methylation of BM and PB samples were observed (p < 0.001).
The mean relative blast cell numbers of BM from pre-B and common ALL patients were 89.9% ± 6.2% and 91.7% ± 5.8%, respectively (Fig. 1B). Thus, in BM aspirates at diagnosis the relative leukaemic blast cell number was generally higher than 76%. However, the PLA2R1 promoter methylation levels varied between 59–97%, excluding one patient who was tested with 82% blasts at a slightly lower PLA2R1 promoter methylation of 31%.
Corresponding PB samples exhibited, on average, approximately half the relative blast cell number of BM aspirates with 46.8% ± 32.3% (p = 0.032) and 48.5% ± 35.8% (p < 0.001) for pre-B and common ALL patients, respectively. In BM aspirates, all samples exhibited both strongly elevated relative blast cell numbers and increased levels of PLA2R1 promoter methylation. A single exception was one patient with common ALL exhibiting 98% leukaemic blast cells but only 9.3% PLA2R1 promoter methylation (Figs. 2 and 3).
Figure 2.

PLA2R1 promoter methylation and blast cell occurrence during childhood ALL treatment protocol. Box plots consist of the median as ‘center value’, the 25th and 75th percentiles as box edges, and the 10th and 90th percentiles as whisker boundaries. (A) Percentage of PLA2R1 promoter methylation was analysed using droplet digital PCR. (B) The relative blast cell number (number of blast cells in relation to the number of total leukocytes in %) was determined using light microscopic and flow cytometric analysis. Data were obtained for BM aspirates (A1, B1) and PB (A2, B2) from children with B cell ALL at diagnosis (n = 18), after 15 (n = 29) and 33 days (n = 22), and before protocol M (n = 29). Dashed lines indicate the 97.5th percentile of the PLA2R1 promoter methylation in the healthy control group.
Figure 3.

Linear correlations between PLA2R1 promoter methylation and blast cell occurrence measured at the time of diagnosis and during childhood ALL treatment protocol. The scatter blots show the relative blast cell numbers plotted against the PLA2R1 methylation percentages determined in BM aspirates (A) and PB samples (B) at the time of diagnosis (white dots) and following ALL treatment protocol (black dots). The percentage of PLA2R1 promoter methylation was defined using ddPCR. The relative blast cell number (number of blast cells in relation to the number of total leukocytes in %) was determined using light microscopic and flow cytometric analyses. Significant positive correlations between the relative blast cell number and the PLA2R1 methylation percentage were detected for BM aspirates (r = 0.92, p < 0.001, n = 98) and PB samples (r = 0.91, p < 0.001, n = 98). The patient with high relative blast cell numbers and low PLA2R1 promoter methylation at diagnosis is indicated by bold circles for BM and PB samples. Dashed lines indicate the 97.5th percentile of the PLA2R1 promoter methylation in the healthy control group.
PLA2R1 promoter methylation and blast cell occurrence during ALL treatment protocol
During the treatment of patients, both the relative leukaemic blast cell number and the PLA2R1 methylation degree decreased in parallel in PB and BM aspirates (Fig. 2A,B). PLA2R1 promoter methylation and leukaemic blast cell number were analysed in BM aspirates and PB samples of children with B-cell ALL at defined timepoints of the ALL treatment protocol (Fig. 2). A representative example of PLA2R1 promoter methylation quantitation by ddPCR and corresponding PLA2R1 methylation percentages during the course of ALL treatment is shown in Supplementary Fig. 2. Two major FAM-positive subfractions were detected, indicating the presence of both homogenously and heterogeneously methylated epialleles identified by probe mismatching, resulting in different FAM signal amplitudes, as recently demonstrated23. Both the relative leukaemic blast cell number and the PLA2R1 methylation percentages significantly decreased during the first 15 days of treatment compared to initial measurements at diagnosis. Mean PLA2R1 promoter methylation of BM and PB samples decreased from 69.3% ± 20.9% to 11.8% ± 4.5% (p < 0.001, Fig. 2A1) and from 42.1% ± 25.0% to 11.4% ± 3.7% (p < 0.001), respectively (Fig. 2A2). The relative blast cell numbers decreased from 89.9% ± 6.7% to 5.0% ± 9.8% (p < 0.001, Fig. 2B1) and from 43.2% ± 34.3% to 0.4% ± 1.5% (p < 0.001, Fig. 2B2) in BM and PB samples, respectively. Both parameters declined continuously during ongoing ALL treatment. Minimal values were reached before the start of protocol M with 2.7% ± 1.2% or 7.7% ± 2.5% PLA2R1 methylation and 0% or 0.2% ± 0.7% leukaemic blasts in BM aspirates or PB samples, respectively.
In conclusion, the correlation between PLA2R1 promoter methylation and relative blast cell number was analysed in BM aspirates and PB samples at the time of diagnosis and during childhood ALL treatment protocol (Fig. 3). Both BM aspirates and PB samples exhibited significant positive correlations with correlation coefficients of 0.93 (p < 0.001) and 0.91 (p < 0.001), respectively.
PLA2R1 promoter methylation was increased in patients with ALL relapse
Five leukaemia relapses occurred during the present study period, comprising four B cell ALL cases and one patient with AML. Two representative examples of ALL relapse and the single case of AML relapse are presented in Supplementary Fig. 3. In BM aspirates with ALL and AML relapse, both PLA2R1 promoter methylation percentages and leukaemic blast cell numbers were strongly elevated in the range of 68.3% ± 6.9% PLA2R1 methylation (p < 0.001 compared to control group) and 87.2% ± 8.7% leukaemic blast cells. PB samples of ALL and AML relapse also exhibited an increase in PLA2R1 methylation (27.1% ± 13.5%, p = 0.001) and relative blast cell numbers (21.3% ± 27.2%) in comparison to the control group. In BM and PB samples, PLA2R1 methylation declined below the methylation level of the control group during the following ALL and AML treatment protocol (Supplementary Fig. 3).
PLA2R1 promoter methylation as biomarker for ALL risk stratification
The selection of the ALL treatment procedure and subsequent treatment success are dependent on the quality of risk stratification. In the present study, patients were subdivided into groups with standard, medium, and high risk according to the AIEOP-BFM ALL 2009 guidelines and the PLA2R1 promoter methylation on day 15 of the ALL induction treatment was evaluated as biomarker for risk stratification (Fig. 4). The mean PLA2R1 promoter methylation percentage of the standard and medium risk group were 10.0 ± 3.4 and 10.8 ± 3.5, respectively. PLA2R1 promoter methylation of the high risk group was significantly increased with 20.2 ± 3.9 compared to the standard (p = 0.002) and medium risk group (p < 0.005; Fig. 4).
Figure 4.
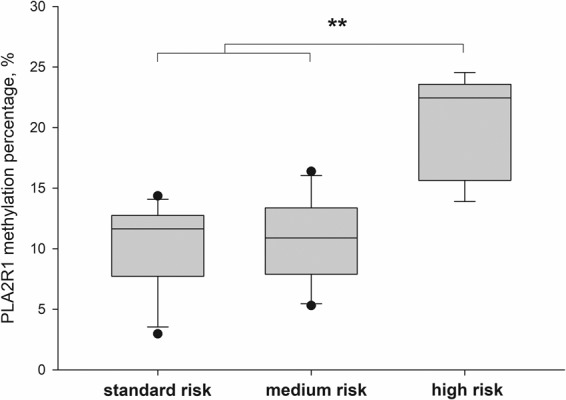
PLA2R1 promoter methylation as biomarker for ALL risk stratification. Box plots consist of the median as ‘center value’, the 25th and 75th percentiles as box edges, and the 10th and 90th percentiles as whisker boundaries. The percentage of PLA2R1 promoter methylation was analysed using droplet digital PCR. Data were obtained for BM aspirates from children with B cell ALL at day 15 of ALL induction treatment. Patients were subdivided into groups with standard (n = 11), medium (n = 15), and high risk (n = 5) according to the AIEOP-BFM ALL 2009 guidelines. The symbol **Indicates significant differences with p < 0.01 between marked cohorts.
Confirmation of the differential PLA2R1 promoter methylation by Illumina Infinium 450k Human DNA methylation data analysis
The PLA2R1 promoter section analysed in the present study is represented by three probes of the Illumina Infinium 450k Human DNA methylation Beadchip array (Supplementary Fig. 1). To validate our ddPCR results, three different GEO datasets (GSE4903124, GSE6340925, and GSE5847726) were analysed with regard to the methylation status of the distinct PLA2R1 promoter region analysed in the present study. GSE49031 investigated the DNA methylation in paediatric ALL and included BM and PB samples of primary paediatric ALL taken at diagnosis (n = 764), remission (n = 86), and relapse (n = 32). Fractionated PB cells from healthy blood donors (n = 51) were also analysed. The methylation status of all PLA2R-associated Illumina probes is illustrated as heatmap in Supplementary Fig. 4 and as box plots in Supplementary Fig. 5. In BM ALL samples, PLA2R1 promoter methylation was increased significantly at diagnosis (52% ± 22%) compared to corresponding samples at remission (10% ± 2%, p < 0.001). ALL relapse samples exhibited a significantly increased PLA2R1 methylation of 65% ± 23% in comparison to samples taken from patients in remission (p < 0.001) and also compared to primary BCP-ALL (52% ± 22%), p < 0.001) and T-ALL (53% ± 24%), p = 0.02) samples (Supplementary Fig. 5A1). Moreover, PLA2R1 methylation in PB ALL samples increased significantly compared to PB samples from healthy controls and their different cell fractions (p < 0.001, Supplementary Fig. 5A2).
Divergent methylation patterns of healthy progenitor cells were investigated in the GSE63409 and GSE58477 datasets and compared to leukaemic stem and blast cells of patients with AML (Supplementary Fig. 5B,C). Different types of normal BM-derived progenitor cells showed similar levels of PLA2R1 promoter methylation of approximately 10%. By contrast, leukaemic progenitor cells exhibited a significantly higher degree of PLA2R1 promoter methylation within the range of 30–50% (GSE63409: p < 0.001 for leukaemic stem cells or leukaemic blasts vs. healthy progenitor cells; GSE58477: p = 0.002 for normal CD34 + vs. leukaemic blasts).
Additionally, analyses of Illumina 450k methylation27 and gene expression data28 from six childhood B-ALL and eight childhood T-ALL cell lines demonstrated that the three Illumina 5’-CpG sites associated with the analysed ddPCR amplicon were strongly methylated. Simultaneously, the PLA2R1 expression was completely or at least notably suppressed in all analysed cell lines (Supplementary Table 2).
Transfection-based overexpression of PLA2R1 in Jurkat cells
To further investigate the effect of different PLA2R1 expression levels in acute childhood leukaemia, human lymphocyte acute leukaemia (Jurkat) cells, which exhibit a high degree of PLA2R1 promoter hypermethylation and an associated silenced receptor expression19, were analysed in vitro.
Jurkat cells were transfected with a PLA2R1 plasmid vector (Jurkat-PLA2R1) to overexpress PLA2R1. Results were compared to control vector transfected Jurkat cells (Jurkat-Ctrl) (Fig. 5). Jurkat-Ctrl and Jurkat-PLA2R1 cells showed comparable levels of β-actin gene expression and actin protein synthesis indicated by quantitative real-time PCR and western blot analysis, respectively. A strong re-expression of PLA2R1 was demonstrated at mRNA and protein levels in Jurkat-PLA2R1 cells, whereas in Jurkat-Ctrl cells, no PLA2R1 gene expression and protein synthesis were detectable (Fig. 5A,B; uncropped western blotting displayed in Supplementary Fig. 6).
Figure 5.

Cell proliferative behaviour in PLA2R1-transfected Jurkat cells (Jurkat-PLA2R1) compared to control vector-transfected Jurkat cells (Jurkat-Ctrl). (A) The amount of PLA2R1-mRNA was determined using RT-qPCR with β-actin as reference gene. Results are shown as mean ± SD of three independent experiments (biological n = 3) with two technical replicates. Bar graphs represent the ratio of PLA2R1 and β-actin gene expression. Symbol # indicates that PLA2R1 expression in Jurkat-Ctrl cells was not detected after 45 PCR cycles and therefore set to zero. (B) The level of PLA2R1 protein expression was assessed using western blotting with actin as reference protein and human recombinant PLA2R1 as positive control. A cropped representative blot section (from four independent experiments) is shown (biological n = 4, full-length blot is presented in Supplementary Fig. 6). (C–E) Results are the means ± SD of three independent experiments (biological n = 3) with two technical replicates. 2 × 105 Jurkat cells were seeded in 6-well plates and trypan blue-negative (C) and -positive Jurkat cells (D) were counted during four consecutive days. (E) Apoptosis was stimulated with hydrogen peroxide for 24 h and determined by Annexin-V-Fluorescein/Hoechst 33258 staining using flow cytometry analysis. Levels of significance are defined as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
PLA2R1 expression influences the proliferative behaviour of Jurkat cells
The proliferation rate of transfected Jurkat cells was assessed using cell growth curve analysis with trypan blue staining of dead cells (Fig. 5C,D). The relative cell number of Jurkat-PLA2R1 was significantly decreased in comparison to Jurkat-Ctrl cells after two days of culturing (p < 0.001) and observed differences increased continuously over the investigation period (Fig. 5C). The maximum change in Jurkat-PLA2R1 cell proliferation was reached after four days, with 33% reduction compared to Jurkat-Ctrl cells (p < 0.001). Furthermore, the number of dead cells was significantly increased in Jurkat-PLA2R1 cells after two days of culture compared to Jurkat-Ctrl cells (p = 0.015), reaching a 4.5-fold higher difference after four days (p = 0.002, Fig. 5D).
Moreover, the transfected Jurkat cells were stained with Annexin-V-Fluorescein/Hoechst 33258 and analysed by flow cytometry to investigate the underlying mechanism of cell death (Fig. 5E). Representative flow cytometry results of transfected Jurkat cells are presented in Supplementary Fig. 7. Jurkat-PLA2R1 exhibited 31% fewer healthy cells than Jurkat-Ctrl (p = 0.003), whereas the fractions of early (p = 0.003) and late apoptotic (p = 0.004) and necrotic cells (p = 0.003) were significantly increased in Jurkat-PLA2R1 compared to Jurkat-Ctrl cells after induction of cell death with hydrogen peroxide.
Discussion
The present study establishes that in addition to adult patients with AML19, the PLA2R1 promoter is also hypermethylated, both in BM aspirates and PB samples from paediatric patients with primary and relapsed acute lymphoblastic leukaemia. Furthermore, lower PLA2R1 promoter methylation coincided with decreases in blast cell numbers in BM and PB during ALL induction treatment. Significant positive correlations between PLA2R1 promoter methylation and relative blast cell numbers were detected in BM and PB samples during complete ALL treatment protocol. Therefore, our data suggests that leukaemic blasts are the origin of PLA2R1 hypermethylation in BM and PB samples.
Although the number of blasts was above 76% in all analysed BM aspirates at diagnosis, PLA2R1 promoter methylation levels varied between 31% and 97%. This data suggests that not all BM cells defined as blasts had methylated PLA2R1 promoter sequences. The observed data indicates BM blast cell heterogeneity, which is consistent with previous data29–31. Moreover, normal BM precursors of mature B-lymphocytes may be present within the analysed aspirates. The precursors share many morphological and immunophenotypic features with neoplastic lymphoblasts of B-ALL32. According to our analysis of Illumina Infinium 450k Human DNA methylation data, normal BM precursors exhibit only marginal PLA2R1 promoter methylation (Supplementary Fig. 5). Therefore, a different proportion of precursors may predispose BM blasts to greater variation in PLA2R1 methylation compared to PB. Further studies involving cell sorting experiments are required to verify this hypothesis.
Taken together, our findings support the hypothesis, initially established from the obtained data in adult patients with AML19, that the determination of PLA2R1 methylation may be also a sensitive marker for treatment efficacy and monitoring of minimal residual disease in childhood ALL. PLA2R1 methylation can be of special interest in patients lacking a common leukaemia subtype classification if no other molecular biomarkers are available. One example of ALL relapse in a patient without ALL-specific molecular genetic markers is shown in Supplementary Fig. 3A (left diagram). In this case unknown ALL-specific genetic markers limited the monitoring of minimal residual disease and therapy effectivity. Generally, PLA2R1 promoter methylation was found to be strongly elevated at diagnosis in BM and PB samples but decreased rapidly during ALL induction treatment mostly falling below the methylation degree of healthy controls. Furthermore, PLA2R1 promoter methylation was significantly increased in the group of high risk patients compared to non-high risk groups at day 15 of the ALL induction treatment. Risk stratification according to the AIEOP-BFM ALL 2009 guidelines is a multi-parameter approach, involving immunohistochemical as well as molecular biological analyses at different points in time (until week 12) of the ALL treatment protocol. Thus, PLA2R1 promoter methylation may be beneficial to identify high risk patients relatively easily, enabling early adaptions to the ALL treatment protocol. To what extend monitoring of minimal residual disease by PLA2R1 promoter methylation is superior to well established methods using clonal markers like IgG-heavy chain or T-cell receptor rearrangements will be the next step of our analysis. In this context it must be kept in mind that unlike chromosomal alterations, gene mutations and gene rearrangements, promoter hypermethylation is reversible and may be altered by therapeutic intervention. There are existing approaches, including the introduction of epigenetic modifying agents into leukaemia treatment protocols33, and PLA2R1 promoter methylation may be an indicator to predict the effectiveness of such treatment strategies19.
In this cohort of paediatric patients, one patient with common ALL exhibited strongly increased relative leukaemic blast cell numbers together with a lack of increased PLA2R1 promoter methylation in both BM and PB samples at diagnosis, suggesting that PLA2R1 promoter hypermethylation is a frequent (95%, 19 out of 20 B-cell ALL patients), but not indispensable event during childhood leukaemogenesis. These findings are consistent with the work of Amin et al., suggesting the existence of a subset of AML and ALL patients with PLA2R1-expressing leukaemic blast cells34. This may correspond to the aforementioned ALL patient exhibiting PLA2R1 promoter methylation degree below the cutoff of the healthy cohort.
The obtained in vitro data from PLA2R1-transfected Jurkat cells suggest that blast cells with silenced PLA2R1 expression may escape apoptosis and necrosis more efficiently than the receptor expressing cells. These findings indicate that PLA2R1 has a tumour-suppressive effect in acute leukaemia. We previously provided evidence of PLA2R1’s tumour-suppressive role in solid tumours investigating the prostate cancer cell line LNCaP in in vivo xenograft models35. Re-expression of the receptor was also discussed and shown to have a tumour-suppressive function in the mammary cancer cell line MDA-MB-453, mediated by JAK2/oestrogen-related receptor-α signalling and the induction of mitochondrial apoptosis pathways15,16,35. It may be possible to activate similar pro-apoptotic signalling pathways in PLA2R1-transfected Jurkat cells. However, this suggestion requires further investigation.
To further evaluate the relevance of our results, we searched the methylation data published by Nordlund et al. using the Illumina Infinium 450k Human DNA methylation Beadchip array24. Our analysis confirmed an increased methylation level of the PLA2R1 promoter in BM and PB samples of patients with childhood ALL compared to matched samples of patients in remission or healthy blood donors. Furthermore, our analysis suggested an increase of PLA2R1 promoter methylation in BM aspirates of patients with ALL relapse compared to primary samples. This is in line with the strongly elevated PLA2R1 promoter methylation in cases of childhood ALL relapse as investigated in the present study.
Moreover, the analysis of Illumina DNA methylation results from patients with AML demonstrated that isolated leukaemic progenitor cells exhibited high degrees of PLA2R1 promoter methylation, whereas normal progenitor cells showed only minor PLA2R1 methylation (Supplementary Fig. 5). These results are consistent with our conclusion that leukaemic blast cells are the origin of the increased PLA2R1 methylation observed in this study of paediatric patients with ALL.
A limitation of our study is the selection of the Jurkat T-ALL cell line as the cell model for our transfection experiments, as it may limit the significance of the in vitro results to children diagnosed with B-ALL. Therefore, further investigation is warranted into whether the tumour-suppressive behaviours of PLA2R1 re-expression will be repeated in B-ALL cell lines and primary blast cells. Using isolated blast cells, it will be possible to determine the extent to which the PLA2R1 gene is actually silenced in blasts of childhood ALL patients at the mRNA and protein levels. In the current study, we have shown that the three Illumina 5′-CpG sites associated with the analysed ddPCR amplicon were strongly methylated and that PLA2R1 expression was completely or notably suppressed in six childhood B-ALL and eight T-ALL cell lines, including the investigated Jurkat cell line (Supplementary Table 2). The reproducibility of these analyses was also supported by the data observed in U937, PC-3, and LNCaP cells. These results were consistent with our previous sequencing data for Jurkat and U937 cells, as well as for the prostate cancer cell lines18,19,35.
Conclusion
The PLA2R1 promoter was found to be hypermethylated in BM aspirates and PB samples of patients diagnosed with different childhood ALL subtypes, which makes the PLA2R1 methylation a suitable biomarker for the monitoring of treatment response in ALL patients. A decline in the leukaemic blast cell numbers together with decreasing PLA2R1 promoter methylation during ongoing ALL treatment suggest that the overall increase of PLA2R1 methylation in BM and PB samples at diagnosis is caused by leukaemic blast cells. Furthermore, the data of this study demonstrated that PLA2R1 promoter methylation may be a useful marker for disease monitoring, risk stratification, and the evaluation of ALL treatment success. This is especially relevant to patients lacking known ALL-specific molecular genetic markers for minimal residual disease monitoring or to predict the effectiveness of epigenetic modifying agents in ALL treatment. In vitro analysis showed that the PLA2R1 re-expression and up-regulation of protein synthesis in transfected Jurkat cells had a negative impact on cellular proliferative behaviour, suggesting an important tumour-suppressive role of PLA2R1 in childhood ALL.
Material and Methods
Sample collection
This study included 44 patients with B-lymphoblastic ALL. Additionally, a single patient with acute myeloid leukaemia (AML; 13 years old male) was assessed for comparison. The B-cell ALL cohort consisted of five cases with pre-B ALL (four females and one male) and 39 cases with common ALL (15 females, 24 males). The average age was 3.0 ± 1.1 years (range 1.1 to 4.0 years) and 7.5 ± 4.4 (range 1.7 to 17.0 years), respectively. EDTA-bone marrow and peripheral EDTA-blood samples were collected at routine examinations. EDTA-peripheral blood was collected from 20 healthy individuals (8 females and 12 males) with average ages of 9.4 ± 4.2 years (range 3.3 to 16.2 years), serving as controls. For risk stratification, bone marrow aspirates from day 15 of the ALL induction treatment were subdivided into groups with standard (n = 11), medium (n = 15), and high risk (n = 5) according to the AIEOP-BFM ALL 2009 guidelines based on chromosomal rearrangements present at diagnosis, prednisolone treatment response at day 8 and overall treatment response assessed in bone marrow aspirates at day 15 of the ALL treatment protocol. An overview of patients and healthy control samples included in the present study is presented in the Supplementary Table 1. The relative blast cell numbers (number of blast cells in relation to the number of total leukocytes in %) were routinely quantified at diagnosis and during ALL and AML treatment protocol by light microscopy and flow cytometry analysis. All patients and their legal guardians provided written informed consent for the study. The study was approved by the Ethical Board of the University Hospital of Dresden. All methods were performed in accordance with the relevant guidelines and regulations.
Extraction of genomic DNA and bisulfite modification
Genomic DNA was isolated from peripheral blood (PB) and bone marrow (BM) aspirate cells as described36 and precipitated in ethanol. Concentration of isolated DNA was measured using the Quantifluor dsDNA-System (Promega GmbH, Mannheim, Germany) according to the manufacturer’s protocol. Bisulfite modification was conducted with 500 ng of isolated genomic DNA using the EZ DNA Methylation Kit (Zymo Research Europe GmbH, Freiburg, Germany).
Digital droplet PCR of bisulfite-modified DNA
The methylation status of the PLA2R1 promoter and down-stream regions −700 to +1340 bp relative to the transcription start site (TSS) was previously analysed in different leukaemic cell lines and in PB samples as well as BM aspirates of adult leukaemic patients via methylation-sensitive high-resolution melting19. Three different 5′-CpG sites (−546 bp, −548 bp and −551 bp relative to TSS) were selected by maximum methylation differences between healthy individuals and leukaemic patient samples/cell lines to represent the methylation status of the distinct region −644 bp to −477 bp from the TSS of PLA2R119,23 (Supplementary Fig. 1). Digital droplet PCR protocol was performed as described elsewhere23 and according to the manufacturer’s instructions (Bio-Rad GmbH, München, Germany). Binding of dual-labelled probes was used to simultaneously quantify methylated (5′-FAM-CCCAACTACTCCGCGACGCAA-3′-BHQ1) and unmethylated (5′-HEX-CCCAACTACTCCACAACACAA-3′-BHQ1) DNA fragments. The applied primer pair 5′-GGG GTA AGG AAG GTG GAG AT-3′ and 5′-ACA AAC CAC CTA AAT TCT AAT AAA CAC-3′ (168 bp product length) was designed to anneal to methylation-independent sections of the DNA. Data were processed using the QuantaSoft software version 1.6.6.0320 (Bio-Rad GmbH). The PLA2R1 methylation percentage was calculated using the number of methylated copies in relation to the sum of methylated and unmethylated PLA2R1 copies in %.
Cell culture and incubation
Human T-lymphocyte acute leukaemia (Jurkat) cells were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany) and cultured as previously described19,35. Transfected Jurkat cells were cultured in RPMI-1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat-inactivated fetal calf serum (Thermo Fisher Scientific) and 500 mg/ml G418 Sulfate (Thermo Fisher Scientific). Cell lines were frequently tested for mycoplasma contamination (MycoAlert™ Mycoplasma Detection Kit, Lonza).
Transfection
According to our previous studies with lymphocytic Jurkat cell line19, a fully methylated PLA2R1 promoter was detected using sequencing analysis in conjunction with PLA2R1 gene silencing. The methylation resulted in a lack of PLA2R1 expression at both mRNA and protein level. Furthermore, a strong re-expression of PLA2R1 was observed after azacitidine (Vidaza) treatment of Jurkat cells in vitro. Therefore, we selected Jurkat cell line as a relevant cell model for our transfection experiments. This established cell line is extensively used as cell model to investigate ALL20–22.
Transfection of Jurkat cells was performed as described elsewhere35 utilizing ViaFectTM Transfection Reagent (Promega, Madison, WI, USA) and an optimized expression vector for phospholipase A2 receptor 1 (NP_031392.3; GenScript, Piscataway, NJ, USA), or a control vector (GenScript). The transfected cells were selected using 500 mg/mL G418 Sulfate and surviving cells were used for subsequent experiments.
RNA isolation and reverse transcription quantitative polymerase chain reaction (RT-qPCR)
RNA isolation and RT-qPCR were described elsewhere35. In brief, RNA was isolated using QIAzol Lysis Reagent (QIAGEN) and miRNeasy Mini Kit with on-column DNase digestion (QIAGEN). Reverse transcription protocol involved incubation of 100 ng RNA template and MuLV Reverse Transcriptase (Thermo Fisher Scientific) for 45 min at 42 °C and 5 min at 99 °C with a final reaction volume of 20 µL. Quantitative PCR was conducted using the Rotor-Gene Q cycler and Rotor-Gene SYBR Green PCR Kit (Qiagen). PCR conditions involved 5 min at 95 °C followed by 45 cycles of 5 s at 95 °C, and 10 s at 59 °C. β-actin was used as reference gene. RNA isolation and RT-qPCR were carried out using three separate cell culture passages, each measured in duplicate (biological n = 3, technical n = 2).
Protein isolation, SDS-PAGE and Western Blot
Protein isolation from cell culture experiments, SDS-PAGE, and Western Blot were described elsewhere35. The method involves cell lysis with CellLytic M lysis buffer (Sigma-Aldrich), protein separation using ClearPAGE 10% sodium dodecylsulfate polyacrylamide gels (C.B.S. Scientific), and sample transfer to PVDF membranes (Fisher Scientific). For detection of PLA2R1, primary recombinant rabbit monoclonal antibody (AB; ab211573, Abcam, Cambridge, UK) and a secondary horseradish peroxidase conjugated goat anti-rabbit IgG (sc-2004, Santa Cruz) were used. Actin was detected using primary mouse monoclonal AB (MAB1501R, Merck Millipore, Darmstadt, Germany) and secondary horseradish peroxidase conjugated goat anti-mouse IgG (sc-2005, Santa Cruz). Protein isolation, SDS-PAGE and Western Blot were repeated three times using separate cell culture passages (biological n = 4).
Analysis of proliferation and apoptosis/necrosis
For cell growth curve analysis, 2 × 105 transfected Jurkat cells were cultured over four days. Numbers of living and dead cells were determined on the second, third and fourth day by trypan blue staining and light microscopic analysis.
Cell death was stimulated with 100 µM hydrogen peroxide (Sigma-Aldrich) in RPMI-1640 medium without fetal calf serum for 24 h. The cells were stained with the Annexin-V-FLUOS staining kit (Sigma-Aldrich) according to the manufacturer’s protocol. Necrotic cells were stained with Hoechst 33258 (Thermo Fisher Scientific) for 30 s and analysed immediately afterwards using flow cytometry to avoid the staining of healthy cells.
Data analysis
‘Center values’ were defined as means with standard deviation as error indication. Normal distribution was analysed using the Shapiro-Wilk test. Variance was estimated using F-test. Data were analysed by two-tailed and unpaired Student’s t test (normally distributed, homoscedastic) or Mann-Whitney Rank Sum test (non-normally distributed) to calculate the indicated p values. Differences were considered significant at p < 0.05. Levels of significance were defined and indicated as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
The Illumina Infinium 450 k Human DNA methylation Beadchip array data was calculated as normalized β-values from four different Gene Expression Omnibus (GEO) datasets. The specific normalization methods were described previously (GSE4903124, GSE5847725, GSE6340926, and GSE6837927). For the GSE49031 dataset, methylation raw data were normalized using peak-based correction, whereas GSE58477, GSE63409, and GSE68379 datasets were normalized using appropriate processing software (Illumina® GenomeStudio® for the first and minfi Bioconductor package for the remaining two datasets). RefSeq gene annotations for Illumina 5′-CpG sites were based on the Human Methylation 450 k manifest file version 1.2 (May 23, 2013). Illumina probes with PLA2R1 annotation were filtered. The remaining probes associated with PLA2R1 were again filtered according to the latest Illumina annotation file37, excluding probes with INDELs, annotated single-nucleotide polymorphism (SNP) at the single-base extension or CpG sites, and non-unique subsequences within the probe sequence. Four out of 18 Illumina 5′-CpG sites were excluded based on the aforementioned criteria. According to the annotated UCSC gene region feature categories, the Illumina Infinium 450 k Human DNA methylation Beadchip array includes five CpG sites within the body of PLA2R1 as well as five CpG sites within 0–200 bases (TSS200), and four CpG sites within 200–1500 bases (TSS1500) upstream of the transcriptional start site. The three different CpG sites associated with the analysed 168 bp ddPCR amplicon are associated with the latter category. Average methylation was estimated as mean β-values for the three different 5′-CpG sites (cg12991125, cg20257553, and cg24235037), that are located within the analysed ddPCR amplicon (Supplementary Fig. 1).
For analysis of gene expression, processed data of leukaemic cell lines were provided by the Cancer Cell Line Encyclopedia (CCLE) project28. In brief, for large insert non-strand specific RNA Sequencing, samples were processed according to Illumina Tru Seq RNA Sample Preparation protocol with 200 ng of total RNA as starting material. Illumina sequencing was done on the Illumina HiSeq. 2000 or HiSeq. 2500. RNA sequencing results are shown as TPM (Transcripts Per Kilobase Million).
Ethics approval and consent to participate
All patients and their legal guardians provided written informed consent for the study. The study was approved by the Ethical Board of the Universxity Hospital of Dresden.Name of the registry: Eudra-CT; trial registration number: 2007-004270-43; date of the registration: 03/12/2010; https://www.clinicaltrialsregister.eu/ctr-search/trial/2007-004270-43/AT.
Consent for publication
All patients and their legal guardians provided written informed consent for the publication of this study.
Supplementary information
Acknowledgements
The authors are grateful to Romy Adler, technician for her expert technical assistance. This work was supported by a grant from Bundesministerium für Bildung und Forschung (BMBF) and Forschungszentrum Jülich GmbH (031B0098C, Germany) (to M.M.).
Author contributions
M.F., K.G., D.T., C.J., and M.M. analysed and interpreted the patient data regarding the haemato-oncological disease. M.F., B.N., and M.M. analysed and interpreted the in vitro data of transfected Jurkat cells. M.F., B.L., O.S., M.S., and M.M. contributed to the study design. M.F. and M.M. were major contributors in writing the manuscript. B.L., O.S., and M.S. had advisory function regarding the writing of the manuscript. All authors read and approved the final manuscript.
Data availability
The datasets generated and analysed during the current study are not publicly available due to protection of privacy of the participants of the study but are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-65825-0.
References
- 1.Steliarova-Foucher E, et al. International incidence of childhood cancer, 2001–10. A population-based registry study. The Lancet Oncology. 2017;18:719–731. doi: 10.1016/S1470-2045(17)30186-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noone A. M. et al (eds). SEER Cancer Statistics Review. Available at https://seer.cancer.gov/csr/1975_2015/ (1975–2015).
- 3.Ranjbar R, et al. The importance of miRNAs and epigenetics in acute lymphoblastic leukemia prognosis. Journal of cellular physiology. 2019;234:3216–3230. doi: 10.1002/jcp.26510. [DOI] [PubMed] [Google Scholar]
- 4.Gatta, G. et al. Rare cancers are not so rare. The rare cancer burden in Europe. European journal of cancer (Oxford, England: 1990) 47, 2493–2511; (2011). [DOI] [PubMed]
- 5.Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA: a cancer journal for clinicians. 2014;64:83–103. doi: 10.3322/caac.21219. [DOI] [PubMed] [Google Scholar]
- 6.Stein Anthony, Franklin Janet L., Chia Victoria M., Arrindell Deborah, Kormany William, Wright Jacqueline, Parson Mandy, Amouzadeh Hamid R., Choudhry Jessica, Joseph Guiandre. Benefit–Risk Assessment of Blinatumomab in the Treatment of Relapsed/Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Drug Safety. 2018;42(5):587–601. doi: 10.1007/s40264-018-0760-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alvarnas JC, et al. Acute Lymphoblastic Leukemia, Version 2.2015. J Natl Compr Canc Netw. 2015;13:1240–1279. doi: 10.6004/jnccn.2015.0153. [DOI] [PubMed] [Google Scholar]
- 8.Greaves M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nature reviews. Cancer. 2018;18:471–484. doi: 10.1038/s41568-018-0015-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng S, et al. Hypermethylation of the 5′ CpG island of the FHIT gene is associated with hyperdiploid and translocation-negative subtypes of pediatric leukemia. Cancer research. 2004;64:2000–2006. doi: 10.1158/0008-5472.CAN-03-2387. [DOI] [PubMed] [Google Scholar]
- 10.Wong IH, Ng MH, Huang DP, Lee JC. Aberrant p15 promoter methylation in adult and childhood acute leukemias of nearly all morphologic subtypes. Potential prognostic implications. Blood. 2000;95:1942–1949. doi: 10.1182/blood.V95.6.1942. [DOI] [PubMed] [Google Scholar]
- 11.Matsushita C, et al. Aberrant methylation in promoter-associated CpG islands of multiple genes in relapsed childhood acute lymphoblastic leukemia. Oncology reports. 2004;12:97–99. [PubMed] [Google Scholar]
- 12.Ramos Kristie N, Ramos Irma N, Zeng Yi, Ramos Kenneth S. Genetics and epigenetics of pediatric leukemia in the era of precision medicine. F1000Research. 2018;7:1104. doi: 10.12688/f1000research.14634.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohara O, Ishizaki J, Arita H. Structure and function of phospholipase A2 receptor. Progress in lipid research. 1995;34:117–138. doi: 10.1016/0163-7827(94)00009-B. [DOI] [PubMed] [Google Scholar]
- 14.Augert A, et al. The M-type receptor PLA2R regulates senescence through the p53 pathway. EMBO reports. 2009;10:271–277. doi: 10.1038/embor.2008.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Augert A, et al. PLA2R1 kills cancer cells by inducing mitochondrial stress. Free radical biology & medicine. 2013;65:969–977. doi: 10.1016/j.freeradbiomed.2013.08.177. [DOI] [PubMed] [Google Scholar]
- 16.Griveau A, et al. The PLA2R1-JAK2 pathway upregulates ERRα and its mitochondrial program to exert tumor-suppressive action. Oncogene. 2016;35:5033–5042. doi: 10.1038/onc.2016.43. [DOI] [PubMed] [Google Scholar]
- 17.Vindrieux D, et al. PLA2R1 mediates tumor suppression by activating JAK2. Cancer research. 2013;73:6334–6345. doi: 10.1158/0008-5472.CAN-13-0318. [DOI] [PubMed] [Google Scholar]
- 18.Sukocheva Olga, Menschikowski Mario, Hagelgans Albert, Yarla Nagendra Sastry, Siegert Gabriele, Reddanna Pallu, Bishayee Anupam. Current insights into functions of phospholipase A2 receptor in normal and cancer cells: More questions than answers. Seminars in Cancer Biology. 2019;56:116–127. doi: 10.1016/j.semcancer.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Menschikowski M, et al. Aberrant methylation of the M-type phospholipase A(2) receptor gene in leukemic cells. BMC cancer. 2012;12:576. doi: 10.1186/1471-2407-12-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abraham RT, Weiss A. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nature reviews. Immunology. 2004;4:301–308. doi: 10.1038/nri1330. [DOI] [PubMed] [Google Scholar]
- 21.Chim CS, Wong ASY, Kwong YL. Epigenetic inactivation of the CIP/KIP cell-cycle control pathway in acute leukemias. American journal of hematology. 2005;80:282–287. doi: 10.1002/ajh.20503. [DOI] [PubMed] [Google Scholar]
- 22.Sak K, Everaus H. Established Human Cell Lines as Models to Study Anti-leukemic Effects of Flavonoids. Current genomics. 2017;18:3–26. doi: 10.2174/1389202917666160803165447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menschikowski M, et al. Identification and Quantification of Heterogeneously-methylated DNA Fragments Using Epiallele-sensitive Droplet Digital Polymerase Chain Reaction (EAST-ddPCR) Cancer genomics & proteomics. 2018;15:299–312. doi: 10.21873/cgp.20088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nordlund J, et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome biology. 2013;14:r105. doi: 10.1186/gb-2013-14-9-r105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung N, Dai B, Gentles AJ, Majeti R, Feinberg AP. An LSC epigenetic signature is largely mutation independent and implicates the HOXA cluster in AML pathogenesis. Nature communications. 2015;6:8489. doi: 10.1038/ncomms9489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qu Y, et al. Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics. 2014;9:1108–1119. doi: 10.4161/epi.29315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iorio F, et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell. 2016;166:740–754. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hassanein NM, Alcancia F, Perkinson KR, Buckley PJ, Lagoo AS. Distinct expression patterns of CD123 and CD34 on normal bone marrow B-cell precursors (“hematogones”) and B lymphoblastic leukemia blasts. American journal of clinical pathology. 2009;132:573–580. doi: 10.1309/AJCPO4DS0GTLSOEI. [DOI] [PubMed] [Google Scholar]
- 30.Hoffmann MH, et al. Clinical impact of leukemic blast heterogeneity at diagnosis in cytogenetic intermediate-risk acute myeloid leukemia. Cytometry. Part B, Clinical cytometry. 2012;82:123–131. doi: 10.1002/cyto.b.20633. [DOI] [PubMed] [Google Scholar]
- 31.Baersch G, et al. Durchflusszytometrische Charakterisierung von Reifungsvorgängen und stammzellnahen Zellpopulationen bei der B-Vorläuferzell-ALL im Kindesalter. Klinische Padiatrie. 1996;208:160–167. doi: 10.1055/s-2008-1046467. [DOI] [PubMed] [Google Scholar]
- 32.Longacre TA, et al. Hematogones: a multiparameter analysis of bone marrow precursor cells. Blood. 1989;73:543–552. doi: 10.1182/blood.V73.2.543.543. [DOI] [PubMed] [Google Scholar]
- 33.Burke MJ, Bhatla T. Epigenetic modifications in pediatric acute lymphoblastic leukemia. Frontiers in pediatrics. 2014;2:42. doi: 10.3389/fped.2014.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amin R, et al. Phospholipase A2 receptors in human leukemic blasts. Leukemia & lymphoma. 2011;52:908–909. doi: 10.3109/10428194.2010.551160. [DOI] [PubMed] [Google Scholar]
- 35.Friedemann M, et al. Diverse effects of phospholipase A2 receptor expression on LNCaP and PC-3 prostate cancer cell growth in vitro and in vivo. Oncotarget. 2018;9:35983–35996. doi: 10.18632/oncotarget.26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic acids research. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou W, Laird PW, Shen H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic acids research. 2017;45:e22. doi: 10.1093/nar/gkw967. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analysed during the current study are not publicly available due to protection of privacy of the participants of the study but are available from the corresponding author on reasonable request.