

Abstract
The reaction of 5,15-diHpETE with human 5-lipoxygenase (LOX), human platelet 12-LOX and human reticulocyte 15-LOX-1 was investigated to determine the reactivity and relative rates of producing lipoxins (LXs). 5-LOX does not react with 5,15-diHpETE, although it can produce LXA4 when 15-HpETE is the substrate. In contrast, both 12-LOX and 15-LOX-1 react with 5,15-diHpETE, forming specifically LXB4. For 12-LOX and 5,15-diHpETE, the kinetic parameters are kcat = 0.17 s-1 and kcat/KM = 0.011 μM-1s-1 (106-fold and 1600-fold lower than for 12-LOX oxygenation of AA, respectively). On the other hand, for 15-LOX-1 the equivalent parameters are kcat = 4.6 s−1 and kcat/KM = 0.21 μM-1s-1 (3-fold higher and similar to that for 12-HpETE formation by 15-LOX-1 from AA, respectively). This contrasts with the complete lack of reaction of 15-LOX-2 with 5,15-diHpETE (Biochemistry 55, 2832–2840, 2016). Our data indicate that 12-LOX is markedly inferior to 15-LOX-1 in catalyzing the production of LXB4 from 5,15-diHpETE. Platelet aggregation was inhibited by the addition of 5,15-diHpETE, with an IC50 of 1.3 μM, however, LXB4 did not significantly inhibit collagen-mediated platelet activation up to 10 μM. In summary, LXB4 is the primary product of 12-LOX and 15-LOX-1 catalysis if 5,15-diHpETE is the substrate, with 15-LOX-1 being 20-fold more efficient than 12-LOX. LXA4 is the primary product with 5-LOX, but only if 15-HpETE is the substrate. Approximately equal proportions of LXA4 and LXB4 are produced by 12-LOX, but only if LTA4 is the substrate, as described previously (Biochimica et Biophysica Acta 1133, 223–234, 1992).
Graphical Abstract
Introduction:
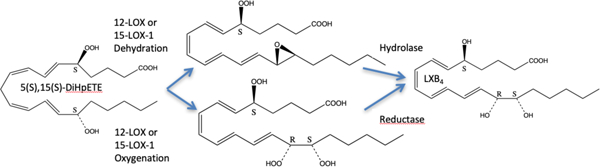
Lipoxins (LXs) were discovered in 19841, 2 and are a class of specialized pro-resolving mediators (SPMs) derived from arachidonic acid (AA), which are involved in inflammation resolution. The first two LXs discovered were lipoxinA4 (LXA4) and lipoxinB4 (LXB4), with the epi-lipoxins, 15-epi-LXA4 and 15-epi-LXB4, being discovered later.3 The initial step of acute inflammation is to recruit macrophages with the generation of leukotrienes (LTs) and hydroxyeicosatetraenoic acids (HETEs). Eventually, SPMs, such as LXs, are generated via transcellular pathways to reverse the inflammatory response and initiate cellular repair.4 Researchers have investigated the biosynthetic pathway for the production of LXs and the generally accepted biosynthesis of LXs requires two distinct enzymatic reactions with AA. The first step is the oxygenation of AA to form a hydroperoxide, which is then converted to an epoxide. This can be accomplished by either 5-LOX or 15-LOX, generating either the 5,6-epoxide or the 14,15-epoxide, respectively. The second step is oxygenation at the other end of the molecule from the epoxide, adding a hydroperoxide moiety. There is then a subsequent hydrolysis of the epoxide and reduction of the hydroperoxide, creating either the 5,6,15-trihydroxy- or the 5,14,15-trihydroxy-eicosatetraenoate, depending on the initiating LOX isozyme (Figure 1).5, 6 The specific pathway depends on the cell type involved, either with one cell type that has both LOXs, or a transcellular biosynthetic pathway, which involves the transfer of an intermediate between two cell types. Another biosynthetic pathway for LXs involves 5-LOX in neutrophils and 12-LOX in platelets. The hypothesis is that 5-LOX generates LTA4 in the neutrophil, which is then transferred to the platelet, where 12-LOX subsequently abstracts a hydrogen atom from C13 of LTA4, with oxygen attacking C15. The epoxide ring is subsequently opened by a hydrolase, generating a delocalized cation, which is attacked by water, generating either LXA4 with a C6 attack or LXB4 with a C14 attack (Figure 1).7, 8
Figure 1: Biosynthetic pathways for LX production.
The carbon whose hydrogen atom is abstracted is listed next to the LOX isozyme, such as C7, C10 or C13. For the conversion of 5,15-DiHpETE to the two lipoxins, the LOX isozyme is not specified because that is the subject of this publication.
Another pathway to making LXs, involves 5,15-diHpETE as an intermediate. A hydrogen atom is abstracted from C10 of 5,15-diHpETE, generating a delocalized radical, which can convert either the C5 hydroperoxide or the C15 hydroperoxide to an epoxide (5,6- or 14,15-epoxide). Subsequent hydrolysis of the expoxide and reduction of the hydroperoxide generates either the 5,6,15-trihydroxy- or the 5,14,15-trihydroxy-eicosatetraenoates. Like the cation intermediate discussed above, both LXA4 and LXB4 can be generated in this manner since the resulting radical is delocalized over 9 carbons due to the full conjugation of the tetraene system (Figure 1).9 The reactivity of this chemical step is enhanced by the fact that 5,15-diHpETE is an “activated” intermediate. The energy required to abstract the C10 hydrogen atom from 5,15-diHpETE is markedly lower than the hydrogen atom from AA or 5-hydroperoxyeicosatetraenoic acid (5-HpETE), due to the greater conjugation of its intermediate, 9-carbons for 5,15-diHpETE versus 5-carbons for AA.9 Therefore, the ease of hydrogen atom abstraction from 5,15-diHpETE could enhance the rate of either LXA4 or LXB4 biosynthesis.
This last biosynthetic pathway is supported by fact that the LX intermediate, 5,15-diHpETE, is observed in a variety of biological samples. One of the earliest observations of 5,15-diHpETE was in activated porcine leukocytes, when either AA or 15-HpETE were added as precursors.10 5,15-diHpETE was also observed with isolated porcine 5-LOX, which reacted with 15-HpETE, at a 30% rate of that with AA.11 5,15-diHpETE could then be further modified to LXB4 with purified rabbit reticulocyte 15-LOX-1 (r15-LOX-1), presumably due to an epoxide intermediate.12 r15-LOX-1 could also convert 15-HETE to LXB4, which was due to double oxygenation at C5 and C15, as evidenced by labeled O2 being found on both carbons.12, 13 Porcine leukocyte 12-LOX also converts 5,15-diHpETE to LXB4, which was deduced by the observation of four isomers of LXB4, due to the non-enzymatic hydrolysis of 5-OH,14,15-LTA4.14 LXB4 was also produced by incubation of 15-HpETE with human leukocytes, with labeled O2 incorporating into only C5, and not C14, indicating the presence of a hydrolyzed 14,15-epoxide.15 5,15-diHpETE can also be generated by the addition of AA to human eosinophils, where both 15-LOX-1 and 5-LOX are present.16
Previously, we have shown that human epithelial 15-LOX-2 does not react with 5,15-diHpETE and thus it is not implicated in producing LXs though this intermediate.9 In the present study, we expand this investigation into the biosynthetic pathway of LXs generation from 5,15-diHpETE by comparing the relative activities of human 5-LOX, human platelet 12-LOX and human reticulocyte 15-LOX-1. These results will help establish a better understanding of the relative roles of these three LOX isozyme in the biosynthesis of LXs from 5,15-diHpETE.
Methods
Chemicals.
The lipid mass spectrometry standards, 5S,15S-dihydroxy-6E,8Z,11Z,13E-eicosatetraenoic acid (5,15-diHETE), 5S,6R,15S-trihydroxy-7E,9E,11Z,13E-eicosatetraenoic acid (LipoxinA4 (LXA4)), and 5S, 14R,15S-trihydroxy-6E,8Z,10E,12E-eicosatetraenoic acid (LipoxinB4 (LXB4)), were purchased from Cayman Chemical. Arachidonic acid (AA) was purchased from Nu Chek Prep, Inc. and used to synthesize 5S-hydroxy-6E,8Z,11Z,14Z-eicosatetraenoic acid (5-HETE), 13S-hydrperoxy-9Z,11E-octadecadienoic acid (13-HPODE), 5S,15S-hydroxy-6E,8Z,11Z,13E-eicosatetraenoic acid (5,15-diHETE), 5S-hydro-15S-peroxy-6E,8Z,11Z,13E-eicosatetraenoic acid (5OH,15-HpETE), and 5S,15S-dihydroperoxy-6E,8Z,11Z,13E-eicosatetraenoic acid (5,15-diHpETE). Synthesis of 5-HETE, 13HPODE, and 5,15-diHpETE were performed as previously described with 5-LOX and AA, 15-LOX-1 and LA, 15-HPETE and 5-LOX, respectively.9, 17,18 The synthesis of 5,15-diHETE was performed as done for 5,15-diHpETE, with the exception that trimethylphosphite was added in 2:1 molar excess as a reductant prior to the HPLC purification. 5OH,15-HpETE was synthesized from 5-HETE as follows. 60μM of 5-HETE was reacted in 1 L of 100mM NaBorate, pH 9.2 and 4 mg of lipoxygenase from soybean (Sigma). This reaction was monitored at 254 nm to completion, quenched with 1% (v/v) glacial acetic acid, extracted with 1 L of DCM, and evaporated to dryness. The 5OH,15-HpETE was then purified isocratically via high performance liquid chromatography (HPLC) on a Higgins Haisil Semi-preparative (5μm, 250mm x 10mm) C18 column with 45:55 of 99.9% acetonitrile, 0.1% acetic acid and 99.9% water, 0.1% acetic acid. Purity was assessed via liquid chromatography-mass spectrometry to be greater than 90%.
Expression and Purification of 15-LOX-1, 12-LOX, and 5-LOX.
Overexpression and purification of wild-type human reticulocyte 15-lipoxygenase-1 (15-LOX-1), platelet 12-lipoxygenase (12-LOX), and 5-lipoxygenase (5-LOX) were performed as previously described.17, 19, 20 The purity of 15-LOX-1 and 12-LOX were assessed by SDS gel to be greater than 85%, and metal content was assessed on a Finnigan inductively-coupled plasma-mass spectrometer (ICP-MS), via comparison with iron standard solution. Cobalt-EDTA was used as an internal standard. 5-LOX was isolated as an impure ammonium sulfate precipitant from E. coli expression due to its dramatic loss of activity upon isolation.
Product Analysis of LOX isozyme reactions with 5,15-diHpETE, 5-OH,15-HpETE, and 5,15-diHETE.
15-LOX-1 (120 nM), 12-LOX (250 nM), or 5-LOX (25mg of ammonium sulfate precipitated protein) was reacted in 12 mL of 25 mM HEPES, pH 7.5, at ambient temperature, with 10 μM of oxylipin (5,15-diHpETE, 5OH,15-HpETE, or 5,15-diHETE) for one hour and quenched with 0.5% glacial acetic acid and 5 μM of 7-hydroxy-docosahexaenoic acid as an extraction standard. 3 μM of 13-HPODE was added to the reactions with 5,15-diHETE to activate iron as described in.21 Each quenched reactions was extracted with 12 mL of DCM, resuspended in 1 mL methanol and split 2:1, with the 30% portion being reduced with trimethylphosphite, while the other 60% was left unreduced. The samples were then evaporated under a stream of N2 to dryness, reconstituted in 50 μL of methanol, and further diluted with 50 μL of 0.1% formic acid in water prior to UPLC-MS/MS analysis. Control reactions without enzymes were also conducted and used for background subtraction, ensuring oxylipin degradation products were removed from analysis. Chromatographic separation was performed on a Dionex UltiMate 3000 UHPLC with a C18 column (Phenomenex Kinetex, 1.7 µm, 150mm x 2.1mm). The autosampler was held at 4° C and injection volume was 90 uL. Mobile phase A consisted of water with 0.1% (v/v) formic acid and mobile phase B was acetonitrile with 0.1% formic acid. Flow rate was 0.400 mL/min. The initial condition (30% B) was maintained for 2.3 minutes. Mobile phase B was then ramped to 65% over 28.7 minutes, held at 65% for 1 minute, ramped to 100% over 0.1 min, held at 100% for 7 minutes, and finally returned to 30% to equilibrate for 7 minutes. The chromatography system was coupled to a Dionex Ultimate-3000 DAD and Velos Pro linear ion trap (Thermo Scientific) for UV and mass spectrum analysis. Analytes were ionized via heated electrospray ionization with −4.0 kV spray voltage, 60, 10, and 0 arbitrary units for sheath, auxiliary and sweep gas, respectively. The RF amplitude of the S-Lens was 49%, and the probe and capillary temperatures were 50° C and 380° C, respectively. All analyses were performed in negative ionization mode at Normal resolution setting. MS2 was performed at 35% normalized collision energy in a targeted manner with a mass list containing the following m/z ratios ± 0.1: 319.2, 335.2, 343.2, 351.2, 367.2, 383.2, and 399.2. Products were identified by matching retention times, UV spectra and fragmentation patterns to known standards. Or in the cases where MS standards were not available, structures were deduced from comparison to known and theoretical fragments.
Steady State Kinetics of 5-LOX, 12-LOX and 15-LOX-1 with 5,15-diHpETE, 12-LOX with 5,15-diHETE.
15-LOX-1 and 5-LOX reactions were performed, at ambient temperature, in a 1 cm2 quartz cuvette containing 2 mL of 25 mM HEPES, pH 7.5 with substrate (AA, 15HpETE, or 5,15-diHpETE). 12-LOX reactions were also performed in a 1 cm2 quartz cuvette containing 2 mL of 25 mM HEPES with substrate (AA, 5,15-diHpETE, or 5,15-diHETE), and the pH was 8.0. Additionally, 12-LOX reactions with 5,15-diHETE contained 3 μM 13-HPODE and 5-LOX reactions contained 200 µM ATP. AA concentrations were varied from 0–29 μM, 15-HpETE concentrations were varied from 0–80 µM, 5,15-diHpETE concentrations were varied from 0–45 μM, and 5,15-diHETE concentrations were varied from 0–130 μM. 5-LOX reactions were compared using 10 µM substrate for AA, 15-HpETE, and 5,15-diHpETE with product formation per time analyzed on UV-Vis as well as LC/MS. Concentration of AA was determined by measuring the amount of 15-HpETE produced from complete reaction with soybean lipoxygenase-1 (sLO-1). Concentrations of 5,15-diHpETE and 5,15-diHETE were determined by absorbance at 247 nm. Reactions were initiated by the addition 15-LOX-1 (120 nM) or 12-LOX (250 nM) or 5-LOX (approx. 200 nM of crude AS preparation) and were monitored on a Perkin-Elmer Lambda 45 UV/VIS spectrophotometer. Product formation was determined by the change in absorbance at 234 nm for 15-HpETE/5HpETE (ε234nm = 25,000 M−1 cm−1), 5,15-diHpETE (ε254nm = 21,90000 M−1 cm−1),9 and 302 nm for lipoxins (ε234nm = 50,000 M−1 cm−1). KaleidaGraph (Synergy) was used to fit initial rates (at less than 20% turnover), as well as the second order derivatives (kcat/KM) to the Michaelis-Menten equation for the calculation of kinetic parameters.
5,15-diHpETE and LXB4 titration into human platelets.
The University of Michigan Institutional Review Board approved all research involving human volunteers. Washed human platelets (250 µL at 3.0 ×108 platelets/mL) were dispensed into glass cuvettes and incubated with 0–10 µM of either LXB4 or 5,15-diHpETE for 10 minutes at 37°C. Oxylipin-treated platelets were then stimulated with 0.25 µg/mL of collagen (Chrono-log), stirring at 1100 rpm and 37°C, in a Chrono-log Model 700D lumi-aggregometer and platelet aggregation was recorded for six minutes.
In order to determine the biosynthetic products of 5,15-diHpETE ex vivo, platelets were isolated from human whole blood via serial centrifugation and adjusted to 3.0 × 108 platelets/mL in Tyrode’s buffer (10 mM HEPES, 12 mM NaHCO3, 127 mM NaCl, 5 mM KCl, 0.5 mM NaH2PO4, 1 mM MgCl2, and 5 mM glucose), as previously published.22 One ml of platelets was incubated with varying concentrations of 5,15-diHpETE (0–20 µM) or vehicle (DMSO) for 10 minutes at 37° C, and then the platelets were pelleted by centrifugation at 1000 x g for 1 minute. The supernatant was transferred to a fresh tube and snap frozen. Oxylipins were extracted and analyzed via UPLC-MS/MS as described previously,9 with the following exceptions. The m/z transitions for 5,15-diHETE, LTB4-d4, RvD2-d5, and LipoxinB4 were 335.2→115, 339.2→197, 380.5→141, and 351.2→221, respectively.
Results and Discussion
Lipoxin synthesis from 5,15-diHpETE, 5-OH,15-HpETE, and 5,15-diHETE.
To study the conversion of 5,15-diHpETE to LX through product identification, 5,15-diHpETE was reacted with human 5-LOX, 12-LOX, and 15-LOX-1. For the reaction of 5-LOX with 5,15-diHpETE, no activity was detected at 302 nm (the absorbance of conjugated lipoxins) and LXA4 and LXB4 were both undetected via LC-MS. This data indicates that 5,15-diHpETE is a poor substrate for 5-LOX in vitro. In order to determine the relative turnover of 5,15-diHpETE with 5-LOX, the rate of AA, 15-HpETE and 5,15-diHpETE were compared under the same reaction conditions. 5-LOX converted AA to 5-HpETE at 5 ng/sec and 15-HpETE to 5,15-diHpETE at 0.5 ng/sec, but there was no conversion of 5,15-diHpETE to LXA4 observed (the detection limit of the experiment is 0.0003 ng/sec). This data indicates that the oxidation reaction of 5-LOX with 5,15-diHpETE is more than 1700-fold slower than that with 15-HpETE. This was unexpected because 5-LOX abstracts the C10 hydrogen atom from 5-HpETE to generate leukotriene A4, (LTA4) which is the same hydrogen atom presented by 5,15-diHpETE. In addition, the hydrogen atom on C10 for 5,15-diHpETE is more reactive than that of 15-HpETE, due to conjugation,9 making this lack of reactivity even more noteworthy. However, 5-LOX is able to convert 15-HpETE into LXA4 at a rate of 0.5 ng/sec. This difference in reactivity of 5,15-diHpETE versus 15-HpETE is most likely due to a difference in the rate of substrate capture, as defined by Northrop et al.23, 24 Previously, we have shown that the kcat for generating LTA4 is the same when starting with arachidonic acid (AA) or 5-HpETE. However, the kcat/KM is 10-fold slower for 5-HpETE than AA, due to a slow binding step.17 We propose that 5-LOX’s lack of reactivity with 5,15-diHpETE is due to an even slower binding step for 5,15-diHpETE relative to 5-HpETE, limiting reactivity completely. We therefore propose that 5-LOX produces LXA4 from 15-HpETE by generating 5,15-diHpETE in situ and subsequently generates LXA4 without releasing the 5,15-diHpETE intermediate, similar to the mechanism for LTA4 synthesis.
In contrast, both 12-LOX and 15-LOX-1 reacted with 5,15-diHpETE, manifesting observable rates at 302 nm, which is consistent with LXs being formed. LC-MS analysis of the reduced products confirmed that both 12-LOX and 15-LOX-1 made LXB4 (RT = 8.8 min), but not LXA4 (Figure 2, dashed traces). We also observe LXB4 isoforms, with the all-trans-LXB4 (RT = 8.1 min), 14S-LXB4-epimer (RT = 8.9 min), and the 14S-all-trans-LXB4-epimer (RT = 8.2 min). These products are synthesized by the opening of the epoxide via dehydration, as previously observed with purified rabbit 15-LOX-1 and 15-HpETE as the substrate.12 However, some of these products could also be formed by the competing reaction, oxygenation. In order to differentiate these two reaction mechanisms, the reaction products were analyzed without reductant added during product extraction (Figure 2, solid traces). Both dehydration (i.e. epoxide formation) and oxygenation require abstraction at C10, with migration of the radical to C14. Dehydration occurs upon reaction of the radical with the hydroperoxide in an anti-periplanar orientation, while oxygenation occurs with antarafacial attack of the radical by molecular oxygen. Both reactions require the active site ferrous-water species to reduce the radical intermediate and form the final product. Comparing reduced and un-reduced products can contribute to differentiating the two sets of products, hydroperoxides and alcohols, produced by oxygenation and dehydration, respectively. Analysis of the data clearly demonstrates that hydroperoxides are present, due to the fact that many of the peaks migrated to shorter retention times upon reduction. Specifically, the tri-hydroperoxy species (5,14,15-triHpETE, RT = 13.6 min, Figure 2) is observed, which could only be formed via oxygenation at C14. It should be noted that while we observe the parent mass of 5,14,15-triHpETE (399 m/z, hydrogen loss in the negative mode), we also observe the neutral loss of water from 5,14,15-triHpETE (Supplemental S2), resulting in the most intense ion for 5,14,15-triHpETE at 381 m/z. Additionally, because there are three hydroperoxide moieties, multiple peaks that correspond to the loss of up to three waters upon ionization (Supplemental S1 & S2) are observed. The additional hydroperoxide peaks correspond to both oxygenation products, which have lost an oxygen, most likely due to reduction during the long reaction time in buffer. These products include: 5-OOH,14-OOH,15-OOH-triHpETE (RT = 13.7 min), 5-OOH,14-OH,15-OOH-triHETE, 5-OH,14-OOH,15-OOH-triHETE, 5-OH,14-OH,15-OOH-triHETE, 5-OOH,14-OOH,15-OH-triHETE, and 5-OH,14-OOH,15-OH-triHETE. These products exhibit multiple peaks, which most likely correspond not only to the hydroperoxide position in these molecules, but also to the all-trans, S-epimers, and other isomers resulting from the non-enzymatic epoxide opening at C14 and C15. The non-enzymatic epoxide opening is also supported by comparing the ratio of the sum of all-trans-LXB4, 14S-LXB4 and 14S-all-trans-LXB4 to LXB4 in the reduced samples of 5,15-diHpETE (LxB4 isomers and epimers/LxB4 = 0.91 ± 0.2) and 5,15-diHETE (LxB4 isomers and epimers = 0.19 ± 0.02) as substrates with 12-LOX. The dehydration and oxygenation peaks have been quantified, demonstrating no preference for dehydration over oxygenation, with 12-LOX having a dehydration/oxygenation ratio of 1.3 ± 0.2 and 15-LOX-1 having a ratio of 1.2 ± 0.2 (Figure 2, supplemental S2). Since only LXB4 is produced by 12-LOX and 15-LOX-1 from 5,15-diHpETE, with it being generated either through oxygenation or dehydration (i.e. via the epoxide), the data indicates similar LX reactivities for both LOX isozymes. It should be noted that due to the reactive nature of these di- and tri-hydroperoxides, the data was scanned for Hock rearrangement decomposition, however, no evidence was observed for these products.25
Figure 2: UPLC-UV-Vis chromatogram of h12-LOX (A) and h15-LOX-1 (B) products from 5,15-diHpETE.
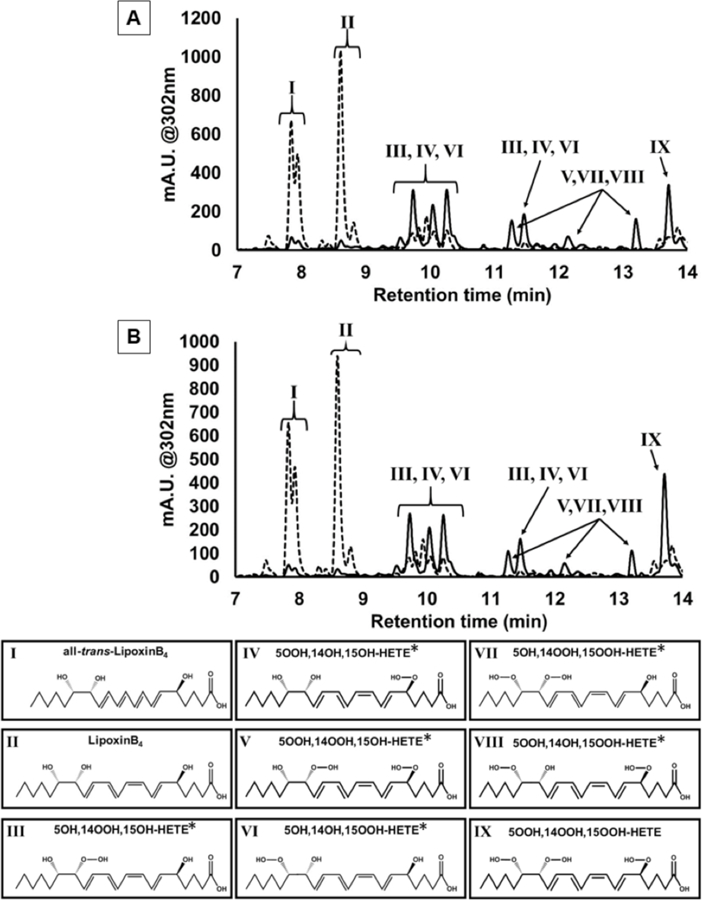
The wavelength of 302 nm is the absorbance maximum for lipoxins (Supplemental S1). At 302 nm, 11 peaks are observed in the unreduced (solid line) reaction that coalesce to 2 clusters of peaks upon reduction (dashed line). Based on the UV-Vis spectrum, retention times, parent masses, and MS/MS spectra (Supplemental S2), the identities of these peaks have been determined to be all-trans-LxB4 and its 14S-epimer at 7.9 min, LxB4 and its 14S-epimer at 8.6 min and 5,14,15-trihydroperoxy-LxB4 at 13.6 min. The peaks at 9.5, 9.8, 10.0, 10.3 and 11.5 min are the monohydroperoxide products and the peaks at 11.3, 12.2, and 13.2 min are the dihydroperoxide products. *Note: although only one double bond stereochemistry is depicted for these compounds, the differing retention times suggest rearrangements. MS/MS spectra contain fragments that indicate the hydroperoxide moiety’s position may differ between these mono- and dihydroperoxy-HETEs.
To eliminate the possibility of epoxide generation, 12-LOX and 15-LOX-1 were reacted with 5,15-diHETE, the reduced form of 5,15-diHpETE, which is incapable of forming epoxides. 12-LOX reacted with 5,15-diHETE producing LxB4 and its isoforms, but 13-HPODE was required. 13-HPODE is a known oxidant of lipoxygenase that reduces lag times and expands the substrate range of lipoxygenases by activating the LOX isozyme when the endogenous hydroperoxide product cannot oxidize the intermediate ferrous ion.18, 26 For this particular reaction, the conversion of 5,15-diHETE to LXB4 implies that 12-LOX oxygenates at C14 because the 14,15-epoxide cannot be formed from the alcohol. The requirement of 13-HpODE is because the ferrous-water intermediate cannot efficiently reduce the hydroperoxide radical at C14, thus releasing the radical intermediate and generating the inactive ferrous-water species. The ferrous-water intermediate is subsequently oxidized by 13-HpODE, generating the active form of 12-LOX. This conclusion is supported by the comparison of the reduced and un-reduced products (Figure 3). The un-reduced reaction manifests a large peak (RT = 9.8 min), which elutes after LxB4 (RT = 8.8 min). This peak has an MS/MS fragmentation pattern of a C14 hydroperoxy moiety (supplemental S3), which becomes LxB4 upon reduction, as seen by its change in retention time and its MS/MS fragmentation (supplemental S3) and confirms an oxygenation reaction on C14. It should be noted that we observe a large reduction in the number of isoforms of LxB4, as compared to the reaction with 5,15-diHpETE as the substrate. This is appropriate given that this is a region- and stereo-specific oxygenation reaction and no epoxide was formed that could undergo non-enzymatic hydrolysis to generate the LxB4 isoforms.
Figure 3: UPLC-UV-Vis chromatogram of h12-LOX products from 5,15-diHETE.
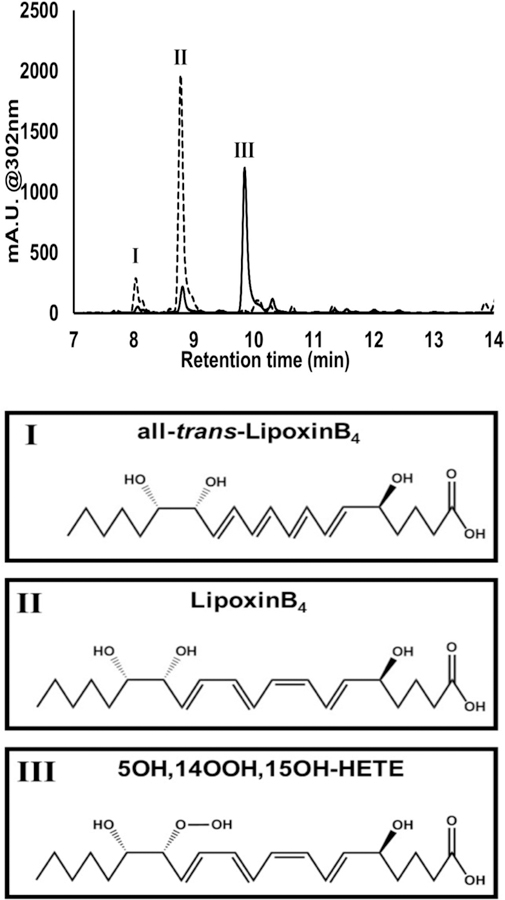
The wavelength of 302 nm is the absorbance maximum for lipoxins (Supplemental S1). At this wavelength, three peaks, at times 8.1, 8.8, and 9.8 min, are observed in the unreduced reaction (solid line) that coalesce to two peaks, 8.1 and 8.8 min, upon reduction (dashed line). Based on the UV-Vis spectrum, retention times, parent mass, and MS/MS spectra (Supplemental S2), the identities of these peaks have been determined to be all-trans-LxB4 at 8.1, LxB4 at 8.8 and 14-hydroperoxy-LxB4 at 9.8 min.
Interestingly, 15-LOX-1 cannot perform an oxygenation reaction with 5,15-diHETE, even with 13-HPODE present, indicating that 15-LOX-1 cannot oxygenate at C14 with 5,15-diHETE as the substrate. This is unexpected since 15-LOX-1 can oxygenate 5,15-diHpETE. Therefore, the hydroxides at C5 and C15 appear to change substrate binding so that either the hydrogen atom abstraction at C10 is prohibited or the oxidation of the active site ferrous-water intermediate to the active ferric-hydroxide moiety by the hydroperoxyl radical is prohibited. This hypothesis is supported by the fact that hydroperoxyl moieties in these eicosanoids behave more hydrophobically than hydroxyl moieties, as evident by their longer retention on a C18 column (Supplemental S1). In addition, 15-LOX-1 will not oxygenate 15-HETE either, but it will oxygenate 15-HpETE, indicating a difference in reactivity with the reduced oxylipin.
It should be reiterated that 15-LOX-1 and 12-LOX do not make LXA4 from 5,15-diHpETE, however, they do make LXB4. This is an interesting observation considering that the radical intermediate of 5,15-diHpETE can potentially form either the 5,6- or 14,15-epoxide. However, the active sites of both 12-LOX and 15-LOX-1 preferentially generate only the 14,15-epoxide. This restricted reactivity could be related to the fact that the oxygen tunnel for both 15-LOX-1 and 12-LOX is located near C14, as observed by the C14 oxygenation products.27, 28 Therefore, the opening to the oxygen tunnel in the active site could provide a dual function. Primarily to oxygenate 5,15-diHpETE but also to provide the space for the 14,15-epoxide to form, thus giving the oxygen tunnel an added functionality.
Steady-state kinetics of 15-LOX-1 and 12-LOX with 5,15-diHpETE and 12-LOX with 5,15-diHETE.
Considering that hydrogen atom abstraction is typically rate-limiting in lipoxygenase reactions,29–31 the steady-state kinetics of 15-LOX-1 and 12-LOX with 5,15-diHpETE revealed an interesting difference between their relative rates. When reacted with AA, 12-LOX abstracts the hydrogen atom at C10, generating more than 95% 12-HpETE.19 On the other hand, 15-LOX-1 abstracts the hydrogen atom at C10 only ≈10% of the time, with C13 being the major target.32–34 We confirmed this result by observing approximately 10% of 12-HpETE and 90% of 15-HpETE being generated from AA reacted with 15-LOX-1. Therefore, 12-LOX has a 78-fold larger kcat/KM than 15-LOX-1 for abstracting the hydrogen atom from C10 of AA and generating 12-HpETE, 18 μM−1s−1 and 0.23 μM−1s−1, respectively (Table 1). It should be noted that these 15-LOX-1 kinetic values for C10 abstraction are estimated from the total rate observed for 15-LOX-1 and its product percentages. This indicates that 12-LOX is more efficient in both product release (kcat) and substrate capture (kcat/KM) than 15-LOX-1 in abstracting a hydrogen atom from C10 on AA (13-fold for kcat and 78-fold for kcat/KM). However, despite the preference of 12-LOX for abstracting at C10 on AA, 15-LOX-1 has both a better product release rate (kcat) and substrate capture rate (kcat/KM) than 12-LOX, for abstracting the C10 hydrogen atom from 5,15-diHpETE (Table 1). For kcat, the 12-LOX rate for C10 abstraction from 5,15-diHpETE is 106-fold less than that of AA (kcat = 0.17 s−1), while the 15-LOX-1 rate is 3.3-fold greater than that of C10 abstraction from AA (kcat = 4.6 s−1). For kcat/KM, the 12-LOX rate for C10 abstraction from 5,15-diHpETE is 1600-fold less than that of AA (kcat/KM = 0.011 μM−1s−1), while the kcat/KM for 15-LOX-1 is the same as that of C10 abstraction from AA (kcat/KM = 0.21 μM−1s−1). Effectively, there is no loss of activity for 15-LOX-1 abstracting the hydrogen atom from C10 of 5,15-diHpETE relative to AA, but 12-LOX has a large loss of activity with 5,15-diHpETE relative to AA, as seen by a significant decrease in kcat and kcat/KM (Table 1). This data indicates that the hydroperoxy moieties on C5 and C15 of 5,15-diHpETE, disrupt its positioning in the 12-LOX active site for C10 abstraction significantly, while not affecting 15-LOX-1 appreciably. It should be noted that for both 12-LOX and 15-LOX-1, only 10% of the total 5,15-diHpETE in the reaction is converted to LXB4 and addition of 12-LOX, 15-LOX-1 or 5,15-diHpETE at 10% conversion did not improve the turnover percentage. This implies that there is some form of suicide product inhibition, which is a common observation in LOX reactions, due to the reactive nature of the radical intermediates, and has previously been observed with 12-LOX and LTA4.35, 36
Table 1:
Steady State Parameters for 15-LOX-1 and 12-LOX reaction with 5,15-diHpETE and 5,15-diHETE
| Enzyme | Substrate | kcat (s−1) | Km (µM) | kcat/Km(µM−1s−1) |
|---|---|---|---|---|
| 12-LOX | AA (C10 abstraction) | 18 ± 0.8 | 1.0 ± 0.1 | 18±2 |
| 15-LOX-1 | AA (C10 abstraction) | 1.4 ± 0.1* | 6 ± 1 | 0.23 ± 0.03* |
| 12-LOX | 5,15-diHpETE | 0.17 ± 0.007 | 14 ± 1 | 0.011 ± 0.007 |
| 15-LOX-1 | 5,15-diHpETE | 4.6 + 0.1 | 23 ± 2 | 0.21 ± 0.01 |
| 12-LOX | 5,15-diHETE** | 0.18 ± 0.02 | 113 ± 19 | 0.0015 ± 0.0001 |
| 15-LOX-1 | 5,15-diHETE** | nr*** | nr | nr |
kcat and kcat/Km values were confirmed daily relative to the kcat and kcat/Km values of AA. The C10 abstraction kinetic values of 15-LOX-1 are 10% of the AA kinetic values, which include both the 15-HpETE and 12-HpETE kinetics (kcat and kcat/Km for AA are 14 ± 1s−1 and 2.3 ± 0.3µM−1 s−1, respectively)
3µM of 13-HPODE is added to activate 12-LOX.
nr = no reaction
As noted above, LX formation from the reduced intermediate, 5,15-diHETE, is only observed with 12-LOX in the presence of 13-HPODE, and no rate was observed for 15-LOX-1. However, the rate of 12-LOX with 5,15-diHETE is extremely slow, manifesting a kcat/KM that is 10-fold less than that seen for 5,15-diHpETE (kcat/KM = 0.0015 μM−1s−1). Since the kcat of 12-LOX with 5,15-diHETE is essentially the same as its kcat for 5,15-diHpETE, this lower rate of substrate capture (kcat/KM) is manifested in a 10-fold increase in KM, (113 ± 19 μM). As discussed above, 12-LOX only oxygenates 5,15-diHETE but it oxygenates and dehydrates 5,15-diHpETE. Since the kcat for both 5,15-diHETE and 5,15-diHpETE are similar, it indicates that the rate of product release for both oxygenation and dehydration are also similar. This implies that the reaction steps after the irreversible hydrogen abstraction step, such as the reduction of the radical intermediate for oxygenation or dehydration by the ferrous-water moiety, are either not rate-limiting or comparable in rate for the two reactions. It is also observed that the rate of substrate capture (kcat/KM)23 for 12-LOX is decreased by a factor of 10 for 5,15-HETE relative to 5,15-HpETE, indicating that there is a rate difference in a reaction step prior to the irreversible hydrogen atom abstraction step, such as substrate binding. As mentioned above, this could be due to its smaller size or the more polar hydroxyl moiety relative to the hydroperoxide, as seen by its RP-HPLC retention time.
5,15-diHpETE and LXB4 titration into human platelets.
To determine whether or not 5,15-diHpETE inhibits platelet activation, platelets from 11 separate donors were treated with varying concentrations of 5,15-diHpETE (0.5–20 μM) or vehicle control (DMSO) prior to stimulation with collagen, a GPVI and α2β1 agonist. Compared to vehicle control, 5,15-diHpETE inhibited platelet aggregation in response to collagen (0.25 µg/mL) stimulation with an IC50 of 1.3 μM (Figure 4). Furthermore, over 90% reduction in collagen-mediated platelet aggregation was observed in platelets treated with a concentration greater than 10 μM of 5,15-diHpETE. The antiplatelet effects of LXB4 were also investigated; however, in contrast to 5,15-diHpETE, LXB4 did not significantly inhibit collagen-mediated platelet activation at the concentrations tested.
Figure 4: 5,15-diHpETE, but not LXB4, inhibits collagen-mediated platelet aggregation.
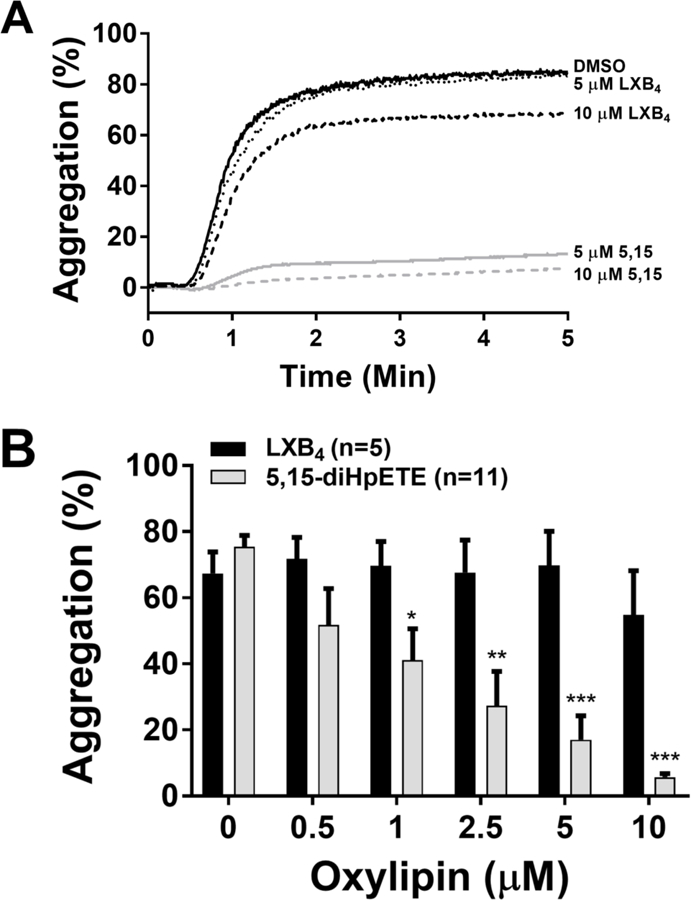
Isolated human platelets were treated with increasing concentrations of either LXB4 or 5,15-diHpETE for ten minutes and then stimulated with collagen (0.25 µg/mL). A) Representative tracings of 5,15-diHpETE or LXB4 treated platelets stimulated with collagen. B) Data represents mean ± S.E.M. Statistical analysis was performed comparing oxylipin concentration to vehicle control using one-way ANOVA with Dunnett’s multiple comparison test. *P<0.05, **P<0.01, ***P<0.001.
With respect to the biosynthetic pathway, human platelets were treated with 5,15-diHpETE and shown to produce LXB4 (Figure 5), but they did not produce LXA4. This is consistent with the in vitro results, where purified 12-LOX only makes LXB4. However, the amount of LXB4 that we observe from 5,15-diHpETE treated platelets is very small compared to the unreacted 5,15-diHpETE (less than 1.2 %), indicating that this is not an efficient pathway for the generation of LXB4. In addition, since LXB4 does not have a strong antiplatelet effect, it is reasonable to conclude that 5,15-diHpETE, and not its downstream product, LXA4, is the active oxylipin species that affects platelet biology in our experiments.
Figure 5: Platelets produce LxB4 from 5,15-diHPETE.
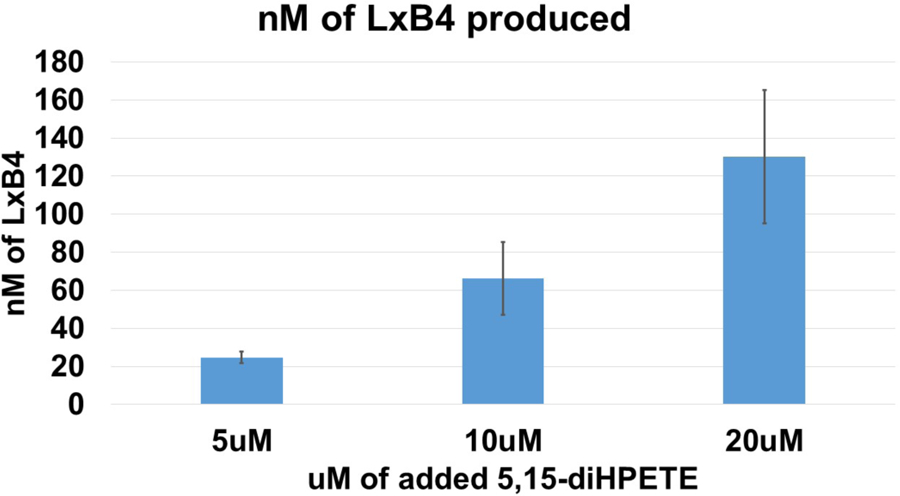
The amount of LxB4 produced by platelets treated with 5, 10 or 20 µM of 5,15-diHPETE are plotted in this histogram. The standard deviation of each value is represented by error bars.
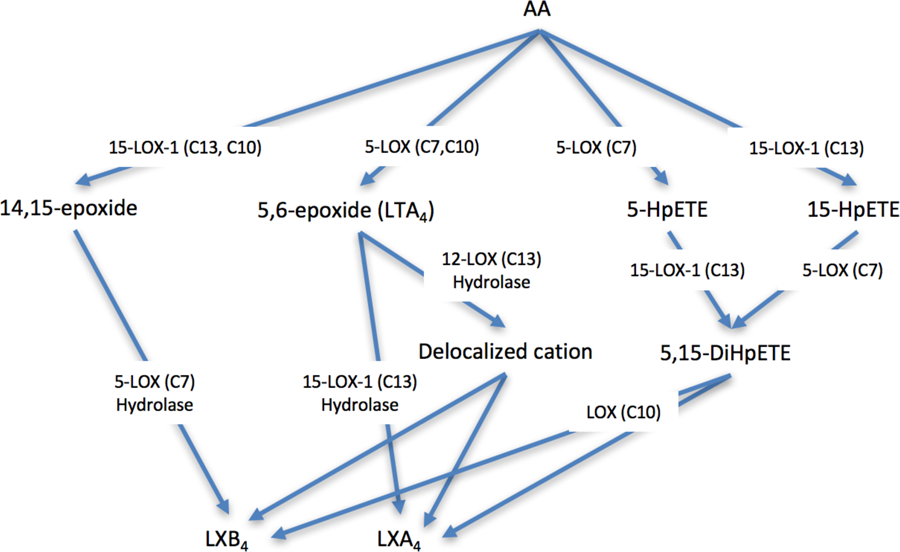
In summary, the reaction of 5,15-diHpETE with various human lipoxygenase isozymes was investigated to determine the relative rates of producing LXs. Previously, it was established that 15-LOX-2 did not react with 5,15-diHpETE, consistent with its enhanced substrate selectivity relative to the other LOX isozymes.9 15-LOX-2 abstracts a hydrogen atom from C13 of AA, but does not abstract from C10, the only available methylene carbon of 5,15-diHpETE. In the current work, it was determined that 5-LOX does not react with 5,15-diHpETE either. However, 5-LOX can produce LXA4 if 15-HpETE is the substrate (Figure 6), indicating that substrate binding is impaired when the hydroperoxide is on both C5 and C15, consistent with our previous work with 5-LOX where oxygenation of the oxylipin affects substrate binding significantly.17 In contrast, both 12-LOX and 15-LOX-1 react with 5,15-diHpETE (Figure 6), albeit at slow rates relative to AA. Surprisingly, 15-LOX-1 has a 27-fold greater kcat and a 20-fold greater kcat/KM than 12-LOX. This result is unusual because C10 is the preferred location for hydrogen abstraction for 12-LOX, so it appears that the active site of 12-LOX is less tolerant of oxygenated fatty acids than 15-LOX-1, which lowers its catalytic rates, as already seen for 5-LOX.17 The low in vitro catalytic rate for 12-LOX with 5,15-diHpETE is corroborated by the fact that when 5,15-diHpETE is added to platelets, LXB4 production is extremely low (1.2 ± 0.2 % total conversion), consistent with the poor in vitro LXB4 production rate. This result is in contrast to the conventional wisdom that assumes 12-LOX is the biosynthetic enzyme for LXs production from 5,15-diHpETE, due to its preference for abstracting a hydrogen atom from C10.8 Nevertheless, LXs are highly active molecules so low production levels in platelets could still have large effects on the cellular response, resulting in 12-LOX still being a potentially biosynthetically relevant LOX isozyme for making LXs. For comparison, Serhan and coworkers determined that human 12-LOX reacted with LTA4, producing both LXA4 and LXB4, with a Vmax of 24.5 nmol/min/mg (or a kcat of 0.031 sec−1) and a Vmax/Km of 3.1 nmol/min/mg/uM (or a kcat/KM of 0.004 sec−1uM−1).35 These values are markedly slower than the current observed rates of 15-LOX-1 with 5,15-diHpETE (150-fold slower for kcat and 50-fold slower for kcat/KM), suggesting that 15-LOX-1 may be a more predominant source of LXs than 12-LOX, depending on the cells involved. Another interesting difference between 5,15-diHpETE and LTA4 as LOX substrates is that with LTA4, both LXA4 and LXB4 are made, but with 5,15-diHpETE as the substrate, only LXB4 is made. Clearly, the mechanisms of LX production are distinct, which could help in the future identification of the source of LX biosynthesis. For example, if 5,15-diHpETE is the substrate, LXB4 is the primary product of 12-LOX and 15-LOX-1 catalysis, whereas LXA4 is the primary product with 5-LOX. However, if LTA4 is the substrate, then 12-LOX generates both LXA4 and LXB4. Therefore, it appears that cells have multiple pathways for making LXs, depending on the available LOX isozyme and the available substrate, 5,15-diHpETE or LTA4. We are currently investigating the molecular mechanism of these reactions with active site mutants in order to gain better insights into the differences between these distinct LX biosynthetic pathways.
Figure 6: Reaction scheme for LXA4 and LXB4 production.
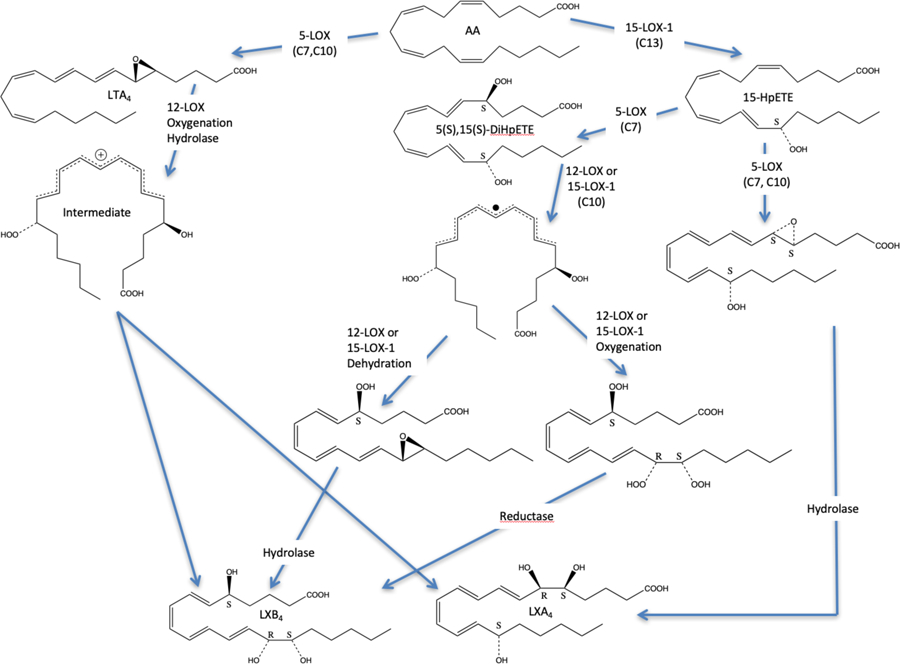
The production of LXA4 and LXB4 is dependent on both the order of LOX reactivity and the nature of the substrate. The carbon atom from which the hydrogen atom is abstracted is indicated below the LOX isozyme label, such as C7, C10 or C13. The central pathway which involves 5(S),15(S)-diHpETE is the subject of this publication.
Supplementary Material
Acknowledgments
FUNDING: NIH R01 GM105671 (MH and TRH) and NIH R01 HL11405 (MH and TRH). NIH F32 HL129491 (BET).
Abbreviations:
- LOX
lipoxygenase
- 15-LOX-1
human reticulocyte 15-lipoxygenase-1
- sLO-1
soybean lipoxygenase-1
- 5-LOX
leukocyte 5-lipoxygenase
- 12-LOX
human platelet 12-lipoxygenase
- GP
glutathione peroxidase
- AA
arachidonic acid
- HETE
hydoxy-eicosatetraenoic acid
- HpETE
hydroperoxy-eicosatetraenoic acid
- diHETEs
dihydroxy-eicosatetraenoic acids
- 5-HETE
5-hydroxy-6E,8Z,11Z,14Z-eicosatetraenoic acid
- 5-HpETE
5-hydro peroxy-6E,8Z,11Z,14Z-eicosatetraenoic acid
- 12-HpETE
12-hydroperoxy-5Z,8Z,11E,14Z-eicosatetraenoic acid
- 15-HpETE
15-hydroperoxy-5Z,8Z,11Z,13E-eicosatetraenoic acid
- 5,15-HETE
5S,15S-dihydroxy-6E,8Z,11Z,13E-eicosatetraenoic acid
- 5,15-diHpETE
5,15-dihydroperoxy-6E,8Z,11Z,13E-eicosatetraenoic acid
- 5,6-diHETE
5S,6R-dihydroxy-7E,9E,11Z,14Z-eicosatetraenoic acid
- LTA4
5S-trans-5,6-oxido-7E,9E,11Z,14Z-eicosatetraenoic acid
- LTB4
5S,12R-dihydroxy-6Z,8E,10E,14Z-eicosatetraenoic acid
- LipoxinA4 (LXA4)
5S,6R,15S-trihydroxy-7E,9E,11Z,13E-eicosatetraenoic acid
- LipoxinB4 (LXB4)
5S,14R,15S-trihydroxy-6E,8Z,10E,12E-eicosatetraenoic acid
Footnotes
Supplemental information. The supplemental figures that are referenced in this article (S1, S2, S3 and S4) along with their corresponding legends are available online. These materials are supplied free of charge at http://pubs.acs.org.
REFERENCES
- [1].Serhan CN, Hamberg M, and Samuelsson B (1984) Trihydroxytetraenes: a novel series of compounds formed from arachidonic acid in human leukocytes, Biochem Biophys Res Commun 118, 943–949. [DOI] [PubMed] [Google Scholar]
- [2].Serhan CN, Hamberg M, and Samuelsson B (1984) Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes, Proc Natl Acad Sci U S A 81, 5335–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Claria J, and Serhan CN (1995) Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions, Proc Natl Acad Sci U S A 92, 9475–9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Serhan CN, Chiang N, and Van Dyke TE (2008) Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators, Nat Rev Immunol 8, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Romano M, Cianci E, Simiele F, and Recchiuti A (2015) Lipoxins and aspirin-triggered lipoxins in resolution of inflammation, Eur J Pharmacol 760, 49–63. [DOI] [PubMed] [Google Scholar]
- [6].Back M, Powell WS, Dahlen SE, Drazen JM, Evans JF, Serhan CN, Shimizu T, Yokomizo T, and Rovati GE (2014) Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7, Br J Pharmacol 171, 3551–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Edenius C, Haeggstrom J, and Lindgren JA (1988) Transcellular conversion of endogenous arachidonic acid to lipoxins in mixed human platelet-granulocyte suspensions, Biochem Biophys Res Commun 157, 801–807. [DOI] [PubMed] [Google Scholar]
- [8].Sheppard KA, Greenberg SM, Funk CD, Romano M, and Serhan CN (1992) Lipoxin generation by human megakaryocyte-induced 12-lipoxygenase, Biochimica et Biophysica Acta 1133, 223–234. [DOI] [PubMed] [Google Scholar]
- [9].Green AR, Barbour S, Horn T, Carlos J, Raskatov JA, and Holman TR (2016) Strict Regiospecificity of Human Epithelial 15-Lipoxygenase-2 Delineates Its Transcellular Synthesis Potential, Biochemistry 55, 2832–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Borgeat P, Picard S, Drapeau J, and Vallerand P (1982) Metabolism of arachidonic acid in leukocytes: isolation of a 5,15-dihydroxy-eicosatetraenoic acid, Lipids 17, 676–681. [DOI] [PubMed] [Google Scholar]
- [11].Ueda N, Kaneko S, Yoshimoto T, and Yamamoto S (1986) Purification of arachidonate 5-lipoxygenase from porcine leukocytes and its reactivity with hydroperoxyeicosatetraenoic acids, J Biol Chem 261, 7982–7988. [PubMed] [Google Scholar]
- [12].Kuhn H, Wiesner R, Alder L, Fitzsimmons BJ, Rokach J, and Brash AR (1987) Formation of lipoxin B by the pure reticulocyte lipoxygenase via sequential oxygenation of the substrate, Eur J Biochem 169, 593–601. [DOI] [PubMed] [Google Scholar]
- [13].Kuhn H, Wiesner R, Lankin VZ, Nekrasov A, Alder L, and Schewe T (1987) Analysis of the stereochemistry of lipoxygenase-derived hydroxypolyenoic fatty acids by means of chiral phase high-pressure liquid chromatography, Anal Biochem 160, 24–34. [DOI] [PubMed] [Google Scholar]
- [14].Ueda N, Yokoyama C, Yamamoto S, Fitzsimmons BJ, Rokach J, Oates JA, and Brash AR (1987) Lipoxin synthesis by arachidonate 12-lipoxygenase purified from porcine leukocytes, Biochem Biophys Res Commun 149, 1063–1069. [DOI] [PubMed] [Google Scholar]
- [15].Serhan CN, Hamberg M, Samuelsson B, Morris J, and Wishka DG (1986) On the stereochemistry and biosynthesis of lipoxin B, Proc Natl Acad Sci U S A 83, 1983–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Morita E, Schroder JM, and Christophers E (1990) Identification of a novel and highly potent eosinophil chemotactic lipid in human eosinophils treated with arachidonic acid, J Immunol 144, 1893–1900. [PubMed] [Google Scholar]
- [17].Smyrniotis CJ, Barbour SR, Xia Z, Hixon MS, and Holman TR (2014) ATP Allosterically Activates the Human 5-Lipoxygenase Molecular Mechanism of Arachidonic Acid and 5(S)-Hydroperoxy-6(E),8(Z),11(Z),14(Z)-eicosatetraenoic Acid, Biochemistry. [DOI] [PMC free article] [PubMed]
- [18].Wecksler AT, Kenyon V, Garcia NK, Deschamps JD, van der Donk WA, and Holman TR (2009) Kinetic and structural investigations of the allosteric site in human epithelial 15-lipoxygenase-2, Biochemistry 48, 8721–8730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ikei KN, Yeung J, Apopa PL, Ceja J, Vesci J, Holman TR, and Holinstat M (2012) Investigations of human platelet-type 12-lipoxygenase: role of lipoxygenase products in platelet activation, J Lipid Res. [DOI] [PMC free article] [PubMed]
- [20].Joshi N, Hoobler EK, Perry S, Diaz G, Fox B, and Holman TR (2013) Kinetic and structural investigations into the allosteric and pH effect on the substrate specificity of human epithelial 15-lipoxygenase-2, Biochemistry 52, 8026–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wecksler AT, Kenyon V, Deschamps JD, and Holman TR (2008) Substrate specificity changes for human reticulocyte and epithelial 15-lipoxygenases reveal allosteric product regulation, Biochemistry 47, 7364–7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tourdot BE, and Holinstat M (2017) Targeting 12-Lipoxygenase as a Potential Novel Antiplatelet Therapy, Trends Pharmacol Sci 38, 1006–1015. [DOI] [PubMed] [Google Scholar]
- [23].Northrop DB (1998) On the meaning of Km and V/K in enzyme kinetics, J. Chem. Ed 75, 1153–1157. [Google Scholar]
- [24].Northrop DB (1999) Rethinking fundamentals of enzyme action, Adv Enzymol Relat Areas Mol Biol 73, 25–55, ix. [DOI] [PubMed] [Google Scholar]
- [25].Gardner HW, and Plattner RD (1984) Linoleate hydroperoxides are cleaved heterolytically into aldehydes by a Lewis acid in aprotic solvent, Lipids 19, 294–299. [DOI] [PubMed] [Google Scholar]
- [26].Kuhn H, Wiesner R, Stender H, Schewe T, Lankin VZ, Nekrasov A, and Rapoport SM (1986) Requirement of monohydroperoxy fatty acids for the oxygenation of 15LS-HETE by reticulocyte lipoxygenase, FEBS Lett 203, 247–252. [DOI] [PubMed] [Google Scholar]
- [27].Knapp MJ, Seebeck FP, and Klinman JP (2001) Steric control of oxygenation regiochemistry in soybean lipoxygenase-1, J Am Chem Soc 123, 2931–2932. [DOI] [PubMed] [Google Scholar]
- [28].Knapp MJ, and Klinman JP (2003) Kinetic Studies of Oxygen Reactivity in Soybean Lipoxygenase-1, Biochemistry 42, 11466–11475. [DOI] [PubMed] [Google Scholar]
- [29].Glickman MH, and Klinman JP (1995) Nature of Rate-Limiting Steps in the Soybean Lipoxygenase-1 Reaction, Biochemistry 34, 14077–14092. [DOI] [PubMed] [Google Scholar]
- [30].Glickman MH, Wiseman JS, and Klinman JP (1994) Extremely Large Isotope Effects in the Soybean Lipoxygense-Linoleic Acid Reaction, J. Am. Chem. Soc 116, 793–794. [Google Scholar]
- [31].Segraves EN, and Holman TR (2003) Kinetic investigations of the rate-limiting step in human 12- and 15-lipoxygenase, Biochemistry 42, 5236–5243. [DOI] [PubMed] [Google Scholar]
- [32].Bryant RW, Bailey JM, Schewe T, and Rapoport SM (1982) Positional specificity of a reticulocyte lipoxygenase. Conversion of arachidonic acid to 15-S-hydroperoxy-eicosatetraenoic acid, J Biol Chem 257, 6050–6055. [PubMed] [Google Scholar]
- [33].Kuhn H, Barnett J, Grunberger D, Baecker P, Chow J, Nguyen B, Bursztynpettegrew H, Chan H, and Sigal E (1993) Overexpression, Purification and Characterization of Human Recombinant 15-Lipoxygenase, Biochim. Biophys. Acta 1169, 80–89. [DOI] [PubMed] [Google Scholar]
- [34].Kuhn H (2000) Structural basis for the positional specificity of lipoxygenases, Prostaglandins Other Lipid Mediat 62, 255–270. [DOI] [PubMed] [Google Scholar]
- [35].Romano M, Chen XS, Takahashi Y, Yamamoto S, Funk CD, and Serhan CN (1993) Lipoxin synthase activity of human platelet 12-lipoxygenase, Biochem J 296 (Pt 1), 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kishimoto K, Nakamura M, Suzuki H, Yoshimoto T, Yamamoto S, Takao T, Shimonishi Y, and Tanabe T (1996) Suicide Inactivation of Porcine Leukocyte 12-Lipoxygenase Associated with Its Incorporation of 15-Hydroperoxy-5,8,11,13-Eicosatetraenoic Acid Derivative, Biochimica et Biophysica Acta - Lipids & Lipid Metabolism 1300, 56–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.