

Abstract
Endoplasmic reticulum stress (ERS), arising from the loss of dynamic balance in endoplasmic reticulum function under stress and inflammation, has been implicated in the progression of sepsis. Multiple organ failure caused by sepsis still has a high mortality rate, of which the heart is one of the more damaged organs. In this research, a rat model of sepsis was set up by cecal ligation and puncture (CLP); serum myocardial enzyme levels were measured using an automated biochemical analyzer, inflammatory cytokine levels were measured by ELISA kit, and cardiac histology and cardiomyocyte apoptosis were measured by hematoxylin and eosin (H&E) staining and Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay to assess the extent of myocardial damage. Western blot was used to detect expression of related proteins. The results showed that serum myocardial enzymes and pro-inflammatory factors were elevated in septic rats, and the increase was most significant in the CLP 24 h group. At the same time, the myocardium of septic rats had a histopathologic abnormality. After CLP, levels of endoplasmic reticulum stress related protein were upregulated. After 12 and 24 hours, the density of apoptotic cells in the myocardium of CLP-treated rats increased significantly, and the expression of apoptosis-related proteins changed significantly. This suggests that the unfolded protein response occurs during sepsis and causes damage to the heart muscle. Endoplasmic reticulum stress-mediated apoptotic signaling pathway is one of the causes of cardiac injury caused by sepsis, and may be a key to clinical prevention of cardiac dysfunction caused by sepsis.
Keywords: Sepsis, endoplasmic reticulum stress, myocardial injury
Introduction
Sepsis is one of the leading causes of death in intensive care patients worldwide, and the in-hospital mortality rate of severe sepsis can be as high as 20-30% [1]. Sepsis can evolve into multiple organ dysfunction syndrome (MODS). As an important organ of the human body, the heart is most vulnerable to damage, leading to a poor prognosis. Studies have shown that, myocardial injury occurs early in sepsis [2], and the degree of injury is positively correlated with mortality. Severe myocardial injury may be an independent risk factor for death in patients with septic shock. Although significant development has been made in understanding the pathology of sepsis in the past decade, little is known about the mechanisms of heart injury caused by sepsis, so it is important to explore the mechanisms of heart injury associated with sepsis.
The endoplasmic reticulum (ER) is a subcellular organelle responsible for facilitating protein folding and assembly, and is involved in several other physiologic activities. Under stress and inflammation, ER loses its functional homeostasis, also called Endoplasmic reticulum stress (ERS). During ERS, the unfolded protein response (UPR) is activated to restore ER function back to normal. However, if the stress is prolonged or beyond the regulatory capacity of UPR, apoptosis is triggered, leading to cell damage, and eventual death. Increasing evidence suggests ER stress to be involved in the pathogenesis of sepsis [3]. Endoplasmic reticulum stress has been confirmed during sepsis. Ma et al. [4] found that the increased rate of spleen lymphocyte apoptosis in septic mice may be related to ER. Qian et al. [5] have shown that ER-related proteins were significantly expressed in the liver of sepsis rats, and inhibition of endoplasmic reticulum stress can reduced liver cell apoptosis. However, it is unclear whether sepsis damages the heart through the endoplasmic reticulum stress-mediated apoptosis signaling pathway.
Therefore, in this study, we used cecal ligation puncture (CLP) to induce a rat model of sepsis, and the role of endoplasmic reticulum stress signaling pathway in sepsis-induced cardiac injury was also studied.
Material and methods
Materials
GRP78, Cleaved Caspase-12 and GAPDH antibodies were from Abcam (Cambridge, MA, USA; no. ab21685, ab62484 and ab181602, respectively). CHOP, Bcl-2, and Bax antibodies were from Proteintech Group, Inc. (Wuhan, China; cat. no. 15204-1-AP, 26593-1-AP, and 60267-1-Ig). TUNEL apoptosis detection kit, was from Roche Applied Science (Rotkreuz, Switzerland; cat. no. 11684817910). Rat TnI and Inflammatory factor ELISA kits were from Elabscience Biotechnology Co., Ltd. (Wuhan, China; cat. no. E-EL-R1253c, E-EL-R0016c, E-EL-R0015c, and E-EL-R0019c).
Animals
Adult male Sprague Dawley rats (weighing 245.15 ± 10.21 g) were from the Animal Center of Xinjiang Medical University (Urumqi, China). Rats were housed in a normal environment, in groups of ten, free to eat and drink ad libitum. Intraperitoneal injection of 10% chloral hydrate (300 mg/kg) was used to anesthetize rats for surgery. All experimental protocols in the present study follow the ARRIVE guidelines and AVMA euthanasia guidelines 2013.
Establishment of sepsis model
Cecal ligation puncture (CLP) was performed in rats to establish an experimental model of sepsis. All rats were allowed to fast for 12 h before surgery, and had free access to water. At first, the rats were anesthetized and fixed. Under sterile conditions, they were cut along the midline of the abdomen to expose the cecum. Immediately, the middle of the cecum was ligated with a thin thread and the end of the cecum was pierced with an 18-gauge needle. A small amount of stomach contents were squeezed out, expressed through the cecum and it was sutured. Cecal puncture was not performed in the sham group, and the remaining steps were the same. Finally, rats were fluid resuscitated with 0.9% sodium chloride saline (subcutaneous injection, 50 ml/kg). After the operation, the rats were returned to their cages and observed regularly.
Experimental grouping
Four groups of animals were used in this research (n = 10 in each group). They were the sham operation group (sham), which only received laparotomy; the sepsis 6-hour group (CLP 6 h) which underwent CLP and was executed at 6 hours; the sepsis 12-hour group (CLP 12 h) which underwent CLP and was executed at 12 hours; the 24-hour sepsis group (CLP 24 h) which underwent CLP and was executed at 24 hours. According to this procedure, all rats could drink and eat freely, and were anesthetized again after 6 hours, 12 hours, and 24 hours, and blood was taken through the rat’s abdominal aorta. After coagulation, the blood samples were centrifuged (3,500×g, 15 minutes) and the clear supernatant was collected. A portion of each serum sample was submitted for testing for cardiac enzymes and inflammatory factors. The remaining samples were stored in liquid nitrogen for subsequent experiments. Heart tissues were obtained after sacrificing rats by cervical dislocation. A portion of myocardial samples was fixed in paraformaldehyde (4%) for histomorphologic analysis and apoptosis detection, and the remainder was used to detect the expression of related proteins by western blot.
Measurement of myocardial enzymes and cardiac TnI (cTnI)
Serum CK-MB and LDH levels were detected by an automatic biochemical analyzer (Modular DPP H7600; Roche Diagnostics, Basel, Switzerland). Serum cTnI levels were measured using an ELISA kit.
Determination of inflammatory cytokines
Serum TNFα, Il-6, and Il-10 levels were measured using an ELISA kit, according to the manufacturer’s instructions.
Histologic analysis
The fixed myocardial tissue was dehydrated with graded alcohols, embedded in paraffin, and cut into 4-μm sections. Then the sections were stained with hematoxylin and eosin (H&E) for approximately 2 min. Finally, the sections were observed under an optical microscope at a magnification of 200×.
Western blot analysis
A total of 10 µl of protein samples (40 mg/ml) were separated using 10% SDS-PAGE, followed by their transfer onto a polyvinylidene fluoride membrane. Membranes were incubated with primary antibodies. The membrane was incubated with the corresponding HRP secondary antibody at 37°C for 2 h. The film was made to react with an enhanced chemiluminescence reagent and thereafter developed using an X-ray film. ImageJ software (Java 1.6.0.24; version 1.51k; National Institutes of Health, Bethesda, MD, USA) was used to quantify the band strength.
Analysis of apoptosis rate
Cardiomyocyte apoptosis by TUNEL Apoptosis Detection Kit: The stained cells were observed under a fluorescence microscope. The apoptotic cells in the tissue section showed green fluorescence under fluorescence microscope while the nucleus appeared blue.
Statistical analysis
All data were analyzed by SPSS 20.0 software (IBM Corp., Armonk, NY, USA). They were expressed as the mean ± SE, and represented at least three independent experiments. Differences between two groups were analyzed using one-way ANOVA, followed by a post hoc analysis. Significance was set at P < 0.05.
Results
Confirmation of the rat model of sepsis
To study the myocardial damage in septic rats, we employed the cecal ligation puncture (CLP) model [14]. Most rats were conscious 2 h after CLP surgery, and gradually showed poor activity, cold, dull, and upright fur at 6 h, and eventually stopped drinking. Four rats developed difficulty in breathing, and reaction time was slow. After the model was generated, the rats were placed in cages and kept under observation. There were 6 observation points: every 2 h after modeling, and 5-min observation each time. The mortality rates in the CLP 12 h and CLP 24 h groups were 20% (n = 10) and 40% (n = 10), respectively, whereas it was 0% in both sham and CLP 6 h groups. Rats in the sham group had normal diet and exercise, and showed no anorexia and vertical fur. Rats in the CLP 6 h group started to develop anorexia and vertical fur, while symptoms in the CLP 12 h group worsened. Rats in the CLP 24 h group performed the worst. Table 1 shows specific mortality rates for each group.
Table 1.
Comparative mortality rates of rats in each group
| Group | Mortality rate, % |
|---|---|
| Sham | 0 |
| CLP 6 h | 0 |
| CLP 12 h | 20 |
| CLP 24 h | 40 |
Heart injury in sepsis
Heart injury occurred after CLP surgery (Figures 1 and 2). As shown in Figure 1, there was no significant difference in serum levels of myocardial enzymes between the sham group and the CLP 6 h group (P > 0.05). Compared with the sham group, the serum myocardial enzymes and troponin levels in the CLP-treated group increased significantly at 12 hours and reached the highest at 24 hours (P < 0.05, Figure 1). Compared with the control group, the levels of serum inflammatory factors (such as TNF-α, II-6, and Il-10) were significantly increased in the group treated with CLP (P < 0.05; Figure 2). The levels of TNF-α and Il-6 in the CLP-treated group reached the highest in 24 hours (P < 0.05). At the same time, the level of IL-10 in the CLP-treated group also increased with time (P < 0.05) (P < 0.05, Figure 2).
Figure 1.
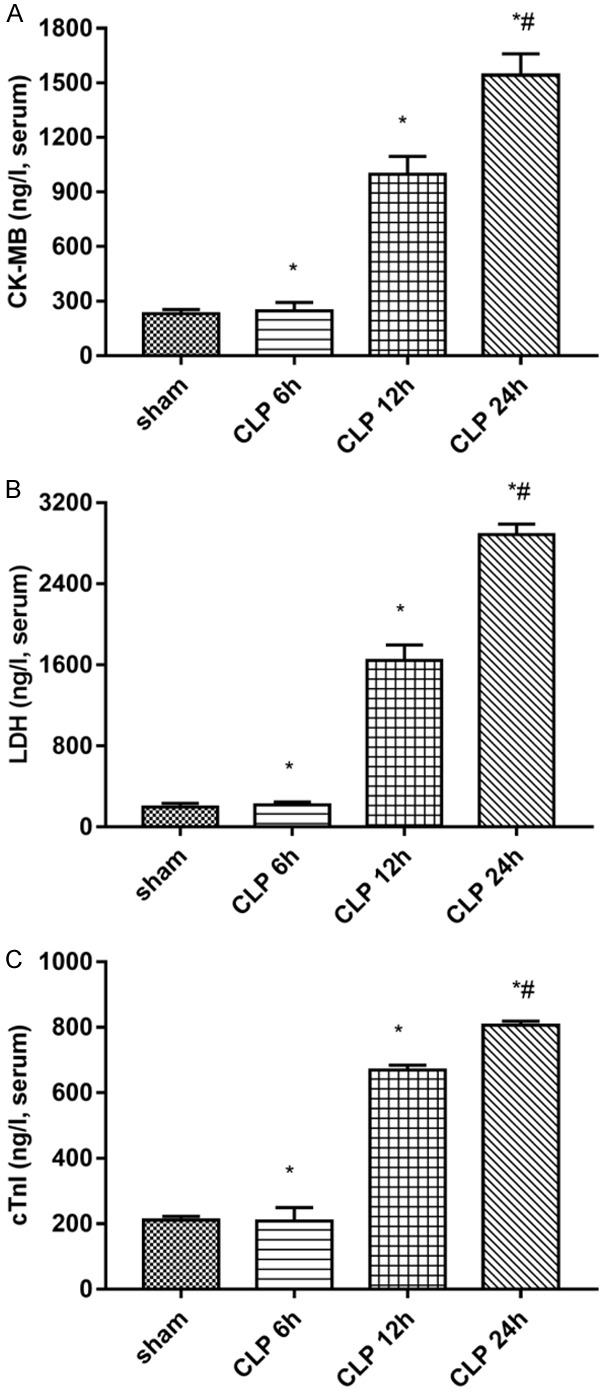
Change in the serum levels of (A) CK-MB, (B) LDH, and (C) cTnI in rats with sepsis. Data are presented as mean ± standard error of the mean. *P < 0.05 vs. sham group; #P < 0.05 vs. CLP group. (CLP = cecal ligation puncture).
Figure 2.
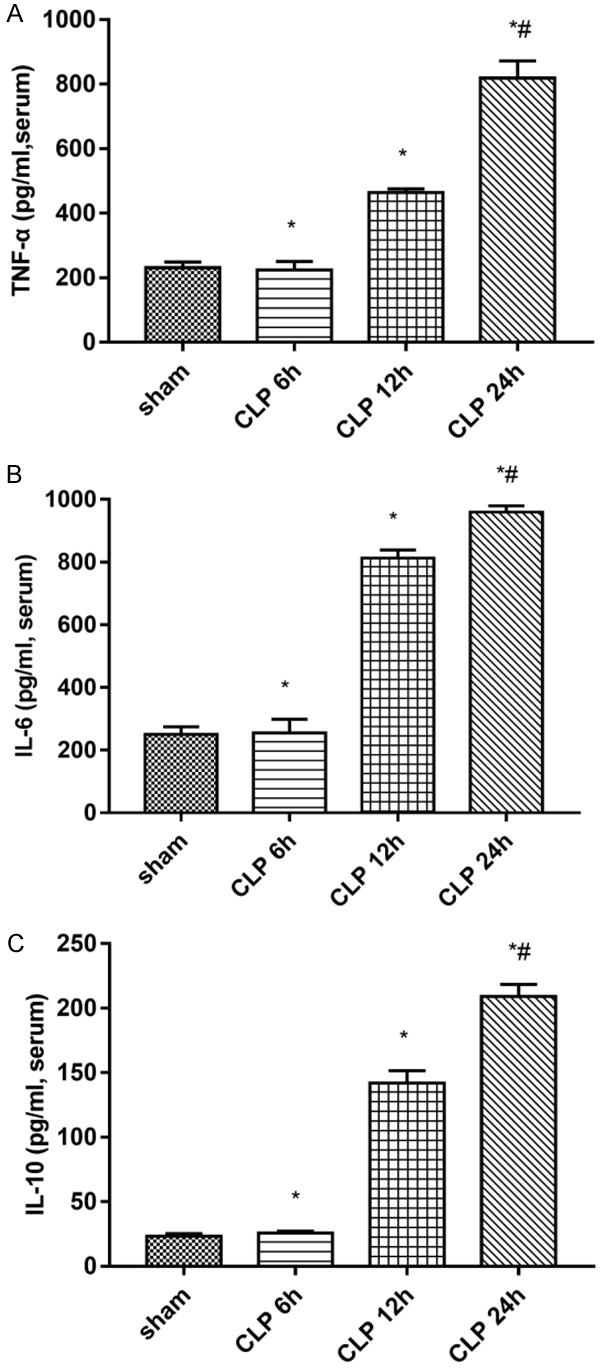
Change in myocardial tissue levels of (A) TNF-α, (B) IL-6, and (C) IL-10 in rats with sepsis. Data are presented as the mean ± standard error of the mean. *P < 0.05 vs. sham group; #P < 0.05 vs. CLP group. (CLP = cecal ligation puncture).
Histological analysis
H&E staining showed the sham groups to have normal morphology of myocardial fibers, regular arrangement of cardiac cells, and no abnormality in the stroma and microvessels (Figure 3). However, in the CLP group, myocardial cells were disordered, myocardial fibers were fractured, and some myocardium showed vacuolar changes and inflammatory cell infiltration. These changes started in the CLP 6 h group and worsened over time (Figure 3).
Figure 3.
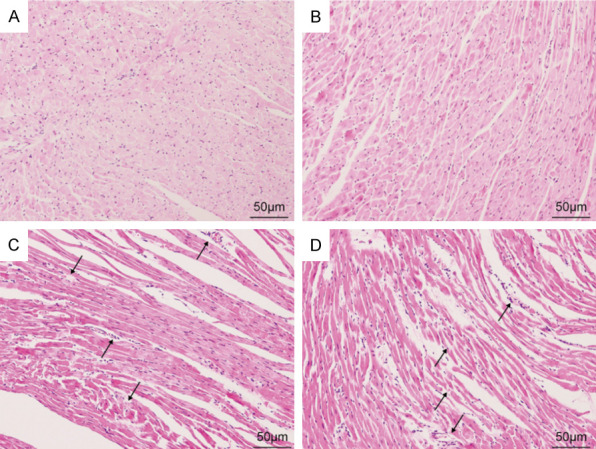
H&E staining showed the morphology of each group of cardiomyocytes (magnification 200×); (A) sham, (B) CLP 6 h, (C) CLP 12 h, (D) CLP 24 h group. (CLP = cecal ligation puncture).
Cardiomyocytes apoptosis in sepsis rats
We detected cardiomyocyte apoptosis by TUNEL analysis and western blot (Figure 4). The apoptotic rate in the CLP group was significantly higher than that in the sham group. At 12 h after CLP, the rate of apoptotic cells increased significantly, and further increased with time. The difference was statistically significant (Figure 4A, 4B). Western blot analysis was employed to determine the levels of Bcl-2 and Bax. Compared to the sham group, CLP operation raised the level of Bax and lowered that of Bcl-2. Compared to the CLP 12 group, Bax in the CLP 24 group increased further, while Bcl-2 levels decreased (Figure 4C, 4D).
Figure 4.
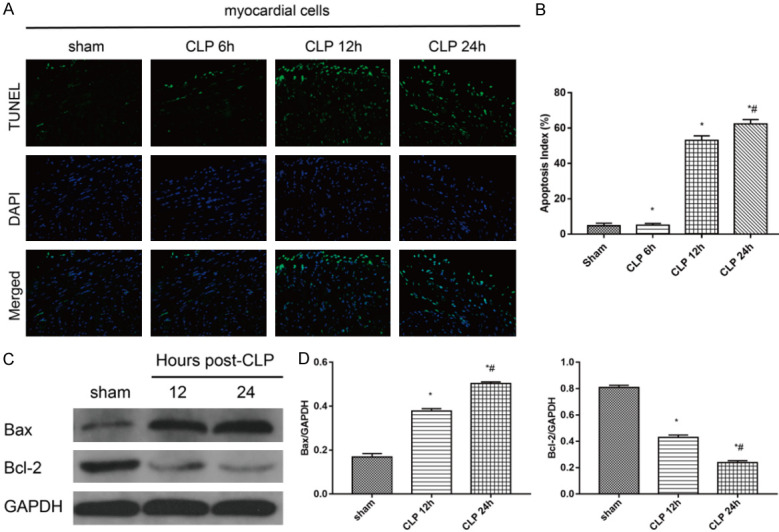
Apoptosis detected by TUNEL assay (magnification, 200×) and western blot. A. Apoptotic cells were detected by a TUNEL assay. Apoptotic cells were marked with green fluorescence and the nuclei of cells were stained by blue fluorescence (DAPI). B. The apoptotic index was calculated for each subgroup (*P < 0.05 vs. sham group; #P < 0.05 vs. CLP group). C. Protein expression of Bcl-2 and Bax were detected by western blot analysis. D. Statistical quantification of Bcl-2 and Bax expression level. Intensity of each band was quantified by densitometry, and data were normalized to the GAPDH signal. The quantified data are expressed as the mean ± standard error. (*P < 0.05 vs. sham group; #P < 0.05 vs. CLP group). (CLP = cecal ligation puncture).
ER stress is triggered in the heart in progression of sepsis
Sepsis can lead to ERS enhancement; ERS-related proteins mainly include GRP 78, CHOP, and Cleaved caspase-12. In this study, we examined their expression, as shown in Figure 5.
Figure 5.
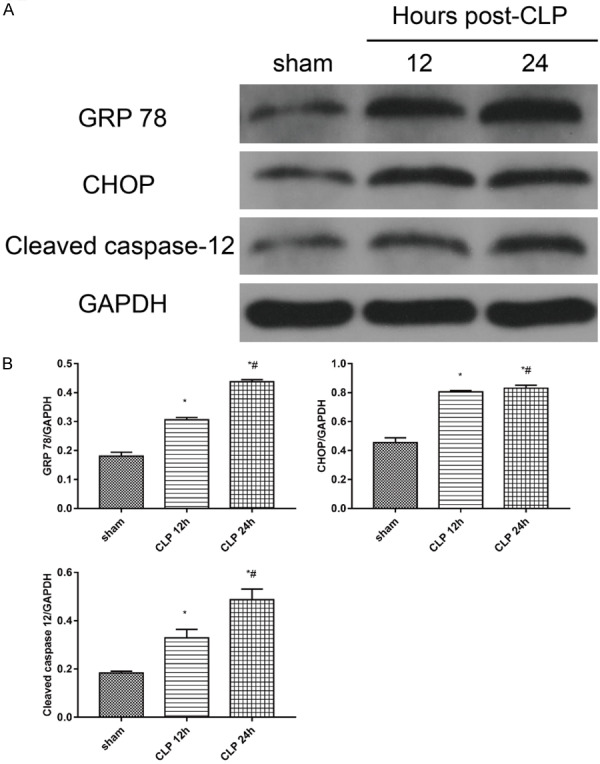
Expression of ERS-related proteins; A. Protein expressions of GRP 78, CHOP, and Cleaved caspase-12 were detected by western blot. B. Statistical quantification of GRP 78, CHOP and Cleaved caspase-12 expression level. The intensity of each band was quantified by densitometry, and data were normalized to the GAPDH signal. Quantified data are expressed as the mean ± standard error. *P < 0.05 vs. sham group; #P < 0.05 vs. CLP group. (CLP = cecal ligation puncture).
Our results suggested the expression of GRP 78, CHOP, and Cleaved caspase-12 to be significantly increased in the CLP group compared to that in the sham group (P < 0.05). The expression of GRP 78, CHOP, and Cleaved caspase-12 protein was upregulated after CLP 12 h, and further increased with time (P < 0.05) (Figure 5).
Discussion
Multiple organ failure caused by sepsis is the main reason for the poor prognosis of patients in the intensive care unit. Although extensive studies have been conducted on myocardial injury caused by sepsis, the underlying mechanisms remain unclear, possibly involving endoplasmic reticulum stress, mitochondrial dysfunction, and oxidative stress injury [6]. Therefore, it has been the focus of medical research to clarify the mechanism of myocardial injury, caused by sepsis, and to prevent early sepsis-induced cardiac dysfunction. CLP is considered to be the gold standard for establishing a sepsis model [7]. In this experiment, we successfully established an animal model of sepsis by CLP. We used male Sprague-Dawley rats of similar age to ensure homogeneity of the subjects.
CK-MB and LDH are myocardial enzymes commonly used in clinical practice [8,9]. cTnI is a myocardial structural protein with high specificity and sensitivity. It is a specific indicator for myocardial damage [10]. We collected serum from all rats and performed biochemical tests on their cardiac markers. The results suggest that myocardial enzymes and troponin in sepsis rats increased from 6 hours and gradually increased with time. Previous studies have shown that inflammatory factor expression increases in patients with sepsis, which is related to poor prognosis [11]. The results of this study indicate that serum proinflammatory cytokines TNF-α and IL-6 concentrations in rats receiving CLP also increased from 6 hours, while the concentration gradient of anti-inflammatory cytokine IL-10 also increased. This is consistent with previous research [12]. These results indicate that sepsis causes damage to rat myocardium through reactions such as inflammation and apoptosis starting at 6 hours, and it gradually increases with time.
The occurrence of ERS is related to many cellular stress factors, which can disturb the function of the endoplasmic reticulum, causing the accumulation of unfolded and misfolded proteins in the endoplasmic reticulum. Studies have shown that ER stress is related to the pathogenesis of various cardiovascular diseases [13-16]. However, the mechanism of cardiac dysfunction due to sepsis remains unknown. The glucose regulator protein 78 (GRP78) is an ER-related chaperone protein. Under physiologic conditions, it binds to three cellular signaling pathways that respond to unfolded protein response (UPR). Nevertheless, if unfolded protein accumulates in the ER lumen, GPR78 dissociates from the ER stress sensor and binds to the unfolded protein, causing it to activate and trigger the UPR. In this way, the ability of ER to fold protein is increased, and the protein with wrong folding is significantly reduced, which plays a role in alleviating ERS. Thus, it is one of the marker proteins of endoplasmic reticulum stress [17-19]. In this research, compared with the sham group, the expression of GRP78 in the myocardium of CLP rats was increased, indicating that sepsis caused endoplasmic reticulum stress, and UPR occurred simultaneously to restore ER homeostasis and improve myocardial cell survival. The endoplasmic reticulum protects cells adaptively by activating unfolded protein responses. However, when stress exceeds its adaptive range, the signal changes from pre-survival to pre-apoptotic. CHOP plays an important role as a transcription factor in endoplasmic reticulum stress-mediated apoptosis [20]. During pathologic conditions or microbial infection, CHOP expression rapidly rises, and apoptosis is activated. Previous studies have found that overexpression of CHOP promotes apoptosis, and ER induces apoptosis through CHOP [21-23]. In our research, we found a significant increase in the rate of myocardial apoptosis in septic rats, while CHOP levels in the CLP group increased significantly. These results indicate that CHOP plays a key role in the endoplasmic reticulum stress-induced cardiomyocyte apoptosis in sepsis rats. At the same time, studies have shown that caspase-12 is closely related to ER stress-mediated cell death [24,25]. Activation of caspase-12 triggers an ER-specific caspase cascade, which leads to cell death. Our data indicate that cleaved caspase 12 expression is significantly increased in the CLP group, demonstrating that it is involved in endoplasmic reticulum stress-mediated cardiomyocyte death in septic rats. In short, the increased expression of CHOP and cleaved caspase-12 proteins indicates that ER-mediated proapoptotic responses cause myocardial cells to die and cause myocardial damage. The above results prove that myocardial apoptosis is closely related to heart injury, resulting from CHOP and caspase-12 in endoplasmic reticulum stress-mediated cell death. We believe that cardiomyocyte apoptosis caused by ERS may accelerate sepsis-induced heart injury.
Sepsis is a disease with a complex pathogenesis. Therefore, septic heart injury may involve multiple pathways of cell death. Our research shows that ER-mediated apoptotic pathways are an important component of cardiomyocyte apoptosis in widespread sepsis. However, sepsis-induced myocardial injury is caused by a variety of mechanisms, and whether there is mutual interference between these mechanisms remains to be elucidated. This will facilitate further clinical trials to explore new treatments.
In sum, our experiment successfully established a rat model of sepsis, and its test results indicate that endoplasmic reticulum stress is associated with septic heart injury. ER stress-induced apoptosis pathways are involved in cardiomyocyte apoptosis in septic rats. Further research on this mechanism may reveal effective interventions in the pathway to prevent cardiac dysfunction caused by sepsis.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No. 81860336).
Disclosure of conflict of interest
None.
References
- 1.Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, Angus DC, Reinhart K International Forum of Acute Care Trialists. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med. 2016;193:259–272. doi: 10.1164/rccm.201504-0781OC. [DOI] [PubMed] [Google Scholar]
- 2.Pulido JN, Afessa B, Masaki M, Yuasa T, Gillespie S, Herasevich V, Brown DR, Oh JK. Clinical spectrum, frequency, and significance of myocardial dysfunction in severe sepsis and septic shock. Mayo Clin Proc. 2012;87:620–628. doi: 10.1016/j.mayocp.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan MM, Yang WL, Wang P. Endoplasmic reticulum stress in sepsis. Shock. 2015;44:294–304. doi: 10.1097/SHK.0000000000000425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma T, Han L, Hu WQ. Study of the role of endoplasmic reticulum stress mediated apoptosis signal pathway in sepsis-induced splenic lymphocyte apoptosis. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2009;21:48–50. [PubMed] [Google Scholar]
- 5.Qian WJ, Cheng QH. Endoplasmic reticulum stress-mediated apoptosis signal pathway is involved in sepsis-induced liver injury. Int J Clin Exp Pathol. 2017;10:9990–9997. [PMC free article] [PubMed] [Google Scholar]
- 6.Y-Hassan S, Settergren M, Henareh L. Sepsis-induced myocardial depression and takotsubo syndrome. Acute Card Care. 2014;16:102–9. doi: 10.3109/17482941.2014.920089. [DOI] [PubMed] [Google Scholar]
- 7.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–6. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dahlin LG, Kågedal B, Nylander E, Olin C, Rutberg H, Svedjeholm R. Early identification of permanent myocardial damage after coronary surgery is aided by repeated measurements of CK-MB. Scand Cardiovasc J. 2002;36:35–40. doi: 10.1080/140174302317282366. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Chen M. Fentanyl ameliorates severe acute pancreatitis-induced myocardial injury in rats by regulating NF-κB signaling pathway. Med Sci Monit. 2017;23:3276–3283. doi: 10.12659/MSM.902245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mair J. Cardiac troponin I and troponin T: are enzymes still relevant as cardiac markers? Clin Chim Acta. 1997;257:99–115. doi: 10.1016/s0009-8981(96)06436-4. [DOI] [PubMed] [Google Scholar]
- 11.Ma X, Chang W, Zhang C, Zhou X, Yu F. Staphylococcal Panton-Valentine leukocidin induces pro-inflammatory cytokine production and nuclear factor-kappa B activation in neutrophils. PLoS One. 2012;7:e34970. doi: 10.1371/journal.pone.0034970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Cheng Q, Li J, He Y, Tian P, Xu C. Significance of hydrogen sulfide in sepsis-induced myocardial injury in rats. Exp Ther Med. 2017;14:2153–2161. doi: 10.3892/etm.2017.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res. 2010;107:1071–1082. doi: 10.1161/CIRCRESAHA.110.227819. [DOI] [PubMed] [Google Scholar]
- 14.Xu J, Zhou Q, Xu W, Cai L. Endoplasmic reticulum stress and diabetic cardiomyopathy. Exp Diabetes Res. 2012;2012:827971. doi: 10.1155/2012/827971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glembotski CC. Endoplasmic reticulum stress in the heart. Circ Res. 2007;101:975–984. doi: 10.1161/CIRCRESAHA.107.161273. [DOI] [PubMed] [Google Scholar]
- 16.Cominacini L, Mozzini C, Garbin U, Pasini A, Stranieri C, Solani E, Vallerio P, Tinelli IA, Fratta Pasini A. Endoplasmic reticulum stress and Nrf2 signaling in cardiovascular diseases. Free Radic Biol Med. 2015;88:233–242. doi: 10.1016/j.freeradbiomed.2015.05.027. [DOI] [PubMed] [Google Scholar]
- 17.Lin PY, Liu HJ, Chang CD, Chen YC, Chang CI, Shih WL. Avian reovirus S1133-induced apoptosis is associated with Bip/GRP79-mediated Bim translocation to the endoplasmic reticulum. Apoptosis. 2015;20:481–490. doi: 10.1007/s10495-015-1085-5. [DOI] [PubMed] [Google Scholar]
- 18.Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J Biol Chem. 2001;276:33869–33874. doi: 10.1074/jbc.M102225200. [DOI] [PubMed] [Google Scholar]
- 19.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 20.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 21.Zhao D, Liu Y, Liu X, Li T, Xin Z, Zhu X, Wu X, Liu Y. HBV suppresses thapsigargin-induced apoptosis via inhibiting CHOP expression in hepatocellular carcinoma cells. Oncol Lett. 2017;14:4403–4409. doi: 10.3892/ol.2017.6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, Ushio Y, Mori M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- 23.Uzi D, Barda L, Scaiewicz V, Mills M, Mueller T, Gonzalez-Rodriguez A, Valverde AM, Iwawaki T, Nahmias Y, Xavier R, Chung RT, Tirosh B, Shibolet O. CHOP is a critical regulator of acetaminophen-induced hepatotoxicity. J Hepatol. 2013;59:495–503. doi: 10.1016/j.jhep.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 24.Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann N Y Acad Sci. 2003;1010:186–194. doi: 10.1196/annals.1299.032. [DOI] [PubMed] [Google Scholar]
- 25.Shiraishi H, Okamoto H, Yoshimura A, Yoshida H. ER stress-induced apoptosis and caspase-12 activation occurs downstream of mitochondrial apoptosis involving Apaf-1. J Cell Sci. 2006;119:3958–3966. doi: 10.1242/jcs.03160. [DOI] [PubMed] [Google Scholar]