

Abstract
Mitochondrial injury and depolarization are primary events in acetaminophen hepatotoxicity. Previous studies have shown that restoration of mitochondrial function in surviving hepatocytes, which is critical to recovery, is at least partially accomplished via biogenesis of new mitochondria. However, other studies indicate that mitochondria also have the potential to spontaneously repolarize. Although repolarization was previously observed only at a sub-hepatotoxic dose of acetaminophen, we postulated that mitochondrial repolarization in hepatocytes outside the centrilobular regions of necrosis might contribute to recovery of mitochondrial function following acetaminophen-induced injury. Our studies utilized longitudinal intravital microscopy of millimeter-scale regions of the mouse liver to characterize the spatio-temporal relationship between mitochondrial polarization and necrosis early in acetaminophen-induced liver injury. Treatment of male C57BL/6J mice with a single intraperitoneal 250 mg/kg dose of acetaminophen resulted in hepatotoxicity that was apparent histologically within 2 hours of treatment, leading to 20 and 60-fold increases in serum aspartate aminotransferase and alanine aminotransferase, respectively, within 6 hours. Intravital microscopy of the livers of mice injected with rhodamine123, TexasRed-dextran, propidium iodide and Hoechst 33342 detected centrilobular foci of necrosis within extended regions of mitochondrial depolarization within 2 hours of acetaminophen treatment. Although regions of necrosis were more apparent 6 hours after acetaminophen treatment, the vast majority of hepatocytes with depolarized mitochondria did not progress to necrosis, but rather recovered mitochondrial polarization within 6 hours. Recovery of mitochondrial function following acetaminophen hepatotoxicity thus involves not only biogenesis of new mitochondria, but also repolarization of existing mitochondria. These studies also revealed a spatial distribution of necrosis and mitochondrial depolarization whose single-cell granularity is inconsistent with the hypothesis that communication between neighboring cells plays an important role in the propagation of necrosis during the early stages of APAP hepatotoxicity. Small islands of healthy, intact cells were frequently found surrounded by necrotic cells, and small islands of necrotic cells were frequently found surrounded by healthy, intact cells. Time-series studies demonstrated that these “islands”, consisting in some cases of single cells, are persistent; over a period of hours, injury does not spread from individual necrotic cells to their neighbors.
Keywords: acetaminophen hepatotoxicity, mitochondria, hepatocytes, intravital microscopy, multiphoton microscopy
Introduction
Since its commercial introduction in 1955, acetaminophen (APAP, N-acetyl-p-aminophenol, paracetamol) has become one the most widely used analgesics. Although APAP is generally safe when used as recommended, APAP hepatotoxicity, resulting from accidental or intentional overdose, is nonetheless the most common cause of acute liver failure(Ramachandran and Jaeschke, 2018).
APAP hepatotoxicity results from the accumulation of a highly-reactive toxic metabolite, N-acetyl-p-benzoquinone imine (NAPQI)(Hinson et al., 2010; Ramachandran and Jaeschke, 2018; Yuan and Kaplowitz, 2013). At therapeutic doses, APAP is rapidly metabolized in the liver into glucuronidated and sulfated metabolites that are rapidly excreted. The small amount of remaining APAP is either secreted directly or is oxidized to form NAPQI, which is nearly instantaneously detoxified by conjugation with glutathione (GSH). However, at high doses, the glucuronidation and sulfation pathways become saturated, increasing cellular levels of APAP and NAPQI. The accumulation of NAPQI depletes GSH, further accelerating NAPQI accumulation to toxic levels(Mitchell et al., 1973b) and exacerbating oxidative stress(Hinson et al., 2010; Reid et al., 2005). NAPQI reacts relatively indiscriminately with proteins (via sulfhydryl groups), so it is difficult to single out specific protein targets that are critical to hepatotoxicity. However, multiple studies indicate that adducts formed with mitochondrial proteins are critical to APAP hepatotoxicity in rodents and humans(Hu et al., 2016; McGill et al., 2012a; McGill et al., 2012b; Moles et al., 2018; Ramachandran and Jaeschke, 2017). NAPQI adducts of mitochondrial proteins induce the formation of additional reactive oxygen species, resulting in the activation of JNK, whose translocation to mitochondria triggers the mitochondrial permeability transition (MPT), and mitochondrial depolarization(Hanawa et al., 2008; Hu et al., 2016; Ramachandran and Jaeschke, 2017; Win et al., 2011; Xie et al., 2015; Yan et al., 2010).
Restoration of mitochondrial function among surviving hepatocytes is a critical component of recovery from APAP-induced liver injury. Previous studies have demonstrated that, in hepatocytes surrounding regions of necrosis, mitochondrial function is restored via a process of mitochondrial biogenesis(Ni et al., 2013), resulting in a slow recovery of electron chain activity that occurs over the 24 to 72 hour period following APAP administration(Du et al., 2017b). However, mitochondrial function may also be restored via spontaneous repolarization of mitochondria in surviving hepatocytes (Hu et al., 2016). Occurring much faster than mitochondrial biogenesis, mitochondrial repolarization was found to be complete within 24 hours of APAP treatment.
Mitochondrial repolarization is generally discussed as a phenomenon occurring following sub-toxic exposure to APAP(Moles et al., 2018; Ramachandran et al., 2018), since reversible depolarization was observed only at a dose that caused no necrosis or increase in serum ALT. However, mitochondrial repolarization might, like mitochondrial biogenesis, be a component of recovery in a sub-population of hepatocytes surrounding the foci of necrosis. The effects of APAP on hepatocytes are not uniform, but vary in a way that is believed to reflect metabolic zonation of the lobule such that NAPQI exposure and toxicity are higher in pericentral hepatocytes than in periportal hepatocytes (Anundi et al., 1993; Mitchell et al., 1973a; Roberts et al., 1991). The spatial distribution of APAP-induced stress suggests that hepatotoxic doses of APAP should result in regions of centrilobular necrosis surrounded by regions of hepatocytes whose mitochondria initially depolarize but subsequently recover polarization. Since the capacity for repolarization would be expected to decline with time in stressed hepatocytes, repolarization in these regions might be expected to occur within the first few hours of APAP exposure.
To explore the spatial and temporal relationship between mitochondrial polarization and necrosis during the early stages of APAP-mediated liver injury we used intravital microscopy to monitor millimeter-scale regions of the mouse liver over the 2-6 hour period following treatment with a hepatotoxic dose of APAP. These studies identify a complex, dynamic pattern of injury and recovery. Within two hours of APAP treatment, three distinct populations of hepatocytes were observed, (1) A centrilobular population of necrotic hepatocytes, (2) A periportal population of unaffected hepatocytes and (3) A large population of intact hepatocytes with partially depolarized mitochondria surrounding the necrotic foci. Rather than progressing to necrosis, the majority of cells in this last population rapidly recovered mitochondrial polarization, such that it approached normal levels within 6 hrs of APAP administration.
Materials and methods
Reagents
Fluorescent probes used for intravital microscopy studies included 70 KDa TexasRed-dextran, Hoechst 33342 trihydrochloride trihydrate, propidium iodide (PI) and rhodamine123 (Thermo Fisher Scientific, Oregon, United States). TexasRed dextran was dialyzed in 0.9% of saline prior to use.
Animals
All animal experiments were approved and conducted according to the Institutional Animal Care and Use Committee guidelines of Indiana University, and adhered to the guide for the care and use of animals(National Research Council (U.S.). Committee for the Update of the Guide for the Care and Use of Laboratory Animals. et al., 2011). Studies were conducted using 7-10 week old male C57BL/6J mice (23-30 grams, Jackson Labs). Mice were acclimated for one week prior the studies in the Laboratory Animal Resource Center of Indiana University. Mice were maintained on a diet of Teklad 4% mouse/rat diet and water ad libitum. Twelve hours before APAP administration, mice were moved to clean cages without food. The day of the study mice received an IP injection of APAP (dose of 250 mg/kg) or saline vehicle (at a volume of 0.2 ml/10g body weight), and were then returned to cages with food. Mice were then analyzed over time as described below
Liver histopathology and analysis
Following euthanasia, mouse livers were perfused with warm saline, removed in toto and weighed. Three to four pieces from left lateral, right or left medial, right lateral, and/or caudate lobes of the liver were excised and fixed in 10% neutral-buffered formalin. Fixed liver tissues were paraffin embedded, sectioned (4-5 μm), and stained by hematoxylin and eosin (H&E) for the evaluation of pathological changes.
Measurement of APAP and metabolites
APAP parent compound and metabolites (including APAP-Glutathione, APAP-Cysteine, APAP-Glucuronide, APAP-Sulfate and APAP-n-acetylcysteine) in serum and liver samples were determined using high-performance liquid chromatography (HPLC), as described previously (Dai et al., 2006) with modifications. Briefly, liver samples (50 mg) were homogenized in 9 volumes (or 450 μl) of ice-cold HPLC-grade methanol. Serum samples (20 μl) were diluted in 9 volumes of ice-cold HPLC-grade methanol. Following centrifugation at 20,000 g at 4°C for 5 min, the resulting supernatant was mixed with 1/10 volume of 35% perchloric acid to precipitate protein. After another centrifugation step, the supernatant was collected and mixed with an equal volume of mobile phase (consisting of 1% acetic acid and 20% MeOH). 10 μl of the mixture was injected into the HPLC system for separation and detection. The HPLC system was equipped with a Waters 2695 separation module with a Supelcosil LC-18-DB, 150mmx4.6mm, 3 μm column, and a Waters 996 Photodiode Array Detector. The concentrations of APAP and its metabolites were calculated using the corresponding standard curves. The results were expressed as ng/ul for serum sample or ng/mg (adjusted to the weight of liver tissue) for liver samples.
Hepatic enzyme activity
The serum hepatic enzymes, alanine aminotransferase (ALT) and aspartate aminotransferase (AST), were measured immediately following serum separation from whole blood using Infinity™ ALT and AST kits (Thermo scientific, Middletown, VA) with a UV microplate reader (Tecan Infinite M200Pro, San Jose, CA).
Animal preparation for intravital microscopy
Intravital microscopy studies were conducted at the indicated times after IP administration of APAP (250 mg/kg bw) or vehicle using approaches previously described(Babbey et al., 2012; Dunn and Ryan, 2017; Ryan et al., 2018; Ryan et al., 2014). Surgical preparation of the liver for intravital microscopy was begun an hour prior to imaging. Mice were placed on an induction chamber to be anesthetized with 3-4 % isoflurane mixed with 0.8%-1% O2. The plane of anesthesia was monitored by evaluation of responses to toe pinches. Once anesthetized, the mouse was transferred to an anesthesia nose cone and moved to a surgery table equipped with a heating pad for the maintenance of body temperature. Body temperature was monitored by a rectal thermometer and maintained at 37°C. Approximately 50 microliters of Hoechst 33342 solution was then injected IP to a dose of 2-6 mg/kg.
The mouse was shaved in the abdominal ventral area below the sternum to expose the liver and in the right ventral of the neck region for the jugular cannulation. A jugular cannula (PE tubing with a 30 gauge tip) was then placed for reagent injections. Approximately 400 microliters of a mixed solution of TexasRed-Dextran and rhodamine123 (R123) was injected IV (doses of 80-120 mg/kg and 0.24-0.36 mg/kg, respectively). After two minutes, the vein catheter was flushed with 50 microliters of saline. For longitudinal studies extending over hours, additional R123 was injected on the microscope stage as necessary to compensate for p-glycoprotein-mediated secretion(Annaert and Brouwer, 2005; Liu, 2012).
After placement of the jugular catheter, the liver was exposed for imaging. A 2×2 piece of gauze moistened in 0.9% saline was placed in the abdominal ventral area. The liver was exposed via a 6mm incision below the sternum, and the tissue crushed at the incision site with hemostats to minimize bleeding. The muscle layer was then crushed with hemostats and another 6 mm incision was made, exposing the liver. The bottom of the left lateral lobe of the liver was gently squeezed free of the body and laid onto the moistened gauze. A drop of cyanoacrylate glue was applied to the tape on the bottom of a Willco coverslip-bottomed dish (GWST-5040, Warner Instruments, Hamden, CT). The mouse was then placed over the coverslip, taking care to prevent the liver from contacting the cyanoacrylate glue. As necessary the gauze paid was remoistened and the mouse was moved to the microscope stage for imaging. Animal temperature was monitored using a rectal thermometer and maintained using an objective heater, heating pad and heat lamp.
Intravital multiphoton microscopy
The studies described here were performed using an upright Leica TCS SP8 high-speed multiphoton and confocal imaging system, adapted for inverted use via an objective inverter (LSM Tech, Etters, PA). All imaging was conducted using a Leica HCX IR APO 25X 0.95 NA W objective lens. IR illumination was provided by a MaiTai HP titanium-sapphire laser tuned to 800 nm. Fluorescence emissions were collected in 3 non-descanned detectors, with emission bandpass filters of 380-500 nm (blue), 500-550 nm (green) and 565-605 nm (red). Image volumes were typically collected as multi-panel mosaics of 512 × 512 × 20 voxel image volumes. Images were collected at a zoom factor of 1, with image planes collected every 1.5 microns, providing voxel dimensions of 0.87 × 0.87 × 1.5 microns. Five minutes before each mosaic image acquisition ~50ul (1-1.5 mg/kg) propidium iodide (PI) was injected intravenously. Mosaic image volumes were then collected every 20-40 minutes for the next ~2 hours.
Supplementary Figure 1 shows an example of multiphoton fluorescence images collected from the liver of a living mouse following sequential injection of four fluorescent probes. The first column of images, collected following IV injection of R123 and PI show two types of cells. The first are healthy cells that have excluded PI and show strong fluorescence of R123 that has accumulated in polarized mitochondria(Rehman et al., 2011; Theruvath et al., 2008). The second are necrotic cells, cells whose membranes have been breached, leading to cellular penetration by the membrane-impermeant nuclear probe, PI(Hu et al., 2016; Rehman et al., 2011; Theruvath et al., 2008). As expected, these cells lack R123 fluorescence, consistent with the absence of mitochondrial polarization. The second column of images shows the same field following IV injection of the membrane permeant nuclear probe, Hoechst 33342, which labels the nuclei of all cells. The third column of images shows the same field following IV injection of TexasRed dextran, which labels both the vasculature and the cytosol of the necrotic cells (Li et al., 2011). We used this combination of fluorescent probes to functionally identify three types of hepatocytes in intravital microscopy studies.
Healthy hepatocytes - cells with strong green R123 mitochondrial fluorescence (Rehman et al., 2011; Theruvath et al., 2008) that have excluded membrane impermeant PI and dextran.
Necrotic hepatocytes - cells whose plasma membrane has lost integrity, so that their nuclei are labeled not only with Hoechst 33342, but also with PI (Hu et al., 2016; Rehman et al., 2011; Theruvath et al., 2008) and their cytosols are labeled with TexasRed dextran(Li et al., 2011). Necrotic hepatocytes are also characterized by the absence of R123 fluorescence.
Stressed hepatocytes - cells that exclude membrane impermeant PI and dextran, but show diminished green R123 mitochondrial fluorescence.
Image analysis and image processing
Quantitative image analysis was conducted using Metamorph image processing software (Molecular Devices, Downington, PA). Centrilobular repolarization of mitochondria was measured on the basis of the recovery of R123 fluorescence in depolarized regions as follows. Depolarized regions, apparent as broad, centrilobular regions of diminished R123 fluorescence, were identified visually. For each mouse, 4 to 10 such regions were identified. For each region, depolarization was quantified as the ratio of the mean R123 fluorescence measured in a 50-pixel-diameter region located within the darkened region to that in a corresponding 50-pixel-diameter region located nearby, but outside the darkened region. Examples of regions used to measure depolarization in one study are shown in Supplementary Figure 2A. Depolarization was quantified as a ratio in this way as it provides a metric of depolarization that is relatively insensitive to differences in tissue depth and differences in overall R123 labeling between studies. To obtain corresponding measurements for vehicle-treated mice, which showed no visible regions of centrilobular depolarization, we quantified ratios of the fluorescence measured in pericentral regions to fluorescence measured in nearby periportal regions, using sinusoidal structure to identify lobular regions. Fluorescence ratios were measured for between 4 and 10 periportal/pericentral region pairs for each vehicle-treated mouse.
To quantify repolarization over time, ratios were measured for measurement regions placed on the mosaic collected at the first time point, and then for these same regions placed on the mosaics collected at subsequent time points (typically 3-4 mosaics, collected over two hours). As necessary, individual measurement regions were adjusted laterally to accommodate slight shifts in the tissue occurring over time. To avoid bias, measurement regions were delineated in the mosaic collected at the first time point without knowledge of changes occurring at subsequent time points. Supplementary Figures 2A–D show examples of regions used to quantify repolarization over a two hour interval in one study. Repolarization data presented in Figure 7C represent combined measurements obtained from 7 mice, 3 that were imaged 2-4 hrs after acetaminophen treatment and 4 that were imaged 4-6 hrs after acetaminophen treatment. Mitochondrial repolarization in necrotic cells was quantified similarly, except that fluorescence ratios were calculated from measurements obtained from regions located within individual necrotic and healthy cells.
The areas of necrosis and mitochondrial depolarization were quantified as the fraction of the total area of an image mosaic occupied by dextran-loaded cells and the fraction of the total area occupied by the centrilobular regions of clearly diminished R123 fluorescence, respectively. In both cases, the affected regions were outlined manually.
Mosaic image volumes were assembled using the Leica Suite X software. In order to reconstruct a single image of a single layer of hepatocytes across the mosaic, multiple focal planes from the original data were imported as layers in Adobe Photoshop (Adobe, Mountain View, CA) and combined in such a way that (1) ensured that the same focal plane was reproduced throughout the mosaic and (2) minimized presentation of regions that were distorted by tissue motion. Quantitative analysis was conducted using raw image data, but micrograph images presented in figures were contrast enhanced in a way that preserved the visibility of both the dim and bright structures of the original images. Images in figures were processed, assembled and annotated using Adobe Photoshop. Graphs and summary statistics were generated using Kaleidagraph (Synergy Software, Reading, PA).
Results
APAP model of hepatotoxicity
Following IP administration of 250 mg/kg, APAP was rapidly metabolized, primarily to APAP-glucuronide, which peaked at 30 minutes in the serum and at 1 hour in the liver (Figure 1). The concentrations of APAP and its metabolites declined to undetectable or nearly undetectable levels within 4 hours in both serum and liver. APAP-mediated hepatotoxicity was evident in the abrupt appearance of aspartate aminotransferase and alanine aminotransferase in the serum 4 hours after treatment, subsequently peaking at 6 hours, then declining for the following 42 hours. As expected, histological evidence of injury appears somewhat earlier; centrilobular injury characteristic of APAP-mediated hepatotoxicity appears within 2 hours of treatment (Figures 1 D–I). Despite the extensive injury, which is apparent beyond 48 hours, no mortality was observed in the 40 mice treated with 250 mg/kg APAP.
Figure 1 – Biochemical analyses of APAP metabolism and hepatotoxicity in male C57BL/6J mice.
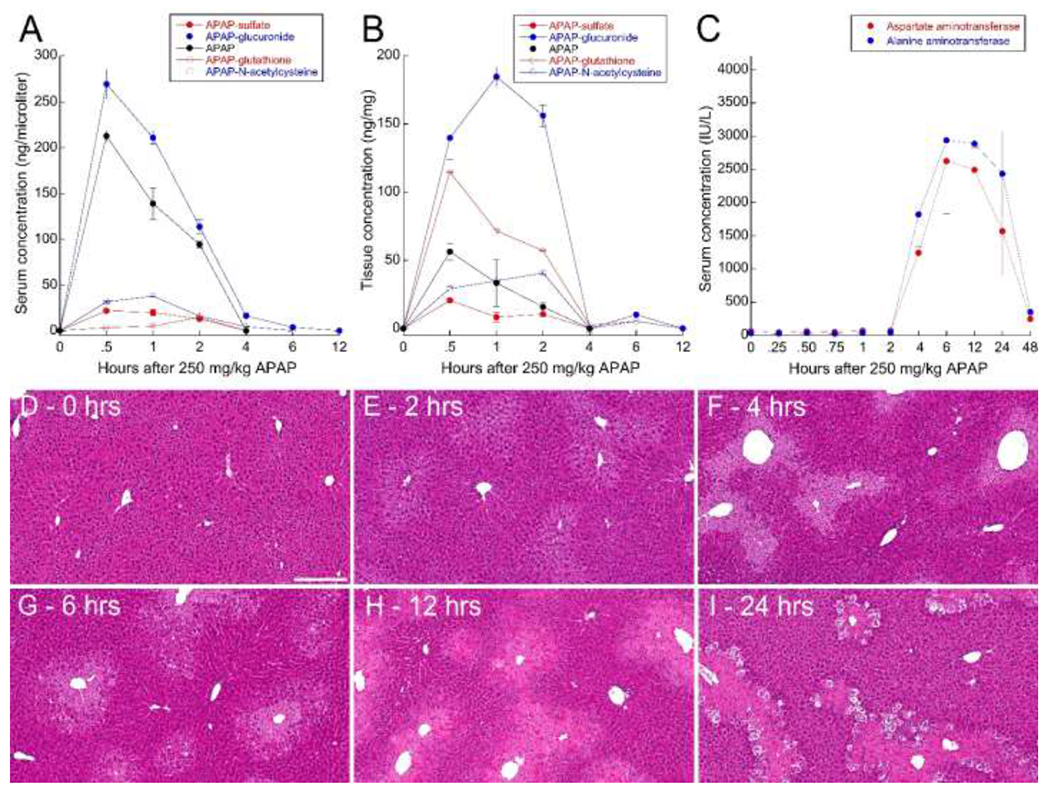
Serum (A) and liver (B) concentrations of APAP (solid black) and APAP metabolites APAP-glucuronide (solid blue), APAP-N-acetylcysteine (open blue), APAP-sulfate (solid red) and APAP-glutathione (open red). C – Serum concentrations of aspartate aminotransferase (red) and alanine aminotransferase (blue) over time after administration of 250 mg/kg APAP. Error bars represent standard errors (N=5 for all studies). D-I – Hematoxylin-eosin stained liver tissue from mice sacrificed at the indicated times after treatment with 250 mg/kg APAP. Scale bar represents length of 200 microns.
Intravital multiphoton microscopy of APAP hepatotoxicity
Intravital microscopy studies were conducted to characterize the relationship between hepatocellular necrosis and mitochondrial dysfunction following treatment with 250 mg/kg APAP. In order to capture regions large enough to encompass the spatial variability of injury, large-scale images were collected as mosaics of 9-16 image volumes, each collected at subcellular resolution. Tissue motion resulting from respiration can make intravital microscopy of the liver challenging. Although the effects of respiration can be avoided through the use of a respirator which is turned off during intervals of image collection(Hu et al., 2016), this approach is incompatible with the extended time needed to collect mosaic image volumes. Although tissue motion can be minimized by stretching the liver or directly gluing it to a coverslip, we elected to minimally constrain the liver, in order to ensure normal perfusion and physiology. As a consequence, all studies showed strong microvascular flow throughout their duration, in some cases at the cost of some tissue motion. We minimized the effects of tissue motion by collecting images at a high rate, and by reconstructing single plane images from 3D volumes (see Methods).
Prior to imaging, mice were injected intravenously with Hoechst 33342, which labels all nuclei, propidium iodide (PI), which labels the nuclei of permeabilized necrotic cells (Hu et al., 2016; Rehman et al., 2011; Theruvath et al., 2008), rhodamine123 (R123), which labels functional mitochondria (Hu et al., 2016; Rehman et al., 2011; Theruvath et al., 2008) and 70 kDa TexasRed dextran, a membrane-impermeant probe of the vasculature, as well as necrotic cells(Li et al., 2011). Figure 2A shows an example of an image mosaic collected from the liver of a mouse injected with vehicle. As expected of a healthy liver, the image shows relatively homogenous green R123 fluorescence, reflecting strong mitochondrial function throughout the region. This image also shows the bright autofluorescence of vitamin A in fat droplets of stellate cells, which appears white, due to its broad emission spectrum(Wang et al., 2015). The image shows little evidence of necrosis; in the 1.2 × 1.2 mm region, there are perhaps 4 permeabilized cells, which can be distinguished by their PI-labeled nuclei and by the cytosolic penetration of the large molecular-weight vascular probe, TexasRed-dextran. Previous studies have likewise observed that large molecular probes label necrotic cells following IV injection (Pires et al., 2014).
Figure 2 –
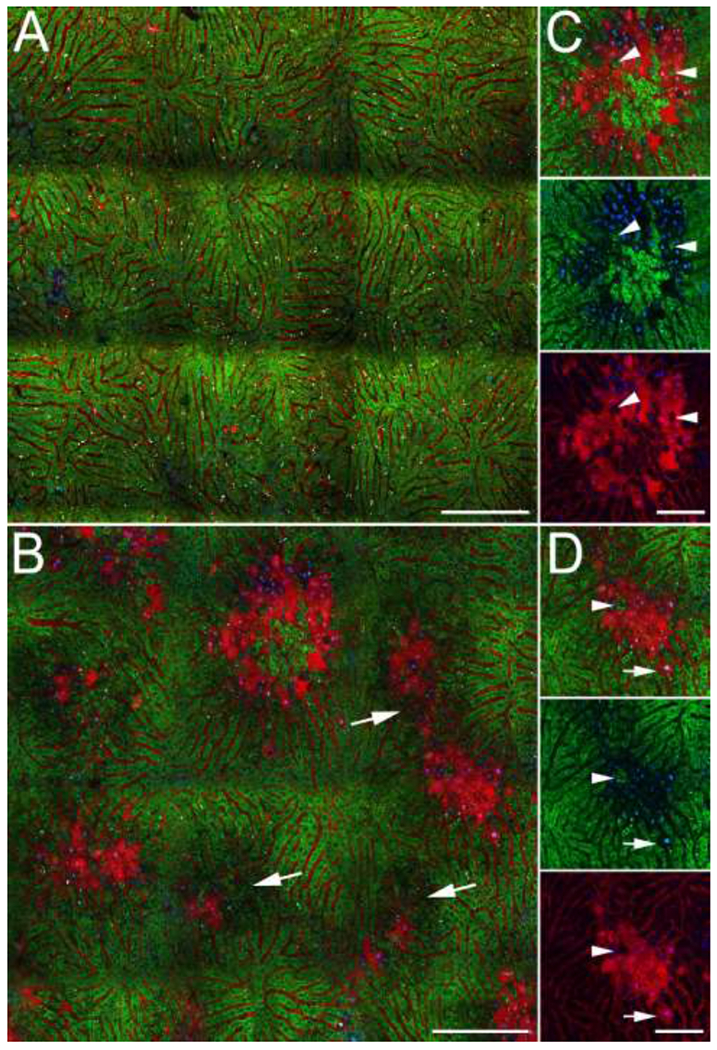
Intravital microscopy of the livers of live mice following intravenous injection of TexasRed dextran (red), PI (red), Hoechst 33342 (blue) and R123 (green). (A) Single plane reconstruction from a mosaic of image volumes collected from mouse 6.5 hours after IP injection of vehicle. (B) Single plane reconstruction from a mosaic of image volumes collected from a mouse 5 hours after IP injection of 250 mg/kg APAP. Arrows indicate regions of diminished R123 fluorescence. (C, D) Subregions of the field shown in Panel A. Three-channel image is shown at top, and below that, images in which the red and green signals have been removed, to better show the absence of mitochondrial R123 and penetration of PI and TexasRed dextran into permeabilized (necrotic) cells. Arrowheads indicate healthy cells (impermeable with strong mitochondrial R123 fluorescence) surrounded by necrotic cells (permeabilized with undetectable mitochondrial R123 fluorescence). Arrow indicates a necrotic cell (permeabilized with undetectable mitochondrial fluorescence) surrounded by intact, healthy cells (impermeable with strong mitochondrial R123 fluorescence). Scale bars represent a length of 200 microns (Panes A and B) or 50 microns (panels C and D).
In contrast, an image mosaic collected from a mouse 5 hours after IP injection of 250 mg/kg APAP displays widespread centrilobular necrosis, reflected in the bright red fluorescence of PI and dextran in permeabilized cells (Figure 2B). As expected, these permeabilized cells completely lack R123 fluorescence, reflecting the absence of mitochondrial polarization (Figures 2C and 2D). Although the appearance of this field is generally consistent with the histology images shown in Figure 1, the regions of necrosis appear less extensive, likely due to differences in the areas of the liver sampled by intravital microscopy and histology. The effects of light scattering limit intravital microscopy to optical sections less than 100 microns from the surface of the liver, a depth that is difficult to section for histopathologic evaluation. For centrilobular injuries such as those induced by APAP treatment, intravital microscopy thus may sample the periphery of injured regions whose full extent is better captured at depth in histological sections. In some mice, we found injured regions only at tissue depths just beyond those that can be effectively imaged by multiphoton microscopy (data not shown).
However, the images of injured regions of the liver lobule captured using intravital microscopy provide physiological information beyond that provided by histology, revealing an unforeseen level of complexity in the spatial relationship between necrotic and healthy cells. Many necrotic regions occur in the middle of larger regions of diminished mitochondrial polarization, as defined by their diminished R123 fluorescence. The images shown in panels C and D demonstrate that, as expected, permeabilized cells completely lack R123 fluorescence, reflecting the absence of mitochondrial function in necrotic cells. However, immediately adjacent to these necrotic hepatocytes are intact cells with reduced, but detectible levels of R123 fluorescence, representing what we have designated as “stressed” hepatocytes.
In addition, the distribution of healthy and necrotic cells was surprisingly granular; with cells with strong R123 fluorescence frequently seen within large areas of necrotic cells (panels C and D). Conversely, individual necrotic cells were frequently found amidst healthy cells (panels B and D). This granularity also demonstrates that the necrotic regions are well-perfused; intravenous dextran and R123 are delivered to cells even in the centers of the necrotic regions.
Longitudinal studies of APAP hepatotoxicity
The images shown in Figure 2 raise several questions as to the trajectory of injury. First, models of APAP-mediated liver injury generally identify mitochondrial depolarization as an early event of hepatocyte injury that precedes the loss of plasma membrane integrity (necrosis)(Hinson et al., 2010; Moles et al., 2018; Ramachandran and Jaeschke, 2017; Yuan and Kaplowitz, 2013). The eventual fate of hepatocytes whose mitochondria are partially depolarized is unclear. Will they recover or continue on to necrosis? Will the areas of necrosis grow to the boundaries of the regions of hepatocytes with diminished mitochondrial function? Will the regions of hepatocytes with depolarized mitochondria grow with time? Some models of APAP-mediated liver injury invoke the role of “neighbor” effects, in which necrosis spreads to the neighbors of necrotic cells via the release of toxic factors into the extracellular environment or through gap junctions (Naiki-Ito et al., 2010; Patel et al., 2012; Saito et al., 2014). Will the healthy and/or stressed cells that are surrounded by necrotic cells become necrotic themselves? In order to address these questions, we developed an approach in which injured regions of the liver could be repeatedly imaged over time.
Preliminary studies were conducted to determine the effect of repeated multiphoton fluorescence microscopy on the physiology of the liver. Regions of the liver imaged seven times over the course of two hours showed no discernible increase in rates of necrosis and no alteration in microvascular function. (Supplementary Figure 3A). Consistent with previous studies(Liu, 2012), overall levels of R123 in hepatocytes declined with time, likely due to p-glycoprotein-mediated secretion(Annaert and Brouwer, 2005). However, side-by-side comparison of a field imaged 7 times with an adjacent field imaged only once demonstrates that repeated imaging had no detectible effect on levels of R123 fluorescence (Supplementary Figure 3B), directly demonstrating that repeated imaging did not compromise polarization, nor result in significant photobleaching. To compensate for secretion in subsequent longitudinal studies, additional R123 was administered as needed.
Figure 3 shows results of studies of two mice in which the same regions of the livers were imaged 4.5 and 6 hours after IP administration of 250 mg/kg APAP. In order to characterize both established injured regions as well as new injury foci, we chose fields that included both extensively and modestly injured regions at 4.5 hours (panels A-D). As in the liver of the APAP-treated mouse shown in Figure 2, both fields include regions of necrosis surrounded by regions of intact cells with reduced mitochondrial polarization. At the level of the entire field, the area of injury modestly increased during the period of imaging; the area of necrosis increased by 11% in the field shown in panels A/B and by 5% in the field shown in panels C/D.In contrast, the area of the regions of mitochondrial depolarization decreased slightly in both experiments, decreasing by 7% in the field shown in panels A/B and by 15% in the field shown in panels C/D.
Figure 3 – Images of the livers of living mice collected sequentially over the period from 4.5 to 6 hours after APAP treatment.
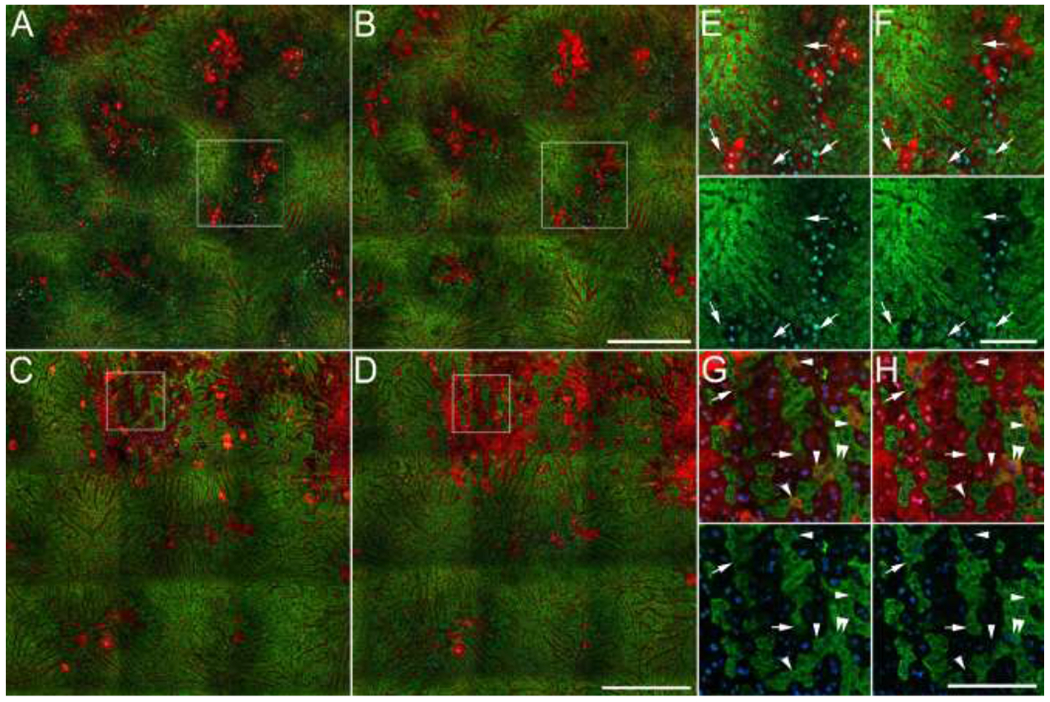
A, C – Fields collected 4.5 hours after administration of 250 mg/kg APAP (B, D) – Same fields collected 6 hours after APAP administration. Panels E and F show 2X magnifications of a region from the middle-right part of the field shown in panels A and B, respectively. Arrows indicate groups of hepatocytes in which mitochondrial polarization (R123 fluorescence) recovers over the interval. Panels G and H show 3X magnifications of indicated regions of the fields shown in panels C and D, respectively. Arrows indicate hepatocytes in which mitochondrial polarization (R123 fluorescence) recovers over the interval. Arrowheads in panel G indicate permeabilized cells with functional mitochondria at 4.5 hrs, cells whose cytosol was labeled with both fluorescent dextran and R123. In most of these cells, mitochondrial polarization is lost prior to the second time point (panel H). Double arrowheads in panels G and H indicate a hepatocyte that lost plasma membrane integrity during the interval. As described in Methods, additional R123 was injected as necessary to accommodate the continuous systemic clearance of R123 (an organic anion transport substrate). Since overall R123 fluorescence levels varied between the two time points of each study, green levels were adjusted in these figures such the peak intensities are similar at the two time points. Scale bars represent a length of 300 microns in panels A-D, and a length of 100 microns in panels E-H.
The fate of individual cells is more readily appreciated in the high-magnification images of individual channels of fluorescence presented in Figures 3E–H. These images reveal three functional categories of hepatocytes: 1. Healthy hepatocytes – cells that have excluded dextran and PI (intact cells) with strong R123 fluorescence, 2. Stressed hepatocytes - intact cells with weak R123 fluorescence and 3. Necrotic hepatocytes - permeabilized cells with no detectible R123 fluorescence.
We anticipated that time-series studies would reveal examples of hepatocytes in which mitochondrial depolarization was a first step to necrosis, i.e. hepatocytes in which diminished R123 fluorescence led to subsequent labeling dextran and PI, reflecting the loss of plasma membrane integrity. However, inspection of hundreds of stressed hepatocytes with compromised R123 labeling revealed none in which mitochondrial depolarization was followed by the subsequent loss of plasma membrane integrity. Instead, we found multiple examples of cells with R123 fluorescence that was initially diminished but subsequently increased to levels equivalent to the brightest cells in the field (indicated with arrows in panels E-H). These results suggest that hepatocytes with compromised mitochondrial function 4.5 hours after APAP treatment are more likely to recover mitochondrial polarization than to progress to necrosis. However, the potential for mitochondrial repolarization appears to be lost once a stressed hepatocyte has lost plasma membrane integrity; hepatocytes with cytosolic dextran or PI were never observed to regain mitochondrial polarization.
While nearly all of the hepatocytes labeled with dextran or PI completely lacked R123 labeling, we found a few examples of permeabilized cells with functional mitochondria, cells whose cytosol was labeled with both fluorescent dextran and R123 (indicated with arrowheads in Figures 3G and H). Since, as mentioned above, we never observed recovery of R123 fluorescence in a permeabilized cell, and most of these cells subsequently lost R123 fluorescence over the next 1.5 hours, cells labeled with both dextran and R123 may represent a transient stage in the progression of necrosis. These observations suggest that plasma membrane integrity may be lost prior to complete loss of mitochondrial polarization, although the very low incidence of cells labeled with both R123 and dextran suggests that mitochondrial depolarization and plasma membrane disintegration may occur almost simultaneously. Consistent with this, Figures 3G and H show a single cell (indicated with double-arrowheads) that lost both plasma membrane integrity and mitochondrial polarization over the 1.5 hr interval.
As in Figure 2, these images show a remarkable heterogeneity in the cellular effects of APAP that is not apparent in H&E histology. Within the injured regions we find multiple examples of individual necrotic cells among healthy and stressed cells and examples of individual healthy or stressed cells among necrotic cells. The time-series shown in Figure 3 demonstrates that these “islands” are persistent; over a period of two hours, injury does not spread from necrotic cells to their neighbors. Indeed, in many cases mitochondrial polarization recovers in stressed hepatocytes that are surrounded by necrotic cells (arrows in Figures 3G–I). Taken together, these observations are inconsistent with the idea that the transfer of toxic materials from necrotic cells to their neighbors plays a significant role in the early progression of APAP-mediated injury.
In order to characterize the early effects of APAP on mitochondrial function, images were collected from mice as soon as 2 hours after APAP administration (Figure 4). As compared with images collected 4.5-6 hours after APAP, necrosis was less extensive, but the regions of reduced R123 fluorescence were more pronounced and abundant. Quantifications of images collected from 4 animals showed that 40.2 ± 5% of the imaged area was depolarized 2-2.5 hours after APAP administration, whereas only 2.3 ± 0.2% of the field was necrotic.
Figure 4 – Images of the livers of living mice collected sequentially over the period from 2 to 4 hours after APAP treatment.
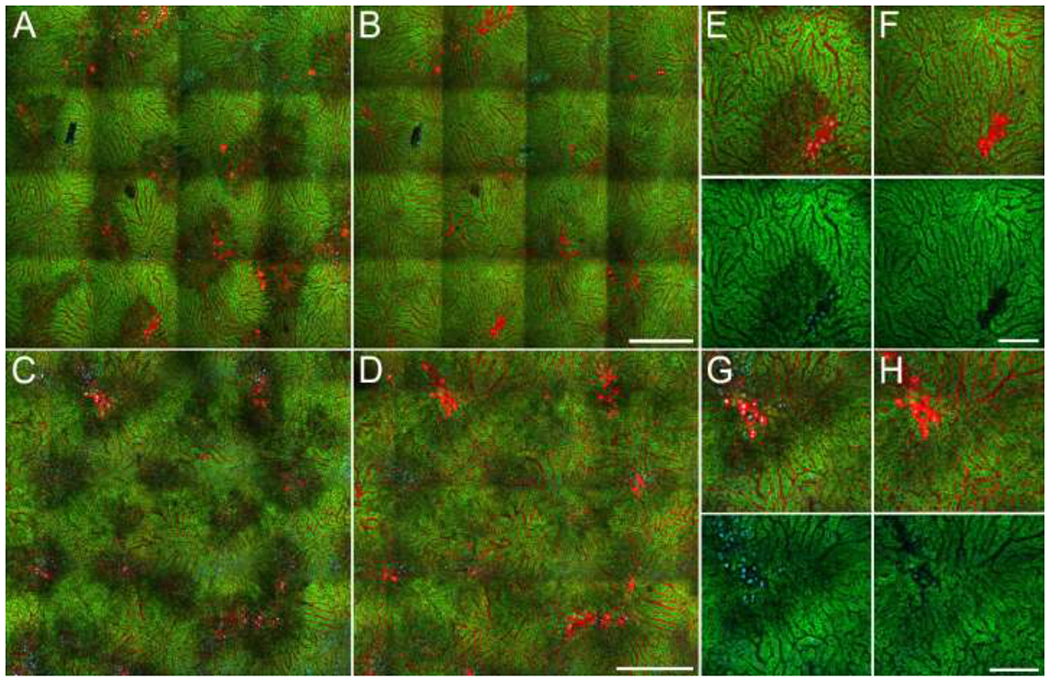
A, C – Fields collected 2 and 2.5 hours, respectively after administration of 250 mg/kg APAP. B, D – Same fields collected 4 hours after APAP administration. E, F – Enlarged region from top left of images shown in panels A and B, respectively. G, H - Enlarged region from the top left of the images shown in panels C and D, respectively. Green levels were adjusted identically for the two time points of each study. Scale bars represent a length of 300 microns in panels A-D, and a length of 100 microns in panels E-H.
As in the images collected between 4.5 and 6 hours after APAP administration, patches of necrotic cells were generally found in the midst of the large regions of intact cells with diminished mitochondrial function (stressed hepatocytes). Images collected from these same fields 4 hours after treatment showed that, rather than progressing to necrosis, hepatocytes in these regions subsequently regained mitochondrial polarization, in some cases to levels similar to those of the unaffected regions. The high magnification fields (Figures 4E–H) show examples of necrotic cells in the midst of intact, stressed hepatocytes that, rather than progressing to necrosis themselves, regain mitochondrial polarization over the next 2 hours. As in the studies conducted at later time points, these studies provide no evidence that necrotic cells have a negative impact on their immediate neighbors.
In order to quantify changes in mitochondrial polarization, we identified depolarized regions in the first field of each time series, blinded to the changes occurring in subsequent time series. For each region, we then quantified the ratio of R123 fluorescence within the depolarized region to that of an immediately adjacent unaffected region (as described in Methods). We then measured R123 fluorescence ratios in these same regions at each subsequent time point. Results of the analysis of the two fields shown in Figure 4 demonstrate a significant recovery of mitochondrial polarization in both studies (Figures 5A and 5B). For comparison, each figure also includes ratiometric measurements obtained from necrotic cells; as expected these cells show no increase in R123 fluorescence over the interval. Figure 5C shows the combined results obtained from studies of seven APAP-treated mice (3 conducted 2-4 hours after APAP treatment and 4 conducted 4-6 hours after APAP treatment). Although the bulk of the recovery occurs within 4 hours of treatment, R123 ratios continue to increase over time, so that ratios in recovering cells, which are significantly depressed during the first few hours, increase to the point that they are statistically indistinguishable from vehicle-treated mice within 6 hours.
Figure 5 – Recovery of mitochondrial polarization in hepatocytes following APAP treatment.
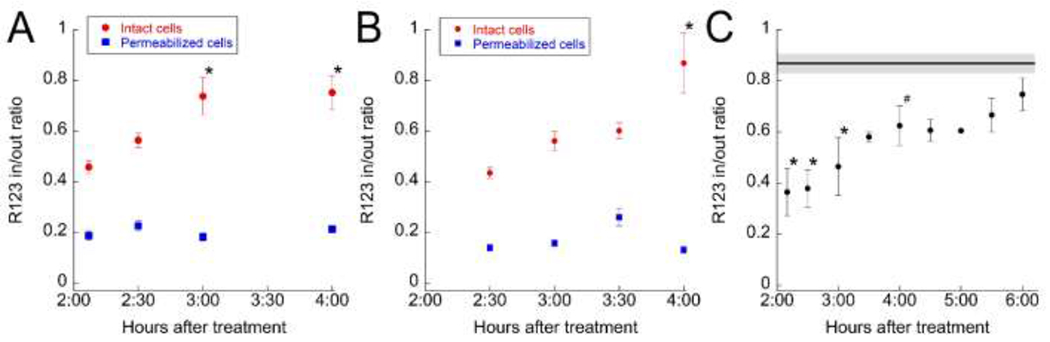
(A) Graph of the ratios of R123 fluorescence in depolarized regions to those in the adjacent unaffected regions over time for the study shown in Figures 5A and 5B. Red circles – ratios of measurements obtained from non-necrotic, depolarized regions to those in the adjacent unaffected regions (means of 6 regions). * - Differs significantly from 2 hr mean P < .01, ANOVA with Dunnett’s multiple comparisons test. Blue squares – ratios of measurements from permeabilized cells to those of cells in the adjacent unaffected regions (18 cells from 5 regions). (B) Graph of the ratios of R123 fluorescence in depolarized regions to those in the adjacent unaffected regions over time for the study shown in Figures 5C and 5D. Red circles – ratios of measurements obtained from non-necrotic, depolarized regions to those in the adjacent unaffected regions (means of 8 regions). * - Differs significantly from 2.5 hr mean, P < .01, ANOVA with Dunnett’s multiple comparisons test. Blue squares – ratios of measurements from permeabilized cells to those of cells in the adjacent unaffected regions (11 cells from 4 regions). (C) Combined analysis of results from 7 APAP-treated mice (each point represents the mean of between 2 and 5 mice, with between 4 and 10 regions for each mouse). Line across top represents mean ratio of vehicle treated mice (plus or minus standard error) determined from the pericentral/periportal fluorescence ratios measured for 7 vehicle-treated mice, with between 4 and 5 regions for each mouse. * - Differs significantly from vehicle P < .01, # - Differs significantly from vehicle P < .05, ANOVA with Dunnett’s multiple comparisons test.
Discussion
The studies described here were designed to characterize the spatio-temporal relationship between mitochondrial depolarization and necrosis in the early stages of APAP-induced liver injury. Mitochondrial injury is a primary event in APAP hepatotoxicity and the restoration of mitochondrial function among surviving hepatocytes is critical to recovery(Du et al., 2017b; Hu et al., 2016; McGill et al., 2012a; McGill et al., 2012b). The long-term restoration of mitochondrial function is accomplished through mitochondrial biogenesis(Du et al., 2017b), occurring over the 24-72 hour period following APAP administration. However, hepatocytes have the potential to recover mitochondrial function prior to mitochondrial biogenesis; previous studies have demonstrated that hepatocytes in mice treated with a dose of APAP insufficient to cause detectible injury experience mitochondrial depolarization within 6 hours of treatment, but that mitochondrial polarization is subsequently regained within 24 hrs (Hu et al., 2016). Based upon the assumption that APAP-induced stress decreases with distance from the centrilobular foci of necrosis, we postulated that mitochondrial repolarization might be a factor in recovery even at hepatotoxic doses of APAP, and that recovery would manifest itself in a pattern of hepatocytes experiencing a reversible loss of mitochondrial polarization surrounding centrilobular regions of necrosis.
Consistent with our prediction, intravital microscopy studies of mice treated with a hepatotoxic dose of APAP demonstrated that APAP induces a rapid partial depolarization of hepatocyte mitochondria in large regions surrounding centrilobular foci of necrosis but, rather than progressing to necrosis, the vast majority of affected hepatocytes regain mitochondrial polarization, such that it approaches normal levels within 6 hours of APAP administration.
The mechanism underlying the recovery of mitochondrial polarization is unclear, but the timeframe, occurring between 2 and 6 hrs after APAP administration, appears to be inconsistent with mitochondrial biogenesis, which was found to restore electron chain activity over the 24-72 hour period following APAP treatment(Du et al., 2017b; Ni et al., 2013). The rapid recovery of R123 fluorescence is thus likely to reflect repolarization of individual mitochondria, rather than generation of a new population of mitochondria. The mechanism underlying reversible depolarization of mitochondria is incompletely understood, but may reflect induction of a reversible mitochondrial permeability transition (MPT) via transient activation of c-jun N-terminal kinase (JNK)(Hu et al., 2016) or a JNK-independent loss of function preceding the MPT(Hanawa et al., 2008), possibly resulting from oxidative stress(Reid et al., 2005).
Our observations of mitochondrial repolarization occurring in the context of hepatotoxic injury differ from those of Hu et al. who found no evidence of mitochondrial repolarization in mice treated at hepatotoxic doses of APAP(Hu et al., 2016). Although the experimental approach of these studies is very similar to that used in our studies (similar dose, same mouse strain, intravital multiphoton microscopy using the same fluorescent probes), we believe that these differences reflect critical differences in experimental design. First, our studies characterized an earlier period of time (2-6 hours vs 6-12 hours after APAP treatment). Second, our longitudinal approach allowed us to characterize the same field of cells over time, to unequivocally detect transient depolarization and to identify the recovery of mitochondrial potential in individual cells. Third, our studies were conducted over much larger areas, enhancing our ability to detect recovery in a spatially-variable injury process.
Based upon common models of APAP-induced liver injury, we had anticipated that our longitudinal studies would detect a chronology of injury in which mitochondrial depolarization would precede the loss of plasma membrane integrity. We were surprised to find that, in exhaustive examinations of hundreds of individual hepatocytes, we never observed an example of an intact cell with depolarized mitochondria that subsequently lost its plasma membrane integrity (i.e., became accessible to either propidium iodide or TexasRed dextran). Rather, we found a few examples of cells that appeared to lose plasma membrane integrity prior to a complete, and irreversible loss of mitochondrial polarization. The speed with which this occurred, along with the rarity of its observation suggest that the irreversible mitochondrial depolarization and the loss of plasma membrane integrity occur nearly simultaneously.
These studies also revealed a granular distribution of necrosis that is inconsistent the hypothesis that communication between neighboring cells plays an important role in either the spread of injury(Naiki-Ito et al., 2010; Patel et al., 2012; Saito et al., 2014) or protection(Saito et al., 2014) between cells during the early stages of APAP hepatotoxicity. Throughout these studies we find examples of individual healthy, intact cells surrounded by necrotic cells, and individual necrotic cells surrounded by healthy, intact cells. Time-series studies demonstrate that these “islands” are persistent; over a period of hours, injury does not spread from individual necrotic cells to their neighbors. Indeed, in many cases mitochondrial polarization recovers in hepatocytes surrounded by necrotic cells. These results are consistent with studies demonstrating that APAP disrupts tight junctions, even at very low doses(Gamal et al., 2017).
The ability to resolve multiple probes at sub-cellular resolution over time in a living animal makes intravital microscopy uniquely capable of detecting changes in the physiology of individual cells in vivo, a capability that was critical to our detecting and characterizing mitochondrial repolarization early in APAP hepatotoxicity. However, intravital microscopy is limited in certain respects. A significant limitation is that the area of the liver that can be imaged at sub-cellular resolution is relatively small. The inverse relation between resolution and field size limits the horizontal dimension of fields collected at high-resolution, and light scatter in liver tissue limits the depth to which multiphoton fluorescence excitation images can be collected to a depth of less than 100 microns. Although we addressed this issue by assembling large mosaics from multiple contiguous image volumes, our studies were nonetheless limited to volumes less than 1/20th of a cubic millimeter, a volume that compromised our ability to capture the progression of large-scale necrosis. The spatial variability of APAP-induced liver injury is such that regions of extensive necrosis are only sporadically encountered at the depths accessible to intravital microscopy. While it is relatively easy to browse around the liver and find large necrotic regions 4 hours after APAP treatment, the likelihood of selecting a field 2 hours after APAP treatment where one of these regions will subsequently develop is very small. The fact that the image volumes we collected 2-4 hours after APAP did not capture the formation of the large necrotic regions that we found in volumes collected 4-6 hours after APAP suggests that the volumes collected at 2 hours were too small to reliably include regions that subsequently progressed to major necrotic injury. While intravital microscopy is ideally suited to characterizing physiological changes occurring in individual cells in regions of drug-induced injury, it must be combined with complementary histological and biochemical techniques, to interpret the cellular observations in the context of the global effects on liver function.
An additional issue is that in vivo labeling of cells depends upon systemic processes of delivery and clearance. Unlike studies of cultured cells, where one can maintain a constant extracellular concentration, probes such as R123, Hoechst and propidium iodide must be replenished to maintain cellular labeling in the setting of continuous systemic clearance. While this can be accomplished by continuous infusion, we elected to inject R123 and propidium prior as needed over the course of our longitudinal studies, to limit adverse effects that might accrue over a 2 hour imaging session. The dependence on vascular delivery also means that studies of pathological conditions must be interpreted with an appreciation that a probe’s fluorescence may reflect not only the physiological readout of interest, but also alterations in perfusion and/or transport. In this regard, we were pleased to find that our studies did not appear to be compromised by loss of perfusion to injured regions; fluorescent dextran accessed cells throughout regions of necrosis and nearly every necrotic region included individual cells with strong R123 fluorescence.
In summary, while mitochondrial function may be restored by biogenesis of new mitochondria occurring over a period of 3 days following APAP treatment(Du et al., 2017b; Ni et al., 2013), our studies demonstrate that for many, if not most, hepatocytes, mitochondrial function is largely restored within the first 6 hours of APAP treatment through rapid repolarization of existing mitochondria. Given the critical role of mitochondrial function in recovery, mechanisms underlying mitochondrial damage and recovery would seem to be ideal targets for therapeutic interventions for APAP-induced liver injury. Striking therapeutic results have been obtained from agents that promote replenishment of mitochondria through biogenesis(Du et al., 2017b) or act to preserve mitochondrial viability by reducing oxidative stress(Du et al., 2017a; Du et al., 2015; Du et al., 2016a; Du et al., 2016b; Xie et al., 2015), inhibiting MPT(Hu et al., 2016) or inhibiting JNK signaling upstream of MPT(Hu et al., 2016; Huo et al., 2017; Xie et al., 2015). Indeed, given its central role in drug hepatotoxicity(Ramachandran et al., 2018; Yuan and Kaplowitz, 2013), the mitochondrion may be a promising target for therapeutics designed to treat drug-induced liver injury in general.
Supplementary Material
Acknowledgments
These studies were supported by funding from the National Institutes of Health (U01 GM111243 and P30 DK079312). Intravital microscopy was conducted at the Indiana Center for Biological Microscopy. Methods of intravital microscopy are based upon refinements of techniques previously developed by Jennifer Ryan.
Abbreviations:
- APAP
acetaminophen
- NAPQI
N-acetyl-p-benzoquinone imine
- GSH
glutathione
- MPT
mitochondrial permeability transition
- AST
alanine aminotransferase (ALT) and aspartate aminotransferase (AST)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Annaert PP, and Brouwer KL. 2005. Assessment of drug interactions in hepatobiliary transport using rhodamine 123 in sandwich-cultured rat hepatocytes. Drug Metab Dispos. 33:388–394. [DOI] [PubMed] [Google Scholar]
- Anundi I, Lahteenmaki T, Rundgren M, Moldeus P, and Lindros KO. 1993. Zonation of acetaminophen metabolism and cytochrome P450 2E1-mediated toxicity studied in isolated periportal and perivenous hepatocytes. Biochem Pharmacol. 45:1251–1259. [DOI] [PubMed] [Google Scholar]
- Babbey CM, Ryan JC, Gill EM, Ghabril MS, Burch CR, Paulman A, and Dunn KW. 2012. Quantitative intravital microscopy of hepatic transport. Intravital. 1:10. [Google Scholar]
- Dai G, He L, Chou N, and Wan YJ. 2006. Acetaminophen metabolism does not contribute to gender difference in its hepatotoxicity in mouse. Toxicol Sci. 92:33–41. [DOI] [PubMed] [Google Scholar]
- Du K, Farhood A, and Jaeschke H. 2017a. Mitochondria-targeted antioxidant Mito-Tempo protects against acetaminophen hepatotoxicity. Arch Toxicol. 91:761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, McGill MR, Xie Y, Bajt ML, and Jaeschke H. 2015. Resveratrol prevents protein nitration and release of endonucleases from mitochondria during acetaminophen hepatotoxicity. Food Chem Toxicol. 81:62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Ramachandran A, and Jaeschke H. 2016a. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol. 10:148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Ramachandran A, McGill MR, Mansouri A, Asselah T, Farhood A, Woolbright BL, Ding WX, and Jaeschke H. 2017b. Induction of mitochondrial biogenesis protects against acetaminophen hepatotoxicity. Food Chem Toxicol. 108:339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Ramachandran A, Weemhoff JL, Chavan H, Xie Y, Krishnamurthy P, and Jaeschke H. 2016b. Editor’s Highlight: Metformin Protects Against Acetaminophen Hepatotoxicity by Attenuation of Mitochondrial Oxidant Stress and Dysfunction. Toxicol Sci. 154:214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KW, and Ryan JC. 2017. Using quantitative intravital multiphoton microscopy to dissect hepatic transport in rats. Methods. 128:40–51. [DOI] [PubMed] [Google Scholar]
- Gamal W, Treskes P, Samuel K, Sullivan GJ, Siller R, Srsen V, Morgan K, Bryans A, Kozlowska A, Koulovasilopoulos A, Underwood I, Smith S, Del-Pozo J, Moss S, Thompson AI, Henderson NC,Hayes PC, Plevris JN, Bagnaninchi PO, and Nelson LJ. 2017. Low-dose acetaminophen induces early disruption of cell-cell tight junctions in human hepatic cells and mouse liver. Sci Rep. 7:37541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, and Kaplowitz N. 2008. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem. 283:13565–13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson JA, Roberts DW, and James LP. 2010. Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol:369–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Ramshesh VK, McGill MR, Jaeschke H, and Lemasters JJ. 2016. Low Dose Acetaminophen Induces Reversible Mitochondrial Dysfunction Associated with Transient c-Jun N-Terminal Kinase Activation in Mouse Liver. Toxicol Sci. 150:204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y, Win S, Than TA, Yin S, Ye M, Hu H, and Kaplowitz N. 2017. Antcin H Protects Against Acute Liver Injury Through Disruption of the Interaction of c-Jun-N-Terminal Kinase with Mitochondria. Antioxid Redox Signal. 26:207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FC, Huang GT, Lin CJ, Wang SS, Sun TL, Lo SY, Lo W, Chiou LL, Dong CY, and Lee HS.2011. Apical membrane rupture and backward bile flooding in acetaminophen-induced hepatocyte necrosis. Cell Death Dis. 2:e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Thorling C, Jin L and Roberts M. 2012. Intravital multiphoton imaging of rhodamine 123 in the rat liver after intravenous dosing. Intravital. 1:54–59. [Google Scholar]
- McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, and Jaeschke H. 2012a. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 122:1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Williams CD, Xie Y, Ramachandran A, and Jaeschke H. 2012b. Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol Appl Pharmacol. 264:387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JR, Jollow DJ, Potter WZ, Davis DC, Gillette JR, and Brodie BB. 1973a. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J Pharmacol Exp Ther. 187:185–194. [PubMed] [Google Scholar]
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, and Brodie BB. 1973b. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther. 187:211–217. [PubMed] [Google Scholar]
- Moles A, Torres S, Baulies A, Garcia-Ruiz C, and Fernandez-Checa JC. 2018. Mitochondrial-Lysosomal Axis in Acetaminophen Hepatotoxicity. Front Pharmacol. 9:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naiki-Ito A, Asamoto M, Naiki T, Ogawa K, Takahashi S, Sato S, and Shirai T. 2010. Gap junction dysfunction reduces acetaminophen hepatotoxicity with impact on apoptotic signaling and connexin 43 protein induction in rat. Toxicol Pathol. 38:280–286. [DOI] [PubMed] [Google Scholar]
- National Research Council (U.S.). Committee for the Update of the Guide for the Care and Use of Laboratory Animals., Institute for Laboratory Animal Research (U.S.), and National Academies Press (U.S.) 2011. Guide for the care and use of laboratory animals. National Academies Press,, Washington, D.C: xxv, 220 p. [Google Scholar]
- Ni HM, Williams JA, Jaeschke H, and Ding WX. 2013. Zonated induction of autophagy and mitochondrial spheroids limits acetaminophen-induced necrosis in the liver. Redox Biol. 1:427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SJ, Milwid JM, King KR, Bohr S, Iracheta-Vellve A, Li M, Vitalo A, Parekkadan B, Jindal R, and Yarmush ML. 2012. Gap junction inhibition prevents drug-induced liver toxicity and fulminant hepatic failure. Nat Biotechnol. 30:179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires DA, Marques PE, Pereira RV, David BA, Gomides LF, Dias AC, Nunes-Silva A, Pinho V, Cara DC, Vieira LQ, Teixeira MM, and Menezes GB. 2014. Interleukin-4 deficiency protects mice from acetaminophen-induced liver injury and inflammation by prevention of glutathione depletion. Inflamm Res. 63:61–69. [DOI] [PubMed] [Google Scholar]
- Ramachandran A, and Jaeschke H. 2017. Mechanisms of acetaminophen hepatotoxicity and their translation to the human pathophysiology. J Clin Transl Res. 3:157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, and Jaeschke H. 2018. Acetaminophen Toxicity: Novel Insights Into Mechanisms and Future Perspectives. Gene Expr. 18:19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Visschers RGJ, Duan L, Akakpo JY, and Jaeschke H. 2018. Mitochondrial dysfunction as a mechanism of drug-induced hepatotoxicity: current understanding and future perspectives. J Clin Transl Res. 4:75–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman H, Sun J, Shi Y, Ramshesh VK, Liu Q, Currin RT, Lemasters JJ, and Zhong Z. 2011. NIM811 prevents mitochondrial dysfunction, attenuates liver injury, and stimulates liver regeneration after massive hepatectomy. Transplantation. 91:406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid AB, Kurten RC, McCullough SS, Brock RW, and Hinson JA. 2005. Mechanisms of acetaminophen-induced hepatotoxicity: role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J Pharmacol Exp Ther. 312:509–516. [DOI] [PubMed] [Google Scholar]
- Roberts DW, Bucci TJ, Benson RW, Warbritton AR, McRae TA, Pumford NR, and Hinson JA. 1991. Immunohistochemical localization and quantification of the 3-(cystein-S-yl)-acetaminophen protein adduct in acetaminophen hepatotoxicity. Am J Pathol. 138:359–371. [PMC free article] [PubMed] [Google Scholar]
- Ryan J, Morgan RE, Chen Y, Volak LP, Dunn RT 2nd, and Dunn KW. 2018. Intravital Multiphoton Microscopy with Fluorescent Bile Salts in Rats as an In Vivo Biomarker for Hepatobiliary Transport Inhibition. Drug Metab Dispos. 46:704–718. [DOI] [PubMed] [Google Scholar]
- Ryan JC, Dunn KW, and Decker BS. 2014. Effects of chronic kidney disease on liver transport: quantitative intravital microscopy of fluorescein transport in the rat liver. Am J Physiol Regul Integr Comp Physiol. 307:R1488–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito C, Shinzawa K, and Tsujimoto Y. 2014. Synchronized necrotic death of attached hepatocytes mediated via gap junctions. Sci Rep. 4:5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theruvath TP, Snoddy MC, Zhong Z, and Lemasters JJ. 2008. Mitochondrial permeability transition in liver ischemia and reperfusion: role of c-Jun N-terminal kinase 2. Transplantation. 85:1500–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liang X, Mohammed YH, Thomas JA, Bridle KR, Thorling CA, Grice JE, Xu ZP, Liu X, Crawford DH, and Roberts MS. 2015. Real-time histology in liver disease using multiphoton microscopy with fluorescence lifetime imaging. Biomed Opt Express. 6:780–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Win S, Than TA, Han D, Petrovic LM, and Kaplowitz N. 2011. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J Biol Chem. 286:35071–35078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Ramachandran A, Breckenridge DG, Liles JT, Lebofsky M, Farhood A, and Jaeschke H. 2015. Inhibitor of apoptosis signal-regulating kinase 1 protects against acetaminophen-induced liver injury. Toxicol Appl Pharmacol. 286:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan HM, Ramachandran A, Bajt ML, Lemasters JJ, and Jaeschke H. 2010. The oxygen tension modulates acetaminophen-induced mitochondrial oxidant stress and cell injury in cultured hepatocytes. Toxicol Sci. 117:515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, and Kaplowitz N. 2013. Mechanisms of drug-induced liver injury. Clin Liver Dis. 17:507–518, vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.