

Key Points
Question
What is the effect of convalescent plasma therapy added to standard treatment, compared with standard treatment alone, on clinical outcomes in patients with severe or life-threatening coronavirus disease 2019 (COVID-19)?
Finding
In this randomized clinical trial that included 103 patients and was terminated early, the hazard ratio for time to clinical improvement within 28 days in the convalescent plasma group vs the standard treatment group was 1.40 and was not statistically significant.
Meaning
Among patients with severe or life-threatening COVID-19, convalescent plasma therapy added to standard treatment did not significantly improve the time to clinical improvement within 28 days, although the trial was terminated early and may have been underpowered to detect a clinically important difference.
Abstract
Importance
Convalescent plasma is a potential therapeutic option for patients with coronavirus disease 2019 (COVID-19), but further data from randomized clinical trials are needed.
Objective
To evaluate the efficacy and adverse effects of convalescent plasma therapy for patients with COVID-19.
Design, Setting, and Participants
Open-label, multicenter, randomized clinical trial performed in 7 medical centers in Wuhan, China, from February 14, 2020, to April 1, 2020, with final follow-up April 28, 2020. The trial included 103 participants with laboratory-confirmed COVID-19 that was severe (respiratory distress and/or hypoxemia) or life-threatening (shock, organ failure, or requiring mechanical ventilation). The trial was terminated early after 103 of a planned 200 patients were enrolled.
Intervention
Convalescent plasma in addition to standard treatment (n = 52) vs standard treatment alone (control) (n = 51), stratified by disease severity.
Main Outcomes and Measures
Primary outcome was time to clinical improvement within 28 days, defined as patient discharged alive or reduction of 2 points on a 6-point disease severity scale (ranging from 1 [discharge] to 6 [death]). Secondary outcomes included 28-day mortality, time to discharge, and the rate of viral polymerase chain reaction (PCR) results turned from positive at baseline to negative at up to 72 hours.
Results
Of 103 patients who were randomized (median age, 70 years; 60 [58.3%] male), 101 (98.1%) completed the trial. Clinical improvement occurred within 28 days in 51.9% (27/52) of the convalescent plasma group vs 43.1% (22/51) in the control group (difference, 8.8% [95% CI, −10.4% to 28.0%]; hazard ratio [HR], 1.40 [95% CI, 0.79-2.49]; P = .26). Among those with severe disease, the primary outcome occurred in 91.3% (21/23) of the convalescent plasma group vs 68.2% (15/22) of the control group (HR, 2.15 [95% CI, 1.07-4.32]; P = .03); among those with life-threatening disease the primary outcome occurred in 20.7% (6/29) of the convalescent plasma group vs 24.1% (7/29) of the control group (HR, 0.88 [95% CI, 0.30-2.63]; P = .83) (P for interaction = .17). There was no significant difference in 28-day mortality (15.7% vs 24.0%; OR, 0.59 [95% CI, 0.22-1.59]; P = .30) or time from randomization to discharge (51.0% vs 36.0% discharged by day 28; HR, 1.61 [95% CI, 0.88-2.95]; P = .12). Convalescent plasma treatment was associated with a negative conversion rate of viral PCR at 72 hours in 87.2% of the convalescent plasma group vs 37.5% of the control group (OR, 11.39 [95% CI, 3.91-33.18]; P < .001). Two patients in the convalescent plasma group experienced adverse events within hours after transfusion that improved with supportive care.
Conclusion and Relevance
Among patients with severe or life-threatening COVID-19, convalescent plasma therapy added to standard treatment, compared with standard treatment alone, did not result in a statistically significant improvement in time to clinical improvement within 28 days. Interpretation is limited by early termination of the trial, which may have been underpowered to detect a clinically important difference.
Trial Registration
Chinese Clinical Trial Registry: ChiCTR2000029757
This randomized trial compares the effects of convalescent plasma therapy with standard care vs standard care alone on time to clinical improvement among patients with severe or life-threatening COVID-19 disease in China.
Introduction
Since December 2019, coronavirus disease 2019 (COVID-19), caused by SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), has spread rapidly around the world, with high rates of transmission and substantial mortality. COVID-19 symptoms can range from mild, self-limited respiratory disease to severe progressive pneumonia, multiple organ failure, and even death.1,2 To date, there is no effective treatment or vaccine to contain the disease.
Convalescent plasma therapy has been used to treat patients with infections using plasma collected from recovered patients.3 This approach has been evaluated in the treatment of SARS,4,5 Middle East respiratory syndrome (MERS),6 and Ebola,7 but not well studied and with no definitive results. Recently, case series from China reported improved outcomes after COVID-19 convalescent plasma transfusion.8,9 The US Food and Drug Administration (FDA) recently approved the emergency use of convalescent plasma for patients with severe or life-threatening COVID-19.10 Although the use of convalescent plasma shows promise, the evidence supporting its use in the treatment of COVID-19 remains limited, and thus its use remains investigational.
In addition, due to the limited understanding of the mechanism and precise therapeutic components of convalescent plasma, there is no standardization or evidence-based rationale for donor selection, quality control of convalescent plasma, or recipient transfusion indications for convalescent plasma. This may help to explain the varied therapeutic effects of convalescent plasma seen in a variety of infectious diseases. To address these issues, the World Health Organization issued a guideline on the use of convalescent plasma in a pandemic, advocating for standardization in donor selection and convalescent plasma quality control to maximize therapeutic potency.11
The objective of this randomized clinical trial was to evaluate the efficacy and adverse effects of convalescent plasma added to standard treatment, compared with standard treatment alone, for patients with severe or life-threatening COVID-19 using a standardized approach in donor selection and convalescent plasma quality control.
Methods
This study was a collaborative effort by the Institute of Blood Transfusion of the Chinese Academy of Medical Sciences; Union Hospital of Tongji Medical College of Huazhong University of Science and Technology; the Guanggu District Maternal and Child Health Hospital of Hubei Province; Tongji Hospital of Tongji Medical College of Huazhong University of Science and Technology; General Hospital of the Central Theater Command of the People's Liberation Army; Wuhan Red Cross Hospital; Wuhan Asia Heart Hospital; and Wuhan Pulmonary Hospital.
This study was approved by the ethics committee of the Institute of Blood Transfusion of the Chinese Academy of Medical Sciences and the ethics committees of the participating hospitals. Written informed consent was obtained from all study participants or their legal representatives. The trial protocol and statistical analysis plan are available in Supplement 1 and Supplement 2.
Participants
Patients were recruited from 7 medical centers. The study recruitment was from February 14, 2020, to April 1, 2020. Follow-up was completed on April 28, 2020.
Inclusion Criteria
Inclusion criteria were the following: (1) signed informed consent; (2) aged at least 18 years; (3) COVID-19 diagnosis based on polymerase chain reaction (PCR) testing; (4) positive PCR result within 72 hours prior to randomization; (5) pneumonia confirmed by chest imaging; (6) clinical symptoms meeting the definitions of severe or life-threatening COVID-19; (7) acceptance of random group assignment; (8) hospital admission; (9) willingness to participate in all necessary research studies and be able to complete the study follow-up; and (10) no participation in other clinical trials, such as antiviral trials, during the study period.
Severe COVID-19 was defined as respiratory distress (≥30 breaths/min; in resting state, oxygen saturation of 93% or less on room air; or arterial partial pressure of oxygen (Pao2)/fraction of inspired oxygen (Fio2) of 300 or less. Life-threatening COVID-19 was defined as respiratory failure requiring mechanical ventilation; shock; or other organ failure (apart from lung) requiring intensive care unit (ICU) monitoring.
Exclusion Criteria
Exclusion criteria were the following: (1) pregnancy or lactation; (2) immunoglobulin allergy; (3) IgA deficiency; (4) preexisting comorbidity that could increase the risk of thrombosis; (5) life expectancy less than 24 hours; (6) disseminated intravascular coagulation; (7) severe septic shock; (8) Pao2/Fio2 of less than 100; (9) severe congestive heart failure; (10) detection of high titer of S protein–RBD-specific (receptor binding domain) IgG antibody (≥1:640); (11) other contraindications as determined by the patient’s physicians; and (12) participation in any antiviral clinical trials for COVID-19 within 30 days prior to enrollment.
Randomization
Potential study participants were screened for eligibility 72 hours prior to study randomization. Patients were randomly assigned via computer-generated random numbering (1:1) to receive standard treatment coupled with convalescent plasma transfusion or standard treatment alone (control group) (Figure 1). The randomization was stratified based on the severity of COVID-19 (severe or life-threatening) and a randomization schedule was generated using block randomization with block size of 4 for each type of COVID-19 by SAS software. Patients and clinicians were not masked to treatment assignment.
Figure 1. Patient Enrollment and Treatment Assignment.

PCR indicates polymerase chain reaction; CHF, congestive heart failure; COVID-19, coronavirus disease 2019.
Procurement of Convalescent Plasma
In brief, patients with a laboratory-confirmed COVID-19 diagnosis, who had fully recovered and been discharged from the hospital for more than 2 weeks, were recruited. Convalescent plasma–specific donor screening and selection were based on the following criteria: age of 18 through 55 years, suitable for blood donation, initially diagnosed with COVID-19 but with 2 negative PCR test results from nasopharyngeal swabs (at least 24 hours apart) prior to hospital discharge, discharged for more than 2 weeks from the hospital, and no persisting COVID-19 symptoms. Convalescent plasma collection was performed based on routine plasma collection procedures via plasmapheresis. The plasma products were prepared as fresh-frozen plasma. COVID-19 convalescent plasma was collected and processed at the Wuhan Blood Center.
S-RBD–specific IgG antibody titer was measured for convalescent plasma products and reported as the following: less than 1:160, 1:160, 1:320, 1:640, 1: 1280, or greater than 1:1280. There was a positive correlation between the SARS-CoV-2 viral neutralization titer and the S-RBD–specific IgG titer (r = 0.622, P = .03). A serum neutralization titer of 1:80 is approximately equivalent to a titer of 1:1280 for S-RBD–specific IgG. To ensure the therapeutic potency of the convalescent plasma, only the plasma units with an S-RBD–specific IgG titer of at least 1:640 were used for this study.
Additional details regarding plasma preparation process can be found in the eMethods in Supplement 3, and the preparation requirements of convalescent plasma used were similar to the recently updated FDA recommendations.10
Convalescent Plasma Transfusion
The transfusion dose of COVID-19 convalescent plasma was approximately 4 to 13 mL/kg of recipient body weight. ABO type of the convalescent plasma transfused was compatible with the patient’s ABO type. In addition, the convalescent plasma was crossmatched with the patient’s red blood cells to ensure compatibility. Convalescent plasma transfusion was administered at approximately 10 mL for the first 15 minutes, which was then increased to approximately 100 mL per hour with close monitoring. Adjustments in the infusion rates were allowed based on the patient’s risk for volume overload and tolerance, at the discretion of the treating physicians. No premedication was given before convalescent plasma transfusion.
Standard Treatment
Standard treatment consisted of symptomatic control and supportive care for COVID-19, mostly based on the evolving Chinese national COVID-19 treatment guidelines and hospital practice.12 Possible treatments included antiviral medications, antibacterial medications, steroids, human immunoglobulin, Chinese herbal medicines, and other medications.
Outcome Measures
The primary end point was time to clinical improvement within a 28-day period. Clinical improvement was defined as patient discharge or a reduction of 2 points on a 6-point disease severity scale.13 The scale was defined as follows: 6 points, death; 5 points, hospitalization plus extracorporeal membrane oxygenation (ECMO) or invasive mechanical ventilation; 4 points, hospitalization plus noninvasive ventilation or high-flow supplemental oxygen; 3 points, hospitalization plus supplemental oxygen (not high-flow or noninvasive ventilation); 2 points, hospitalization with no supplemental oxygen; 1 point, hospital discharge.
Patient discharge criteria included body temperature returned to normal for longer than 3 days, respiratory symptoms significantly improved without the need for oxygen support, and 2 consecutive negative PCR test results from nasopharyngeal swabs at least 24 hours apart.
Secondary clinical outcomes were as follows: (1) 28-day mortality, including analysis of time from randomization to death; (2) duration of hospitalization, including analyses of time from randomization to discharge, time from admission to discharge, and 28-day discharge rates; and (3) conversion of nasopharyngeal swab viral PCR results from positive at baseline to negative at follow-up assessed at 24, 48, and 72 hours. Once nasopharyngeal swab viral PCR testing yielded negative results 2 times consecutively, no further testing was performed.
A post hoc analysis was added to compare rates of improvement at days 7, 14, and 28.
This clinical trial was an open-label, randomized clinical study. To avoid assessment bias, the evaluation of clinical outcomes was performed by an investigator who was blind to the study group allocation.
Statistical Analysis
The original sample size was determined to be 100 for each group, which would provide 80% power, with a 2-sided significance level of α = .05, to detect an 8-day change for the convalescent plasma group in time to clinical improvement, assuming that this would be 20 days in the control group and 60% of the patients would reach clinical improvement.
Unless otherwise stated, analyses were performed based on the full analysis set, which is defined as the set of all randomized patients who received at least one treatment specified in the trial. Statistical analysis was performed on randomly assigned treatment groups. Continuous variables were summarized by presenting the median and interquartile range (IQR) for the total number of patients who contributed values. Categorical variables were summarized by presenting the frequency and proportion of patients in each category. Time-to-event data were analyzed using the Kaplan-Meier method, and the median time to event and corresponding 95% CI were calculated. For the cases in which more than 50% of patients were censored and therefore the median time to event was indeterminate, the restricted mean survival time would be used for post hoc analysis.
For the primary end point of time to clinical improvement, death, withdrawal, and crossover between groups before day 28 were considered to be right-censored at day 28, and otherwise would be considered to be right-censored at the last observation date. Hazard ratios (HRs) with 95% CIs were calculated using Cox proportional hazards models. Three Cox proportional hazards models were fitted in this study. We referred to the model that included only the treatment group as the unadjusted model. The model that included disease severity (severe or life-threatening) and treatment group is referred to as model 1, and the model that further considered the interaction between disease severity and treatment group is referred to as model 2. Study sites were considered as a random effect in these models. Proportionality hazard assumption was assessed for treatment group and disease severity by extending the Cox models to include the corresponding time-dependent covariates.14 If the coefficient of the time-dependent covariate was statistically significant, the proportionality hazard assumption would be considered to be violated.
A per-protocol analysis was performed for the primary end point as a sensitivity analysis. The per-protocol set was defined as the set of all randomized patients who received at least one treatment specified in the trial and who had no significant protocol violations that affected the efficacy evaluation.
Treatment effects for secondary end points were assessed using odds ratios and HRs with 95% CIs for the discrete variables and time-to-event data, respectively. For the analyses of time from randomization to discharge, time from randomization to death, and length of stay, the definition of censoring was consistent with the primary end point. Missing data for secondary outcomes and adverse events were not imputed. Only observed values were used for data analysis and presentation.
Subgroup analyses of efficacy were performed according to disease severity. Interactions between treatment group and disease severity group were tested using model 2.
Statistical analyses were performed with SAS software, version 9.4. Statistical significance was defined using a 2-sided significance level of α = .05. Because of the potential for type I error due to multiple comparisons, findings for analyses of secondary end points should be interpreted as exploratory.
Early Study Termination
Due to the containment of the COVID-19 epidemic in Wuhan, China, the numbers of patients with COVID-19 decreased in late March 2020. No new cases were reported in Wuhan for 7 consecutive days after March 24 (data from the National Health Commission of the People’s Republic of China).15 The last patient enrolled in this study was on March 27, 2020, and for the next 3 days, we were not able to recruit more patients and did not have any recruitment targets. After discussion with the expert committee of the Institute of Blood Transfusion, the study was terminated by the sponsor (Chinese Academy of Medical Sciences) and the leading primary investigator on April 1, 2020, with a total of 103 patients enrolled. There was no interim or preliminary data review prior to making this decision.
Results
Study Population
A total of 103 patients were enrolled in this randomized clinical trial. They were assigned to either the convalescent plasma group or the control group in a 1:1 ratio and were categorized as follows: 23 patients in the convalescent plasma group and 22 patients in the control group had severe COVID-19, and 29 patients in the convalescent plasma group and 29 patients in the control group had life-threatening COVID-19. Of these, 1 patient with life-threatening disease in the convalescent plasma treatment group withdrew from the study and 1 patient with life-threatening disease in the control group received convalescent plasma transfusion after randomization (protocol violation). Thus, 103 patients were included in the full analysis set and 101 patients were included in the per-protocol set (Figure 1).
The median age was 70 years (IQR, 62-78 years) among all patients, 71 years (IQR, 66-82 years) for patients with severe COVID-19, and 69 years (IQR, 61-76 years) for patients with life-threatening COVID-19. Of the patients included in the study, 60 (58.3%) were men, and the percentages of men with severe and life-threatening COVID-19 were 53.3% and 62.1%, respectively. A total of 89.2% of the patients had a normal body temperature at the time of participation, and the median body temperature was 36.5 °C (IQR, 36.2-36.7 °C) (Table 1 and eTable 1 and eTable 2 in Supplement 3).
Table 1. Baseline Demographics and Clinical Characteristics of All Patients With COVID-19a.
| Convalescent plasma group (n = 52) |
Control group (n = 51) |
|
|---|---|---|
| Demographic and clinical characteristics | ||
| Age, median (IQR), y | 70 (62-80) | 69 (63-76) |
| Sex, No. (%) | ||
| Male | 27 (51.9) | 33 (64.7) |
| Female | 25 (48.1) | 18 (35.3) |
| Allergy history, No. (%)b | 6 (11.5) | 5 (9.8) |
| Coexisting diseases, No. (%)c | ||
| Hypertension | 29 (55.8) | 27 (52.9) |
| Cardiovascular disease | 14 (26.9) | 12 (23.5) |
| Cerebrovascular disease | 11 (21.2) | 7 (13.7) |
| Diabetes | 9 (17.3) | 12 (23.5) |
| Liver disease | 5 (9.6) | 5 (9.8) |
| Cancer | 3 (5.8) | 0 |
| Kidney disease | 2 (3.9) | 4 (7.8) |
| Laboratory valuesd | ||
| Body temperature, median (IQR), °C | 36.5 (36.2-36.7) [n = 52] | 36.4 (36.2-36.8) [n = 50] |
| ≥37.3 °C, No. (%) | 4/52 (7.7) | 7/50 (14.0) |
| Respiratory rate >24/min, No. (%) | 11/52 (21.2) | 7/49 (14.3) |
| Heart rate >100/min, No. (%) | 13/52 (25.0) | 8/50 (16.0) |
| Systolic blood pressure >140 mm Hg, No. (%) | 10/52 (19.2) | 15/50 (30.0) |
| White blood cell count, median (IQR), cells/μL | 7590 (6300-11 460) | 7160 (6130-11 200) |
| White blood cell count categories, No. (%) | ||
| <4000/μL | 5 (9.6) | 4 (7.8) |
| 4000-10 000/μL | 31 (59.6) | 29 (56.9) |
| >10 000/μL | 16 (30.8) | 18 (35.3) |
| Neutrophil count, median (IQR), cells/μL | 6050 (4350-9940) | 5310 (4280-9920) |
| Neutrophil count categories, No. (%) | ||
| <1800/μL | 0 | 3 (5.9) |
| 1800-6300/μL | 27 (51.9) | 26 (51.0) |
| >6300/μL | 25 (48.1) | 22 (43.1) |
| Lymphocyte count, median (IQR), cells/μL | 830 (570-1420) | 800 (500-1370) |
| Lymphocyte count categories, No. (%) | ||
| <1000/μL | 32 (61.5) | 32 (62.8) |
| ≥1000/μL | 20 (38.5) | 19 (37.3) |
| Platelet count, median (IQR), ×103/μL | 164.5 (99.0-248.0) | 214.0 (138.0-274.0) |
| Platelet count categories, No. (%) | ||
| <100 × 103/μL | 13 (25.0) | 7 (13.7) |
| ≥100 × 103/μL | 39 (75.0) | 44 (86.3) |
| CRP, median (IQR), mg/L | 20.40 (5.13-65.60) [n = 49] | 8.87 (1.73-40.32) [n = 48] |
| >5 mg/L, No. (%) | 37/49 (75.5) | 29/48 (60.4) |
| IL-6, median (IQR), pg/mL | 16.62 (5.76-73.68) [n = 44] | 21.67 (5.10-64.00) [n = 35] |
| >7 pg/mL, No. (%) | 32/44 (72.7) | 25/35 (71.4) |
| Prothrombin time, median (IQR), s | 13.50 (12.00-15.20) [n = 51] | 13.30 (12.35-14.15) [n = 48] |
| APTT, median (IQR), s | 34.00 (28.80-41.80) [n = 49] | 34.25 (30.05-45.00) [n = 48] |
| Thrombin time, median (IQR), s | 16.45 (14.60-18.20) [n = 46] | 15.80 (15.00-17.70) [n = 48] |
| Fibrinogen, median (IQR), mg/dL | 386 (293-471) [n = 50] | 400 (329-512) [n = 48] |
| D-dimer, median (IQR), μg/mL | 1.88 (0.91-4.78) [n = 47] | 2.23 (0.79-5.21) [n = 46] |
| >0.2 μg/mL, No. (%) | 45/47 (95.7) | 43/46 (93.5) |
| ALT, median (IQR), U/L | 35.05 (22.25-55.90) [n = 52] | 28.50 (18.95-59.50) [n = 48] |
| ALT categories, No. (%) | ||
| ≤50 U/L | 36/52 (69.2) | 33/48 (68.8) |
| >50 U/L | 16/52 (30.8) | 15/48 (31.3) |
| AST, median (IQR), U/L | 28.50 (20.95-42.00) [n = 52] | 24.50 (19.10-33.50) [n = 48] |
| AST categories, No. (%) | ||
| ≤40 U/L | 35/52 (67.3) | 40/48 (83.3) |
| >40 U/L | 17/52 (32.7) | 8/48 (16.7) |
| Urea nitrogen, median (IQR), mg/dL | 20.36 (14.34-28.07) [n = 50] | 20.08 (16.22-32.97) [n = 49] |
| Urea nitrogen categories, No. (%) | ||
| <5.0 mg/dL | 0/50 | 0/49 |
| 5.0-19.9 mg/dL | 23/50 (46.0) | 24/49 (49.0) |
| >19.9 mg/dL | 27/50 (54.0) | 25/49 (51.0) |
| Serum creatinine, median (IQR), mg/dL | 0.75 (0.60-0.89) [n = 50] | 0.83 (0.62-1.04) [n = 49] |
| Serum creatinine categories, No. (%) | ||
| ≤1.5 mg/dL | 46/50 (92.0) | 47/49 (95.9) |
| >1.5 mg/dL | 4/50 (8.0) | 2/49 (4.1) |
Abbreviations: ALT, alanine aminotransferase; APTT, activated partial thromboplastin time; AST, aspartate aminotransferase; COVID-19, coronavirus disease 2019; CRP, C-reactive protein; IQR, interquartile range.
SI conversion factors: To convert D-dimer to nmol/L, multiply values by 5.476; to convert urea nitrogen to mmol/L, multiply values by 0.357; to convert creatinine to μmol/L, multiply values by 88.4.
The values shown were based on total number of patients who contributed values.
History of allergy to certain allergens, including food, medicine, etc.
Details of coexisting diseases were collected from medical records.
The vital signs and laboratory values are the last available values within 72 hours prior to randomization. The laboratory values selected were associated with the clinical status and factors that may affect convalescent plasma therapy. The values used for categorization of laboratory values are local divisions of low, normal, and high values.
The median interval between the onset of symptoms and randomization was 30 days (IQR, 20-39 days) overall, 33 days (IQR, 22-43 days) for patients with severe disease, and 26 days (IQR, 20-36 days) for patients with life-threatening disease. There were 5 patients with severe disease and 3 patients with life-threatening disease who had an interval between the onset of symptoms and randomization that was fewer than 14 days (Table 2).
Table 2. Patients’ Clinical Status at Randomization and Medications Receiveda.
| All patients | Convalescent plasma group (n = 52) | Control group (n = 51) |
|---|---|---|
| Time between symptom onset and randomization, median (IQR), d | 27 (22-39) [n = 49] | 30 (19-38) [n = 48] |
| ≤14 d, No. (%) | 3/49 (6.1) | 5/48 (10.4) |
| >14 d, No. (%) | 46/49 (93.9) | 43/48 (89.6) |
| Interval between symptom onset and admission, median (IQR), d | 12 (5-20) [n = 49] |
10 (6-16) [n = 48] |
| 6-Point scale at study day 1, No. (%) | ||
| 2- Hospitalization, no supplemental oxygen | 1/51 (2.0) | 1/50 (2.0) |
| 3- Hospitalization, requiring supplemental oxygen (not high-flow or noninvasive ventilation) | 15/51 (29.4) | 15/50 (30.0) |
| 4- Hospitalization, requiring noninvasive ventilation and/or high-flow supplemental oxygen | 21/51 (41.2) | 23/50 (46.0) |
| 5- Hospitalization, requiring extracorporeal membrane oxygenation and/or invasive mechanical ventilation | 14/51 (27.5) | 11/50 (22.0) |
| Medications used after randomization | ||
| Antiviral | 41/46 (89.1) | 44/49 (89.8) |
| Antibacterial | 38/46 (82.6) | 39/49 (79.6) |
| Chinese herbal medicine | 26/46 (56.5) | 30/49 (61.2) |
| Steroids | 21/46 (45.7) | 16/49 (32.7) |
| Antifungal | 15/46 (32.6) | 13/49 (26.5) |
| Human immunoglobulin | 13/46 (28.3) | 11/49 (22.5) |
| Interferon | 12/46 (26.1) | 7/49 (14.3) |
The values shown were based on total number of patients who contributed values. Details of medications used were provided in eTable 7 in Supplement 3.
Overall and within disease severity strata, the convalescent plasma and control groups were similar in terms of demographic characteristics, baseline laboratory results, and distribution on the 6-point disease severity scale at baseline, with the exception of systolic blood pressure in the patients with severe COVID-19 and sex in the patients with life-threatening COVID-19 (eTable 1 and eTable 2 in Supplement 3). Additional details regarding patients’ clinical status at the time of randomization and additional medications received are provided in Table 2 and eTable 3, eTable 4, and eTable 7 in Supplement 3. For patients in the convalescent plasma group, median plasma infusion volume was 200 mL (IQR, 200-300 mL), and 96% received a single dose of plasma infusion.
Primary Clinical Outcome
For all patients combined, there was no significant difference in the primary outcome of time to clinical improvement within 28 days: 51.9% (27/52) in the convalescent plasma group vs 43.1% (22/51) in the control group (difference, 8.8% [95% CI, −10.4% to 28.0%]; HR, 1.40 [95% CI, 0.79-2.49]; P = .26). Results for the per-protocol population were not materially different (eTable 5 in Supplement 3). Among those with severe disease, the primary outcome occurred in 91.3% (21/23) vs 68.2% (15/22) (HR, 2.15 [95% CI, 1.07-4.32]; P = .03). Among those with life-threatening disease, the primary outcome occurred in 20.7% (6/29) vs 24.1% (7/29) (HR, 0.88 [95% CI, 0.30-2.63]; P = .83) (P for interaction = .17) (Table 3, Figure 2, and eTable 6 in Supplement 3). For all proportional hazards models, the proportionality assumptions were met.
Table 3. Primary and Secondary Clinical Outcomes at Day 28a.
| Convalescent plasma group (n = 52) | Control group (n = 51) | Absolute difference (95% CI)b | Effect estimate (95% CI) | P valuec | |
|---|---|---|---|---|---|
| All patients | |||||
| Primary clinical outcome | |||||
| Time to clinical improvement, median (IQR),dd | 28.00 (13.00-Indeterminate) | Indeterminate (18.00-Indeterminate) | −2.15 (−5.28 to 0.99) | HR, 1.40 (0.79-2.49) | .26 |
| Clinical improvement rate, No./total (%)e | |||||
| At day 7 | 5/52 (9.6) | 5/51 (9.8) | −0.2% (−11.6% to 11.2%) | OR, 0.98 (0.27-3.61) | .97 |
| At day 14 | 17/52 (32.7) | 9/51 (17.6) | 15.0% (−1.4% to 31.5%) | OR, 2.27 (0.90-5.71) | .08 |
| At day 28 | 27/52 (51.9) | 22/51 (43.1) | 8.8% (−10.4% to 28.0%) | OR, 1.42 (0.65-3.09) | .37 |
| Secondary clinical outcomes | |||||
| Discharge rate at 28 d, No./total (%) | 26/51 (51.0) | 18/50 (36.0) | 15.0% (−4.1% to 34.1%) | OR, 1.85 (0.83-4.10) | .13 |
| Time from randomization to discharge, median (IQR), dd | 28.00 (13.00-Indeterminate) | Indeterminate (19.00-Indeterminate) | −2.43 (−5.56 to 0.69) | HR, 1.61 (0.88-2.95) | .12 |
| Time from hospitalization to discharge, median (IQR),dd | 41.00 (31.00-Indeterminate) | 53.00 (35.00-Indeterminate) | −11.95 (−26.33 to 2.43) | HR, 1.68 (0.92-3.08) | .09 |
| Mortality at 28 d, No./total (%) | 8/51 (15.7) | 12/50 (24.0) | −8.3% (−23.8% to 7.2%) | OR, 0.59 (0.22-1.59) | .30 |
| Time from randomization to death, median (IQR), dd | Indeterminate | Indeterminate (26.00-Indeterminate) | 0.52 (−2.10 to 3.14) | HR, 0.74 (0.30-1.82) | .52 |
| Viral nucleic acid negative rate, No./total (%) | |||||
| At 24 h | 21/47 (44.7) | 6/40 (15.0) | 29.7% (11.7% to 47.7%) | OR, 4.58 (1.62-12.96) | .003 |
| At 48 h | 32/47 (68.1) | 13/40 (32.5) | 35.6% (15.9% to 55.3%) | OR, 4.43 (1.80-10.92) | <.001 |
| At 72 h | 41/47 (87.2) | 15/40 (37.5) | 49.7% (32.0% to 67.5%) | OR, 11.39 (3.91-33.18) | <.001 |
| Patients with severe disease | |||||
| Primary clinical outcome | |||||
| Time to clinical improvement, median (IQR), dd | 13.00 (9.00-21.00) | 19.00 (15.00-Indeterminate) | −4.94 (−9.33 to −0.54) | HR, 2.15 (1.07-4.32) | .03 |
| Clinical improvement rate, No./total (%)e | |||||
| At day 7 | 3/23 (13.0) | 4/22 (18.2) | −5.1% (−26.3% to 16.1%) | OR, 0.68 (0.13-3.43) | .70 |
| At day 14 | 14/23 (60.9) | 6/22 (27.3) | 33.6% (6.3% to 60.9%) | OR, 4.15 (1.18-14.59) | .02 |
| At day 28 | 21/23 (91.3) | 15/22 (68.2) | 23.1% (−3.9% to 50.2%) | OR, 4.90 (0.89-26.97) | .07 |
| Secondary clinical outcomes | |||||
| Discharge rate at 28 d, No./total (%) | 21/23 (91.3) | 15/22 (68.2) | 23.1% (−3.9% to 50.2%) | OR, 4.90 (0.89-26.97) | .07 |
| Time from randomization to discharge, median (IQR), dd | 13.00 (10.00-16.00) | 19.00 (11.00-Indeterminate) | −4.09 (−8.44 to 0.27) | HR, 1.97 (1.00-3.88) | .05 |
| Time from hospitalization to discharge, median (IQR), d | 32.00 (26.00-40.00) | 41.00 (30.00-53.00) | −9.38(−23.63 to 4.88) | HR, 1.74 (0.89-3.41) | .11 |
| Mortality at 28 d, No./total (%) | 0/23 | 2/22 (9.1) | −9.1% (−25.6% to 7.4%) | .23 | |
| Time from randomization to death, median (IQR), dd | Indeterminate | Indeterminate (26.00-Indeterminate) | 1.42 (−0.88 to 3.71) | HR, 0.00 | >.99 |
| Viral nucleic acid negative rate, No./total (%) | |||||
| At 24 h | 7/21 (33.3) | 2/17 (11.8) | 21.6% (−9.1% to 52.2%) | OR, 3.75 (0.66-21.20) | .15 |
| At 48 h | 13/21 (61.9) | 6/17 (35.3) | 26.6% (−4.2% to 57.4%) | OR, 2.98 (0.79-11.25) | .10 |
| At 72 h | 19/21 (90.5) | 7/17 (41.2) | 49.3% (22.7% to 75.9%) | OR, 13.57(2.36-77.95) | .001 |
| Patients with life-threatening disease | |||||
| Primary clinical outcome | |||||
| Time to clinical improvement, median (IQR), dd | Indeterminate | Indeterminate | 0.23 (−3.11 to 3.57) | HR, 0.88 (0.30-2.63) | .83 |
| Clinical improvement rate, No./total (%)e | |||||
| At day 7 | 2/29 (6.9) | 1/29 (3.4) | 3.4% (−11.4% to 18.3%) | OR, 2.07 (0.18-24.23) | >.99 |
| At day 14 | 3/29 (10.3) | 3/29 (10.3) | 0.0% (−19.1% to 19.1%) | OR, 1.00 (0.18-5.42) | >.99 |
| At day 28 | 6/29 (20.7) | 7/29 (24.1) | −3.4% (−24.9% to 18.0%) | OR, 0.82 (0.24-2.83) | .75 |
| Secondary clinical outcomes | |||||
| Discharge rate at 28 d, No./total (%) | 5/28 (17.9) | 3/28 (10.7) | 7.1% (−14.7% to 28.9%) | OR, 1.81 (0.39-8.44) | .71 |
| Time from randomization to discharge, median (IQR), dd | Indeterminate | Indeterminate | −0.80 (−3.74 to 2.14) | HR, 1.77 (0.42-7.40) | .44 |
| Time from hospitalization to discharge, median (IQR), dd | Indeterminate (46.00-Indeterminate) | Indeterminate | −4.61 (−15.07 to 5.85) | HR, 1.90 (0.45-8.04) | .38 |
| Mortality at 28 d, No./total (%) | 8/28 (28.6) | 10/28 (35.7) | −7.1% (−31.5% to 17.2%) | OR, 0.72 (0.23-2.22) | .57 |
| Time from randomization to death, median (IQR), dd | Indeterminate (22.00-Indeterminate) | Indeterminate (15.00-Indeterminate) | −0.04 (−3.86 to 3.77) | HR, 0.86 (0.34-2.17) | .74 |
| Viral nucleic acid negative rate, No./total (%) | |||||
| At 24 h | 14/26 (53.8) | 4/23 (17.4) | 36.5% (11.8% to 61.1%) | OR, 5.54 (1.47-20.86) | .01 |
| At 48 h | 19/26 (73.1) | 7/23 (30.4) | 42.6% (17.3% to 68.0%) | OR, 6.20 (1.79-21.46) | .003 |
| At 72 h | 22/26 (84.6) | 8/23 (34.8) | 49.8% (25.9% to 73.7%) | OR, 10.31(2.63-40.50) | <.001 |
Abbreviations: HR, hazard ratio; IQR, interquartile range; OR, odds ratio.
The values shown were based on total number of patients who contributed values. The primary clinical outcome was analyzed by primary analysis set. Times to outcomes in the secondary clinical outcomes were analyzed by primary analysis set. Indeterminate events could not be calculated due to the low percentage of outcome events.
The absolute differences of time to clinical improvement, times to discharge, and times from randomization to death were calculated based on restricted mean survival time.
P value was calculated by Cox regression, χ2 test, or Fisher exact test.
Medians and quartiles of time to clinical improvement, times to discharge, and times from randomization to death could not be determined because too few patients had reached improvement or discharge by the end of the study. By the end of the study, for all, severe, and life-threatening patients, respectively, 27 (51.9%), 21 (91.3%), and 6 (20.7%) were clinically improved in the convalescent plasma group; 22 (43.1%), 15 (68.2%), and 7 (24.1%) were clinically improved in the control group; 26 (51.0%), 21 (91.3%), and 5 (17.9%) were discharged in the convalescent plasma group; 18 (36.0%), 15 (68.2%), and 3 (10.7%) were discharged in the control group; 8 (15.7%), 0, and 8 (28.6%) died in the convalescent plasma group; and 12 (24.0%), 2 (9.1%), and 10 (35.7%) died in the control group.
These analyses were developed post hoc to better illustrate disease progression.
Figure 2. Time to Clinical Improvement in Patients With COVID-19.
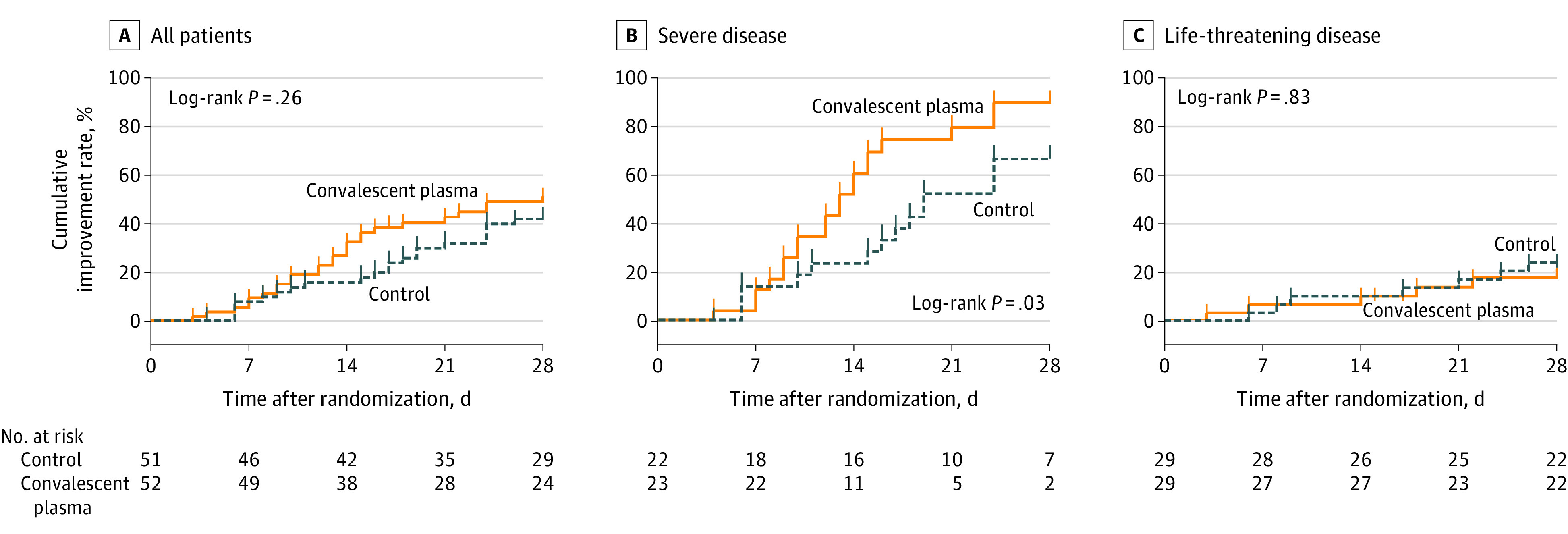
The cumulative improvement rate is the percentage of patients who experienced a 2-point improvement or were discharged alive from the hospital. Ticks on the curves indicate censored events. All patients who did not reach clinical improvement were observed for the full 28-day period or until death. COVID-19 indicates coronavirus disease 2019.
The median (IQR) follow-up times for the convalescent plasma group and control group, respectively, were 15 (10-28) days and 24 (13-28) days overall; 13 (10-16) and 18.5 (11-26) days among those with severe COVID-19; and 28 (12-28) and 26 (15-28) days among those with life-threatening COVID-19.
Secondary Clinical Outcomes
There was no significant difference in the secondary outcome of 28-day mortality (15.7% in the convalescent plasma group vs 24.0% in the control group; OR, 0.59 [95% CI, 0.22-1.59]; P = .30). There was also no significant difference in the time from randomization to death between the convalescent group and the control group (HR, 0.74 [95% CI, 0.30-1.82]; P = .52) (Table 3). Among patients with severe disease, no patients died in the convalescent plasma group, while 2 patients (9.1%) died in the control group. Among patients with life-threatening disease, 8 patients (28.6%) died in the convalescent plasma group, while 10 patients (35.7%) died in the control group.
There was also no significant difference in the secondary outcome of time from randomization to discharge (51.0% in the convalescent plasma group vs 36.0% in the control group were discharged by day 28; HR, 1.61 [95% CI, 0.88-2.95]; P = .12). The 28-day discharge rate of the convalescent plasma group reached 51%, among which the discharge rate of those with severe disease in the convalescent plasma group reached 91.3%.
At 24, 48, and 72 hours, the rates of negative SARS-CoV-2 viral PCR in the convalescent plasma group were all significantly higher than that of the control group (44.7% vs 15.0%, P = .003 at 24 hours; 68.1% vs 32.5%, P < .001 at 48 hours; 87.2% vs 37.5%, P < .001 at 72 hours) (Table 3 and eFigure 1 in Supplement 3). Among patients with severe disease, the rate of negative viral PCR at 72 hours was significantly higher in the convalescent plasma group compared with the control group, but there was no significant difference at 24 hours and 48 hours. Among patients with life-threatening disease, there was a statistically significant difference in favor of the convalescent plasma group at 24, 48, and 72 hours.
Post hoc Analysis
The clinical improvement rates overall and by disease severity subgroups at various time points during the study are presented in Table 3.
Adverse Events
Two participants reported transfusion-related adverse events following convalescent plasma transfusion. One patient in the severe COVID-19 group developed chills and rashes within 2 hours of transfusion but recovered fully after treatment with dexamethasone and promethazine. This was determined to be a definite nonsevere allergic transfusion reaction and also a probable nonsevere febrile nonhemolytic transfusion reaction. The other patient, who was in the life-threatening COVID-19 group, presented with shortness of breath, cyanosis, and severe dyspnea within 6 hours of transfusion. The patient was given dexamethasone, aminophylline, and other supportive care immediately and gradually improved after 2 hours. This was determined to be possible severe transfusion-associated dyspnea.
Discussion
In this randomized clinical trial of patients with severe or life-threatening COVID-19, there was no significant difference in the time to clinical improvement between patients who received convalescent plasma transfusion therapy combined with standard treatment vs those who received standard treatment alone. There was also no significant difference in secondary outcomes of 28-day mortality or time from randomization to discharge. Convalescent plasma treatment was associated with higher rates of negative SARS-CoV-2 viral PCR results from nasopharyngeal swabs at 24, 48, and 72 hours, demonstrating that the convalescent plasma treatment was associated with antiviral activity in patients with COVID-19.
Plasma transfusions can result in transfusion-related adverse events including febrile and allergic transfusion reactions, transfusion-associated dyspnea, hypotensive transfusion reaction, to hemolytic transfusion reactions, septic transfusion reaction, transfusion-related acute lung injury, and transfusion-associated circulatory overload. In this study, most patients tolerated convalescent plasma transfusions well. There were 2 cases of reported transfusion-associated adverse events. One case was determined to be a definite nonsevere allergic transfusion reaction and also a probable nonsevere febrile nonhemolytic transfusion reaction, and the other case was determined to be possible severe transfusion-associated dyspnea. The rate is somewhat higher than the general rate of reactions associated with plasma transfusion, possibly due to the small sample size and active surveillance.16
In the subgroup analysis by disease severity, there was a signal of possible clinical benefit for convalescent plasma among patients with severe COVID-19. However, because the test for interaction by disease severity was not statistically significant, the findings for the severe and life-threatening subgroups should not be interpreted as different. Given the early study termination, it is possible that the study was underpowered to detect a statistically significant interaction, and a larger trial of convalescent plasma focusing on patients with severe COVID-19 may be warranted.
Patients with COVID-19 who were recruited in this study were heterogeneous with regard to the duration and severity of the illness. The possible antiviral activity with convalescent plasma treatment in patients aged 60 to 80 years and after 14 days of disease onset is notable. To our knowledge, no other antiviral treatments have demonstrated therapeutic effects in this age group or this late in the course of the disease. However, the convalescent plasma treatment was given at least 14 days after the onset of symptoms in most cases, and it is not known whether earlier convalescent plasma treatment could have resulted in better clinical outcomes. Further studies are needed to optimize patient selection and timing of convalescent plasma treatment.
While the use of convalescent plasma has been investigated and used many times in the past,3,17 its use has not been well studied. It is notable that for most studies of the use of convalescent plasma,4,5,18 there was lack of standardization and procedure control with regard to the donor selection process and the nature or level of antibodies in convalescent plasma units. This may explain the varied therapeutic effects seen across a variety of diseases or even across patients with the same disease. The guidance on convalescent plasma issued by the World Health Organization11 highlighted the importance of standardized processes and laboratory testing–based quality control of convalescent plasma units, the selection of clinical indications, as well as program deployment to recruit adequate numbers of donors and maintain sufficient inventory.
One of the potential strengths of this study was the controlled process for donor selection and convalescent plasma quality control. A predefined process was used to enroll donors for convalescent plasma collection. Only units with a high titer of S-RBD–specific IgG antibody were used for convalescent plasma treatment in this study.
Limitations
This study has several limitations. First, the sample size was small and the study was terminated early. It is possible that the study was underpowered to detect a clinically important benefit of convalescent plasma therapy. Second, the median time between the onset of symptoms and randomization was 30 days, and it is unclear whether earlier treatment would have resulted in greater benefit. Third, this was an open-label study and the primary outcome was based to some degree on physicians’ clinical management decisions. Fourth, the use of standard therapy was allowed in both groups and was not protocolized, which could have potentially influenced outcomes. Fifth, the relatively short 28-day time frame of the study follow-up may have precluded the observation of clinical improvement in patients with severe diseases, especially life-threatening COVID-19, as they may take longer time to respond and recover. Sixth, plasma was not used for the control group, which would have been a more ideal control group, making blinded design possible. Seventh, the study findings should be interpreted cautiously given that practices may vary from country to country, and hospital to hospital, such as the types of standard treatment, supportive care, and thresholds for intubation and hospital admission. The outcomes of this study may be related to a combination of many factors, such as the quality of the convalescent plasma products in terms of potency, the selection of the patients (severe and life-threatening COVID-19), and the timing of convalescent plasma transfusion.
Conclusion
Among patients with severe or life-threatening COVID-19, convalescent plasma therapy added to standard treatment, compared with standard treatment alone, did not significantly improve the time to clinical improvement within 28 days. Interpretation is limited by early termination of the trial, which may have been underpowered to detect a clinically important difference.
Trial protocol
Trial SAP
eMethods.
eFigure 1. Accumulative viral nucleic acid negative rate at 24, 48 and 72 hours
eFigure 2. Survival curves in patients with COVID-19
eTable 1. Baseline demographics and clinical characteristics of patients with severe COVID-19
eTable 2. Baseline demographics and clinical characteristics of patients with life-threatening COVID-19
eTable 3. Clinical status at randomization and medications received of patients with severe COVID-19
eTable 4. Clinical status at randomization and medications received of the patients with life-threatening COVID-19
eTable 5. Primary clinical outcome of patients with COVID-19 in per-protocol set
eTable 6. Cox regression analysis of primary and secondary clinical outcomes
eTable 7. Antiviral and steroids used during study period
Data sharing statement
References
- 1.Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061-1069. doi: 10.1001/jama.2020.1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239-1242. doi: 10.1001/jama.2020.2648 [DOI] [PubMed] [Google Scholar]
- 3.Garraud O, Heshmati F, Pozzetto B, et al. Plasma therapy against infectious pathogens, as of yesterday, today and tomorrow. Transfus Clin Biol. 2016;23(1):39-44. doi: 10.1016/j.tracli.2015.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng Y, Wong R, Soo YO, et al. Use of convalescent plasma therapy in SARS patients in Hong Kong. Eur J Clin Microbiol Infect Dis. 2005;24(1):44-46. doi: 10.1007/s10096-004-1271-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeh KM, Chiueh TS, Siu LK, et al. Experience of using convalescent plasma for severe acute respiratory syndrome among healthcare workers in a Taiwan hospital. J Antimicrob Chemother. 2005;56(5):919-922. doi: 10.1093/jac/dki346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arabi YM, Hajeer AH, Luke T, et al. Feasibility of using convalescent plasma immunotherapy for MERS-CoV infection, Saudi Arabia. Emerg Infect Dis. 2016;22(9):1554-1561. doi: 10.3201/eid2209.151164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kraft CS, Hewlett AL, Koepsell S, et al. ; Nebraska Biocontainment Unit and the Emory Serious Communicable Diseases Unit . The use of TKM-100802 and convalescent plasma in 2 patients with Ebola virus disease in the United States. Clin Infect Dis. 2015;61(4):496-502. doi: 10.1093/cid/civ334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen C, Wang Z, Zhao F, et al. Treatment of 5 critically ill patients with COVID-19 with convalescent plasma. JAMA. 2020;323(16):1582-1589. doi: 10.1001/jama.2020.4783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duan K, Liu B, Li C, et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc Natl Acad Sci U S A. 2020;117(17):9490-9496. doi: 10.1073/pnas.2004168117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Recommendations for Investigational COVID-19 Convalescent Plasma. US FDA. Published May 1, 2020. Accessed May 26, 2020. https://www.fda.gov/vaccines-blood-biologics/investigational-new-drug-ind-or-device-exemption-ide-process-cber/recommendations-investigational-covid-19-convalescent-plasma
- 11.Position Paper on Use of Convalescent Plasma, Serum or Immune Globulin Concentrates as an Element in Response to an Emerging Virus. In: Network WBR, ed. 2017. Accessed April 18, 2020. https://www.who.int/bloodproducts/brn/en/
- 12.National Health Commission of the People’s Republic of China Covid-19 treatment plan (trial version 6). Accessed April 20, 2020. http://www.nhc.gov.cn/yzygj/s7653p/202002/8334a8326dd94d329df351d7da8aefc2/files/b218cfeb1bc54639af227f922bf6b817
- 13.Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395(10236):1569-1578. doi: 10.1016/S0140-6736(20)31022-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleinbaum DG. Evaluating the proportional hazards assumption In: Survival Analysis. Statistics in the Health Sciences. Springer; 1996:183-184. [Google Scholar]
- 15.Daily update on covid-19. National Health Commission of the People’s Republic of China. Accessed May 24, 2020. http://www.nhc.gov.cn/xcs/yqtb/202003/f01fc26a8a7b48debe194bd1277fdba3.shtml [DOI] [PMC free article] [PubMed]
- 16.Annual SHOT (Serious Hazards of Transfusion) Report 2018. Published July 2019. Accessed May 25, 2020. https://www.shotuk.org/
- 17.Beigel JH, Aga E, Elie-Turenne MC, et al. ; IRC005 Study Team . Anti-influenza immune plasma for the treatment of patients with severe influenza A: a randomised, double-blind, phase 3 trial. Lancet Respir Med. 2019;7(11):941-950. doi: 10.1016/S2213-2600(19)30199-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soo YO, Cheng Y, Wong R, et al. Retrospective comparison of convalescent plasma with continuing high-dose methylprednisolone treatment in SARS patients. Clin Microbiol Infect. 2004;10(7):676-678. doi: 10.1111/j.1469-0691.2004.00956.x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial protocol
Trial SAP
eMethods.
eFigure 1. Accumulative viral nucleic acid negative rate at 24, 48 and 72 hours
eFigure 2. Survival curves in patients with COVID-19
eTable 1. Baseline demographics and clinical characteristics of patients with severe COVID-19
eTable 2. Baseline demographics and clinical characteristics of patients with life-threatening COVID-19
eTable 3. Clinical status at randomization and medications received of patients with severe COVID-19
eTable 4. Clinical status at randomization and medications received of the patients with life-threatening COVID-19
eTable 5. Primary clinical outcome of patients with COVID-19 in per-protocol set
eTable 6. Cox regression analysis of primary and secondary clinical outcomes
eTable 7. Antiviral and steroids used during study period
Data sharing statement